

Abstract
Sunitinib is a potent anti-cancer scaffold that acts as a VEGFR-2 inhibitor. Although the scaffold exhibits potent anti-cancer activity, it is cardiotoxic and also induces hypothyroidism. The current research aims to optimize the Sunitinib for cardio-toxicity and thyro-toxicity by scaffold hopping approach using the admetSAR server. The server has optimized the physico-chemical properties of Sunitinib, which were contributing to the cardiotoxicity and thyro-toxicity. The library of the optimized compounds was further screened by the molecular docking studies and results were validated by the MD simulation and DFT analysis for VEGFR-2 inhibition. Compounds 163 and 432 exhibited the highest affinity to VEGFR-2 receptor with minimal cardiotoxicity and thyro-toxicity. These two compounds could be the starting point for the further discovery of angiogenic inhibitors.
Supplementary Information
The online version contains supplementary material available at 10.1007/s40203-022-00125-1.
Keywords: Sunitinib, VEGFR-2, Cardiotoxicity, Thyro-toxicity, Docking
Introduction
Cancer is the topmost disease in today’s era; causing more than 8,000,000 deaths in recent years (Sung et al. 2021; Debnath and Ganguly 2016). According to WHO, more than 10,000,000 new cancer incidences are diagnosed globally every year out of which 50% are in developed countries. According to GLOBACAN 2020 report, overall 19,300,000 new cancer incidences of various types are reported and more than 50% of deaths occurred in 2020 all over the world (Santos et al. 2019). In addition to this, the detection of novel cancer types is a warning bell to public health. Although there is an increasing number of anticancer therapies, they have not helped appropriately to decrease the risk of cancer cases. Novel chemotherapeutic agents have a less pharmacological effect but severe adverse reactions are produced (Mahmoud et al. 2020). All these points make thrust area in the development of novel anticancer drug candidates which will have a minimal adverse reaction (Ammar et al. 2018). Most of the current chemotherapeutic agents have toxic properties, hence researchers need to find new agents that are selectively killing or preventing the proliferation of tumor cells without being necessarily toxic. (Singh et al. 2010; Zhao and Adjei 2015). The group of tyrosine kinases is enzymes that catalyze the transfer of adenosine triphosphate's phosphate group to targeted proteins (Jiao et al. 2018). Based on the vital function performed in regulatory processes at a cellular level, the enzymes are classified as-
Receptor tyrosine kinases
Non-receptor tyrosine kinases (Arora and Scholar 2005).
Receptor tyrosine kinases include broad ranges of endogenous molecules such as growth factors including vascular endothelial growth factor, fibroblast growth factor, endothelial growth factor, adhesion molecules, proteinases, extracellular protein matrix, transcription factor, and signaling molecules (Simons 2012). There are significant number of human malignancies, in case of gliomas and carcinomas overexpression in vascular endothelial growth factors (Wang et al. 2018). Similar observation contributed to developing a hypothesis about VEGF produced by malignant tissue which promotes paracrine multiplication of blood vessels along with a proliferation of endothelial tissue accelerating malignant growth by improved blood circulation (Bruce and Tan 2011). As a consequence, isomers of VEGF and receptors associated with tyrosine kinase (VEGFRs) are considered desirable proteins for inhibition of angiogenesis (Peng et al. 2017; Hui et al. 2011). From past decades, numerous novel candidates were discovered for angiogenic inhibition of VEGFR as a prime target (Gingrich et al. 2003). Sunitinib was approved by USFDA in 2006 as a multitargeted tyrosine kinase inhibitor containing tumor and angiogenetic inhibitory properties (Cui et al. 2014; Shah et al. 2016). Although the drug is well tolerated and has a promising pharmacological safety profile, it does have some unique side effects that are not seen in other cancer treatments and must be addressed. Commonly observed adverse effects of Sunitinib are cardiovascular toxicity (hypertension, decline in left ventricular ejection fraction), dermatologic toxicity (rash, hand-foot syndrome), and endocrine toxicity (hypothyroidism and adrenal insufficiency) (Buda-Nowak et al. 2017; Le Tourneau et al. 2007; Ayllon et al. 2011; Zhang et al. 2014; Kappers et al. 2010). Hence, captivating amendment of scaffold for the reduction in virulent properties of the existing treatment and to address this issue, we have optimized the structure of the Sunitinib by minimizing the physico-chemical properties which contribute for the cardiotoxicity and thyro-toxicity by admetSAR tool.
