

Abstract
Introduction
The National Institute on Aging Late‐Onset Alzheimer's Disease Family Based Study (NIA‐LOAD FBS) was established to study the genetic etiology of Alzheimer's disease (AD).
Methods
Recruitment focused on families with two living affected siblings and a third first‐degree relative similar in age with or without dementia. Uniform assessments were completed, DNA was obtained, as was neuropathology, when possible. Apolipoprotein E (APOE) genotypes, genome‐wide single nucleotide polymorphism (SNP) arrays, and sequencing was completed in most families.
Results
APOE genotype modified the age‐at‐onset in many large families. Novel variants and known variants associated with early‐ and late‐onset AD and frontotemporal dementia were identified supporting an international effort to solve AD genetics.
Discussion
The NIA‐LOAD FBS is the largest collection of familial AD worldwide, and data or samples have been included in 123 publications addressing the genetic etiology of AD. Genetic heterogeneity and variability in the age‐at‐onset provides opportunities to investigate the complexity of familial AD.
1. INTRODUCTION
In 2020, 5.8 million Americans were living with Alzheimer's disease (AD). By 2050, this number is projected to grow to 13.9 million people, nearly 3.3% of the US population. 1 The etiology of AD remains unclear, but there is substantial evidence that AD is a highly heritable disorder. The National Institute on Aging Late‐Onset Alzheimer's Disease Family Based Study (NIA‐LOAD FBS) is the largest collection of multiplex AD families recruited and longitudinally assessed worldwide. Since 2003, the central goal of this cohort has been to broadly support genetic research by making clinical and genomic data and biological samples rapidly available to the AD research community to support the development of therapies to treat or prevent AD. The use of multiplex families to understand genetic susceptibility to AD began with early‐onset forms. These studies led to the discovery of rare and highly penetrant mutations in autosomal dominant AD: APP 2 , PSEN1, 3 , 4 and PSEN2 5 , 6 Nearly 60% of individuals with early‐onset AD have other affected family members, 7 and of these, approximately 13% inherit the mutations in an autosomal dominant manner 8 While variants in these genes have explained only a small proportion of the occurrence of familial, early‐onset AD, they formed the basis for the development of novel therapies now being investigated in AD regardless of age at onset of disease 9
Genetic research on the more common late onset form of AD was transformed by the work of Pericak‐Vance and Bebout 10 using a novel linkage strategy in multiply affected families with late‐onset AD. They were the first to identify a locus on chromosome 19. 10 This led to the discovery of the association between the apolipoprotein E (APOE) ε4 allele and the increased risk of AD as well as the reduced risk of AD with the APOE ε2 allele. 11 , 12 The association was later confirmed in almost all ethnic groups, but with slightly weaker effects among those of African ancestry. 13 Overall, the population attributable risk of the APOE ε4 allele has been estimated at 20% 14 Nonetheless, the accumulating evidence indicates that late‐onset AD is multifactorial with high heritability, 70% or higher, implying that other genes and risk factors also contribute to its etiology.
Genetic investigations typically require time and effort to ascertain samples for genetic studies. While it was presumed that recruitment of unrelated patients and controls would be a faster and more efficient way to find risk alleles than families, there are many scientific and practical advantages for recruiting families for genetic research. The observed incidence rates for AD are estimated to be three to five times higher in multiplex families. 15 ) Multiplex families may be enriched in disease‐causing genetic variants, allowing penetrance to be studied while minimizing the effects of population stratification. Familial enrichment is particularly valuable for rare variants (minor allele frequency [MAF] < 0.01), where population‐level studies have little power. Family members are generally eager to participate in such research and often have shared environments and parents of origin. Moreover, they also provide the opportunity to assess interaction between alleles at the same locus and to detect epistasis due to the high penetrance.
The NIA‐LOAD FBS has contributed to nearly every genetic study of AD over the past two decades, and with the advent of novel strategies to understand how genetic variation leads to this disease, it will continue to do so. Here, we describe the cohort, the variable range of age‐at‐onset across and within families, the effects of APOE on the risk of AD across ethnic groups, and the genetic heterogeneity and highlight the broad availability of clinical and genetic data and biological samples.
2. METHODS
2.1. Participant recruitment
Families assumed to be multiply affected by AD by history or autopsy were recruited from sites across the United States (Figure S1 in supporting information). The criteria for study entry included two living affected siblings, 60 years of age or older, and a third living relative, similar in age, with or without dementia. We also included families with deceased affected siblings, as long as frozen brain tissue was available for affected members. Families in which participants were symptomatic, but did not meet criteria for AD, were still included with the commitment for follow‐up. Healthy controls without a family history of AD in a first‐degree relative, who were 55 years of age or older were also recruited. Participants demonstrated the full range of cognitive function, including normal cognition and dementia (Table 1). The clinical diagnosis of AD was required to meet established research criteria. 16
TABLE 1.
NIA‐LOAD family‐based study: clinical data and diagnoses
| Clinical diagnoses 16 | N (%) |
| Probable AD | 2672 (27.6%) |
| Possible AD | 395 (4.1%) |
| Other dementias a | 160 (1.7%) |
| Dementia by family report only | 227 (2.3%) |
| Unaffected family members | 4587 (47.4%) |
| Assessments | N |
| Cognitive | 8610 (89%) |
| NPI‐Q | 3187 (33%) |
| Cardiovascular and other risk factor questionnaire | 6553 (68%) |
Note: Percentages are based on the total number of family members 9682 individuals.
