

Abstract
INTRODUCTION:
The APOE ε2 and ε4 alleles have beneficial and adverse impacts on Alzheimer’s Disease (AD), respectively, with incomplete penetrance, which may be modulated by other genetic variants.
METHODS:
We examined whether the associations of the APOE alleles with other polymorphisms in the genome can be sensitive to AD-affection status.
RESULTS:
We identified associations of the ε2 and ε4 alleles with 314 and 232 polymorphisms, respectively. Of them, 35 and 31 polymorphisms had significantly different effects in AD-affected and unaffected groups, suggesting their potential involvement in the AD pathogenesis by modulating the effects of the ε2 and ε4 alleles, respectively. Our survival-type analysis of the AD risk supported modulating roles of multiple group-specific polymorphisms. Our functional analysis identified gene enrichment in multiple immune-related biological processes, e.g., B cell function.
DISCUSSION:
These findings suggest involvement of local and inter-chromosomal modulators of the effects of the APOE alleles on the AD risk.
Keywords: Neurodegenerative Diseases, Dementia, Aging, GWAS, Cox Regression, Age at the Onset, Sex-Specific Associations, Genetic Modulators, Genetic Polymorphisms
1. BACKGROUND
Apolipoprotein E (APOE) gene that encodes a protein involved in lipids transport and metabolism1 has been widely studied in the past decades due to its broad functional implications and potential roles in various traits2, such as Alzheimer’s disease (AD)3–5, vascular dementia6, dementia with Lewy bodies7, coronary artery diseases8,9, cerebrovascular accidents10–12, Parkinson’s disease-associated dementia13, frontotemporal lobar degeneration14, malignancies15, immune/inflammatory responses and autoimmune disorders16–18, and longevity19,20. The APOE gene has three main alleles, i.e., ε2, ε3, and ε4. The ε4 allele encoded by the minor allele of rs429358 single-nucleotide polymorphisms (SNP) is considered as the strongest single genetic risk factor for AD, which is associated with AD in various populations4,21. The ε2 allele encoded by the minor allele of rs7412 shows beneficial associations with AD4,22, but the understanding of its potential protective role is tempered due to, in part, its small population frequency and the diminished number of AD cases among ε2 carriers.
Despite decades of research, the role of the APOE gene and its neighboring region in AD development is not entirely clear because of uncertainty about how to treat genetic variants from this region. For example, while most of the field tends to consider the role of the ε4 allele itself, the role of more complex structures such as haplotypes with variants from different genes in the APOE region is also widely emphasized23–27. The complex role of the APOE region variants, as well as the other variants from the entire genome, in AD has been supported by environmental28 and evolutionary27,29 studies.
In this study, we examined the associations of the ε2 and ε4 alleles with other SNPs in the human genome. We leveraged a strategy, which can partly address heterogeneity in genetic predisposition to AD by examining these allele-SNP associations in the AD-affected and unaffected subjects separately. We evaluated whether these associations in the AD-affected and unaffected subjects were different. Our analyses aimed to better understand genetic modulators of contributions of the APOE alleles to AD pathogenesis, especially variants outside of the neighboring genes in the APOE 19q13.3 region. These analyses identified a large number of promising ε2- and ε4-associated loci, both within and outside the APOE region, in the AD-affected, unaffected, or both groups. We found a subset of these loci in which associations of the ε2 and ε4 alleles with the other SNPs were statistically different in the AD-affected and unaffected groups. These findings suggest the roles of interactions of the ε2- and ε4 alleles with SNPs from specific loci spread throughout the entire genome in AD pathogenesis.
2. METHODS
2.1. Study participants
We used data on individuals of European ancestry from five studies: Cardiovascular Health Study (CHS)30, Framingham Heart Study (FHS)31,32, Late Onset Alzheimer’s Disease Family Study (LOADFS) from the National Institute on Aging (NIA)33, whole genome sequencing (WGS) data from Alzheimer’s Disease Sequencing Project (ADSP-WGS)34,35, and three cohorts from the NIA Alzheimer’s Disease Centers (ADCs), which are a part of the Alzheimer’s Disease Genetics Consortium (ADGC)36. The overlapping ADSP-WGS participants with the other datasets were excluded. The APOE genotypes in CHS and LOADFS were determined based on their genotypes at rs429358 and rs7412 loci. The ADGC, ADSP-WGS, and FHS have directly reported the APOE genotypes for recruited subjects. AD patients were directly identified by the ADGC, ADSP-WGS, FHS, and LOADFS researchers primarily based on the neurologic exam criteria37,38. In CHS, AD-affected subjects were determined using the International Classification of Disease codes, ninth revision (i.e., code: 331.0). Basic information on 6136 AD-affected and 10555 unaffected subjects is presented in Table S1.
2.2. Genotype data and quality control (QC)
Genetic data in the selected five studies were from the array-based (i.e., ADGC, CHS, FHS, and LOADFS) or whole-genome sequencing (i.e., ADSP-WGS) platforms. First, we imputed SNPs to harmonize about 2.5 million of variants to facilitate cross-platform analyses39. Then, we performed QC using PLINK package40 to filter out low-quality data including: imputed SNPs with r2<0.7 (in ADGC, CHS, FHS, and LOADFS), SNPs/subjects with missing rates >5%, SNPs with minor allele frequencies <1% or PHardy-Weinberg<1E-06, and SNPs/subjects/families with Mendel error rates >2% in family-based datasets (i.e., ADSP-WGS, FHS, and LOADFS). The QC process resulted in 1904013, 1844347, 1695409, 1541793, 1829245 SNPs in ADGC, ADSP-WGS, CHS, FHS, and LOADFS, respectively.
2.3. Two-stage genetic analysis
Design.
We used two variables as outcomes in our analysis. One outcome included carriers of the ε2ε2 and ε2ε3 AD-protective genotype (herein referred to as the ε2 allele) as cases, and the other included carriers of the ε4ε4 and ε3ε4 AD-risk genotype (herein referred to as the ε4 allele) as cases. The same ε3ε3 genotype was used as a reference in each outcome. The analyses leveraged a two-stage approach. The first stage was designed to examine associations of the ε2 and ε4 alleles with the other SNPs in the genome in AD-affected (AD) and unaffected (NAD) groups of subjects separately. At stage two, we examined group-specific effects by evaluating the differences in associations of the ε2 and ε4 alleles with the other SNPs in the AD-affected and unaffected subjects, which were selected at stage one.
Stage one: Genome-wide association study (GWAS).
Additive genetic models were fitted separately in each dataset to associate the ε2 or ε4 alleles with the other SNPs in the genome. The models were adjusted for fixed-effects covariates, including the top five principal components of genetic data, sex, age/birth year, and ADC cohorts (in ADGC), as well as random-effects family structure (in ADSP-WGS, FHS, and LOADFS). The logistic regression models were fitted using GENESIS R package41,42. The GWAS results from these five datasets were combined using a fixed-effects inverse-variance meta-analysis implemented in GWAMA package43.
We used two GWAS strategies. First, following the discovery-replication strategy, we selected two independent sets of data. One set (referred to as nonADGC) included data from ADSP-WGS, CHS, FHS, and LOADFS, and the other set (referred to as ADGC) was represented by the ADGC cohort. These datasets were used as the discovery and replication sets interchangeably. In other words, results of the meta-analysis of the GWAS statistics from the nonADGC studies were used as the discovery set and ADGC as the replication set, and vice versa. The second strategy was to pool the results from all five datasets (i.e., nonADGC+ADGC samples) using meta-regression. We selected promising SNPs for stage two from the associations attained genome-wide (P<5E-08) or suggestive-effect (5E-08≤ P<5E-06) significance in: (i) the discovery dataset and had the same effect direction and P<0.05 in the replication dataset, and vice versa, and (ii) the meta-analysis of all five datasets.
Stage two: Group-specific analysis
Group-specific analysis provides quantitative metric to identify AD or NAD group-specific associations. This metric is necessary because significance of the association in one group and the lack of significance in the other group does not automatically guarantee significant difference of the associations between these groups. We quantified the differences in the associations between these groups by fitting an interaction model with a SNP-by-AD status term in the pooled sample of AD and NAD subjects for each SNP selected at stage one. Significant findings from the interaction model were identified after Bonferroni correction for the number of SNPs selected at stage one.
2.4. Analysis of the role of sex
To examine the potential role of sex as a modulator of the associations of the ε2 and ε4 alleles with group-specific SNPs, we fitted the same models as in our stage-two analysis with an additional SNP-by-sex interaction term using GENESIS R package41,42.
2.5. Analysis of the AD risk
To examine whether the group-specific SNPs identified at stage two can modulate the impact of the APOE alleles on the AD risk, we performed survival-type analysis using Cox regression model. We evaluated the main effects of the ε2-coding rs7412 or ε4-coding rs429358 and each group-specific SNP, along with their interactions. We used age at onset (AAO) of AD as a time variable. As in our GWAS analysis, we fitted additive genetic models and included the same fixed- and random-effects adjustments. These analyses were performed using coxme (for family-based studies) and survival R packages44,45.
2.6. Functional enrichment analysis
To make biological sense of the observed statistical associations, we examined gene enrichment in bio-functions (defined by “molecular and cellular function” and “physiological system development and function” categories) using the Ingenuity Pathway Analysis (IPA) tool (QIAGEN Inc., https://www.qiagenbioinformatics.com/products/ingenuitypathway-analysis).