Structure-based computational study protocol
Structure-based virtual screening is primarily used computational protocol in the recent span with wide application in drug discovery (Cosconati et al. 2010). The proposed protocol is to recollect an extensive database of prevailing ligands to a recognized minute subset for selection and experimental analysis. The current study attempted to optimize toxicity parameters of Sunitinib. Figure 1 explains the protocol of structure-based virtual screening protocol of Sunitinib. Firstly we optimized Sunitinib in the admetSAR tool for toxicity prediction, followed by the development of a database of Sunitinib analogs which were filtered as per to hERG (Human either-a-go-go inhibition) and TRB (thyroid receptor binding) using the QikProp tool. Amongst the developed compounds, candidates bearing lower cardiotoxicity and decreased affinity to thyroid receptors were selected further for in-silico study.
Fig. 1.

Virtual screening protocol
In silico docking of selected Sunitinib analogs was carried out using Maestro to find out reactivity as VEGFR-2 inhibitor (PDB ID: 4AGD). Compounds exhibiting significant VEGFR-2 inhibitory activity were further optimized for prime MM-GBSA binding free analysis. Further MD simulation (100 ns) and DFT studies were validated and confirmation of the top-scored molecule interaction with the VEGFR-2 enzyme was performed.
Computational specification (hardware and software)
| Sr no. | In-silico tools | Operating system/hardware |
|---|---|---|
| 1 | Schrodinger (Glide and QuickPro module on HP Z2 TOWER workstation (8-core processor) | Linux Ubuntu Enterprise Version 20.4 |
| 2 | VEGFR-2 (crystal structure) PDB ID: 4AGD | RCSB PDB |
| 3 | Prime MM-GBSA approach, the binding free energies, and MD simulation | Academic Desmond |
| 4 | Density functional theory (DFT) | Jaguar module of Schrödinger |
Database (ligand) structure and toxicity prediction
Toxicity prediction for Sunitinib was performed using the admetSAR tool. By using a similar search function provided by the software, 478 structural analogs of Sunitinib were obtained. (Molecules exhibiting fewer hERG values and lowered thyroid-binding affinity range and details of the library are depicted in Supplementary Figure S1). The structures of ligand were generated in the CDX format using ChemBioDraw Ultra 14.0. Before being subjected to ligand preparation in the LigPrep module, first, these ligands were converted to the mol format. Epik, LigPrep were used to desalt and produce all conceivable tautomers and states at pH 7.0, while retaining definite chirality and minimizing ligands using the OPLS 2005 force field. It predicts descriptors that are physically important qualities of candidates in batch or individually (Patel et al. 2015; Vistoli et al. 2008).
Molecular modeling study
Sunitinib with co-crystallized protein of VEGFR-2 (PDB: 4AGD) was obtained from RCSB-PDB (https://www.rcsb.org/structure/4AGD). Protein-PDB files were prepared for analysis using Maestro's Protein Preparation Wizard, which ensured chemical correctness, assigned bond ordering, eliminated water molecules, and added hydrogen for pH 7.0 and termini were capped, and to fill up missing side chains, loops was carried out using Prime. The diminution of the protein structure was done with a default constraint of 0.30 RMSD and the OPLS 2005 force field to complete protein preparation (Schrödinger Release 2008). The receptor grid was created around the co-crystallized Sunitinib, and the binding site was delineated around it. Molecular modeling was performed using the Glide ligand docking module by SP (Standard Precision) mode, where the receptor grid was defined in a folder where named receptor grid generation was performed using the LigPrep tool. And to find out critical interactions, the binding conformations were studied (Halgren et al. 2004; Friesner et al. 2004).
