

Abstract
We previously demonstrated that in Alzheimer’s disease (AD) patients, European apolipoprotein E (APOE) ε4 carriers express significantly more APOEε4 in their brains than African AD carriers. We examined single nucleotide polymorphisms near APOE with significant frequency differences between African and European/Japanese APOE ε4 haplotypes that could contribute to this difference in expression through regulation. Two enhancer massively parallel reporter assay (MPRA) approaches were performed, supplemented with single fragment reporter assays. We used Capture C analyses to support interactions with the APOE promoter. Introns within TOMM40 showed increased enhancer activity in the European/Japanese versus African haplotypes in astrocytes and microglia. This region overlaps with APOE promoter interactions as assessed by Capture C analysis. Single variant analyses pinpoints rs2075650/rs157581, and rs59007384 as functionally different on these haplotypes. Identification of the mechanisms for differential regulatory function for APOE expression between African and European/Japanese haplotypes could lead to therapeutic targets for APOE ε4 carriers.
Keywords: ancestry, apolipoprotein E, massively parallel reporter assays, promoter capture protective variant, regulatory elements
1 |. NARRATIVE
1.1 |. Contextual background
The most significant genetic risk factor for Alzheimer’s disease (AD) is apolipoprotein E (APOE), with the APOE ε4 allele inferring both an increased risk for disease and a decrease in age at onset.1,2 APOE ε4 is thought to contribute almost 40% of the genetic risk for AD in European populations. Yet, there is no therapeutic intervention to reduce this risk for APOE ε4 carriers, nor is there full understanding of how APOE ε4 leads to AD.
However, the risk of developing AD from the APOE ε4 allele varies greatly among populations3 (Figure 1). Despite having the same APOE ε4 gene, nature has found a way to reduce the risk for APOE ε4 carriers in those individuals with African ancestry. This finding suggests that if we could identify the mechanism that protects (or reduces risk in) APOE ε4 carriers of African ancestry from developing AD from APOE ε4, we could use this information to develop therapeutic treatments to slow or prevent AD in APOE ε4 carriers of all ancestries. In this paper, we identify potential areas of the genome that may begin to fulfill this goal.
FIGURE 1.
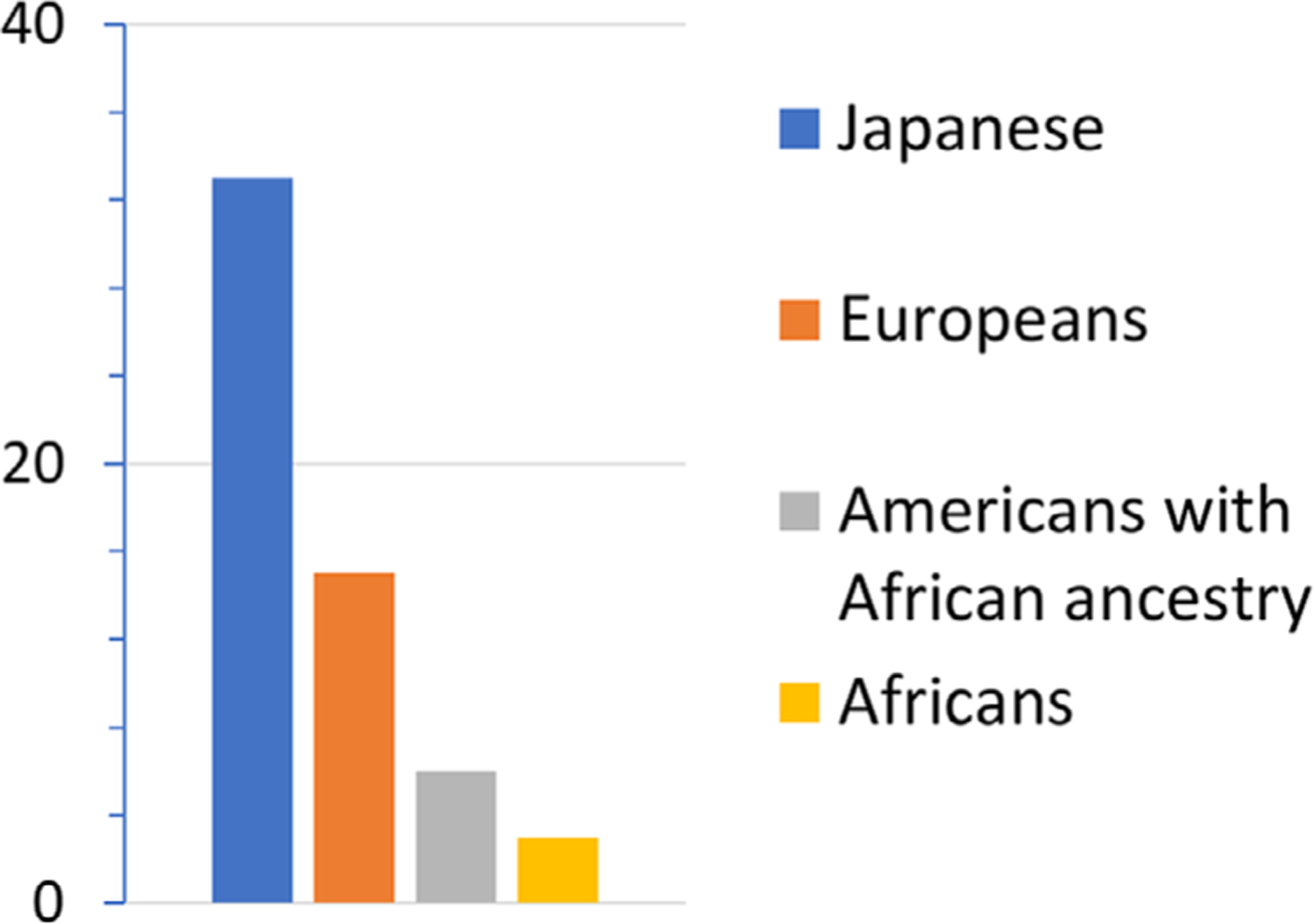
Apolipoprotein E (APOE) ɛ4 risk effects in different populations. Odds ratios for the risk of developing Alzheimer’s disease from APOE ɛ4 compared to APOE ɛ3 for homozygous carriers
Using populations with recent ancestral admixture (African Americans, e.g., African and European ancestries, and Latino/Hispanic populations, e.g., African, European, and Amerindian ancestries), Rajabli et al.4 and later Blue et al.5 demonstrated that this protective mechanism for AD in African APOE ε4 carriers was associated with the ancestral origin of the DNA surrounding APOE ε4, and not elsewhere on the chromosomes. That is, if you inherited the area surrounding the APOE ε4 gene (called the “local ancestry,” or LA) from African ancestors, you have the African risk for AD from APOE ε4, and if you inherited APOE ε4 and its surrounding genetic region from European ancestors, you have the European risk for AD from APOE ε4. However, what the genetic changes are that cause this risk difference for local APOE ε4 ancestry is not known. One major problem is that most genomic studies have been done only in European genomes, thus there is a real gap in knowledge about how differences in genetic sequence among ancestries can affect genomic function and risk.