3. RESULTS
Our stage-one (i.e., GWAS) analyses revealed several associations of the ε2 and ε4 alleles with other SNPs across the genome in AD, NAD, or both groups. Figures S1–S4 show Manhattan and QQ plots for these results in nonADGC, ADGC, and nonADGC+ADGC samples. The genomic control values (i.e., lambda) in these analyses were between 0.972–1.002 in nonADGC samples and 1.010–1.023 in ADGC dataset, indicating adequate control of potential confounding effects of population structure. Next, we discuss the results from the promising associations.
3.1. Associations for the ε2 allele.
The ε2 allele showed promising associations with 29 SNPs in 13 loci in the AD group but not in the NAD group (Table S2), and with 191 SNPs in 16 loci in the NAD group but not in the AD group (Table S3). In the AD group, we identified three (of 29) promising SNPs associated with the ε2 allele in the APOE 19q13.3 locus. In contrast, the vast majority of SNPs identified in the NAD group, 159 of 191 (83.2%), were in the APOE 19q13.3 locus. In addition to these 220 (=29+191) SNPs, the ε2 allele was associated with 94 SNPs (all in the APOE 19q13.3 locus) in both AD and NAD groups (Table S4), totaling 314 promising SNPs in all groups combined.
Differences in the detected associations of SNPs with the ε2 allele in the AD and NAD groups suggested potential group-specific effects, i.e., interactions of SNPs with the AD status. The stage-two analysis identified group-specific associations of the ε2 allele with 35 SNPs in 11 loci, which attained a conservative Bonferroni-adjusted (i.e., all selected SNPs for this test were conservatively considered as independent) significance level of P<1.59×10−4 (i.e., 0.05/314) in the interaction analyses (Tables 1 and S5).
Table 1.
SNP-by-AD status interaction meta-analysis for ε2-associated group-specific SNPs in the pooled samples of AD and NAD groups.
| SNPs | SNP-by-AD Status Interaction Effect | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Rep/Meta | CHR | Locus | Gene | SNP | POS | EA | EAF | N | BETA | SE | P-value | Effects |
| AD-Meta | 1p13.3 | 1 | VAV3 | rs77841908 | 107713714 | T | 0.012 | 5568 | 3.268 | 0.751 | 1.39E-05 | +??++ |
| AD-Meta | 4p12 | 2 | GNPDA2 | rs7667144 | 44988902 | T | 0.240 | 10493 | −0.585 | 0.124 | 2.22E-06 | −−−−− |
| AD-Meta | 4p12 | 2 | GNPDA2 | rs34071858 | 44989232 | G | 0.240 | 10489 | −0.587 | 0.124 | 2.12E-06 | −−−−− |
| AD-Meta | 4p12 | 2 | GNPDA2 | rs7436238 | 44995666 | A | 0.239 | 10477 | −0.585 | 0.124 | 2.35E-06 | −−−−− |
| AD-Meta | 8q24.3 | 3 | MROH5 | rs77186917 | 141434800 | C | 0.047 | 7334 | 1.317 | 0.265 | 6.87E-07 | −+?−+ |
| AD-Rep | 9p24.1 | 4 | RIC1 | rs74364314 | 5640617 | G | 0.016 | 3025 | 2.685 | 0.701 | 1.28E-04 | +???+ |
| AD-Meta | 9p23 | 5 | MPDZ | rs7848179 | 13030037 | A | 0.028 | 10493 | 1.417 | 0.323 | 1.16E-05 | +++−+ |
| AD-Meta | 9p23 | 5 | MPDZ | rs113433603 | 13036364 | T | 0.026 | 10493 | 1.623 | 0.337 | 1.53E-06 | +++−+ |
| NAD-Meta | 10p13 | 6 | FRMD4A | rs3101357 | 14077721 | T | 0.382 | 10492 | 0.459 | 0.111 | 3.82E-05 | −++++ |
| AD-Meta | 11q22.3 | 7 | DDX10 | rs2884183 | 109127412 | T | 0.212 | 10474 | 0.736 | 0.131 | 1.76E-08 | +++++ |
| AD-Meta | 16q24.1 | 8 | IRF8 | rs16882 | 85901967 | C | 0.180 | 10489 | 0.615 | 0.136 | 6.20E-06 | +++++ |
| AD-Meta | 16q24.1 | 8 | IRF8 | rs305082 | 85903372 | C | 0.183 | 10463 | 0.616 | 0.135 | 5.27E-06 | +++++ |
| AD-Meta | 16q24.1 | 8 | IRF8 | rs8052521 | 85925123 | T | 0.248 | 10473 | 0.550 | 0.122 | 7.26E-06 | +++++ |
| AD-Meta | 16q24.1 | 8 | IRF8 | rs880364 | 85925559 | G | 0.249 | 10472 | 0.543 | 0.122 | 9.39E-06 | +++++ |
| AD-Meta | 16q24.1 | 8 | IRF8 | rs880365 | 85925756 | T | 0.246 | 10471 | 0.508 | 0.123 | 3.77E-05 | +++++ |
| AD-Meta | 16q24.1 | 8 | IRF8 | rs880366 | 85925832 | C | 0.248 | 10472 | 0.551 | 0.123 | 6.96E-06 | +++++ |
| AD-Meta | 17q25.1 | 9 | SDK2 | rs9895288 | 73623974 | A | 0.075 | 10490 | 0.902 | 0.198 | 5.16E-06 | +++−+ |
| AD-Meta | 17q25.1 | 9 | SDK2 | rs75122991 | 73639797 | G | 0.067 | 10490 | 0.974 | 0.209 | 3.13E-06 | +++−+ |
| AD-Meta | 17q25.1 | 9 | SDK2 | rs28527783 | 73640957 | T | 0.068 | 10491 | 0.983 | 0.209 | 2.52E-06 | +++−+ |
| AD-Meta | 17q25.1 | 9 | SDK2 | rs76380229 | 73643396 | T | 0.067 | 10489 | 0.989 | 0.209 | 2.23E-06 | +++−+ |
| AD-Meta/NAD-Meta | 19q13.31 | 10 | CEACAM22P | rs144439590 | 44537330 | T | 0.026 | 5568 | 1.896 | 0.470 | 5.56E-05 | +??−+ |
| AD-Rep/NAD-Rep | 19q13.32 | 10 | CEACAM16 | rs11881756 | 44717621 | C | 0.107 | 4791 | 0.964 | 0.193 | 5.75E-07 | ++??+ |
| AD-Rep/NAD-Rep | 19q13.32 | 10 | BCL3 | rs1531517 | 44738916 | A | 0.072 | 7332 | 1.529 | 0.221 | 4.54E-12 | ++?−+ |
| AD-Rep/NAD-Rep | 19q13.32 | 10 | BCL3 | rs4803750 | 44744370 | G | 0.067 | 7326 | 1.535 | 0.224 | 7.56E-12 | ++?−+ |
| AD-Rep/NAD-Rep | 19q13.32 | 10 | BCL3 | rs10401176 | 44750234 | T | 0.117 | 4791 | 0.872 | 0.183 | 1.84E-06 | ++??+ |
| AD-Rep/NAD-Rep | 19q13.32 | 10 | BCL3 | rs62117205 | 44752009 | C | 0.066 | 7334 | 1.589 | 0.225 | 1.74E-12 | ++?−+ |
| AD-Meta/NAD-Meta | 19q13.32 | 10 | NECTIN2 | rs7254892 | 44886339 | A | 0.031 | 2296 | 2.952 | 0.469 | 3.18E-10 | ????+ |
| AD-Rep/NAD-Rep | 19q13.32 | 10 | APOE | rs75627662 | 44910319 | T | 0.082 | 7334 | 2.211 | 0.079 | 2.07E-171 | ++?++ |
| AD-Rep/NAD-Rep | 19q13.32 | 10 | APOE | rs72654473 | 44911142 | A | 0.091 | 7332 | 2.074 | 0.101 | 8.90E-94 | +−?++ |
| AD-Rep/NAD-Rep | 19q13.32 | 10 | APOC1 | rs445925 | 44912383 | A | 0.092 | 7333 | 2.164 | 0.102 | 1.58E-99 | +−?++ |
| AD-Rep/NAD-Rep | 19q13.32 | 10 | APOC1 | rs483082 | 44912921 | T | 0.104 | 7334 | 1.894 | 0.111 | 9.78E-65 | +−?++ |
| AD-Rep/NAD-Rep | 19q13.32 | 10 | APOC1P1 | rs141622900 | 44923535 | A | 0.046 | 3025 | 4.825 | 0.320 | 2.89E-51 | +???+ |
| AD-Meta | 20q13.2 | 11 | ZFP64 | rs6021874 | 52342233 | C | 0.085 | 10486 | 0.865 | 0.184 | 2.66E-06 | −++++ |
| AD-Meta | 20q13.2 | 11 | ZFP64 | rs6021877 | 52345128 | T | 0.085 | 10492 | 0.839 | 0.184 | 4.98E-06 | −++++ |
| AD-Meta | 20q13.2 | 11 | ZFP64 | rs2426435 | 52357570 | A | 0.082 | 10481 | 0.847 | 0.186 | 5.55E-06 | −++++ |
Abbreviations: AD = Alzheimer’s disease-affected group; NAD = Alzheimer’s disease-unaffected group; SNP = single-nucleotide polymorphism; Rep/Meta = SNP identified at the stage one of our two-stage genetic analysis by the discovery-replication strategy (i.e., nonADGC and ADGC as discovery and replication sets or vice versa) or in the meta-analysis of five datasets under consideration (i.e., combined nonADGC and ADGC); CHR = chromosomal region (i.e., cytogenetic band); POS = SNP position based on Human Genome version 38 (hg38); EA = effect allele; EAF = effect allele frequency; N = Number of subjects; BETA and SE = effect size and its standard error; Effects = directions of effects in the ADSP, LOADFS, FHS, CHS, ADGC datasets, respectively.