Binding free energy analyses (Prime MM-GBSA)
Using the Schrödinger suite, default parameters a Prime MM-GBSA module was applied to calculate binding free energies (GBind) for target protein–ligand composites. The properties were analyzed utilizing the Glide pose viewer file, which was further calculated for evaluation of relative binding interactions to receptor (kcal/mol). The binding energies (MM-GBSA) were calculated as free binding energies, stronger binding was represented by a more negative value (Vijayakumar et al. 2014; Patel et al. 2021a, b; Casalvieri et al. 2020).
Molecular dynamics simulations
The Desmond program, which is an explicit solvent MD package accompanying a fixed OPLS 2005 force field was applied to study the MD simulation for top docked protein–ligand composites (Desmond Molecular Dynamics System 2017). The protein–ligand composites were developed by “protein preparation wizard” where predefined SPC (simple point charge) water model along with orthorhombic box shape (size of the box as 10 Å × 10 Å distance). Implementing parameters of system-built, ionic strength was set to an approximate concentration of 0.15 M sodium chloride and was configured in between simulation box and protein atom (10 Å approximately). Energy minimization was performed to bring the system to the local energy level. The model was submitted to 100 ns MD simulation utilizing OPLS_2005 force field. The parameters were Noose-Hover chain thermostat algorithm at 300 k, Martyna-Tobias-Klein barostat algorithm at 1.01325 bar, isoelectric coupling, coulombic cutoff at 0.9 nm. The rest of the parameters were default (Patel et al. 2021a, b; Jin et al. 2020a). The Simulations Interaction Diagram (SID) tool was used to evaluate the ligand-receptor interactions using trajectories of MD simulations.
Computational quantum mechanics calculation by using density functional theory (DFT) analysis
Density functional theory (DFT) is a computational quantum mechanical modeling method for the investigation of the electronic structure of receptors and their interactions with ligands. Three best-hit compounds with Sunitinib were selected for electronic and structural properties. The calculation was performed using the Becke3-Lee–Yang–Parr (B3LYP) method incorporated a 6-31G* basis set into a jaguar module and hybrid DFT with Beckes 3-parameter exchange potential. The parameter calculated which were used to study includes HOMO (highest occupied molecular orbital) and LUMO (lowest unoccupied molecular orbital) energies, as well as electron affinity and electrophilicity index. The molecular electrostatic potential surfaces (MESPs) were obtained from the population analysis calculations. These parameters play a key factor in explaining the magnitude of ligand for interaction in the binding region of the VEGFR-2 enzyme (Bochevarov et al. 2013).
Results and discussion
The sequential filters were used to find out a selective VEGFR-2 enzyme inhibitor based on a virtual screening protocol.
Toxicity prediction was carried out using the admetSAR tool for this optimization of Sunitinib. Sunitinib as VEGFR-2 inhibitors can frequently cause hypertension and hypothyroidism as adverse effects. Hypertension is produced due to VEGF antagonism which decreases the bioavailability of a potent vasodilator (nitric oxide) and leads to constriction of the vasculature, and reduction in renal excretion of sodium ions (Bono et al. 2011). Subclinical hypothyroidism is usually induced by Sunitinib (TSH elevation alone with normal FT4 levels) or overt (TSH elevation and low FT4) but rarely severe. Hypertension associated with Sunitinib can be a strong prognostic marker for efficacious treatment in metastatic renal cell carcinoma (Isidori et al. 2017).
In silico prognostic of the human Ether-à-go-go-Related Gene (hERG) hindrance and thyroid receptor binding affinity through admetSAR toxicity prediction tool produced 478 possible Sunitinib analogs (toxicity values for human Ether-à-go-go-Related Gene < − 0.7396 and thyroid receptor binding < − 0.6869 Resp.), which were filtered to the next stage (Supplementary Table S3). The range of hERG inhibition and TRB of Sunitinib analogs was hERG < − 0.6074 and TRB < − 0.515 respectively. On filtering, we got 295 structural analogs. After screening, 296 compounds including Sunitinib were selected for further consideration.