As the apoE4 protein is similar, if not identical among these populations, other genetic factors in the region other than those producing protein must be driving this protection. This led us to explore and recently publish that the amounts of APOE ε4 expressed in AD brains with European LA are significantly higher (P < 10–318) than those in which the APOE ε4 gene was surrounded by African ancestry. In this study, we sought to identify what genetic changes in regulation could lead to this difference in APOE ε4 expression among local ancestries.
1.2 |. Study approach and findings
To determine those base pairs in the LA region that are significantly different in allele frequency between African and European APOE ε4 genomes, we used sequence data from the 1000 Genomes Project database4 (Figure 2). Next, we focused on the region where these significant variants seem to accumulate, which was located within a predicted regulatory region called a topologically associated domain (TAD) surrounding APOE. TADs are genomic regions with increased cross regulation within its boundaries (for more information on TADs, see Pombo and Dillon6). To determine whether these sequence differences led to differences in regulatory function, we used three different methods of “reporter assays.” In these assays, pieces of DNA, each representing one of the identified sequence differences in regions with potential enhancer activity, are placed in a vector with a “reporter” gene, encompassing a barcode or coding for a fluorescence-producing protein. These vectors are then inserted into a living cell and use the cell’s inherent regulatory transcription components to activate expression of the reporter. How the sequence differences enhance the amount of barcode expressed from the vector or fluorescence emitted by the coded protein can be measured. All fragment assays were tested in three different cell types, Neuron (SH-SY5Y), Microglia (HMC3), and Astrocyte (U-118) lines. The first two reporter assays are based on the technique of massively parallel reporter assay (MPRA, described in more detail below), which allows many reporter assays to be done at once through measurement of the barcodes. Within the MPRA approach, we used two different forms of DNA fragments. The haplotype or polymerase chain reaction (PCR)-based assay used larger DNA fragments (≈850 bp) surrounding the variants we were testing. This is important as often not just one variant, but a cluster of variants can affect regulatory function. We also used MPRA with synthetically made DNA fragments (probe-based), which were much smaller and focused on only a small region surrounding a single variant. This allowed us to determine, in those regions where multiple sequence differences between the ancestries were close together, which variant of the clustered variants were likely driving the functional difference. For the variants that had differential function between the ancestries or failed in the MPRA test, we completed a third approach with single reporter assays as well.
FIGURE 2.

Overview of experimental set-up. A, All variants identified to have differential frequency between European (1000 Genomes CEU) versus African (1000 Genomes YRI) apolipoprotein E (APOE) ɛ4 haplotypes; color-coded for significance; red; P < 1*10–5, orange P < 1*10–4 (corresponding to the top 25% results), rest P < .05. B, All variants identified to have differential frequency between Japanese (1000 Genomes JPT) versus African (1000 Genomes YRI) APOE ɛ4 haplotypes; color-coded for significance; red; P < 1*10–5, orange P < 1*10–4, rest P < .05. C; All 39 designed amplicons for inclusion in the polymerase chain reaction (PCR)-based massively parallel reporter assay (MPRA)
Finally, the question remains if the identified functional areas affect the expression of the APOE gene or some other locus? To determine this, we used a three-dimensional interaction method called “promoter Capture C” (for chromatin conformation capture techniques, see Davies et al.7). In this chromatin conformation capture technique, physical connections between genomic locations and gene promoters are detected, allowing for identification of enhancer-to-promoter interactions. Here we are specifically looking to see if our identified functional regions are physically interacting with the APOE promoter in the cells.
Through these analyses, we have identified regions in the introns 2–3 of TOMM40 (fragments 10 and 13; Figure 3A–C) that are strong candidates to contribute to the difference in APOE ε4 expression between ancestries. The single-variant assays pinpointed the difference in activity in those fragments to be driven by variants rs2075650/rs157581 (European/Japanese allele G-C; fragment 10) and rs59007384 (European allele T; fragment 13; Figure 3D). Rs2075650 and rs59007384 are predicted to significantly affect binding sites for transcription factors SP1 and PLAG, respectively, based on in silico MotifBreakR analysis. The promoter Capture C analyses showed that these functional regions had strong chromatin interaction with the APOE promoter in both glial cell types, but not in neurons (Figure 3E).
FIGURE 3.

Overview of results and regulatory evidence in the region of interest. A, All 39 designed amplicons for inclusion in the polymerase chain reaction (PCR)-based massively parallel reporter assay (MPRA). B, Representation of results of successful fragments in the PCR-based MPRA. C, Representation of results of successful fragments in the luciferase experiments. D, Representation of results of successful variant analyses in the probe-based MPRA. Blue; astrocytes. Green; microglia. Pink; neurons. E, Significant interactions of apolipoprotein E (APOE) promoter within the region of interest based on the promoter Capture C data at 4-DpnII fragment resolution; interactions between promoter and candidate regulatory element (green), interaction between promoters (red). F, Available in silico annotation data; including previously reported enhancers, enhancers and H3K27ac marks identified in prefrontal cortex from the PsychENCODE project, chromHMM tracks from the RoadMap Epigenome Project (primary HMM) and Transcription factor ChIP-Seq cluster data from ENCODE freeze 3; using data from 338 factors in 130 cell types
Therefore, several findings support these regions being important for APOE regulation: (1) They demonstrated functional differences in expression using microglia and astrocytes, but not neurons. This fits any expected regulatory region of APOE, as brain APOE is primarily expressed in astrocytes and microglia, but minimally in neurons. (2) Higher enhancer activity (corresponding to higher expression) was found for the European alleles compared to the African alleles, supporting our previous observations in AD brains between African LA and European LA APOE ε4.8 (3) The promoter Capture C analyses evaluating chromatin interaction within the region of interest showed that these functional regions had strong chromatin interaction with the APOE promoter in both glial cell types, but not in neurons. (4) These variants/regions have been described to be involved in AD before,9–12 but never in context with APOE ε4 or ancestral background.
1.3 |. Limitations and disease implications
The expression of APOE is thought to be complex,13–23 and thus, it is highly likely that there are other mechanisms that control APOE ε4 expression, both at genomic, RNA, and protein levels. We have focused on the sequence differences in the genomic regions closest to APOE, but the ancestral regions found in admixed populations around APOE can be larger in size and may include other genomic regions in its regulation. Alternatively, sequence differences between the two APOE ε4 backgrounds could also influence methylation activity in this region. Although differences in methylation between APOE ε4 and the other APOE statuses have been studied,21–24 the effect of different ancestral backgrounds has not been fully assessed.25 Preliminary data from our studies did not find significant methylation differences involving variants with differential allele frequencies between ancestries.
While we have used multiple approaches to measure regulatory function, these approaches are still artificial, and thus, the next steps will be to test the effect of changing the sequence in these potential regions by using CRISPR techniques in astrocytes, microglia, and neurons derived from inducible pluripotent stem cells from AD patients. For example, we could assess whether repression of APOE expression through CRISPR interference assays in cell lines derived from European APOE ε4 carriers mimic AD-relevant phenotypes of African APOE ε4 carriers. If validated, these cell lines could then serve as models to test molecules as potential therapeutic interventions for treating APOE ε4 carriers by manipulating the gene expression of APOE ε4 in a similar fashion as has occurred in the African carriers of the risk allele. This work also highlights the importance of including all ancestries in research in AD, as differences in risk can provide windows of opportunity to understand the mechanisms of disease.