Most of these SNPs, 22 of 35, were selected at stage one based on promising associations with the ε2 allele only in the AD group, and one SNP (rs3101357 mapped to FRMDF4) was associated with this allele only in the NAD group. These 23 SNPs were mapped to ten genes/loci which were on nine chromosomes outside of the APOE 19q13.3 locus. The other 12 SNPs with group-specific effects had promising significant association signals in both AD and NAD groups. These SNPs were mapped to 7 genes within the APOE 19q13.3 locus. Figure 1 illustrates the effect sizes of the ε2-associated group-specific SNPs from the analyses of nonADGC+ADGC samples in the AD and NAD groups. In general, the magnitudes of the effects were larger in the APOE 19q13.3 locus than in the non-APOE loci in both AD and NAD groups. Also, the magnitudes of effect sizes of the associations of the ε2 allele with each of these 35 SNPs were larger in the AD than NAD group, indicating stronger associations between the ε2 allele and alleles of these SNPs in the AD group. For most of these SNPs, 26 of 35, the effect directions were positive in both AD and NAD groups, indicating stronger associations between their minor alleles and the ε2 allele in the AD group. The remaining 9 of 35 SNPs had different directions of effects in the AD and NAD groups, denoting the opposite patterns of associations between the minor/major alleles of these SNPs and the ε2 allele in the two groups.
Figure 1.
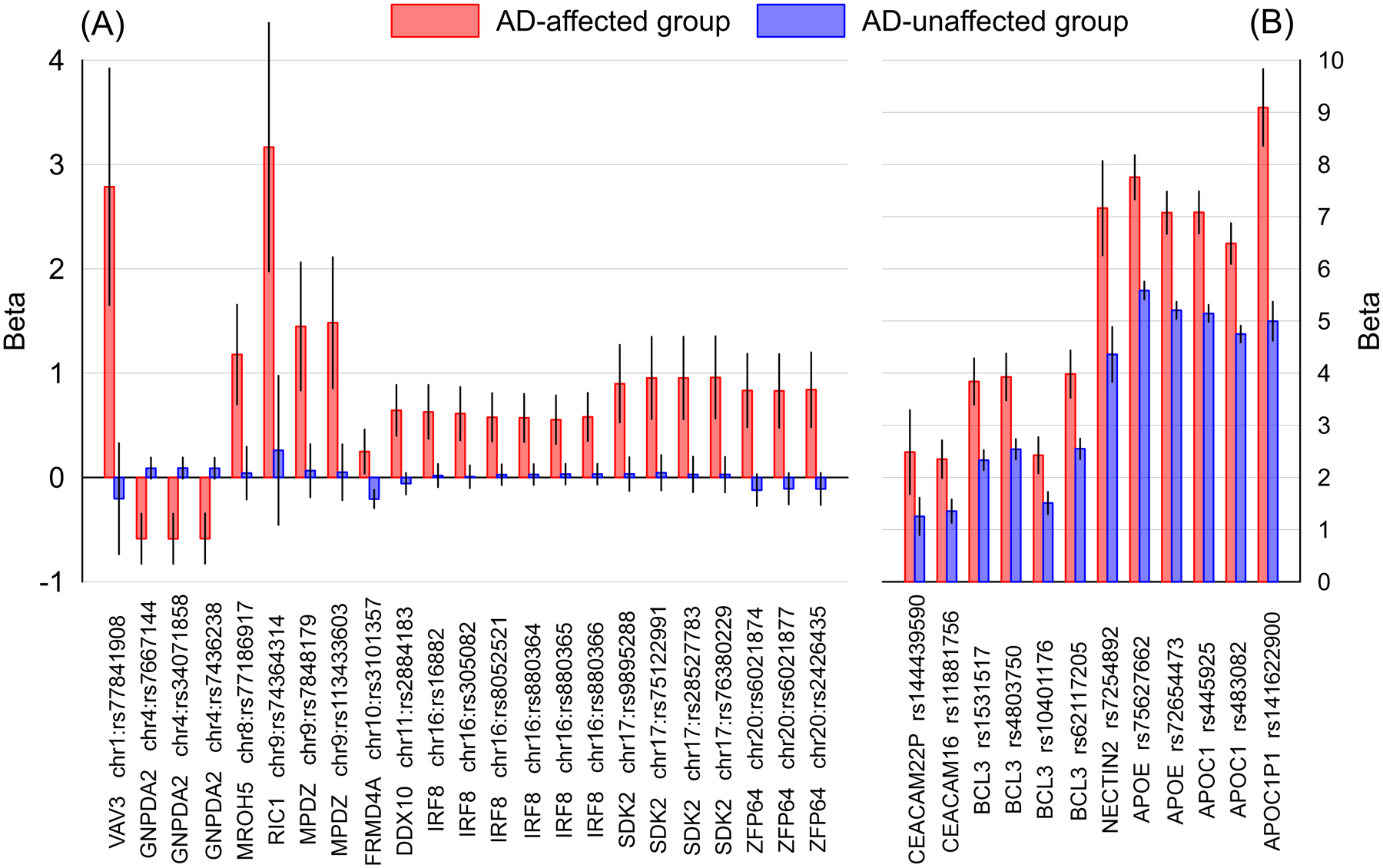
The effect sizes of the ε2-associated group-specific SNPs in Alzheimer’s disease-affected (AD) and unaffected (NAD) groups. (A) SNPs outside of the APOE 19q13.3 locus and (B) SNPs within the APOE 19q13.3 locus. The x-axis shows SNPs and genes identifiers; the y-axis shows the effect sizes (i.e., beta coefficients), red bars indicate the AD group; blue bars indicate the NAD group. Vertical lines show 95% confidence intervals.
3.2. Associations for the ε4 allele.
In the AD group, we identified promising associations of the ε4 allele with 12 SNPs in seven non-APOE loci and 86 SNPs in the APOE 19q13.3 locus, totaling 98 SNPs in eight loci (Table S6). In the NAD group, there were nine promising associations in six non-APOE loci and 14 in the APOE 19q13.3 locus, totaling 23 SNPs in seven loci (Table S7). In addition, there were 111 SNPs (all within the APOE 19q13.3 locus) with promising associations with the ε4 allele in both AD and NAD groups (Table S8). Overall, this analysis identified 232 promising associations with the ε4 allele.
Of them, the stage-two analysis identified group-specific associations of the ε4 allele with 31 SNPs at a conservative Bonferroni-adjusted significance level of P<2.16×10−4 (i.e., 0.05/232) in the fitted interaction models (Tables 2 and S9). Figure 2 illustrates the effect sizes of the ε4-associated group-specific SNPs from the analyses of nonADGC+ADGC samples in the AD and NAD groups. Six SNPs with group-specific effects were associated with the ε4 allele only in the AD (5 SNPs) or NAD (1 SNP) groups. They were mapped to 3 genes in 3 loci, including EXOC3L2 gene in the APOE 19q13.3 locus. All these 6 SNPs had negative effect directions in the AD group and positive ones in the NAD groups, highlighting the opposite patterns of the associations of the ε4 allele and minor/major alleles of these SNPs in the two groups. The other 25 SNPs with group-specific effects (mapped to 5 genes within the APOE 19q13.3 locus) were associated with the ε4 allele in both AD and NAD groups. They had the same directions of effects in both groups (14 positive and 11 negative effects). The effect sizes of SNPs with positive effect directions were larger in the NAD group than the AD group implying stronger associations between the ε4 allele and minor alleles of these SNPs in the NAD group. In contrast, magnitudes of the effects of SNPs with negative directions were larger in the AD group than the NAD group indicating stronger associations between the ε4 allele and major alleles of these SNPs in the AD group. Again, the magnitudes of the effects were mainly larger in the APOE 19q13.3 locus than in the non-APOE loci.
Table 2.