Molecular modeling studies
For the docking studies of ligand into the VEGFR-2 enzyme binding pocket, docking tool, GLIDE, was used. To standardize the docking model, the methodology was validated by calculating RMSD properties of the co-crystalized (internal) ligand and extracted internal ligand of the docked target protein–ligand complex structure, shown in Fig. 2. Glide SP docking was used to evaluate the optimal orientation of the co-crystalized ligand. From the results, the RMSD value of 1.5084 showed that the method was accurate for predicting the binding affinity for unknown ligands. Docking results of top-scoring compounds are given in Table 1 and the remaining is shown in (Supplementary Table S1).
Fig. 2.
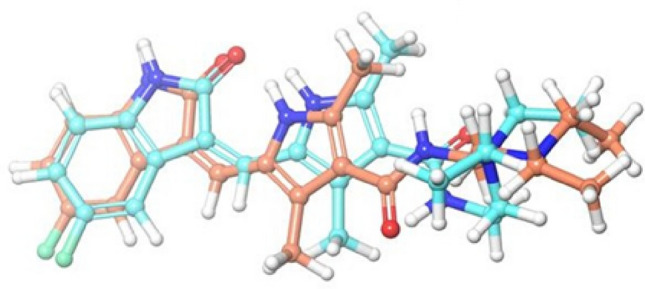
Data obtained in the validation of the molecular docking protocol, Superimposition of perfectly docked ligand Sunitinib for its crystallized conformation obtained from the bioactive complex structure; cyan ligand—original ligand; sky blue—docked ligand
Table 1.