Table 3 shows the diagnoses of other dementias.
Abbreviations: AD, Alzheimer's disease; NIA‐LOAD, National Institute on Aging Late‐Onset Alzheimer's Disease; NPI‐Q, Neuropsychiatric Inventory Questionnaire.
Families were enrolled and consented at individual study sites and completed the following protocols either by in‐person or telephone interview and assessments. The study protocol included a family pedigree, standardized cognitive and psychiatric assessments, as well as a review of AD and cardiovascular risk factors (Table 1). Regular follow‐up visits were used to update the family history and obtain additional clinical information on new family members who had not previously participated. All study data were stored at Columbia University, the coordinating center.
2.2. Cognitive assessment
Standardized cognitive assessments began in 2006, and more than 75% of the cohort has been assessed. 17 , 18 The 15‐minute cognitive battery consisted of seven brief tests that were administered in person or by telephone and assessed: working memory—Digit Span Forward and Backward and Digit Ordering; episodic memory—Immediate and Delayed Recall of story “A” from the Wechsler Memory Scale; and semantic memory—asked persons to name members of two semantic categories (Animals, Vegetables) in separate 1‐minute trials. From these measures, a factor analysis of the seven tests summarizing measures of working memory, declarative memory, episodic memory, semantic memory, and global cognition was developed and is regularly updated. Interviews using different methods, in‐person or by telephone, had little effect on performance and provided operational flexibility and a means of reducing costs and missing data. 17
2.3. Psychiatric interview
The assessment of psychiatric manifestations was added in 2010 and was designed to be completed in an interview with a knowledgeable informant and could be conducted after the participant's death. This approach captured psychotic symptoms that occurred in the prior month, identifying the month during which each symptom was most persistent, and recording the frequency of symptoms. 19 The Neuropsychiatric Inventory Questionnaire (NPI‐Q) 19 , 20 , a widely used, validated informant‐based interview that is used to evaluate neuropsychiatric symptoms in patients with dementia, was selected. The NPI‐Q has good interrater reliability and concurrent validity compared to more structured interviews. 21 , 22
1. RESEARCH IN CONTEXT
Systematic review: The National Institute on Aging Late‐Onset Alzheimer's Disease Family Based Study (NIA‐LOAD FBS) was established as a resource to support research focused on the etiology and early detection of Alzheimer's disease (AD) and to spur therapeutic development. It is one of the largest collections of such families and has been used in genetic research worldwide.
Interpretation: The NIA‐LOAD FBS has recruited 1756 families and obtained and stored DNA. Investigators have completed a genome‐wide single nucleotide polymorphism array, whole exome and whole genome sequencing, and have ascertained neuropathological examinations in 836 individuals. In some large families, the age‐at‐onset varied by as much 22 years suggesting the possibility of genetic or environmental modifying factors. All biological resources and acquired data are shared with researchers.
Future directions: The NIA‐LOAD FBS intends to expand capacity for multiomics research, include recently developed blood‐based biomarkers to add precision to the diagnoses, and expand the ethnic diversity of families recruited.
2.4. Antecedent risk factors
A structured interview was used to collect putative risk and protective factors associated with AD, at baseline and follow‐up to incorporate any changes. The primary focus was on modifying factors such as hypertension, heart diseases (myocardial infarction, congestive heart failure, or other type of heart disease), and diabetes. 23 A stroke assessment captured the number of stroke events. Antecedent (risk or protective) factors were also used in follow‐up by telephone. For participants unable to be interviewed, the person who best knew the patient provided the information. Other factors to be ascertained included history of head injury, smoking, alcohol use, physical activities, and leisure activities. Current medications were queried. In addition, for each medical condition or risk factor endorsed, the age at onset, duration, treatment history, and whether there had been any change in the condition since the prior assessment was ascertained.
2.5. Biological resources collection
Initially, blood, saliva, or frozen brain tissue was collected at baseline and at subsequent visits. All such biological materials were sent to the National Centralized Repository for Alzheimer's Disease and Related Dementias (NCRAD), which extracted and stored DNA. As shown in Table S1 in supporting information, we collected DNA from 7925 participants. In 2018, we expanded blood collection to also obtain peripheral blood mononuclear cells (PBMCs) at follow‐up visits.
2.6. Genotyping and sequencing
Table S1 summarizes available genetic data. Genome‐wide single nucleotide polymorphism (SNP) association study (GWAS) data were obtained in 1340 families (76.3%) in the cohort, which included 7012 (72.4%) of their family members. Whole exome (WES) or whole genome sequencing (WGS) was conducted in 1340 (76.3%) of the families. APOE genotyping was performed in 1260 families (71.8%). The results of all genetic studies have been stored at the National Institute on Aging Genetics of Alzheimer's Disease Data Storage Site (NIAGADS).
2.7. Neuropathological confirmation of diagnosis
At the inception of the study, NIA‐LOAD FBS began preplanning brain donation with all family members to maximize the likelihood that neuropathology would be performed after death. This resulted in a robust brain donation program in which autopsy reports or brain tissue was successfully obtained in 836 individuals (Table 2). In the event an autopsy was performed, and no tissue was available, the autopsy report was requested. Brain autopsy was performed at Columbia University or at one of the study sites, with diagnoses based on the 2012 NIA‐Alzheimer's Association Working Group guidelines. 24 Brain tissues, frozen and fixed, were sent to NCRAD to share with the research community. In 2019, we expanded the use of brain tissue to serve not only as a means of confirming the diagnosis, but also to support functional studies including transcriptomics and DNA methylation studies.