SNP-by-AD status interaction meta-analysis for ε4-associated group-specific SNPs in the pooled samples of AD and NAD groups.
| SNPs | SNP-by-AD Status Interaction Effect | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Rep/Meta | CHR | Locus | Gene | SNP | POS | EA | EAF | N | BETA | SE | P-value | Effects |
| NAD-Meta | 1p31.2 | 1 | LRRC7 | rs1414663 | 69168451 | G | 0.102 | 11576 | −0.423 | 0.108 | 8.60E-05 | −−?−− |
| AD-Meta | 8q24.22 | 2 | KCNQ3 | rs9297847 | 132351982 | T | 0.318 | 15047 | −0.292 | 0.066 | 8.90E-06 | −−−−− |
| AD-Meta | 8q24.22 | 2 | KCNQ3 | rs4458854 | 132360860 | A | 0.338 | 15048 | −0.277 | 0.065 | 2.00E-05 | −−−−− |
| AD-Meta | 8q24.22 | 2 | KCNQ3 | rs7820317 | 132362575 | A | 0.339 | 15044 | −0.265 | 0.065 | 4.58E-05 | −−−−− |
| AD-Meta | 8q24.22 | 2 | KCNQ3 | rs2673604 | 132399360 | C | 0.299 | 15044 | −0.281 | 0.067 | 2.98E-05 | −−−−− |
| AD-Rep/NAD-Rep | 19q13.32 | 3 | NECTIN2 | rs283815 | 44887076 | G | 0.307 | 11576 | −0.274 | 0.052 | 1.54E-07 | −−?−− |
| AD-Rep/NAD-Rep | 19q13.32 | 3 | TOMM40 | rs71352238 | 44891079 | C | 0.245 | 11573 | −0.475 | 0.061 | 4.90E-15 | −−?−− |
| AD-Rep/NAD-Rep | 19q13.32 | 3 | TOMM40 | rs157580 | 44892009 | G | 0.314 | 10506 | −0.316 | 0.071 | 9.68E-06 | ?−?−− |
| AD-Rep/NAD-Rep | 19q13.32 | 3 | TOMM40 | rs2075650 | 44892362 | G | 0.243 | 11570 | −0.553 | 0.061 | 1.17E-19 | −−?−− |
| AD-Rep/NAD-Rep | 19q13.32 | 3 | TOMM40 | rs34095326 | 44892587 | A | 0.184 | 11576 | −0.542 | 0.077 | 1.81E-12 | −−?−− |
| AD-Rep/NAD-Rep | 19q13.32 | 3 | TOMM40 | rs157582 | 44892962 | T | 0.307 | 11574 | −0.282 | 0.052 | 6.53E-08 | −−?−− |
| AD-Rep/NAD-Rep | 19q13.32 | 3 | TOMM40 | rs76366838 | 44896639 | A | 0.048 | 8875 | −0.937 | 0.178 | 1.40E-07 | −−??− |
| AD-Rep/NAD-Rep | 19q13.32 | 3 | TOMM40 | rs8106922 | 44898409 | G | 0.369 | 11559 | −0.395 | 0.062 | 2.38E-10 | −−?−− |
| AD-Rep/NAD-Rep | 19q13.32 | 3 | TOMM40 | rs115881343 | 44899959 | T | 0.050 | 8874 | −0.910 | 0.172 | 1.29E-07 | −−??− |
| AD-Rep/NAD-Rep | 19q13.32 | 3 | TOMM40 | rs1160985 | 44900155 | T | 0.391 | 11576 | −0.435 | 0.061 | 8.42E-13 | −−?−− |
| AD-Rep/NAD-Rep | 19q13.32 | 3 | TOMM40 | rs7259620 | 44904531 | A | 0.388 | 11576 | −0.413 | 0.061 | 1.18E-11 | −−?−− |
| AD-Rep/NAD-Rep | 19q13.32 | 3 | APOE | rs405509 | 44905579 | G | 0.437 | 11572 | −0.286 | 0.063 | 5.06E-06 | −−?−− |
| AD-Rep/NAD-Rep | 19q13.32 | 3 | APOE | rs769449 | 44906745 | A | 0.225 | 11573 | −1.065 | 0.053 | 3.18E-91 | −−?−− |
| AD-Rep/NAD-Rep | 19q13.32 | 3 | APOE | rs769450 | 44907187 | A | 0.372 | 11574 | −0.422 | 0.062 | 1.18E-11 | −−?−− |
| AD-Rep/NAD-Rep | 19q13.32 | 3 | APOE | rs75627662 | 44910319 | T | 0.228 | 11576 | −0.994 | 0.053 | 1.40E-79 | −−?−− |
| AD-Rep/NAD-Rep | 19q13.32 | 3 | APOE | rs72654473 | 44911142 | A | 0.051 | 11572 | −0.770 | 0.147 | 1.84E-07 | −−?−− |
| AD-Rep/NAD-Rep | 19q13.32 | 3 | APOE | rs439401 | 44911194 | T | 0.342 | 11558 | −0.270 | 0.065 | 3.68E-05 | +−?−− |
| AD-Rep/NAD-Rep | 19q13.32 | 3 | APOC1 | rs445925 | 44912383 | A | 0.054 | 11575 | −0.741 | 0.144 | 2.71E-07 | −−?−− |
| AD-Rep/NAD-Rep | 19q13.32 | 3 | APOC1 | rs483082 | 44912921 | T | 0.286 | 11574 | −0.747 | 0.044 | 3.31E-65 | −−?−− |
| AD-Rep/NAD-Rep | 19q13.32 | 3 | APOC1 | rs584007 | 44913221 | A | 0.341 | 11573 | −0.274 | 0.065 | 2.81E-05 | +−?−− |
| AD-Rep/NAD-Rep | 19q13.32 | 3 | APOC1 | rs56131196 | 44919589 | A | 0.287 | 12347 | −0.447 | 0.046 | 1.63E-22 | −−−?− |
| AD-Rep/NAD-Rep | 19q13.32 | 3 | APOC1 | rs4420638 | 44919689 | G | 0.287 | 12332 | −0.451 | 0.046 | 8.56E-23 | −−−?− |
| AD-Rep/NAD-Rep | 19q13.32 | 3 | APOC1 | rs78959900 | 44920379 | A | 0.298 | 5508 | −0.463 | 0.090 | 2.94E-07 | −???− |
| AD-Rep/NAD-Rep | 19q13.32 | 3 | APOC1P1 | rs4803770 | 44924096 | G | 0.325 | 5508 | −0.516 | 0.087 | 3.36E-09 | −???− |
| AD-Meta/NAD-Meta | 19q13.32 | 3 | APOC1P1 | rs71352239 | 44926286 | T | 0.280 | 4450 | −0.434 | 0.107 | 5.33E-05 | ????− |
| AD-Rep | 19q13.32 | 3 | EXOC3L2 | rs11083767 | 45212422 | C | 0.338 | 5508 | −0.418 | 0.097 | 1.61E-05 | −???− |
Please see the description provided below Table 1.
Figure 2.
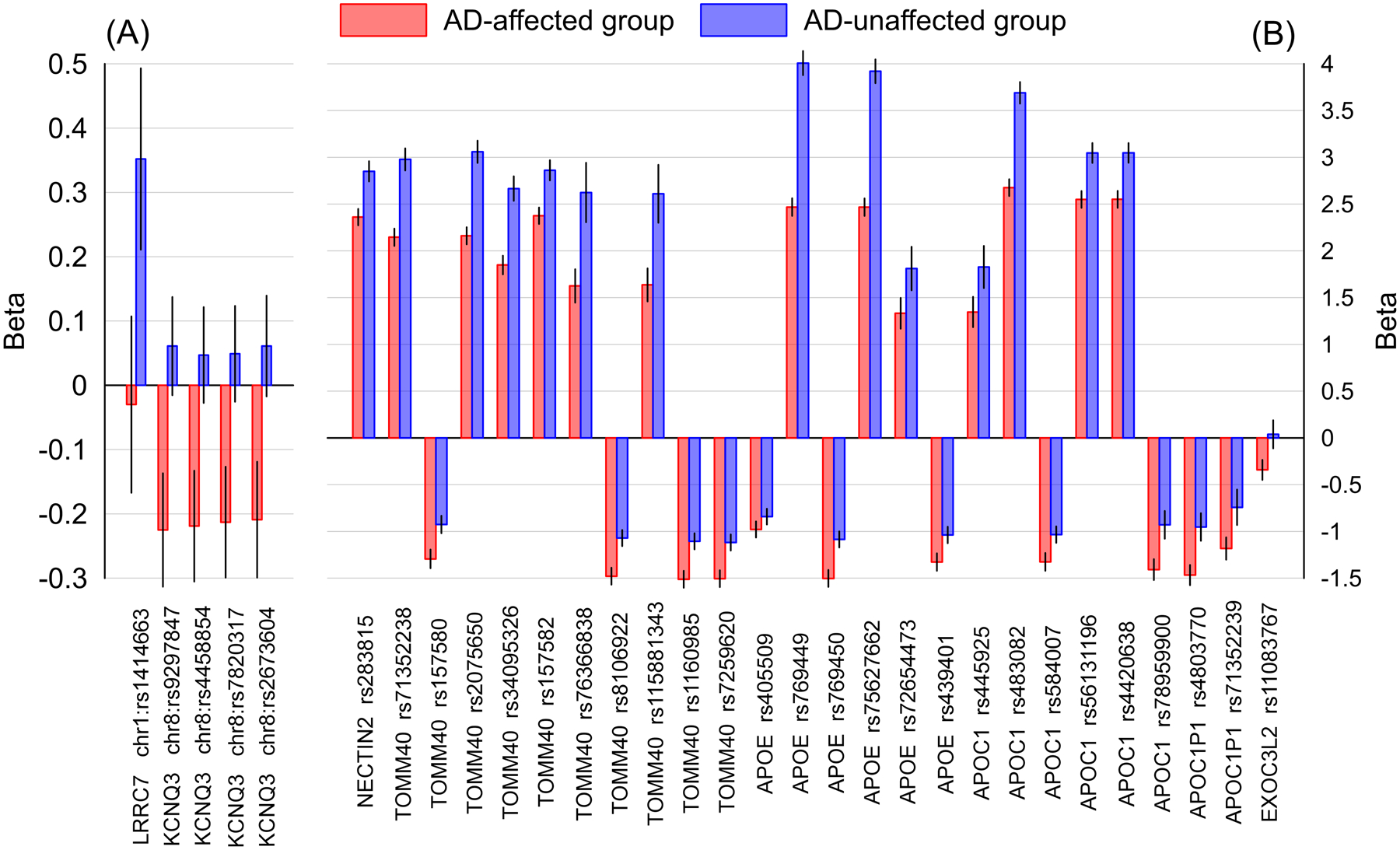
The effect sizes of the ε4-associated group-specific SNPs in Alzheimer’s disease-affected (AD) and unaffected (NAD) groups. (A) SNPs outside of the APOE 19q13.3 locus and (B) SNPs within the APOE 19q13.3 locus. The x-axis shows SNPs and genes identifiers; the y-axis shows the effect sizes (i.e., beta coefficients), red bars indicate the AD group; blue bars indicate the NAD group. Vertical lines show 95% confidence intervals.