Glide SP docking score of top Sunitinib analogs against VEGFR-2 binding site
| Compound code | Docking score (Kcal/mol) | Glide gscore | Glide emodel |
|---|---|---|---|
|
− 10.579 | − 10.579 | − 88.266 |
|
− 10.342 | − 10.342 | − 87.406 |

|
− 10.267 | − 10.274 | − 72.325 |

|
− 10.248 | − 10.248 | − 86.718 |
|
− 10.212 | − 10.28 | − 89.326 |
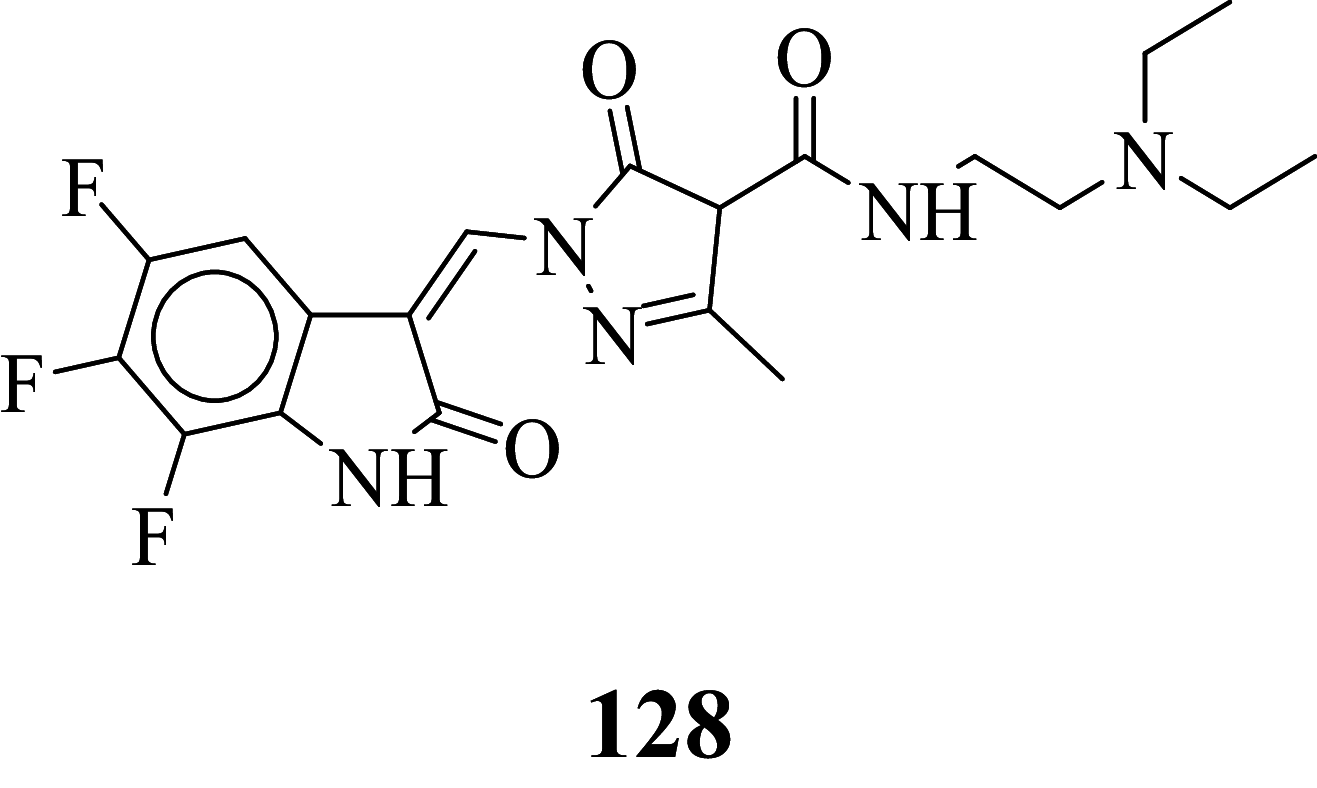