TABLE 2.
NIA‐LOAD family‐based study neuropathology data
| Autopsy | N (%) |
| Total neuropathology/autopsy information | 836 |
| Neuropathology/autopsy reports provided by family | 181 (21.6%) |
| Autopsy by Columbia or study site | 655 (78.3%) |
| Definite AD 24 | 492 (58.9% |
| Non‐AD dementia | 47 (5.6%) |
| No dementia | 116 (13.9%) |
Note: Percentages are based on the total number of individuals with neuropathology or autopsy information.
Abbreviations: AD, Alzheimer's disease; NIA‐LOAD, National Institute on Aging Late‐Onset Alzheimer's Disease.
2.8. Accessible biological samples, and clinical and genetic data
As shown in Figure 1, qualifying families (A) were recruited by the NIA‐LOAD FBS investigators (B). Blood was sent to NCRAD (C), which in turn, supplied biological samples (D) to the Alzheimer's Disease Sequencing Project (ADSP) and to other qualified individual investigators (D) along with phenotype data from the Columbia University Coordinating Center for the NIA‐LOAD FBS. Any new data that were generated by these investigators was required to be shared by sending it to NIAGADS (E) where other investigators could request clinical and genetic results from these studies (F).
FIGURE 1.
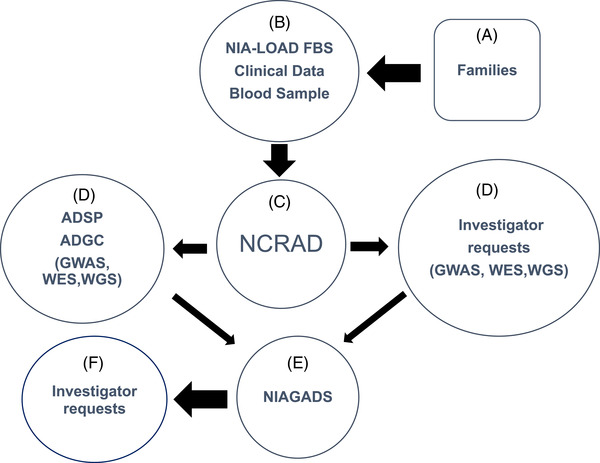
Potentially qualifying families (A) were recruited by the National Institute on Aging Late‐Onset Alzheimer's Disease Family Based Study (NIA‐LOAD FBS) investigators (B). Blood was sent to National Centralized Repository for Alzheimer's Disease and Related Dementias (NCRAD) (C). NCRAD supplied biological samples to the Alzheimer's Disease Sequencing Projects (ADSP) (D) and to other qualified individual investigators (D) along with phenotype data from the Columbia University Coordinating Center. Any new data generated by investigators was required to be shared by sending it to National Institute on Aging Genetics of Alzheimer's Disease Data Storage Site (NIAGADS; E) where other investigators could request clinical and genetic results from these studies (F)
3. RESULTS
3.1. Recruitment and demographics
We recruited 1756 families and acquired data from 9682 family members. The families were diverse: 181 (10.3%) Black; 425 (24.2%) Caribbean Hispanic; 138 (7.8%) self‐reporting as “other or mixed”; and 1012 White, non‐Hispanic (Table S2 in supporting information). Among the affected and unaffected family members, 63% were women, a percentage that differed significantly by ethnic group (72% among Blacks and 66% among Hispanics, 62% among White non‐Hispanics, and 57% the other group, P = .009). Among the 1756 families, 451 (25.7%) had three or more affected family members. Another 31.5% had two affected individuals while 598 (34%) families consisted of only a single affected individual or a single individual who, after clinical evaluation, did not meet criteria for dementia or AD but had cognitive impairment. The proportions of affected to unaffected individuals were similar across the ethnic groups (Table S2). Ultimately, 1343 families with two or more affected individuals meeting research criteria for AD were recruited. Of those, 569 (42.4%) families represented a single generation; 683 (50.8%) were represented by two generations and 91 (6.8%) of families were represented by three generations. Ascertainment of other family members continues for the remaining 413 families.
3.2. Data collection
In the family cohort, 8610 (77.7%) individuals had detailed clinical information including multiple cognitive assessments. The other 22.3% were either too impaired for cognitive testing or were recruited before the clinical assessments were standardized in 2006. Tables 1 and 3 summarize the data collected and the resulting clinical diagnoses. Telephone interviews were initiated early on to make it easier for family members to participate. We conducted in‐person interviews in 7412 (68.5%) and follow‐up interviews in 3349 (30.9%). Because we had alternatives to in‐person assessment in place prior to the COVID‐19 pandemic, we were able to continue to follow family members; however, our ability to recruit new families was limited. Since the inception of the study in 2003, 15.1% of the cohort has been lost to follow‐up without leaving a forwarding address or telephone number, and 2878 (26.6%) died.
TABLE 3.