3.3. The role of sex
We found interactions between sex and each of four and three group-specific SNP in their associations with the ε2 and ε4 alleles, respectively, at P<0.05. Only the interaction of sex with ε2-associated rs445925 (APOC1 variant), however, attained Bonferroni-adjusted significance (P<1.43E-03=0.05/35). All these seven SNPs were within the APOE 19q13.3 locus (Tables S10 and S11).
3.4. Associations with the AD risk
Among 35 ε2-associated group-specific SNPs, our survival-type analysis revealed significant (P<0.05) interaction effects of the ε2-coding rs7412 with 30 SNPs on the AD risk in the ε4-negative sample (Table S12). Twelve of these 30 interactions attained Bonferroni-adjusted significance (P<1.43E-03=0.05/35) (Table 3). All these 12 interactions had positive effect directions and were with SNPs not on chromosome 19. Among 31 ε4-associated group-specific SNPs, we identified significant (P<0.05) interaction effects of the ε4-coding rs429358 with 16 SNPs on the AD risk in the ε2-negative sample (Table S13). Four of them attained Bonferroni-adjusted significance (P<1.61E-03=0.05/31) (Table 3). All these four interactions had negative effect directions and were with SNPs in the APOE locus.
Table 3.
Bonferroni-adjusted significant results from the survival-type analysis of the main and interaction effects of the rs7412 or rs429358 (i.e., ε2- or ε4-encoding SNPs) and the ε2/ε4-associated group-specific SNPs on the AD risks in the pooled samples of AD and NAD groups.
| SNPs | SNP Main Effect | rs7412 or rs429358 Main Effect | Interaction Effect | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CHR | Gene | SNP | POS | EA | EAF | N | BETA | SE | P-value | BETA | SE | P-value | BETA | SE | P-value |
| ε2-associated group-specific SNPs | |||||||||||||||
| 1p13.3 | VAV3 | rs77841908 | 107713714 | T | 0.012 | 5430 | −0.062 | 0.225 | 7.83E-01 | −0.655 | 0.091 | 7.73E-13 | 1.418 | 0.363 | 9.32E-05 |
| 8q24.3 | MROH5 | rs77186917 | 141434800 | C | 0.047 | 6513 | −0.038 | 0.086 | 6.63E-01 | −0.734 | 0.090 | 5.35E-16 | 0.724 | 0.200 | 3.04E-04 |
| 11q22.3 | DDX10 | rs2884183 | 109127412 | T | 0.213 | 6849 | −0.136 | 0.045 | 2.54E-03 | −0.750 | 0.099 | 2.97E-14 | 0.451 | 0.112 | 6.00E-05 |
| 16q24.1 | IRF8 | rs16882 | 85901967 | C | 0.180 | 6853 | −0.110 | 0.048 | 2.09E-02 | −0.743 | 0.095 | 4.84E-15 | 0.441 | 0.115 | 1.32E-04 |
| 16q24.1 | IRF8 | rs305082 | 85903372 | C | 0.183 | 6846 | −0.111 | 0.047 | 1.93E-02 | −0.756 | 0.095 | 2.23E-15 | 0.457 | 0.116 | 8.75E-05 |
| 17q25.1 | SDK2 | rs9895288 | 73623974 | A | 0.078 | 6854 | −0.052 | 0.070 | 4.60E-01 | −0.638 | 0.084 | 3.99E-14 | 0.598 | 0.172 | 5.07E-04 |
| 17q25.1 | SDK2 | rs75122991 | 73639797 | G | 0.069 | 6853 | −0.057 | 0.074 | 4.45E-01 | −0.638 | 0.083 | 2.06E-14 | 0.662 | 0.178 | 1.99E-04 |
| 17q25.1 | SDK2 | rs28527783 | 73640957 | T | 0.069 | 6854 | −0.061 | 0.074 | 4.10E-01 | −0.639 | 0.083 | 1.96E-14 | 0.664 | 0.178 | 1.91E-04 |
| 17q25.1 | SDK2 | rs76380229 | 73643396 | T | 0.069 | 6852 | −0.066 | 0.074 | 3.78E-01 | −0.640 | 0.083 | 1.87E-14 | 0.667 | 0.178 | 1.76E-04 |
| 20q13.2 | ZFP64 | rs6021874 | 52342233 | C | 0.085 | 6855 | −0.152 | 0.067 | 2.34E-02 | −0.665 | 0.086 | 8.45E-15 | 0.611 | 0.155 | 8.23E-05 |
| 20q13.2 | ZFP64 | rs6021877 | 52345128 | T | 0.086 | 6854 | −0.147 | 0.067 | 2.79E-02 | −0.662 | 0.086 | 1.13E-14 | 0.595 | 0.154 | 1.20E-04 |
| 20q13.2 | ZFP64 | rs2426435 | 52357570 | A | 0.083 | 6854 | −0.162 | 0.068 | 1.81E-02 | −0.660 | 0.085 | 1.01E-14 | 0.609 | 0.156 | 9.86E-05 |
| ε4-associated group-specific SNPs | |||||||||||||||
| 19q13.32 | APOE | rs769449 | 44906745 | A | 0.217 | 9932 | 0.352 | 0.068 | 2.19E-07 | 0.698 | 0.045 | 1.17E-54 | −0.120 | 0.037 | 1.24E-03 |
| 19q13.32 | APOE | rs72654473 | 44911142 | A | 0.051 | 9932 | 0.337 | 0.104 | 1.23E-03 | 0.875 | 0.026 | 1.18E-241 | −0.266 | 0.074 | 2.95E-04 |
| 19q13.32 | APOC1 | rs445925 | 44912383 | A | 0.053 | 9934 | 0.322 | 0.103 | 1.83E-03 | 0.877 | 0.026 | 3.84E-242 | −0.257 | 0.073 | 4.70E-04 |
| 19q13.32 | APOC1 | rs483082 | 44912921 | T | 0.277 | 9933 | 0.483 | 0.076 | 2.27E-10 | 0.593 | 0.077 | 1.17E-14 | −0.110 | 0.033 | 9.05E-04 |
Please see the description provided below Table 1. The Bonferroni-adjusted significance thresholds were 1.43E-03 (i.e., 0.05/35) and 1.61E-03 (i.e., 0.05/31) in the ε2 and ε4 analyses, respectively.
3.5. Functional enrichment analysis
The analysis was performed for 15 and seven protein-coding genes (excluding CEACAM22P and APOC1P1 pseudogenes) harboring group-specific SNPs associated with the ɛ2 and ɛ4 alleles, respectively. We found that 32 and four bio-functions were enriched by three or more genes for the ɛ2- and ɛ4-associated SNPs, respectively (Figure 3, Tables S14 and S15), at a false discovery rate adjusted P<0.0546. One of them, activation of leukocytes, was significantly enriched in both sets (Figure 3).
Figure 3.