It has long been a matter of debate whether increasing or decreasing APOE ε4 would have therapeutic effects26,27 citing both gain of function and loss of protective function as possible mechanisms for APOE ε4 effects. Interestingly though, anti-apoE4 immunotherapy in mice has consistently showed improvement of AD phenotypes (i.e., cognition, amyloid beta plaque burden).28–30 More recent functional studies in mice have shown that removal of APOE ε4 in astrocytes is protective against tau-mediated neurodegeneration31 and that the positive effect of repetitive transcranial magnetic stimulation on cognition in milder AD32 is mitigated through reduction of apoE.33 Taken together with the data presented here, these reports support that the increased risk for AD in APOE ε4 on European LA can be ascribed to increased, pathogenic levels of apoE4. Therefore, this gain of toxicity mechanism of APOE ε4 (on European LA specifically but also overall in all carriers) supports the importance of identifying the controlling regions that contribute to the higher expression of APOE ε4 in European ancestry8 and development of therapies targeting the overexpression of APOE ε4.
2 |. CONSOLIDATED RESULTS AND STUDY DESIGN
The LA variants identified having significant differential allele frequencies on the different risk APOE ε4 haplotypes peaked in a small topologically associated domain surrounding APOE (chr19:45375k-45440k)4. We selected all variants that met Bonferroni significance (adjusted P < .05, N = 56) and had a greater than 0.01 allele frequency (Table 1). For the MPRA assay using large PCR-based genome fragments we amplified 39 larger fragments (≈850 bp) encompassing the significant LA variant haplotypes using PCR (Figure 2). The protocol for library creation in an enhancer reporter plasmid backbone was adjusted from Trizzino et al.39 In short, candidate enhancer fragments and barcodes are subsequently cloned into the vector in front and in the 3′UTR of the reporter gene, respectively, to create unique haplotype-to-barcode links within the same plasmid. The PCR-based MPRA was supplemented with single luciferase reporter assays using the same amplicon primers as for the MPRA. In the second MPRA assay, we used shorter, synthetically made probes differing just in alleles for one variant at a time. Combined variant allele and barcode fragments are cloned into the pGL4 plasmid backbone, with subsequent insertion of the reporter gene between them, as previously described by Tewhey et al.40 Differences in proportion of the two haplotypes/alleles in the expressed material (RNA) versus the expected ratio (plasmid DNA library) was assessed by exact binomial test.34
TABLE 1.
Overview included variants in analyses and summarized results
| Fragment | Centered LA SNPs | Location | Allele most common in
ɛ4 |
Enhancer activity (highest
activity, cell type) |
GTEx |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AFR | EUR/JPT | PCR-based MPRA | Luciferase | Probe-based MPRA | Capture C APOE promoter interaction | RoadMap Epigenome | brain | non-brain | MotifbreakR | |||
| 1 | rs34278513 | intron PVRL2 | C | T | Negative | NA | JPT astrocytes | BCAM, PVRL2 | ||||
| 2 | rs3865427 | intron PVRL2 | C | A | NA | JPT neurons | JPT astrocytes | BCAM, PVRL2 | ||||
| 3 | rs71352237* | intron PVRL2 | T | C | NA | JPT neurons | EUR astrocytes | Enhancer hepatocytes, adrenal gland | BCAM, PVRL2 | RARB & RARG | ||
| rs71171301* | intron PVRL2 | – | C | NA | BCAM, PVRL2 | NA | ||||||
| rs34224078* | intron PVRL2 | A | G | Negative | BCAM, PVRL2 | |||||||
| rs35879138* | intron PVRL2 | T | A | Negative | BCAM, PVRL2 | |||||||
| 4 | rs142042446 | intron PVRL2 | – | TAA | Negative | NA | NA | BCAM | ||||
| 5 | rs283808 | intron PVRL2 | C | A | NA | negative | AFR astrocytes | PVRL2 | TOMM40 | |||
| rs283809 | intron PVRL2 | G | A | Negative | PVRL2 | TOMM40 | ||||||
| 6 | rs12972156 | intron PVRL2 | C | G | Negative | NA | Negative | Enhancer hepatocytes | BCAM | TFAIP2A | ||
| rs12972970 | intron PVRL2 | G | A | AFR astrocytes | BCAM | FOXD1 | ||||||
| 7 | rs34342646 | intron PVRL2 | G | A | NA | EUR astrocytes | AFR microglia, astrocytes | BCAM, TOMM40 | ||||
| 8 | rs6857 | 3′UTR PVRL2 | C | T | NA | AFR microglia | – | BCAM, APOC1 | ||||
| 9 | rs71352238 | Up TOMM40 | T | C | NA | NA | AFR astrocytes | Promoter TOMM40 | BCAM, TOMM40 | |||
| 10 | rs2075650* | intron TOMM40 | A | G | EUR microglia | NA | Negative | NPC, astrocytes, microglia | Enhancer brain | BCAM | SP1 | |
| 11 | rs34095326 | intron TOMM40 | G | A | NA | AFR neurons | Negative | NPC, astrocytes, microglia | BCAM | HMGA2 & EHF | ||
| rs34404554 | intron TOMM40 | C | G | AFR astrocytes | Enhancer brain | BCAM | ||||||
| 12 | rs11556505 | exonic TOMM40 | C | T | NA | Negative | Negative | NPC, astrocytes, microglia | Enhancer brain, hepatocytes, adrenal gland | |||
| 13 | rs59007384* | intron TOMM40 | G | T | EUR microglia, astrocytes | EUR microglia | EURmicroglia | NPC, astrocytes, microglia | Enhancer brain, hepatocytes, adrenal gland | APOC1 | PLAG | |
| rs157583 | intron TOMM40 | T | G | Negative | ||||||||
| 14 | rs157584 | intron TOMM40 | C | T | Negative | NA | Negative | NPC, astrocytes, microglia | Enhancer adrenal gland | APOC1P1, APOE, APOC1 | ||
| 15 | rs157585 | intron TOMM40 | C | A | NA | Negative | Negative | NPC, astrocytes, microglia | Enhancer adrenal gland | MAFK | ||
| 16 | rs157588 | intron TOMM40 | T | C | NA | AFR astrocytes | Negative | Enhancer adrenal gland | APOC1P1, APOE | |||
| 17 | rs157590 | intron TOMM40 | C | A | AFR astrocytes | Negative | Negative | Enhancer adrenal gland | APOC1P1, APOE | FEV & EOMES | ||
| 18 | rs205909 | intron TOMM40 | G | T | Negative | NA | Negative | NPC | APOE | CBLC | ||
| 19 | rs542555887 | intron TOMM40 | T | – | NA | NA | NA | APOE | CBLC | |||
| rs491153 | intron TOMM40 | T | C | Negative | CBLC | |||||||