Other forms of dementia
| Clinical diagnosis | N (%) |
| Frontotemporal degeneration | 21 (13%) |
| Parkinson's disease with dementia | 9 (5.6%) |
| Progressive supranuclear palsy | 5 (3.1%) |
| Corticobasal degeneration | 1 (0.6%) |
| Communicating, obstructive, or normal pressure hydrocephalus | 1 (0.6%) |
| Cerebrovascular disease with dementia | 32 (20%) |
| Dementia with Lewy bodies | 28 (17.5%) |
| Primary progressive aphasia | 2 (1.2%) |
| Posterior cortical atrophy | 1 (0.6%) |
| Dementia associated with multiple non‐Alzheimer's etiologies | 28 (17.5%) |
| Dementia of unknown cause | 32 (20%) |
| Total | 160 |
Note: Percentages are based on the 160 individuals with a clinical diagnosis of dementia other than Alzheimer's disease.
Below, we present some of the published and unpublished results from this data collection.
3.3. Age‐at‐onset
The average age of onset in affected family members differed slightly, but significantly, across all ethnic groups ranging from 72 years in Caribbean Hispanics to 74 years in Blacks and White non‐Hispanics (P < .0001). The average age at last visit among unaffected family members differed significantly by ethnic group (66.2 years, Black, 63.9 years, Hispanic vs. 68.7 years, White non‐Hispanic; P < .0001). In general, we observed a very wide range of age‐at‐onset among families especially those with three or more affected members. Individuals with later ages‐at‐onset were significantly less likely to carry an APOE ε4 allele (non‐ε4 44.9% vs. ε4 60.5%) and more likely to carry an APOE ε3 allele (ε3 47.1% vs. ε4 33.3%; Figures SS2a and S2b in supporting information). APOE ε4 had the stronger effects in White non‐Hispanic families when the genotype was homozygous or heterozygous. However, among Caribbean Hispanics and Blacks, the genotypic effects were limited to APOE ε4 homozygotes (Figures S3a‐d in supporting information), indicating that although APOE is not only a robust risk factor for sporadic AD, but also has a role in familial AD.
3.4. Conversion rate to dementia
The annual incidence, or conversion rate, to dementia in the NIA‐LOAD FBS participants ranged from 3% per year between ages 65 and 74 years, 7% per year in those aged 75 to 84 years, and 8% in those 85 years or older. Contrasting these results with the population‐based estimates, the incidence was increased by 2‐fold for NIA‐LOAD FBS (standardized annual incidence ratio, 3.44). 25
3.5. Genome‐wide SNP array
Using data from 992 families, investigators confirmed results seen among unrelated individuals in BIN1, CLU, CR1, and APOE and identified a novel gene CUGBP2 among APOE ε4 carriers. 26 The variants in these families were not statistically significant due to the small sample size but nominal significance was found in CD2AP and EPHA1 and contributed to several large AD meta‐analyses (Figure 2). 27 , 28 , 29 Investigators also created a polygenic risk score constructed from summary statistics from sporadic AD that demonstrated the enrichment of genetic risk variants in these families. 23 , 30
FIGURE 2.
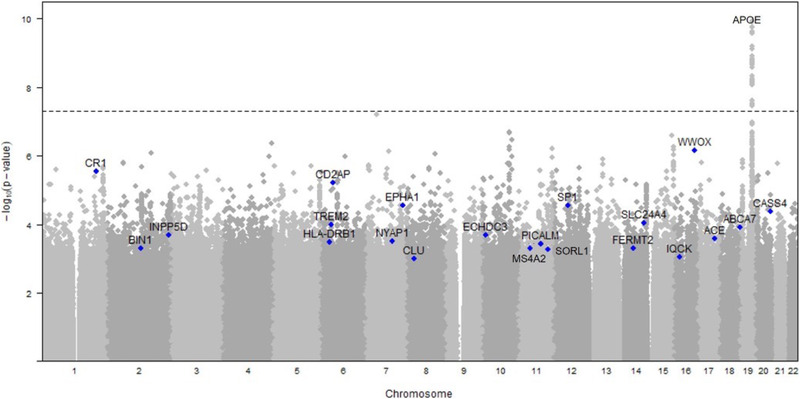
Results of genome‐wide array in the National Institute on Aging Late‐Onset Alzheimer's Disease Family Based Study (NIA‐LOAD FBS). The apolipoprotein E (APOE) locus reached genome‐wide significance in association with Alzheimer's disease (AD). However, a number of other loci, labeled on the Manhattan plot below, were nominally significant. Many of these loci supported several large meta‐analyses that included other cohorts 27 , 28 , 29
3.6. Variant analyses
In this group of families, we also found variants in APP, PSEN1, and PSEN2 usually associated with familial early‐onset forms of AD and MAPT, GRN, and C9orf72 usually associated with frontotemporal dementia (FTD). 31 , 32 , 33 , 34 , 35 The most frequent variants were found were PSEN1 p.A79V (0.46% of the families) and the C9ORF72 expansion (0.48% of the families). Among the probands, 1.82% carried pathogenic variants in APP, PSEN1, and PSEN2, and 1.94% had pathogenic variants in MAPT, GRN, or C9orf72. This suggests that modifying factors may exist, either genetic or environmental, that delay the onset of dementia in these families.