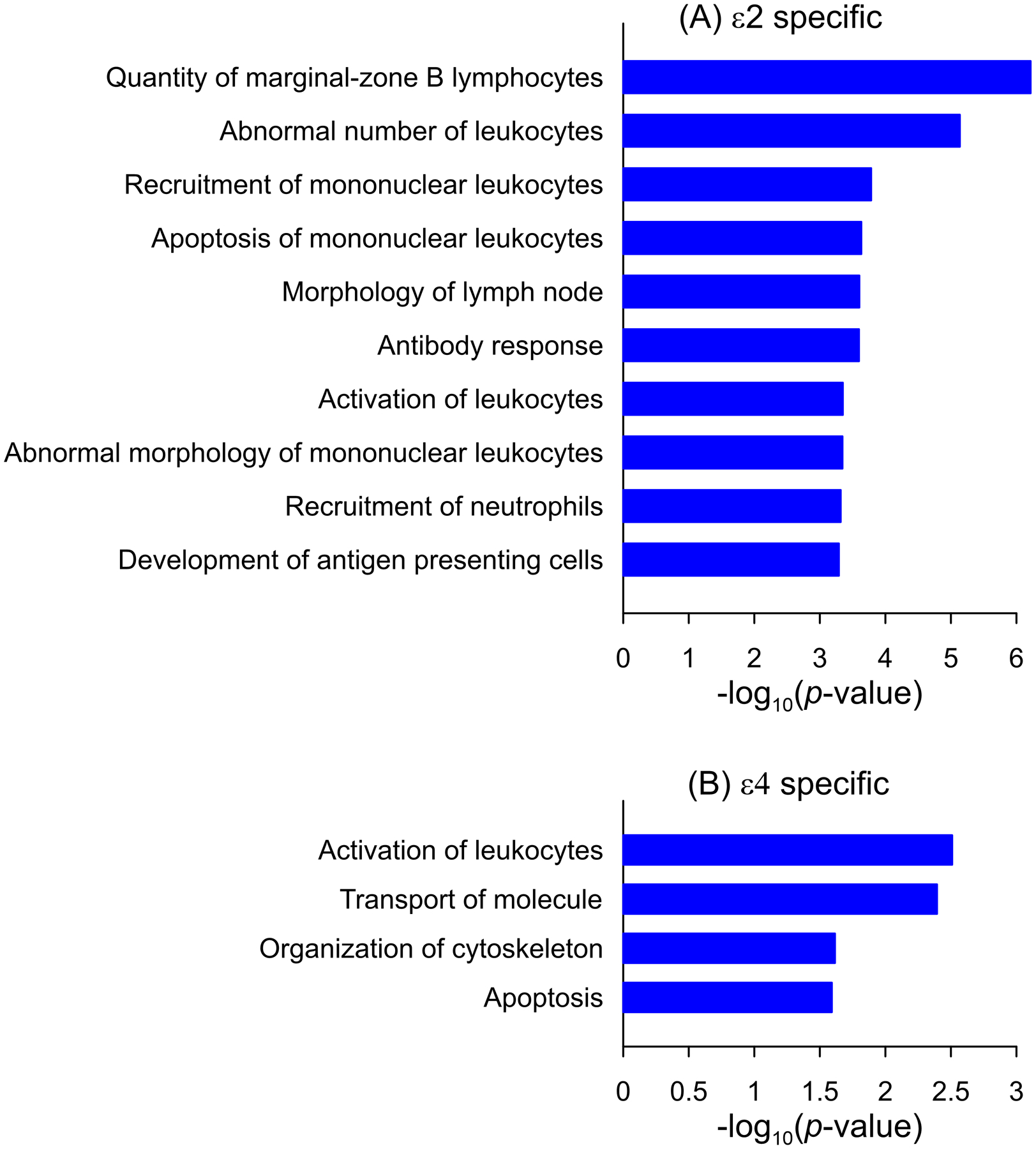
Enrichment of bio-functions. (A) Top-10 bio-functions enriched for genes harboring the ɛ2-associated SNPs. (B) Enrichment of bio-functions for genes harboring the ɛ4-associated SNPs. All bio-functions are significantly enriched at a false discovery rate-adjusted P<0.05.
4. DISCUSSION
In this study, we assumed the APOE alleles as proxies for potential biological processes related to the protection against (ε2 allele) or predisposition to (ε4 allele) cognitive decline. We analyzed the associations of the ε2 and ε4 alleles with other SNPs in the genome to identify genetic variants, which may modulate the effects of these alleles. We were particularly interested in dissecting heterogeneous genetic architecture of AD by identifying associations that can be different between the AD-affected and unaffected subjects. Such differences may indicate genetic modulators of APOE impacts on AD development and partly explain the incomplete penetrance of the APOE alleles47,48.
Our stage-one analysis revealed promising associations of the ε2 and ε4 alleles with 314 and 232 SNPs, respectively. The associations identified only in the AD or NAD groups were with SNPs both within and outside of the APOE 19q13.3 locus (Tables S2, S3, S6, and S7), whereas those identified in both AD and NAD groups were with SNPs within the APOE 19q13.3 locus only (Tables S4 and S8).
Among SNPs with significant association signals only in the AD or NAD groups, our stage-two analysis revealed group-specific effects for 23 and six ε2- and ε4-associated SNPs, respectively (Tables 1, 2, S5, and S9). These SNPs were mapped to 13 genes/loci, of which only EXOC3L2 gene was located within the APOE 19q13.3 locus. The magnitude of the effects of all these SNPs, except rs1414663 (LRRC7 variant), were larger in the AD than NAD group. Almost half of these SNPs had different directions of effects (i.e., opposite patterns of associations between their minor/major alleles and the APOE alleles) in the two groups. Accordingly, the alleles of these SNPs may affect the AD risk by modulating the effects of the ε2 or ε4 alleles. A literature review revealed that polymorphisms in most of the genes harboring these group-specific SNPs were implicated in AD pathology. For instance, SNPs mapped to FRMD4A49 and EXOC3L250,51 were previously associated with AD at the genome-wide significance. Also, a previous study of epistatic associations with AD reported that interactions of SNPs mapped to VAV3, MPDZ, FRMD4A, DDX10, SDK2, ZFP64, and KCNQ3 with SNPs in the other genes were associated with pathological hallmarks of AD such as paired helical filament tau protein, neurofibrillary tangles, and diffuse brain plaques52. Additionally, LRRC7 was previously associated with cognitive performance53. Notably, none of the previously reported SNPs from these genes are in significant LD with SNPs identified in our study in the Caucasian population54.
Among promising SNPs with significant association signals in both AD and NAD groups, 12 and 25 SNPs exhibited group-specific associations with the ε2 and ε4 alleles, respectively (Tables 1, 2, S5, and S9). They were mapped to CEACAM22P, CEACAM16, BCL3, NECTIN2, TOMM40, APOE, APOC1, APOC1P1 genes within the APOE 19q13.3 locus. Four SNPs from the APOE (rs75627662 and rs72654473) and APOC1 (rs445925 and rs483082) genes were associated with both ε2 and ε4 alleles. The magnitudes of the effects for all 12 ε2-associated group-specific SNPs (with positive effects in the AD and NAD groups) and those for 11 of 25 ε4-associated group-specific SNPs (with negative effects in the AD and NAD groups) were larger in the AD than NAD group, indicating stronger associations of the ε2 or ε4 alleles with alleles of these SNPs in the AD group. In contrast, the effect sizes of the remaining 14 of 25 ε4-associated SNPs (with positive effects in the AD and NAD groups) were larger in the NAD than AD group, indicating stronger associations of their minor alleles and the ε4 allele in the NAD group. Hence, the alleles of these 37 SNPs are likely involved in modulating the effects of the ε2 or ε4 alleles on AD risk. Our findings are consistent with the other reports, which emphasize the roles of the complex haplotype structure in the APOE 19q13.3 locus in the AD risk and support the importance of more complex analyses to dissect heterogeneity in genetic architecture of AD26,27,55–59.
Expression quantitative trait loci (eQTLs) that are in high LD (i.e., r2 and/or D’ ≥ 0.8) with several group-specific SNPs were previously reported to alter the expressions APOC1P1, GNPDA2, KCNQ3, NECTIN2, and ZFP64 in brain tissue at P<5E-0660 (Table S16). Since the group-specific SNPs differentially impacted the AD and NAD groups, we suggest the alterations in these genes’ expressions may contribute to the AD pathogenesis.
Our analysis supported the minor role of sex as a modulator of the associations of the ε2 or ε4 allele with group-specific SNPs. Our survival-type analyses revealed a three-time larger number of interactions of the ε2-encoding rs7412, than the ε4-encoding rs429358, with group-specific SNPs (12 vs. 4 interactions) in their associations with the AD risk at Bonferroni-adjusted significance (Table 3). All SNP-ε2 interactions were with 12 SNPs not on chromosome 19, whereas all interactions with the ε4 allele were with four SNPs in the APOE 19q13.3 locus. These interactions imply that the beneficial effect of the ε2 allele (i.e., smaller AD risk, or, equivalently, AAO at older ages, compared to the ε3ε3 carriers) can be significantly modulated by alleles from SNPs spread throughout the entire genome. This study also shows that the adverse effect of the ε4 allele (i.e., larger AD risk or, equivalently, AAO at younger ages, compared to the ε3ε3 carriers) can be significantly modulated by alleles from the other APOE and APOC1 SNPs. While genetic linkage may drive the associations of the ε2/ε4 alleles and local variants (i.e., cis modulators), the functional linkage may underline the roles of, particularly, trans-modulators of the effects of these alleles61.
Our functional enrichment analysis revealed that the genes harboring the group-specific ɛ2-associated SNPs were mainly enriched in inflammation- and immunity-related processes. For example, significantly enriched functions highlighted B and T lymphocytes and phagocytes, such as neutrophils and macrophages, which are involved in antigen-specific (adaptive) and nonspecific (innate) immunity. Immune system and inflammatory responses have been implicated in AD pathogenesis39,62–64. The top term enriched for the ɛ2-associated genes was the quantity of marginal-zone B (MZB) lymphocytes (Figure 3A). It is believed that MZB cells mainly produce IgM antibodies and may regulate autoimmunity65,66. The MZB cells play their vital role in the early antibody reaction to pathogens by mobilizing an optimal response of the innate and adaptive immune systems67,68. Although B cells may have a neuroprotective effect by producing immunoglobulins against amyloid-beta (Aβ)69, murine AD models show that B cells may also influence the formation of Aβ plaques through deposition of immunoglobulins and appear to be enriched in the AD brains64. Interestingly, the inflammatory response-related process, activation of leukocytes, was also at the top for genes harboring the group-specific ɛ4-associated SNPs (Figure 3B). These results suggest inflammation and immunity as mechanisms modulating penetrance of the APOE alleles.