| rs490243 | intron TOMM40 | T | C | Negative | APOE | CBLC | ||||||
| 20 | rs417357 | intron TOMM40 | T | C | Negative | NA | NA | Astrocytes, microglia | APOE | CBLC | ||
| 21 | rs394819 | intron TOMM40 | T | G | Negative | NA | Negative | Astrocytes, microglia | APOE | CBLC | ||
| 22 | rs435380 | up APOE | A | G | Negative | NA | Negative | NPC, astrocytes, neurons | Enhancer brain, adrenal gland; promoter APOE hepatocytes | APOE | CBLC | ZFX |
| rs446037 | up APOE | T | G | Negative | APOE | CBLC | ||||||
| rs434132 | up APOE | G | C | Negative | APOE | CBLC | ||||||
| 23 | rs449647 | up APOE | T | A | NA | Negative | AFR astrocytes | NPC, astrocytes, neurons | Promoter APOE brain, hepatocytes | PVRL2 | ||
| 24 | rs769449 | intron APOE | G | A | Negative | NA | Negative | Promoter APOE brain, hepatocytes | APOC1 | |||
| 25 | rs75627662 | intergenic | C | T | NA | Negative | EUR astrocytes | Enhancer hepatocytes, adrenal gland | APOC1, APOE | many | ||
| rs148353395* | – | del | NA | APOE, CLASRP | CBLC | |||||||
| 26 | rs445925 | up APOC1 | A | G | NA | EUR microglia | Negative | Enhancer all | APOE | PPARG | ||
| rs10414043* | up APOC1 | G | A | Negative | APOC1 | |||||||
| rs7256200* | up APOC1 | G | T | Negative | APOC1 | |||||||
| 27 | rs390082 | up APOC1 | G | T | Negative | NA | Negative | Enhancer all | APOE | |||
| 28 | rs34954997 = rs11568822* | 5′ UTR APOC1 | – | CGTT | AFR microglia | Negative | NA | Promoter APOC1 all | APOC1, APOE | |||
| 29 | rs5117* | intron APOC1 | T | C | NA | NA | Negative | APOC1, APOE | ||||
| 30 | rs389261 | intron APOC1 | A | G | NA | NA | Negative | |||||
| 31 | rs12721046 | intron APOC1 | G | A | NA | NA | EUR astrocytes | |||||
| 32 | rs12721051 | intron APOC1 | C | G | NA | NA | Negative | APOE | ||||
| 33 | rs56131196* | down APOC1 | G | A | Negative | Negative | Negative | APOC1, APOE | ||||
| rs4420638* | down APOC1 | A | G | Negative | APOC1, APOE | |||||||
| 34 | rs157591 | intergenic | A | G | NA | Negative | Negative | Enhancer hepatocytes, adrenal gland | CLASRP | CBLC | ||
| 35 | rs157596 | intergenic | T | C | Negative | NA | Negative | CBLC | ||||
| 36 | rs157597 | intergenic | C | A | NA | negative | NA | Enhancer hepatocytes, adrenal gland | CBLC | |||
| 37 | rs111789331 | intergenic | T | A | NA | AFR neurons | Negative | Enhancer hepatocytes, adrenal gland | ||||
| 38 | rs157599 | intergenic | G | A | NA | Negative | Negative | Enhancer all | ||||
| rs149345 | up APOC1P1 | G | T | Negative | ||||||||
| rs66626994 | up APOC1P1 | G | A | Negative | ||||||||
| 39 | rs10424663 | up APOC1P1 | A | G | Negative | NA | Negative | APOE, CLASRP | CBLC | |||
Abbreviations: APOE, apolipoprotein E; Down, downstream; EUR: European, AFR: African, JPT: Japanese; LA, local ancestry; MPRA, massively parallel reporter assay; NA, not assessed/low coverage/failed; NPC, neuroprogenitor cells; SNP, single nucleotide polymorphism; Up, upstream.
= frequency different general population versus APOE ɛ4 haplotypes.
We performed high resolution (i.e., using the 4 bp-cutter DpnII, as opposed to the more commonly used 6 bp-cutter HindIII) genome-wide promoter-focused Capture C analyses on neuroprogenitor cells (NPCs), astrocyte, microglia, and neuronal cell lines, following the previously described protocol.35 Significant interactions at 4-DpnII fragment resolution were called using CHiCAGO.36
Overall results on enhancer activity—assayed in representatives of microglia, astrocytes, and neurons—across all methods can be found in the overview of Table 1, and are visualized in Figure 3.
Specifically, we identified a significant regulatory region in microglia and astrocytes supported by several levels of evidence. We identified differential activity in microglia and astrocytes between significant LA variant haplotypes for fragments 10 and 13, both located in the first introns of TOMM40 (Table 2). Higher enhancer activity is observed for the European/Japanese APOE ε4 haplotype in fragment 10 (containing two significant LA variant alleles: rs2075650-G and rs34404554G) and the European APOE ε4 haplotype in fragment 13 (containing two significant LA variant alleles: rs59007384-T and rs157583-G; Table 2). Haplotype fragment 13 showed consistently higher activity driven by the significant LA variant rs59007384 European allele T in the PCR-based MPRA and luciferase assays (both glia cell lines), and the probe-based analyses (in microglia), supporting the T allele variant as the driving signal in fragment 13. Besides the two significant LA variants (rs2075650 and rs34404554), European/Japanese and African haplotypes in the PCR-based fragment 10 also contained two additional variants that did not meet Bonferroni significance in the local ancestry comparison (European/Japanese haplotype; rs157580-G and rs157581-T; Table 2). Luciferase analyses of overlapping fragment 11, centering on significant LA variants rs34095326 and rs34404554, did not support differential activity driven by these variants in microglia and astrocytes, suggesting that rs2075650 is the driving factor for the regulatory activity seen with fragment 10. Although two out of three replicates showed increased enhancer activity for the European/Japanese significant LA variant rs2075650-G allele, this effect could not be replicated in the probe-based analyses (Table S1 in supporting information). Rs2075650 and rs157581 are both predicted by the JASPAR transcription factor binding database to be located in transcription factor binding sites and are located within 100 bp of each other, potentially indicating a synergistic effect of these two TAD variants that was missed in the probe-based single variant MPRA. Additionally, according to MotifbreakR, both rs2075650 and rs59007384 are predicted to significantly affect binding sites for SP1 and PLAG, respectively.
TABLE 2.