Although autopsies were not available for all the individuals with FTD pathogenic variants, the neuropathology in some individuals was compatible with a diagnosis of AD with mixed pathologies. 36 C9orf72 expansion has also been associated with several clinical presentations (FTD and amyotrophic lateral sclerosis). The NIA‐LOAD FBS extends these data to other genes and diseases, supporting the previous finding of pathogenic variants in FTD genes among individuals with AD pathology. 37
The NIA‐LOAD FBS dataset was also instrumental in the identification of novel risk variants, and confirmed the large Alzheimer's Disease Genetic Consortium Case‐Control study and in a study using WGS. 26 , 31 Beecham et al. 38 identified rare, damaging variants segregating with disease in the non‐Hispanic White families using WES data that included NOS1AP, RP11‐433J8, ABCA1, and FISP2. A rare‐variant association study identified two known AD genes—FERMT2 and SLC24A4—and a similar approach was used to identify rare variants in the 67 families of Hispanic ancestry. 35 Lambert et al. 27 conducted a meta‐analysis that identified 11 novel gene regions, most notably HLA‐DRB5, HLA‐DRB1, PTK2B, SORL1, SLC24A4 RIN3, and DSG2. Kunkle et al. 28 identified new risk regions that implicated disorders of amyloid, tau, immunity, and lipid processing. Using a slightly different approach, investigators found genes that are strongly expressed in immune‐related tissues and cell types (e.g., microglia), and gene‐set analyses indicated biological mechanisms involving lipid‐related processes and degradation of amyloid precursor proteins. 29 Using a whole exome meta‐analysis, Bis et al. 39 confirmed associations with common and rare variants in several previously established AD genes: ABCA7, APOE, HLA‐DPA1, MS4A6A, PILRA, SORL1, and TREM2 and nominated variants in two new genes: IGHG3 and ZNF655.
3.7. New methods developed for family‐based analyses
He Z, et al. 40 used the NIA‐LOAD FBS to develop a rare variant extension of the generalized disequilibrium test (GDT) to analyze sequence data obtained from nuclear and extended families. The GDT uses genotype differences of all discordant relative pairs to assess associations within a family and combines the single‐variant GDT statistic over a genomic region of interest. They also developed rare variant, non‐parametric linkage methods to analyze data from WGS studies, 41 and subsequently extended the analytic method to include quantitative non‐parametric methods to evaluate sharing of minor alleles. 42 Fernandez et al. 43 examined the performance of several collapsing, variance‐component, and transmission disequilibrium tests across eight different software packages and 22 models using the NIA‐LOAD FBS cohort and developed a method using gene‐based and variant‐based approaches.
3.8. Neuropathological confirmation of diagnosis
In 492 (82%) participants, neuropathology confirmed the diagnosis of AD. Other diagnoses accounted for the remaining 163 with and without dementia (Table 2). Brain tissue is available by request to NCRAD.
4. DISCUSSION
The success of the NIA‐LOAD FBS can be measured by the broad use of the data and samples (Table 4). More than 67,000 biological samples collected as part of the NIA‐LOAD FBS have been distributed to approved investigators. These samples have been analyzed, and the resulting data briefly described above are available through NIAGADS. In total, more than 830 national and international investigators have requested data from the NIA‐LOAD FBS, including the Alzheimer's Disease Genetics Consortium, the Alzheimer's Disease Sequencing Project, the Consortium for Alzheimer's Disease Research, the NIA‐sponsored Alzheimer's Disease Research Centers, and the National Alzheimer's Coordinating Center. Investigators have generated 123 publications from the data and samples (Supplement 4 in supporting information) that include not only genotyping but methods development and assessments of risk to family members.
TABLE 4.
Summary of biological sample and data requests for NIA‐LOAD family‐based study
| Sample requests | N |
| Number of researchers approved for specimens from NCRAD | 56 |
| Number of DNA samples distributed from NCRAD | 67,626 |
| Inclusion in independent efforts | |
| DNA samples in the ADSP White, non‐Hispanic | 27 families; 142 individuals |
| DNA samples in the ADSP Caribbean Hispanic | 31 families; 186 individuals |
| Independently approved researchers for sequencing (WES or WGS) | 421 families; 1655 individuals |
| Approved data requests | |
| dbGaP‐phs000160.v1.p1 (NIA‐LOAD linkage) | 228 |
| dbGaP‐phs000168.v2.p2 (NIA‐LOAD GWAS) | 321 |
| dbGaP‐phs000372.v1.p1 (ADGC & NIA‐LOAD GWAS) | 101 |
| dbGaP‐phs0001572.v7.p4 (ADSP & NIA‐LOAD WGS/WES) | 166 |
| dbGaP‐phs000126.v1.p1 (controls for PD case‐control) | 268 |
| Special analysis group data requests to the ADGC & NIA‐LOAD | 145 |
| NIA Genetics of Alzheimer's Disease Data Storage Site (NIAGADS) | |
| (NG00032, NG00020, NG00033, NG00039, NG00027, NG00015, NG00081, NG00062, NG00036, NG00041, NG00048, NG00058, NG00065, NG00075, NG00076, NG00078, NG00100, NG00067, NG00091) | 111 |
Abbreviations: ADGC, Alzheimer's Disease Genetics Consortium; ADSP, Alzheimer's Disease Sequencing Project; GWAS, genome wide association study; NCRAD, National Centralized Repository for Alzheimer's Disease and Related Dementias; NIA‐LOAD FBS, National Institute on Aging Late‐Onset Alzheimer's Disease Family Based Study; WES, whole exome sequencing; WGS, whole genome sequencing.