Despite the rigor of this study, we acknowledge its limitations. First, although we analyzed five well-known AD datasets, further validation of our findings in larger samples would provide additional strength. Second, the statistical power of the ε2 allele-related analysis may not be optimal due to the small frequency of this allele in the general population of Caucasians and, especially, in cohorts enriched for AD patients. Third, the functional enrichment analysis had an inherent limitation of a relatively small number of protein-coding genes.
In conclusion, our analyses demonstrated that the associations of the APOE ε2 and ε4 alleles with multiple SNPs spread throughout the entire genome are affected by the AD-affection status. We found that 66 SNPs had significantly different effects in the AD-affected and unaffected groups. The group-specific SNPs may modulate the contributions of the ε2 or ε4 alleles to the AD protection or susceptibility. Our survival-type analysis of the AD risk supported modulating roles of multiple group-specific SNPs. Genes harboring the group-specific SNPs were mainly enriched in inflammation- and immune-related biological processes, e.g., B cell function. These findings provide novel insights into the incomplete penetrance of the APOE alleles and suggest involvement of local and inter-chromosomal modulators of their effects on the AD risk.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by Grants from the National Institute on Aging (P01AG043352, R01AG047310, R01AG061853, R01AG065477, and R01AG070488). The funders had no role in study design, data collection and analysis, decision to publish, or manuscript preparation. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This manuscript was prepared using limited access datasets obtained from dbGaP [accession numbers: phs000372.v1.p1 (ADGC), phs000572.v8.p4 (ADSP), phs000287.v5.p1 (CHS), phs000007.v28.p10 (FHS), and phs000168.v2.p2 (LOADFS)] and NIAGADS [accession number: NG00067 (ADSP)]. Please also see the Supporting Acknowledgment in the Supplementary Information File regarding these five datasets.
Footnotes
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
SUPPLEMENTARY MATERIALS
Supplementary Information File: containing Supporting Acknowledgment, Table S1, and Figures S1–S4.
REFERENCES
- 1.Stelzer G, Rosen N, Plaschkes I, et al. The GeneCards suite: from gene data mining to disease genome sequence analyses. Current Protocols in Bioinformatics. 2016;54(1):1.30.1–1.30.33. doi: 10.1002/cpbi.5 [DOI] [PubMed] [Google Scholar]
- 2.Belloy ME, Napolioni V, Greicius MD. A quarter century of APOE and Alzheimer’s disease: progress to date and the path Forward. Neuron. 2019;101(5):820–838. doi: 10.1016/j.neuron.2019.01.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pericak-Vance MA, Bebout JL, Gaskell PC, et al. Linkage studies in familial Alzheimer disease: evidence for chromosome 19 linkage. Am J Hum Genet. 1991;48(6):1034–1050. [PMC free article] [PubMed] [Google Scholar]
- 4.Farrer LA, Cupples LA, Haines JL, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease: a meta-analysis. JAMA. 1997;278(16):1349–1356. [PubMed] [Google Scholar]
- 5.Liu C-C, Liu C-C, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 2013;9(2):106–118. doi: 10.1038/nrneurol.2012.263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yin Y-W, Li J-C, Wang J-Z, et al. Association between apolipoprotein E gene polymorphism and the risk of vascular dementia: a meta-analysis. Neurosci Lett. 2012;514(1):6–11. doi: 10.1016/j.neulet.2012.02.031 [DOI] [PubMed] [Google Scholar]
- 7.Harrington CR, Louwagie J, Rossau R, et al. Influence of apolipoprotein E genotype on senile dementia of the Alzheimer and Lewy body types: significance for etiological theories of Alzheimer’s disease. Am J Pathol. 1994;145(6):1472–1484. [PMC free article] [PubMed] [Google Scholar]
- 8.Bennet AM, Di Angelantonio E, Ye Z, et al. Association of apolipoprotein E genotypes with lipid levels and coronary risk. JAMA. 2007;298(11):1300–1311. doi: 10.1001/jama.298.11.1300 [DOI] [PubMed] [Google Scholar]
- 9.Rasmussen KL. Plasma levels of apolipoprotein E, APOE genotype and risk of dementia and ischemic heart disease: a review. Atherosclerosis. 2016;255:145–155. doi: 10.1016/j.atherosclerosis.2016.10.037 [DOI] [PubMed] [Google Scholar]
- 10.Lanterna LA, Ruigrok Y, Alexander S, et al. Meta-analysis of APOE genotype and subarachnoid hemorrhage: clinical outcome and delayed ischemia. Neurology. 2007;69(8):766–775. doi: 10.1212/01.wnl.0000267640.03300.6b [DOI] [PubMed] [Google Scholar]
- 11.Biffi A, Anderson CD, Jagiella JM, et al. APOE genotype and extent of bleeding and outcome in lobar intracerebral haemorrhage: a genetic association study. Lancet Neurol. 2011;10(8):702–709. doi: 10.1016/S1474-4422(11)70148-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wei LK, Au A, Menon S, et al. Polymorphisms of MTHFR, eNOS, ACE, AGT, APOE, PON1, PDE4D, and ischemic stroke: meta-analysis. J Stroke Cerebrovasc Dis. 2017;26(11):2482–2493. doi: 10.1016/j.jstrokecerebrovasdis.2017.05.048 [DOI] [PubMed] [Google Scholar]
- 13.Tsuang D, Leverenz JB, Lopez OL, et al. APOE ε4 increases risk for dementia in pure synucleinopathies. JAMA Neurol. 2013;70(2):223–228. doi: 10.1001/jamaneurol.2013.600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rubino E, Vacca A, Govone F, De Martino P, Pinessi L, Rainero I. Apolipoprotein E polymorphisms in frontotemporal lobar degeneration: a meta-analysis. Alzheimers Dement. 2013;9(6):706–713. doi: 10.1016/j.jalz.2012.10.013 [DOI] [PubMed] [Google Scholar]
- 15.Kulminski AM, Culminskaya I, Arbeev KG, Ukraintseva SV, Arbeeva L, Yashin AI. Trade-off in the effect of the APOE gene on the ages at onset of cardiocascular disease and cancer across ages, gender, and human generations. Rejuvenation Res. 2013;16(1):28–34. doi: 10.1089/rej.2012.1362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Høgh P, Oturai A, Schreiber K, et al. Apoliprotein E and multiple sclerosis: impact of the epsilon-4 allele on susceptibility, clinical type and progression rate. Mult Scler. 2000;6(4):226–230. doi: 10.1177/135245850000600403 [DOI] [PubMed] [Google Scholar]
- 17.Maehlen MT, Provan SA, de Rooy DPC, et al. Associations between APOE genotypes and disease susceptibility, joint damage and lipid levels in patients with rheumatoid arthritis. PLoS ONE. 2013;8(4):e60970. doi: 10.1371/journal.pone.0060970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tao Q, Ang TFA, DeCarli C, et al. Association of chronic low-grade inflammation with risk of Alzheimer disease in ApoE4 carriers. JAMA Netw Open. 2018;1(6):e183597. doi: 10.1001/jamanetworkopen.2018.3597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abondio P, Sazzini M, Garagnani P, et al. The genetic variability of apoe in different human populations and its implications for longevity. Genes (Basel). 2019;10(3):222. doi: 10.3390/genes10030222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sebastiani P, Gurinovich A, Nygaard M, et al. Apoe alleles and extreme human longevity. J Gerontol A Biol Sci Med Sci. 2019;74(1):44–51. doi: 10.1093/gerona/gly174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Raichlen DA, Alexander GE. Exercise, APOE genotype, and the evolution of the human lifespan. Trends Neurosci. 2014;37(5):247–255. doi: 10.1016/j.tins.2014.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reiman EM, Arboleda-Velasquez JF, Quiroz YT, et al. Exceptionally low likelihood of Alzheimer’s dementia in APOE2 homozygotes from a 5,000-person neuropathological study. Nat Commun. 2020;11(1):667. doi: 10.1038/s41467-019-14279-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roses AD, Lutz MW, Amrine-Madsen H, et al. A TOMM40 variable-length polymorphism predicts the age of late-onset Alzheimer’s disease. Pharmacogenomics J. 2010;10(5):375–384. doi: 10.1038/tpj.2009.69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lutz MW, Crenshaw D, Welsh-Bohmer KA, Burns DK, Roses AD. New genetic approaches to AD: lessons from APOE-TOMM40 phylogenetics. Curr Neurol Neurosci Rep. 2016;16(5):48. doi: 10.1007/s11910-016-0643-8 [DOI] [PubMed] [Google Scholar]
- 25.Zhou X, Chen Y, Mok KY, et al. Non-coding variability at the APOE locus contributes to the Alzheimer’s risk. Nat Commun. 2019;10(1):3310. doi: 10.1038/s41467-019-10945-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kulminski AM, Philipp I, Loika Y, He L, Culminskaya I. Haplotype architecture of the Alzheimer’s risk in the APOE region via co-skewness. Alzheimers Dement (Amst). 2020;12(1):e12129. doi: 10.1002/dad2.12129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kulminski AM, Shu L, Loika Y, et al. APOE region molecular signatures of Alzheimer’s disease across races/ethnicities. Neurobiology of Aging. 2020;87:141.e1–141.e8. doi: 10.1016/j.neurobiolaging.2019.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Finch CE, Kulminski AM. The Alzheimer’s disease exposome. Alzheimers Dement. 2019;15(9):1123–1132. doi: 10.1016/j.jalz.2019.06.3914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nesse RM, Ganten D, Gregory TR, Omenn GS. Evolutionary molecular medicine. J Mol Med. 2012;90(5):509–522. doi: 10.1007/s00109-012-0889-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fried LP, Borhani NO, Enright P, et al. The cardiovascular health study: design and rationale. Ann Epidemiol. 1991;1(3):263–276. doi: 10.1016/1047-2797(91)90005-W [DOI] [PubMed] [Google Scholar]