Results TOMM40 intronic potential enhancer region
| SNPs | PCR-based MPRA (AFR - EUR/JPT
haplotype) |
PCR-based results | Luciferase (AFR EUR/JPT
haplotype) |
Luciferase results | Probe-based results | ChIPs-Equation (Haploreg) | RegulomDB |
MotifBreakR | GTEx | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 10 | 11 | 12 | 11 | 12 | Score | JASPAR | |||||||
| rs157580 | A-G | – | – | Fragment 10; EURmicroglia | – | – | Fragment 10; NA | NA | – | 1f | – | – | APOE skin, APOC1P1 cultured fibroblasts and skin, APOC1 skin |
| rs2075650 | A-G | A-G | – | A-G | – | Negative | – | 1b | RREB1 | SP1 | BCAM tibial nerve | ||
| rs157581 | T-C | C-C | – | C-C | – | NA | – | 2b | ZEB1 | – | APOC1mucosa esophagus | ||
| rs34095326 | G-G | G-A | G-A | Fragment 11; NA | G-A | A-A | Fragment 11; AFR neurons | Negative | POL2 (K562) | 4 | – | HMGA2 & EHF | BCAM tibial nerve |
| rs34404554 | C-G | C-G | C-G | C-G | G-G | AFR astrocytes | POL2 (K562) | 4 | – | – | BCAM tibial nerve | ||
| rs11556505 | – | C-T | C-T | Fragment 12; NA | C-T | C-T | Fragment 12; negative | Negative | POL2 (K562) | 4 | – | – | – |
| 13 | 14 | 13 | |||||||||||
| rs59007384 | G-T | G-T | – | Fragment 13; EUR astrocytes, microglia | G-T | – | Fragment 13; EUR microglia | EUR microglia | GR (A549) | 4 | – | PLAG | APOC1mucosa esophagus and adrenal gland |
| rs157583 | T-G | T-G | – | G-G | – | Negative | GR (A549) | 4 | – | – | – | ||
| rs157584 | C-T | C-T | – | Fragment 14; negative | C-C | – | Fragment 14; NA | Negative | GR (A549) | 4 | – | – | APOE skin and atrial appendage heart, APOC1 atrial appendage heart |
Bold SNPs = significant LA variants, Italic/underline; variants in center of fragment.
Abbreviations: APOE, apolipoprotein E;MPRA, massively parallel reporter assay; PCR, polymerase chain reaction.
Promoter Capture C data in microglia, astrocytes, and NPCs consistently show physical interaction of the region encompassing fragments 10–14 with the APOE promoter. All interactions within the small TAD are shown in Figure 3. Additional available in silico annotation data (Figure 3F) support a regulatory function of this region, with RoadMap Epigenome chromatin mark analyses indicating enhancer activity in the region overlapping with fragments 10–12 (TOMM40 introns 2–3) in AD-relevant brain regions (hippocampus, frontal cortex), and fragment 13 (TOMM40 intron 4) in other brain regions (e.g., cingulate gyrus, substantia nigra). Additionally, H3K27ac peaks from prefrontal cortex tissue in PsychENCODE support active regulatory activity in the TOMM40 introns 1–2 region (overlapping fragments 10–11). No evidence for effect on expression of APOE (or any other gene) was observed in the Genotype-Tissue Expression (GTEx) project for these variants in the brain.
3 |. DETAILED METHODS AND RESULTS
3.1 |. Methods
See supporting information for full details on the library creation, transfection, and RNA extraction experiments.
3.1.1 |. Reporter assays design
APOE lies within a larger well-defined TAD, based on the 3D genome browser data from hippocampus37 and Hi-C data from the Jin lab38 (Figure S1 in supporting information). The significant LA variants peaked in a small region (≈subTAD) surrounding APOE (chr19:45375k-45440k);4 which we selected for further analyses. We selected all variants identified through the 1000 Genomes Project in this subTAD region directly surrounding APOE that met Bonferroni significance in differential frequency on the African versus European or African versus Japanese APOE ε4 haplotype (Bonferroni-adjusted P < .05) and had a greater than 0.01 allele frequency (Table S2 in supporting information). To assess the functional potential of these variants, we designed two different MPRA assays, one using large PCR-based genome fragments and the other using a probe-based design assessing single variants.
In the first assay, we amplified larger fragments (≈850 bp) encompassing the significant LA haplotypes using PCR (PCR-based MPRA; Figures 2, 3 and Table S2). These larger amplicons allow for full inclusion of larger regulatory elements in the fragment, as well as inclusion of multiple variants closely located to each other on the same haplotype permitting inclusion of synergistic signals. This allows the fragments to contain additional LA located on African or European haplotypes that could participate in regulation but did not survive the significance threshold determined by Rajabli et al.4 Fragments 26–27 and 39 overlap with reported enhancer regions Multi-enhancer E1 and 2 (ME1–218,19); B control region (BCR)20 is not included, as it was located outside the region with significant LA variant convergence. Variant or variant haplotype fragments were amplified in African American heterozygous individuals carrying the variants of interest located in the middle of the fragments (primer sequences in Table S3 in supporting information) and cloned into the enhancer reporter vector pGL4.24 (E8421, Promega). Subsequently, a library of 20 bp barcodes was cloned into the 3′UTR of the vector library. This final library was used for the transfection experiments. The variant (haplotype) was linked to a unique barcode using the methodology published in Trizzino et al.39
The PCR-based MPRA results were supplemented by single variant/haplotype reporter assays using the Dual-Glo Luciferase System (Promega Corporation) for confirmation or when MPRA assays failed to generate sufficient data for analyses. For each region, fragments of both haplotypes were amplified using the same primers as the PCR-based MPRA design (Tables S2 and S3) and cloned separately into the pGL4.24 plasmid to test for enhancer activity.
In the second assay, we used shorter, synthetically made probes (probe-based MPRA). This allows for high-throughput inclusion of variants and single variant assessment but is more limited in the inclusion of adjacent variants because the probe size is restricted to ≈200 bp. This approach was used to identify regulatory variants as part of a larger design, including several AD loci, totaling ≈24,000 variants. For each significant LA variant, 180 bp of reference sequence (90 bp on either side of variant) was extracted for the “ref” allele fragment. An alternate (“alt”) allele fragment was created by replacing just the position of interest with the alternate allele. The same variants as used for the PCR-based MPRA design were included in this approach, except for the five indel variants, which were excluded for technical reasons in the synthesis. Using these sequences, DNA probes were synthesized (Twist Biosciences) after which 20 bp barcodes were attached to these fragments by PCR using a barcoded primer library. Combined fragments were cloned into the pGL4 plasmid backbone and sequenced for variant-to-barcode link determination, as previously described by Tewhey et al.40 Once validated, the reporter gene with a minimal promoter was cloned in between the fragments and barcodes.
We performed these reporter assays in immortalized AD-relevant cell lines; neuronal (SH-SY5Y), microglia (HMC3), and astrocyte (U-118) cell lines. All were obtained from ATCC (CRL-2266, CRL-3304, HTB-15) and cultured according to manufacturer’s instructions.
3.1.2 |. MPRA sequencing protocols
For the PCR-based MPRA, we performed 2 × 100 bp paired-end sequencing on the Illumina HiSeq3000- for the determination of barcode to fragment link in the tagmented library. Sequencing was performed at the Center for Genome Technology (CGT) at the John P. Hussman Institute for Human Genomics (HIHG). Using the subassembly method described in Trizzino et al.,39 paired reads were grouped per barcode in read 1 after which sequences of read 2 were used to reassemble the larger fragment and determine variant allele presence. We generated >143 million paired-end reads for both the HindIII or BglII induced libraries of fragment-to-barcode ligated vectors, which totaled >147,000 unique barcodes in either sample. Only barcodes unique across both HindIII and BglII samples and unique for European or African haplotype were used in the further analyses; these amounted to ≈51,000 unique barcodes. We generated on average 19 million, 56 million, and 28 million single-end reads for both replicates of RNA expressed barcodes in microglia, astrocytes, and neurons, respectively.