Multiplex families are more difficult to recruit for research studies, sometimes taking several years to identify all the affected relatives in a single family and complete their phenotyping. Large multiplex families are often thought to be too rare to be relevant. However, such families may be much more common than assumed, and there are families in isolated geographical regions where very large sibships occur. 44 , 45 , 46 Families with multiple affected members can more easily be found and ascertained for such studies. With the increased use of virtual visits, the ability to longitudinally follow these families is becoming easier. These large multiplex families may hold the key to survival without dementia despite carrying a disease‐causing variant.
The ultimate goals of genetic studies of complex diseases, such as AD, are to identify causal biological pathways that might provide targets for therapeutic development. However, diseases such as AD are multigenic and it is likely that there are multiple strategies to accomplish this difficult task. A significant proportion of the heritability remains unexplained but may be due to rare, inherited, moderately penetrant variants unique to individual families.
When the NIA‐LOAD FBS began in 2003, a goal was set to collect as many as 1000 families or more including all appropriate members in existing families and complete follow‐up assessments. Blood samples sent to NCRAD for DNA extraction and storage allowed samples to be broadly available to the research community. Initially, clinical data regarding the diagnoses were limited to the criteria used by each Alzheimer's Disease Center, but a more standardized approach was used to collect clinical information in 2006. Clinical data stored centrally at the Columbia University Coordinating Center was immediately shared along with genetic data with the AD research community. Nineteen dataset accession numbers contain genetic data from the NIA‐LOAD FBS (Table 4).
The ADSP Discovery Extension Phase (2015–2018) included WGS on individuals from multiply affected families provided by the NIA‐LOAD FBS. The ADSP Follow‐Up Study (2018–2023) relies heavily on resources from the NIA‐LOAD FBS and depends on continued longitudinal follow‐up of families and the further collection from 500 families from under‐represented populations. Thus, over the past two decades, the NIA‐LOAD FBS has acted as the key source of biological materials, and genetic and longitudinal clinical data on large multiply affected AD families from ethnically diverse populations.
Phenotypic variation among family members with dementia, including differences in the age of onset and the effects of APOE or the presence of sleep behavior disorder, psychosis, and parkinsonism may be due to interactions with other genes or epigenetic effects. Thus, we began brain transcriptomics and DNA methylation may help to clarify the origin of these differences. Moreover, because vascular risk factors clearly contribute to the pathogenesis of AD, 23 the effect of cerebrovascular pathology continues to be investigated.
The NIA‐LOAD FBS remains a critical component of the overall strategy to tackle AD genetics in the United States that includes the Alzheimer's Disease Genetics Consortium, the Alzheimer's Disease Sequencing Project, the NCRAD, and NIAGADS. Together, these groups have created a large collaborative international effort to combine data worldwide in pursuit of uncovering the relevant genetic risk factors for AD with the hope that patterns may emerge that ultimately will uncover biologic pathways underlying the illness.
CONFLICTS OF INTEREST
The following authors have no relevant conflicts of interest to report: Dolly Reyes, Kelley Faber, and Dr. Chao. Dr. Vardarajan has grants from the National Institute on Aging (NIA) of the National Institutes of Health and the Department of Defense and is a consultant to Kodikaz Therapeutics. Dr. Goate has grants from the NIA/NIH and receives money from Athena Diagnostics for licensing of TDP43 mutations, and has consulted for UK Dementia Research Institute, UK VIB, Katholik University, Leuven, Belgium Center for Molecular Neurology, Antwerp, Belgium Queensland Brain Institute, Brisbane, Australia. Dr. Renton has grants from the NIA/NIH, the Alzheimer's Association, and from the JPB Foundation. Dr. Boeve has grants from the NIH, receives royalties as a co‐editor of a textbook on dementia, and is on the Scientific Advisory Board (SAB) for the Tau Consortium. Dr. Rosenberg has grants from the NIA/NIH and The Zale Foundation and receives license/royalty fees from Elsevier Publishing Inc., Springer Publishing Inc.; payments from Elsevier, Springer and Vitruvian, Inc., and The American Academy of Neurology; and he has a 2009 patent on an Amyloid Beta Gene vaccine. Dr. Tsuang has grants from the NIA/NIH and receives consulting fees from Acadia Pharma and is on the SAB for the Lewy Body Association. Dr. Sweet has grants from the NIA/NIH and National Institute of Mental Health of the NIH. Dr. Bennett has grants from the NIA/NIH and the NeuroVision Institution, and consulting relationships with AbbVie Inc., Takeda Pharma, and Origent Data Sciences. Drs. Cruchaga, Pericak‐Vance, Haines, and Wilson have grants from the NIH. Dr. Foroud has grants from the NIA/NIH, The Department of Defense, and the Michael J. Fox Foundation; is on the SAB for several academic institutions; and receives support from Northwestern University for Continuing Medical Education. Dr. Mayeux has grants from the NIA/NIH and is on the SAB for the Rush Alzheimer's Disease Research Center.