- 31.Dawber TR, Meadors GF, Moore FE. Epidemiological approaches to heart disease: the Framingham study. Am J Public Health Nations Health. 1951;41(3):279–286. doi: 10.2105/ajph.41.3.279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Feinleib M, Kannel WB, Garrison RJ, McNamara PM, Castelli WP. The Framingham offspring study: design and preliminary data. Prev Med. 1975;4(4):518–525. doi: 10.1016/0091-7435(75)90037-7 [DOI] [PubMed] [Google Scholar]
- 33.Lee JH, Cheng R, Graff-Radford N, Foroud T, Mayeux R. Analyses of the national institute on aging late-onset Alzheimer’s disease family study: implication of additional loci. Arch Neurol. 2008;65(11):1518–1526. doi: 10.1001/archneur.65.11.1518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beecham GW, Bis JC, Martin ER, et al. The Alzheimer’s Disease Sequencing Project: study design and sample selection. Neurol Genet. 2017;3(5):e194. doi: 10.1212/NXG.0000000000000194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Crane PK, Foroud T, Montine TJ, Larson EB. Alzheimer’s Disease Sequencing Project discovery and replication criteria for cases and controls: Data from a community-based prospective cohort study with autopsy follow-up. Alzheimers Dement. 2017;13(12):1410–1413. doi: 10.1016/j.jalz.2017.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Naj AC, Jun G, Beecham GW, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet. 2011;43(5):436–441. doi: 10.1038/ng.801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology. 1984;34(7):939–944. doi: 10.1212/wnl.34.7.939 [DOI] [PubMed] [Google Scholar]
- 38.McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s & Dementia. 2011;7(3):263–269. doi: 10.1016/j.jalz.2011.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nazarian A, Yashin AI, Kulminski AM. Genome-wide analysis of genetic predisposition to Alzheimer’s disease and related sex disparities. Alzheimer’s Research & Therapy. 2019;11(1):5. doi: 10.1186/s13195-018-0458-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–575. doi: 10.1086/519795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Conomos MP, Miller MB, Thornton TA. Robust inference of population structure for ancestry prediction and correction of stratification in the presence of relatedness. Genet Epidemiol. 2015;39(4):276–293. doi: 10.1002/gepi.21896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gogarten SM, Sofer T, Chen H, et al. Genetic association testing using the GENESIS R/Bioconductor package. Bioinformatics. Published online July 22, 2019:btz567. doi: 10.1093/bioinformatics/btz567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mägi R, Morris AP. GWAMA: software for genome-wide association meta-analysis. BMC Bioinformatics. 2010;11:288. doi: 10.1186/1471-2105-11-288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Therneau TM. Coxme: A Package for Mixed Effects Cox Models in R. R Package Version 2.2–16; 2020. Accessed August 15, 2021. https://CRAN.R-project.org/package=coxme
- 45.Therneau TM. Survival: A Package for Survival Analysis in R. R Package Version 3.2–13; 2021. Accessed August 15, 2021. https://CRAN.R-project.org/package=survival
- 46.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society Series B (Methodological). 1995;57(1):289–300. doi: 10.2307/2346101 [DOI] [Google Scholar]
- 47.Corder EH, Saunders AM, Strittmatter WJ, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261(5123):921–923. doi: 10.1126/science.8346443 [DOI] [PubMed] [Google Scholar]
- 48.Freudenberg-Hua Y, Freudenberg J, Vacic V, et al. Disease variants in genomes of 44 centenarians. Mol Genet Genomic Med. 2014;2(5):438–450. doi: 10.1002/mgg3.86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lambert J-C, Grenier-Boley B, Harold D, et al. Genome-wide haplotype association study identifies the FRMD4A gene as a risk locus for Alzheimer’s disease. Mol Psychiatry. 2013;18(4):461–470. doi: 10.1038/mp.2012.14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marioni RE, Harris SE, Zhang Q, et al. GWAS on family history of Alzheimer’s disease. Transl Psychiatry. 2018;8(1):99. doi: 10.1038/s41398-018-0150-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jansen IE, Savage JE, Watanabe K, et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat Genet. 2019;51(3):404–413. doi: 10.1038/s41588-018-0311-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang H, Yang J, Schneider JA, De Jager PL, Bennett DA, Zhang H-Y. Genome-wide interaction analysis of pathological hallmarks in Alzheimer’s disease. Neurobiol Aging. 2020;93:61–68. doi: 10.1016/j.neurobiolaging.2020.04.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee JJ, Wedow R, Okbay A, et al. Gene discovery and polygenic prediction from a genome-wide association study of educational attainment in 1.1 million individuals. Nat Genet. 2018;50(8):1112–1121. doi: 10.1038/s41588-018-0147-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Machiela MJ, Chanock SJ. LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics. 2015;31(21):3555–3557. doi: 10.1093/bioinformatics/btv402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Templeton AR, Maxwell T, Posada D, Stengård JH, Boerwinkle E, Sing CF. Tree scanning: a method for using haplotype trees in phenotype/genotype association studies. Genetics. 2005;169(1):441–453. doi: 10.1534/genetics.104.030080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yu C-E, Seltman H, Peskind ER, et al. Comprehensive analysis of APOE and selected proximate markers for late-onset Alzheimer’s disease: patterns of linkage disequilibrium and disease/marker association. Genomics. 2007;89(6):655–665. doi: 10.1016/j.ygeno.2007.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lescai F, Chiamenti AM, Codemo A, et al. An APOE haplotype associated with decreased ε4 expression increases the risk of late onset Alzheimer’s disease. Journal of Alzheimer’s Disease. 2011;24(2):235–245. doi: 10.3233/JAD-2011-101764 [DOI] [PubMed] [Google Scholar]
- 58.Babenko VN, Afonnikov DA, Ignatieva EV, Klimov AV, Gusev FE, Rogaev EI. Haplotype analysis of APOE intragenic SNPs. BMC Neurosci. 2018;19(Suppl 1):16. doi: 10.1186/s12868-018-0413-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kulminski AM, Huang J, Wang J, He L, Loika Y, Culminskaya I. Apolipoprotein E region molecular signatures of Alzheimer’s disease. Aging Cell. 2018;17(4):e12779. doi: 10.1111/acel.12779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.GTEx Consortium. Genetic effects on gene expression across human tissues. Nature. 2017;550(7675):204–213. doi: 10.1038/nature24277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Linghu B, Franzosa EA, Xia Y. Construction of Functional Linkage Gene Networks by Data Integration. In: Mamitsuka H, DeLisi C, Kanehisa M, eds. Data Mining for Systems Biology: Methods and Protocols. Methods in Molecular Biology. Humana Press; 2013:215–232. doi: 10.1007/978-1-62703-107-3_14 [DOI] [PubMed] [Google Scholar]
- 62.Querfurth HW, LaFerla FM. Alzheimer’s Disease. New England Journal of Medicine. 2010;362(4):329–344. doi: 10.1056/NEJMra0909142 [DOI] [PubMed] [Google Scholar]
- 63.Keren-Shaul H, Spinrad A, Weiner A, et al. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell. 2017;169(7):1276–1290.e17. doi: 10.1016/j.cell.2017.05.018 [DOI] [PubMed] [Google Scholar]
- 64.Kim K, Wang X, Ragonnaud E, et al. Therapeutic B-cell depletion reverses progression of Alzheimer’s disease. Nat Commun. 2021;12(1):2185. doi: 10.1038/s41467-021-22479-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Appelgren D, Eriksson P, Ernerudh J, Segelmark M. Marginal-zone B-cells are main producers of IgM in humans, and are reduced in patients with autoimmune vasculitis. Front Immunol. 2018;9:2242. doi: 10.3389/fimmu.2018.02242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Marinkovic D, Marinkovic T. Putative role of marginal zone B cells in pathophysiological processes. Scand J Immunol. 2020;92(3):e12920. doi: 10.1111/sji.12920 [DOI] [PubMed] [Google Scholar]
- 67.Martin F, Kearney JF. Marginal-zone B cells. Nat Rev Immunol. 2002;2(5):323–335. doi: 10.1038/nri799 [DOI] [PubMed] [Google Scholar]
- 68.Cerutti A, Cols M, Puga I. Marginal zone B cells: virtues of innate-like antibody-producing lymphocytes. Nat Rev Immunol. 2013;13(2):118–132. doi: 10.1038/nri3383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Söllvander S, Ekholm-Pettersson F, Brundin R-M, et al. Increased number of plasma B cells producing autoantibodies against Aβ 42 protofibrils in Alzheimer’s disease. Journal of Alzheimer’s Disease. 2015;48(1):63–72. doi: 10.3233/JAD-150236 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.