For the probe-based MPRA, we performed 2 × 150 bp sequencing on the Illumina NovaSeq at the CGT allowing for complete readthrough of the total assembled amplicon harboring the fragment with the variant allele of interest and barcode. We generated close to 6 million paired-end reads of the library without reporter gene, in which 3.5 million unique barcodes were detected across ≈24,000 total included variants, including the 51 non-indel APOE subTAD variants. We generated on average 118 million, 74 million, 94 million, and 164 million single-end reads for all three replicates of DNA and RNA expressed barcodes in neurons, microglia, and astrocytes, respectively. Barcodes matching multiple fragments or opposite alleles in the same fragment within either set-up were excluded from further analyses.
Practically, the one-step incorporation of fragment and barcode to the vector in the probe-based MPRA proved to be significantly more efficient (percentage uniquely linked barcodes versus total barcodes) at identifying unique barcodes than the two-step incorporation in the PCR-based MPRA.
3.1.3 |. Reporter assay statistical analyses
For the MPRA analyses, we performed the exact binomial test to find whether the proportion of the two alleles in the expressed material (RNA-Alt, RNA-Ref) is significantly different from the expected ratio (observed in MPRA library; DNA-Alt, DNA-Ref).34 We implemented analysis using in-house R scripts. Raw data can be found in Table S1. For the PCR-based MPRA, we required at least one out of two replicates to survive multiple testing, with the other meeting P < .05 for confirmation. For the probe-based MPRA, we required at least one out of three replicates to survive multiple testing, with a second replicate also surviving multiple testing, or both replicates meeting P < .05, for confirmation. For the single luciferase assays, results from three independent experiments were evaluated by t test. A P-value of < .05 was considered significant in these analyses.
3.1.4 |. Capture C data
We performed high resolution (i.e., using the 4 bp-cutter DpnII, as opposed to the more commonly used 6 bp-cutter HindIII) genome-wide promoter-focused Capture C analyses on HMC3 cells, primary human astrocytes (NHA, Lonza cat#CC-2565), and healthy control induced pluripotent stem cell (iPSC)-derived NPCs and neuronal cells, following the previously described protocol.35 NPC and neurons were derived from two iPSC lines from control individuals (CHOPWT10 and CHOPWT14) as described in Su et al.41 Libraries were generated in triplicate for each cell type and donor and paired-end sequenced on the Illumina Novaseq 6000 platform (51 bp read length) at the Center of Spatial and Functional Genomics at Children’s Hospital of Philadelphia (CHOP). Reads were preprocessed using the HiCUP pipeline (v0.5.942), with the bowtie aligner and hg19 as the reference genome. Significant interactions at 4-DpnII fragment resolution were called using CHiCAGO.36
The chromatin interactions observed in the promoter Capture C experiments represent a summary of interaction across a pool of cells at any given time point, allowing for short-term differences within a cell population to still be picked up, though larger temporal effects in cell development may be missed.
In silico annotation
For each variant included in the probe-based MPRA design, we evaluated their presence in ChIP-seq transcription factor binding sites (TFBS) from ENCODE,43 in putative enhancer or promoter regions (based on RoadMap Epigenome data44) and in predicted TFBS (using the RegulomeDB JASPAR query45 and MotifbreakR46). Although ENCODE has ChIP-seq data for many transcription factors in many cell types, data on transcription factors in brain cell types is very limited; neural cell (SMC3, CTCF, EP300, MXI1, RAD21), astrocytes (CTCF), SH-SY5Y (GATA2, GATA3), and NPC (CTCF, EZH2). PrimaryHMM results from RoadMap Epigenome project of brain regions (hippocampus, dorsolateral prefrontal cortex, singular and angulate gyrus, substantia nigra, and anterior caudate), as well as adrenal gland and liver cells were evaluated. We used the MotifbreakR HOmo sapiens COmprehensive MOdel COllection (HOCOMOCO47) for transcription factor binding site identification with a P-value significance threshold of P < 5 × 10–5. Further, we evaluated expressive quantitative trait locus status of the variants in brain or non-brain tissues using GTEx. Last, PsychENCODE H3K27ac peak analysis was used to identify putative enhancer regions based on prefrontal cortex tissue.48
3.2 |. Results
Detailed results can be found in Table S1 (MPRA) and Figure S2 (promoter Capture C).
3.2.1 |. Outcomes PCR-based MPRA
We identified 56 LA variants with significant different frequency on European/Japanese versus African APOE ε4 haplotypes, for which we designed 39 amplicons of ≈850 bp in the PCR-based MPRA, which included 11 amplicons with two or more significant LA. Of the 39 projected amplicons in the PCR-based MPRA, three were excluded as we were unable to optimize amplification of an 850 bp fragment in that region centering around the variant(s) of interest due to repetitive elements. After library creation and analyses to identify barcodes uniquely linked the European/Japanese or African haplotypes, we detected 51,000 unique barcodes originally identified in the DNA plasmid library. Of these, we observed ≈17,000, ≈15,800, and ≈14,500 in RNA of microglia, astrocytes, and neurons to be used in the differential expression analyses.
Fragments 2 and 3 showed consistent low incorporation in the library (evidenced by low read numbers in the library of fragment-to-barcode ligated vectors), indicating Gibson assembly for these two fragments did not perform optimally. Of the remaining 34 fragments, we were able to obtain data on unique barcodes for 24 fragments, though 7 of these (15, 16, 23, 31, 32, 36, 38) had low total read counts for the barcodes in the DNA library or expressed RNA samples in the final analyses. For those with sufficient data (Figure 3B/Table 1 overview/Table S1), we did not identify a significant allele difference for 13 fragments (1, 4, 6, 14, 18, 20, 21, 22, 24, 27, 33, 35, and 39), but observed (nominally) significant differences in both replicates for fragments 13, 17 in astrocytes, fragments 10, 13, 28 in microglia. For both fragment 10 and 13, the haplotype with alleles more common on the European ε4 background induce higher expression, whereas for fragments 17 and 28 the alleles more common on the African ε4 background display higher expression.
3.2.2 |. Outcomes luciferase reporter gene analyses
Of 19 fragments tested in all three cell types (Figure 3C/Table 1 overview/Table S1), we observed very high enhancer activity values compared to the base vector with minimal promotor for fragments 2, 28 (all cell types); 3, 5, 11, 15, 25, 39 (microglia); 7 (neurons); 23 (astrocytes); and 37 (astrocytes, neurons). We detected significant differences (P < .05) in enhancer activity driven by opposite haplotypes for fragments 16 in astrocytes; fragments 8, 13 in microglia; and fragments 2, 3, 37 in neurons with > 25% activity level difference. Lower fold difference at P < .05 were detected for fragment 7, 13 in astrocytes; 26, 33 in microglia; and 11 in neurons. Higher enhancer activity of alleles more common on European APOE ε4 was observed for fragments 2, 3, 7, 13, 26, and 33, whereas more common alleles on African APOE ε4 drove higher activity for fragments 2, 8, 11, 16, and 37.
Based on ENCODE ChIP-Seq data indicating many transcription factor binding sites, besides POL2- in fragment 9 overlaying TOMM40’s promoter, we evaluated the effect of haplotypes in this fragment in the context of promoter activity using pGL4.10 as base vector. We observed a significant higher expression in microglia compared to neuronal or astrocyte cell lines overall as expected and a two2fold higher expression level of the haplotype more common on European APOE ε4 (driven by rs71352238 C) than the African APOE ε4 background in all three cell types.