Supporting information
Supporting Information
ACKNOWLEDGMENTS
The NIA‐LOAD FBS supported the collection of samples used in this study through National Institute on Aging (NIA) grants U24AG026395, U24AG021886, R01AG041797, and U24AG056270. Additional families were contributed to the NIA‐LOAD FBS through NIH grants: R01AG028786, R01AG027944, RO1AG027944, RF1AG054074, U01AG052410. We thank contributors, including the Alzheimer's Disease Centers who collected samples used in this study, as well as patients and their families, whose help and participation made this work possible. We also thank Cory G. Weinfeld who helped with the analysis of the effects of APOE variability on age at onset of Alzheimer's disease, and created the related figures in the supplemental data.
Reyes‐Dumeyer D, Faber K, Vardarajan B, et al. The National Institute on Aging Late‐Onset Alzheimer's Disease Family Based Study: A resource for genetic discovery. Alzheimer's Dement. 2022;18:1889–1897. 10.1002/alz.12514
REFERENCES
- 1. 2020 Alzheimer's disease facts and figures. Alzheimers Dement. 2020;16:391‐460. [DOI] [PubMed] [Google Scholar]
- 2. Chartier‐Harlin MC, Crawford F, Houlden H, et al. Early‐onset Alzheimer's disease caused by mutations at codon 717 of the beta‐amyloid precursor protein gene. Nature. 1991;353(6347):844‐846. [DOI] [PubMed] [Google Scholar]
- 3. Schellenberg GD, Bird TD, Wijsman EM, et al. Genetic linkage evidence for a familial Alzheimer's disease locus on chromosome 14. Science. 1992;258(5082):668‐671. [DOI] [PubMed] [Google Scholar]
- 4. Sherrington R, Rogaev EI, Liang Y, et al. Cloning of a gene bearing missense mutations in early‐onset familial Alzheimer's disease. Nature. 1995;375(6534):754‐760. [DOI] [PubMed] [Google Scholar]
- 5. Levy‐Lahad E, Wasco W, Poorkaj P, et al. Candidate gene for the chromosome 1 familial Alzheimer's disease locus. Science. 1995;269(5226):973‐977. [DOI] [PubMed] [Google Scholar]
- 6. Levy‐Lahad E, Wijsman EM, Nemens E, et al. A familial Alzheimer's disease locus on chromosome 1. Science. 1995;269(5226):970‐973. [DOI] [PubMed] [Google Scholar]
- 7. Bekris LM, Yu CE, Bird TD, Tsuang DW. Genetics of Alzheimer disease. J Geriatr Psychiatry Neurol. 2010;23(4):213‐227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Campion D, Dumanchin C, Hannequin D, et al. Early‐onset autosomal dominant Alzheimer disease: prevalence, genetic heterogeneity, and mutation spectrum. Am J Hum Genet. 1999;65(3):664‐670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cummings J, Lee G, Ritter A, Sabbagh M, Zhong K. Alzheimer's disease drug development pipeline: 2020. Alzheimers Dement (N Y). 2020;6(1):e12050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pericak‐Vance MA, Bebout JL. Linkage studies in familial Alzheimer disease: evidence for chromosome 19 linkage. Am J Hum Genet. 1991;48(6):1034‐1050. [PMC free article] [PubMed] [Google Scholar]
- 11. Corder EH, Saunders AM, Strittmatter WJ, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. 1993;261(5123):921‐923. [DOI] [PubMed] [Google Scholar]
- 12. Belloy ME, Napolioni V, Greicius MD. A quarter century of APOE and Alzheimer's disease: progress to date and the path forward. Neuron. 2019;101(5):820‐838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Farrer LA, Cupples LA, Haines JL, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta‐analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA. 1997;278(16):1349‐1356. [PubMed] [Google Scholar]
- 14. Slooter AJ, Cruts M, Kalmijn S, et al. Risk estimates of dementia by apolipoprotein E genotypes from a population‐based incidence study: the Rotterdam Study. Arch Neurol. 1998;55(7):964‐968. [DOI] [PubMed] [Google Scholar]
- 15. Lee JH, Kahn A, Cheng R, et al. Disease‐related mutations among Caribbean Hispanics with familial dementia. Mol Genet Genomic Med. 2014;2(5):430‐437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7(3):263‐269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wilson RS, Leurgans SE, Foroud TM, et al. Telephone assessment of cognitive function in the late‐onset Alzheimer's disease family study. Arch Neurol. 2010;67(7):855‐861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wilson RS, Barral S, Lee JH, et al. Heritability of different forms of memory in the Late Onset Alzheimer's Disease Family Study. J Alzheimers Dis. 2011;23(2):249‐255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hollingworth P, Sweet R, Sims R, et al. Genome‐wide association study of Alzheimer's disease with psychotic symptoms. Mol Psychiatry. 2012;17(12):1316‐1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cummings JL. The Neuropsychiatric Inventory: assessing psychopathology in dementia patients. Neurology. 1997;48(5):S10‐16. Suppl 6. [DOI] [PubMed] [Google Scholar]
- 21. Hamilton M. Development of a rating scale for primary depressive illness. Br J Soc Clin Psychol. 1967;6(4):278‐296. [DOI] [PubMed] [Google Scholar]
- 22. Kaufer DI, Cummings JL, Ketchel P, et al. Validation of the NPI‐Q, a brief clinical form of the Neuropsychiatric Inventory. J Neuropsychiatry Clin Neurosci. 2000;12(2):233‐239. [DOI] [PubMed] [Google Scholar]