3.2.3 |. Outcomes probe-based MPRA analyses
Of the 56 variants in the region of interest, we included all non-indel variants (N = 51) in the probe-based MPRA design. After library creation, barcode linking and detection in all extracted RNA samples, we identified three variants with insufficient coverage in DNA or expression libraries of one or both alleles. Additionally, we observed 2081 barcodes that were uniquely linked to one allele of one variant in the APOE region and were detected in all cell experiments in the differential expression analyses; to be used in the subsequent analyses. We observed significant differences for variants rs34278513 (included in larger fragment 1), rs3865427 (fragment 2), rs71352237 (fragment 3), rs283808 (fragment 5), rs12972970 (fragment 6), rs34342646 (fragment 7), rs71352238 (fragment 9), rs34404554 (fragment 11), rs449647 (fragment 23), rs75627662 (fragment 25) in astrocytes, and rs34342646 (fragment 7), and rs59007384 (fragment 13) in microglia (Figure 3D). Alleles more common on either APOE ε4 haplotype show higher activity; European/Japanese rs34278513 (fragment 1), rs3865427 (fragment 2), rs71352237 (fragment 3), rs59007384 (fragment 13), rs75627662 (fragment 25), and rs12721046 (fragment 31) versus African rs283808 (fragment 5), rs12972970 (fragment 6), rs34342646 (fragment 7), rs71352238 (fragment 9), rs34404554 (fragment 11), and rs449647 (fragment 23).
APOE promoter variants previously reported to influence overall APOE expression were not significantly different in frequency between European or Japanese and African APOE ε4 haplotypes (rs405509/rs440446)4 or did not consistently affect enhancer activity in the current analyses (rs449647; PCR-based MPRA fragment 23 or probe-based MPRA).
Supplementary Material
Research in Context.
Systematic review: The mechanisms by which apolipoprotein E (APOE) ε4 risk is decreased in African ancestry carriers are not understood but may be due to variants with different frequency on African versus European/Japanese local ancestry affecting gene regulation. We reviewed literature regarding the regulatory elements influencing APOE expression.
Interpretation: In this reporter gene assay and promoter Capture C analysis study, the TOMM40 intron 2–3 region showed higher enhancer activity for European/Japanese versus African APOE ε4 haplotypes as well as chromatin interaction with APOE in microglia and astrocytes, supporting a role of this region in the observed differential APOE ε4 risk.
Future directions: Cellular investigation of the relationship between the regulatory elements in the TOMM40 intronic region and APOE ε4 expression in Alzheimer’s disease and ancestry-specific genomic context will be critical to expand upon this work.
ACKNOWLEDGMENTS
We thank Dr. Ryan Tewhey for consulting on the probe-based MPRA library creation. This research was supported by the National Institute on Aging (AG059018 – PI JV; KN, MLV, LW, AG, KC, OO, JY, DD-, AG054074 – PI MP; LW, AG, NH, SR, DD, CGS, FR, DB, JV-, AG072547 – PI MP; JV-, AG057659 – PI MP, HG009658- PI FJ; SZ-, AG057516 – PI SG; MA, AC-) as well as Alzheimer Association (ZEN-19–591585- PI JV; KN, MLV, LW, KC, OO, JY, DD) and BrightFocus (A2018425S – PI JV; KN, LW, AG, FR, JY).
Footnotes
CONFLICTS OF INTEREST
Authors further disclosure support from NIH (KN, LW, FR, SG, AG, JY, CB, DD, MP, JV), State of Florida grants (KN, AG, JY, DMD), Miami Heart Research Institute (LW), Helping Hands for GAND Foundation (JY). Royalties have been received by JV (Duke University) and JY (Elsevier). Consulting fees have been received by AG (Northwestern University) and JV (University of Pennsylvania). SG receives funds from patents (US Patent Number 10,125,395, 2018; US Patent Number 9,926,600, 2018; US Patent Number 10,066,266, 2018; Canada Patent Number 2,714,713, 2018; US Patent Number 10,266,896, 2019). JY has contributed to the Helping Hands for GAND Foundation scientific advisory board without compensation. ML, DVB, KC, OO, NH, and SR declare no further disclosures.
SUPPORTING INFORMATION
Additional supporting information may be found in the online version of the article at the publisher’s website.
REFERENCES
- 1.Corder EH, Saunders AM, Strittmatter WJ, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993;261:921–923. [DOI] [PubMed] [Google Scholar]
- 2.Corder EH, Saunders AM, Risch NJ, et al. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat Genet 1994;7:180–184. [DOI] [PubMed] [Google Scholar]
- 3.Farrer LA, Cupples LA, Haines JL, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 1997;278:1349–1356. [PubMed] [Google Scholar]
- 4.Rajabli F, Feliciano BE, Celis K, et al. Ancestral origin of ApoE epsilon4 Alzheimer disease risk in Puerto Rican and African American populations. PLoS Genet 2018;14:e1007791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blue EE, Horimoto ARVR, Mukherjee S, Wijsman EM, Thornton TA. Local ancestry at APOE modifies Alzheimer’s disease risk in Caribbean Hispanics. Alzheimers Dement 2019;15:1524–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pombo A, Dillon N. Three-dimensional genome architecture: players and mechanisms. Nat Rev Mol Cell Biol 2015;16:245–257. [DOI] [PubMed] [Google Scholar]
- 7.Davies JO, Oudelaar AM, Higgs DR, Hughes JR. How best to identify chromosomal interactions: a comparison of approaches. Nat Methods 2017;14:125–134. [DOI] [PubMed] [Google Scholar]
- 8.Griswold AJ, Celis K, Bussies PL, et al. Increased APOE epsilon4 expression is associated with the difference in Alzheimer’s disease risk from diverse ancestral backgrounds. Alzheimers Dement 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bekris LM, Lutz F, Yu CE. Functional analysis of APOE locus genetic variation implicates regional enhancers in the regulation of both TOMM40 and APOE. J Hum Genet 2012;57:18–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bekris LM, Millard SP, Galloway NM, et al. Multiple SNPs within and surrounding the apolipoprotein E gene influence cerebrospinal fluid apolipoprotein E protein levels. J Alzheimers Dis 2008;13:255–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Potkin SG, Guffanti G, Lakatos A, et al. Hippocampal atrophy as a quantitative trait in a genome-wide association study identifying novel susceptibility genes for Alzheimer’s disease. PLoS One 2009;4:e6501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim S, Swaminathan S, Shen L, et al. Genome-wide association study of CSF biomarkers Abeta1–42, t-tau, and p-tau181p in the ADNI cohort. Neurology 2011;76:69–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bray NJ, Jehu L, Moskvina V, et al. Allelic expression of APOE in human brain: effects of epsilon status and promoter haplotypes. Hum Mol Genet 2004;13:2885–2892. [DOI] [PubMed] [Google Scholar]
- 14.Choi KY, Lee JJ, Gunasekaran TI, et al. APOE promoter polymorphism-219T/G is an effect modifier of the influence of APOE epsilon4 on Alzheimer’s disease risk in a multiracial sample. J Clin Med 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lambert JC, Araria-Goumidi L, Myllykangas L, et al. Contribution of APOE promoter polymorphisms to Alzheimer’s disease risk. Neurology 2002;59:59–66. [DOI] [PubMed] [Google Scholar]
- 16.Roks G, Cruts M, Houwing-Duistermaat JJ, et al. Effect of the APOE-491A/T promoter polymorphism on apolipoprotein E levels and risk of Alzheimer disease: the Rotterdam Study. Am J Med Genet 2002;114:570–573. [DOI] [PubMed] [Google Scholar]
- 17.Laws SM, Hone E, Gandy S, Martins RN. Expanding the association between the APOE gene and the risk of Alzheimer’s disease: possible roles for APOE promoter polymorphisms and alterations in APOE transcription. J Neurochem 2003;84:1215–1236. [DOI] [PubMed] [Google Scholar]
- 18.Shih SJ, Allan C, Grehan S, Tse E, Moran C, Taylor JM. Duplicated downstream enhancers control expression of the human apolipoprotein E gene in macrophages and adipose tissue. J Biol Chem 2000;275:31567–31572. [DOI] [PubMed] [Google Scholar]
- 19.Mak PA, Laffitte BA, Desrumaux C, et al. Regulated expression of the apolipoprotein E/C-I/C-IV/C-II gene cluster in murine and human macrophages. A critical role for nuclear liver X receptors alpha and beta. J Biol Chem 2002;277:31900–31908. [DOI] [PubMed] [Google Scholar]
- 20.Zheng P, Pennacchio LA, Le Goff W, Rubin EM, Smith JD. Identification of a novel enhancer of brain expression near the apoE gene cluster by comparative genomics. Biochim Biophys Acta 2004;1676:41–50. [DOI] [PubMed] [Google Scholar]
- 21.Foraker J, Millard SP, Leong L, et al. The APOE gene is differentially methylated in Alzheimer’s disease. J Alzheimers Dis 2015;48:745–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shao Y, Shaw M, Todd K, et al. DNA methylation of TOMM40-APOE-APOC2 in Alzheimer’s disease. J Hum Genet 2018;63:459–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu CE, Cudaback E, Foraker J, et al. Epigenetic signature and enhancer activity of the human APOE gene. Hum Mol Gen0et 2013;22:5036–5047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mitsumori R, Sakaguchi K, Shigemizu D, et al. Lower DNA methylation levels in CpG island shores of CR1, CLU, and PICALM in the blood of Japanese Alzheimer’s disease patients. PLoS One 2020;15:e0239196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu J, Zhao W, Ware EB, Turner ST, Mosley TH, Smith JA. DNA methylation in the APOE genomic region is associated with cognitive function in African Americans. BMC Med Genomics 2018;11(43):018–03630369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Belloy ME, Napolioni V, Greicius MD. A quarter century of APOE and Alzheimer’s disease: progress to date and the path forward. Neuron 2019;101:820–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Safieh M, Korczyn AD, Michaelson DM. ApoE4: an emerging therapeutic target for Alzheimer’s disease. BMC Med 2019;17(64):019–12991294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luz I, Liraz O, Michaelson DM. An anti-apoE4 specific monoclonal antibody counteracts the pathological effects of apoE4 in vivo. Curr Alzheimer Res 2016;13:918–929. [DOI] [PubMed] [Google Scholar]
- 29.Liao F, Hori Y, Hudry E, et al. Anti-ApoE antibody given after plaque onset decreases Abeta accumulation and improves brain function in a mouse model of Abeta amyloidosis. J Neurosci 2014;34:7281–7292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim J, Eltorai AE, Jiang H, et al. Anti-apoE immunotherapy inhibits amyloid accumulation in a transgenic mouse model of Abeta amyloidosis. J Exp Med 2012;209:2149–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang C, Xiong M, Gratuze M, et al. Selective removal of astrocytic APOE4 strongly protects against tau-mediated neurodegeneration and decreases synaptic phagocytosis by microglia. Neuron 2021;109(1657):1674.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sabbagh M, Sadowsky C, Tousi B, et al. Effects of a combined transcranial magnetic stimulation (TMS) and cognitive training intervention in patients with Alzheimer’s disease. Alzheimers Dement 2020;16:641650. [DOI] [PubMed] [Google Scholar]
- 33.Chen X, Dong GY, Wang LX. High-frequency transcranial magnetic stimulation protects APP/PS1 mice against Alzheimer’s disease progress by reducing APOE and enhancing autophagy. Brain Behav 2020;10:e01740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kalita CA, Moyerbrailean GA, Brown C, Wen X, Luca F, QuASAR-MPRA Pique-RegiR. Accurate allele-specific analysis for massively parallel reporter assays. Bioinformatics 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chesi A, Wagley Y, Johnson ME, et al. Genome-scale Capture C promoter interactions implicate effector genes at GWAS loci for bone mineral density. Nat Commun 2019;10(1260):019–09302-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cairns J, Freire-Pritchett P, Wingett SW, et al. CHiCAGO: robust detection of DNA looping interactions in Capture Hi-C data. Genome Biol 2016;17:127,016–0992-0992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Y, Song F, Zhang B, et al. The 3D Genome Browser: a web-based browser for visualizing 3D genome organization and long-range chromatin interactions. Genome Biol 2018;19(151):018–1519-1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lu L, Liu X, Huang WK, et al. Robust Hi-C maps of enhancer-promoter interactions reveal the function of non-coding genome in neural development and diseases. Mol Cell 2020;79(521):534.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Trizzino M, Park Y, Holsbach-Beltrame M, et al. Transposable elements are the primary source of novelty in primate gene regulation. Genome Res 2017;27:1623–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tewhey R, Kotliar D, Park DS, et al. direct identification of hundreds of expression-modulating variants using a multiplexed reporter assay. Cell 2016;165:1519–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Su C, Argenziano M, Lu S, et al. 3D promoter architecture reorganization during iPSC-derived neuronal cell differentiation implicates target genes for neurodevelopmental disorders. Prog Neurobiol 2021:102000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wingett S, Ewels P, Furlan-Magaril M, et al. HiCUP: pipeline for mapping and processing Hi-C data. F1000Res 2015;4:1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012;489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Consortium RoadmapEpigenomics, Kundaje A, Meuleman W, et al. Integrative analysis of 111 reference human epigenomes. Nature 2015;518:317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fornes O, Castro-Mondragon JA, Khan A, et al. JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res 2020;48:D87–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Coetzee SG, Coetzee GA, Hazelett DJ. MotifbreakR: an R/Bioconductor package for predicting variant effects at transcription factor binding sites. Bioinformatics 2015;31:3847–3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kulakovskiy IV, Vorontsov IE, Yevshin IS, et al. HOCOMOCO: towards a complete collection of transcription factor binding models for human and mouse via large-scale ChIP-Seq analysis. Nucleic Acids Res 2018;46:D252–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang D, Liu S, Warrell J, et al. Comprehensive functional genomic resource and integrative model for the human brain. Science 2018;362. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.