- 23. Tosto G, Bird TD, Bennett DA, et al. The role of cardiovascular risk factors and stroke in familial Alzheimer disease. JAMA Neurol. 2016;73(10):1231‐1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Montine TJ, Phelps CH, Beach TG, et al. National Institute on Aging‐Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol. 2012;123(1):1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vardarajan BN, Faber KM, Bird TD, et al. Age‐specific incidence rates for dementia and Alzheimer disease in NIA‐LOAD/NCRAD and EFIGA families: national Institute on Aging Genetics Initiative for Late‐Onset Alzheimer Disease/National Cell Repository for Alzheimer Disease (NIA‐LOAD/NCRAD) and Estudio Familiar de Influencia Genetica en Alzheimer (EFIGA). JAMA Neurol. 2014;71(3):315‐323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wijsman EM, Pankratz ND, Choi Y, et al. Genome‐wide association of familial late‐onset Alzheimer's disease replicates BIN1 and CLU and nominates CUGBP2 in interaction with APOE. PLoS Genet. 2011;7(2):e1001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lambert JC, Ibrahim‐Verbaas CA, Harold D, et al. Meta‐analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet. 2013;45(12):1452‐1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kunkle BW, Grenier‐Boley B, Sims R, et al. Genetic meta‐analysis of diagnosed Alzheimer's disease identifies new risk loci and implicates Abeta, tau, immunity and lipid processing. Nat Genet. 2019;51(3):414‐430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jansen IE, Savage JE, Watanabe K, et al. Genome‐wide meta‐analysis identifies new loci and functional pathways influencing Alzheimer's disease risk. Nat Genet. 2019;51(3):404‐413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tosto G, Bird TD, Tsuang D, et al. Polygenic risk scores in familial Alzheimer disease. Neurology. 2017;88(12):1180‐1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Barral S, Cheng R, Reitz C, et al. Linkage analyses in Caribbean Hispanic families identify novel loci associated with familial late‐onset Alzheimer's disease. Alzheimers Dement. 2015;11(12):1397‐1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Blue EE, Bis JC, Dorschner MO, et al. Genetic variation in genes underlying diverse dementias may explain a small proportion of cases in the Alzheimer's Disease Sequencing Project. Dement Geriatr Cogn Disord. 2018;45(1‐2):1‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cruchaga C, Haller G, Chakraverty S, et al. Rare variants in APP, PSEN1 and PSEN2 increase risk for AD in late‐onset Alzheimer's disease families. PLoS One. 2012;7(2):e31039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fernandez MV, Kim JH, Budde JP, et al. Analysis of neurodegenerative Mendelian genes in clinically diagnosed Alzheimer Disease. PLoS Genet. 2017;13(11):e1007045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vardarajan BN, Barral S, Jaworski J, et al. Whole genome sequencing of Caribbean Hispanic families with late‐onset Alzheimer's disease. Ann Clin Transl Neurol. 2018;5(4):406‐417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Harms M, Benitez BA, Cairns N, et al. C9orf72 hexanucleotide repeat expansions in clinical Alzheimer disease. JAMA Neurol. 2013;70(6):736‐741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kohli MA, John‐Williams K, Rajbhandary R, et al. Repeat expansions in the C9ORF72 gene contribute to Alzheimer's disease in Caucasians. Neurobiol Aging. 2013;34(5):1519. e1515‐1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Beecham GW, Vardarajan B, Blue E, et al. Rare genetic variation implicated in non‐Hispanic white families with Alzheimer disease. Neurol Genet. 2018;4(6):e286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bis JC, Jian X, Kunkle BW, et al. Whole exome sequencing study identifies novel rare and common Alzheimer's‐Associated variants involved in immune response and transcriptional regulation. Mol Psychiatry. 2020;25(8):1859‐1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. He Z, Zhang D, Renton AE, et al. The rare‐variant generalized disequilibrium test for association analysis of nuclear and extended pedigrees with application to Alzheimer Disease WGS Data. Am J Hum Genet. 2017;100(2):193‐204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhao L, He Z, Zhang D, et al. A rare variant nonparametric linkage method for nuclear and extended pedigrees with application to late‐onset Alzheimer disease via WGS data. Am J Hum Genet. 2019;105(4):822‐835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhao L, Zhang Z, Rodriguez SMB, et al. A quantitative trait rare variant nonparametric linkage method with application to age‐at‐onset of Alzheimer's disease. Eur J Hum Genet. 2020;28(12):1734‐1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fernandez MV, Budde J, Del‐Aguila JL, et al. Evaluation of gene‐based family‐based methods to detect novel genes associated with familial late onset Alzheimer disease. Front Neurosci. 2018;12:209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Athan ES, Williamson J, Ciappa A, et al. A founder mutation in presenilin 1 causing early‐onset Alzheimer disease in unrelated Caribbean Hispanic families. JAMA. 2001;286(18):2257‐2263. [DOI] [PubMed] [Google Scholar]
- 45. Lalli MA, Cox HC, Arcila ML, et al. Origin of the PSEN1 E280A mutation causing early‐onset Alzheimer's disease. Alzheimers Dement. 2014;10(5):S277‐S283. Suppl. e210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lee JH, Cheng R, Vardarajan B, et al. Genetic modifiers of age at onset in carriers of the G206A mutation in PSEN1 with familial Alzheimer disease among Caribbean Hispanics. JAMA Neurol. 2015;72(9):1043‐1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information