|
− 10.086 | − 10.086 | − 85.552 |
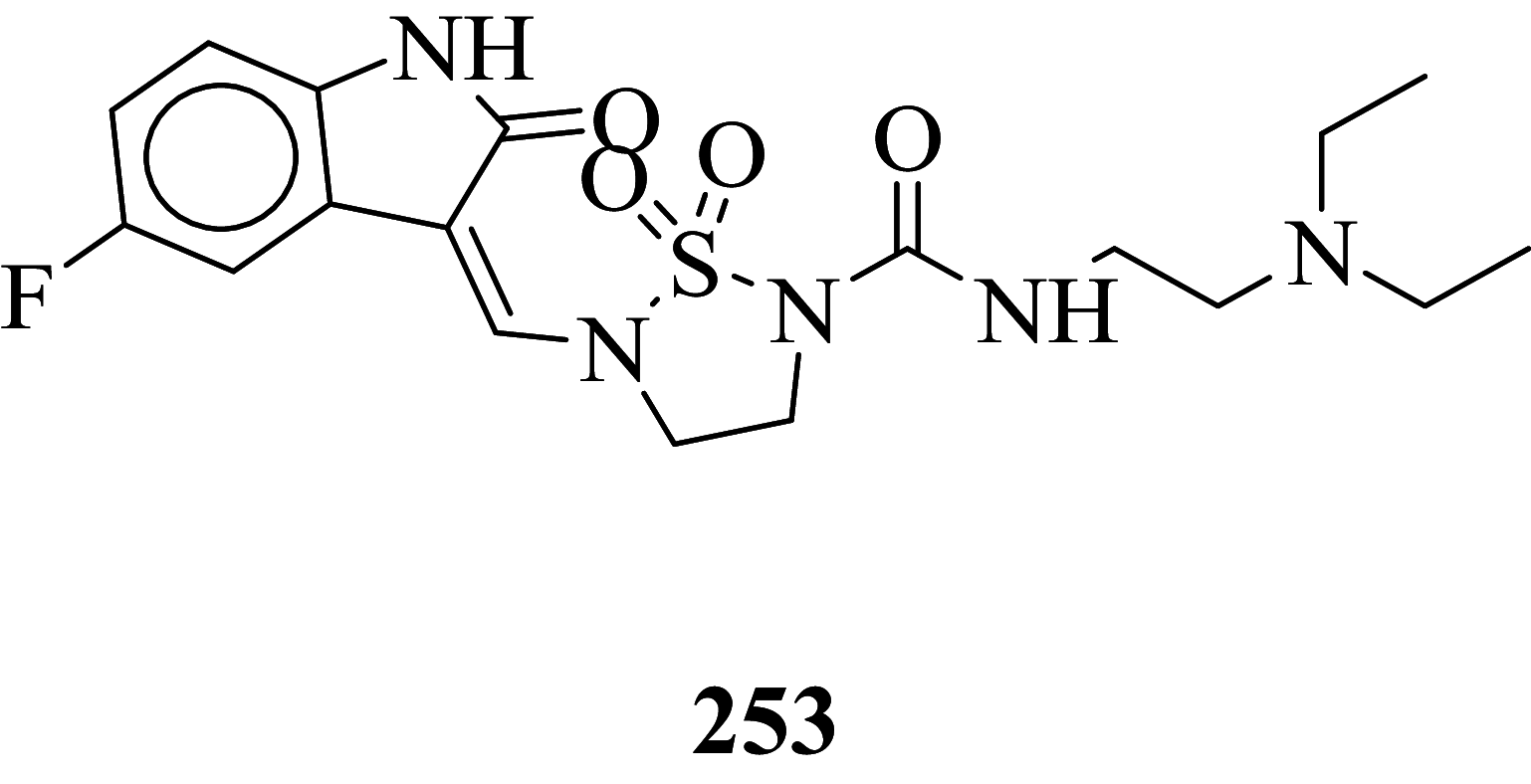
|
− 10.051 | − 10.055 | − 71.582 |
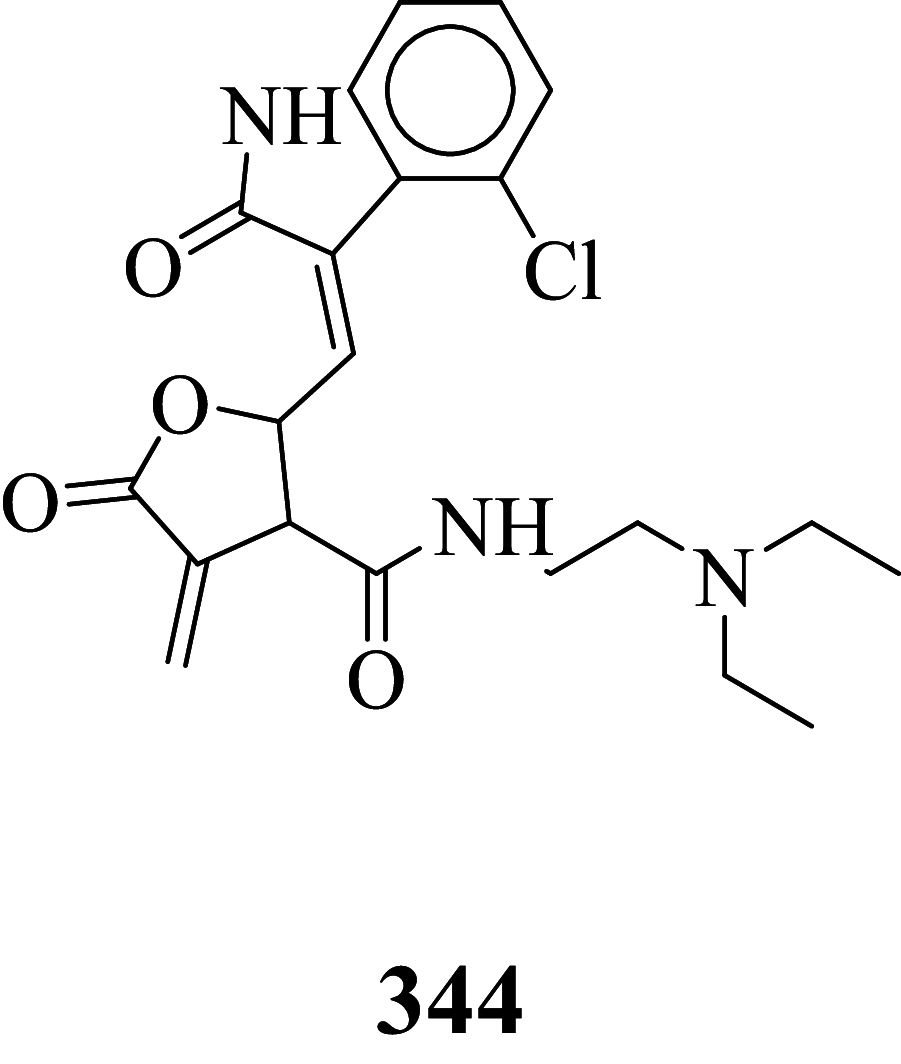
|
− 9.979 | − 9.985 | − 75.258 |
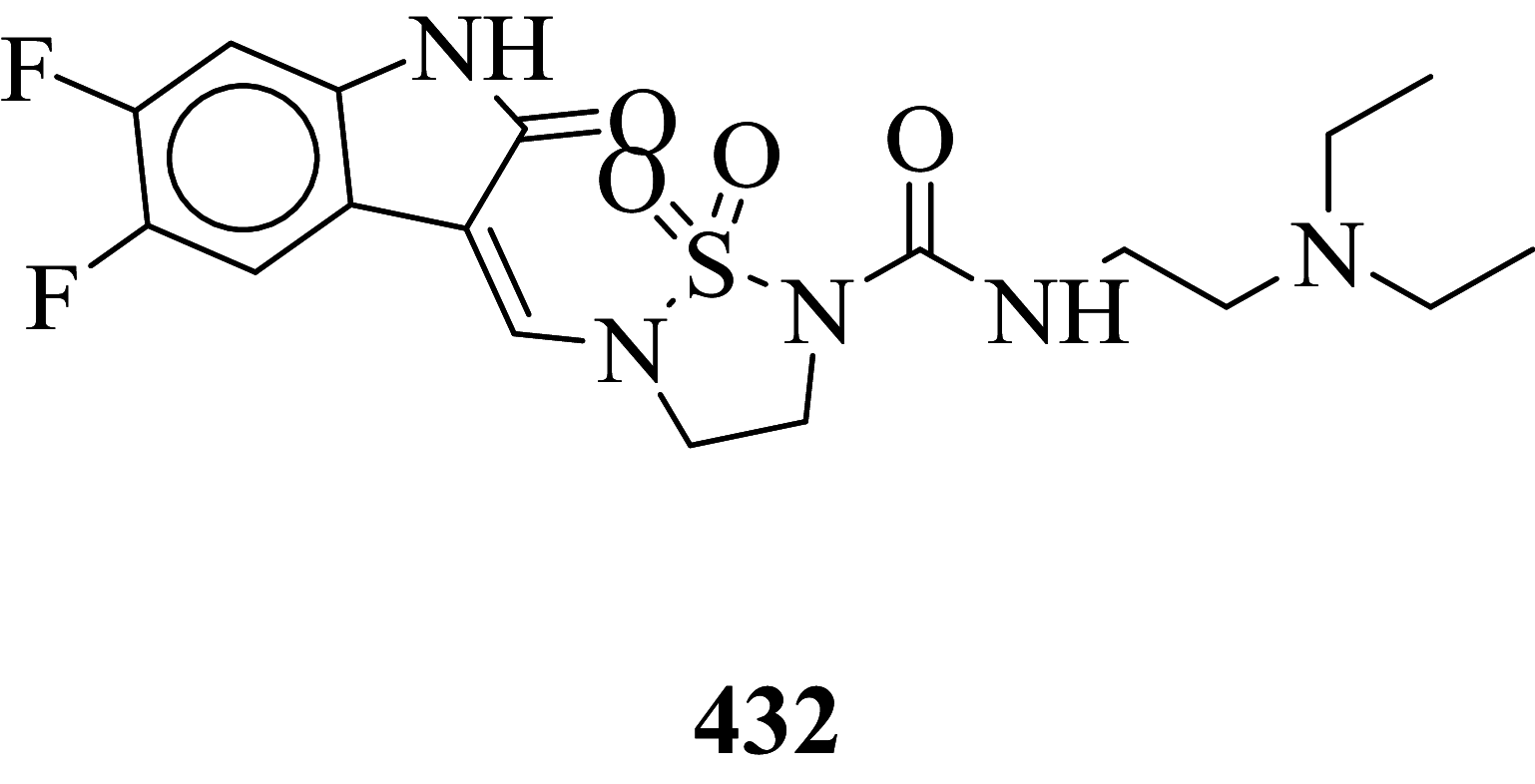
|
− 9.935 | − 9.941 | − 68.104 |
|
− 5.58 | − 5.584 | − 42.956 |
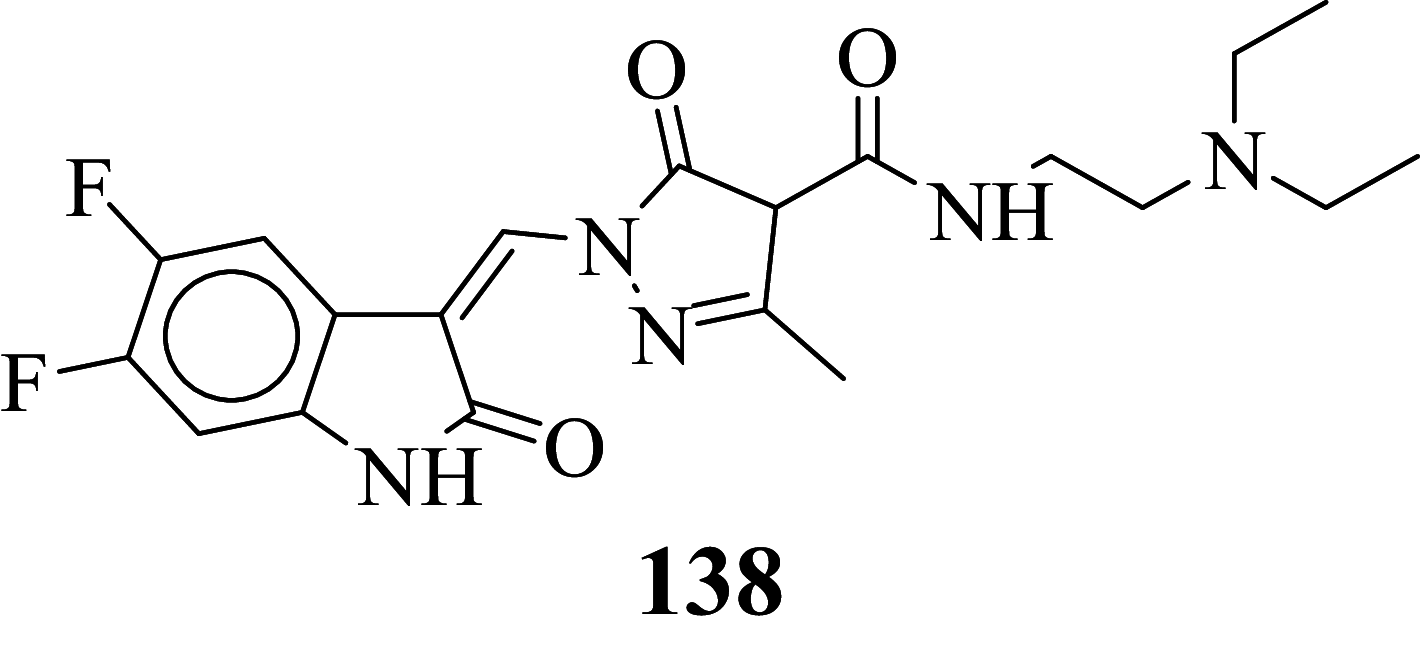
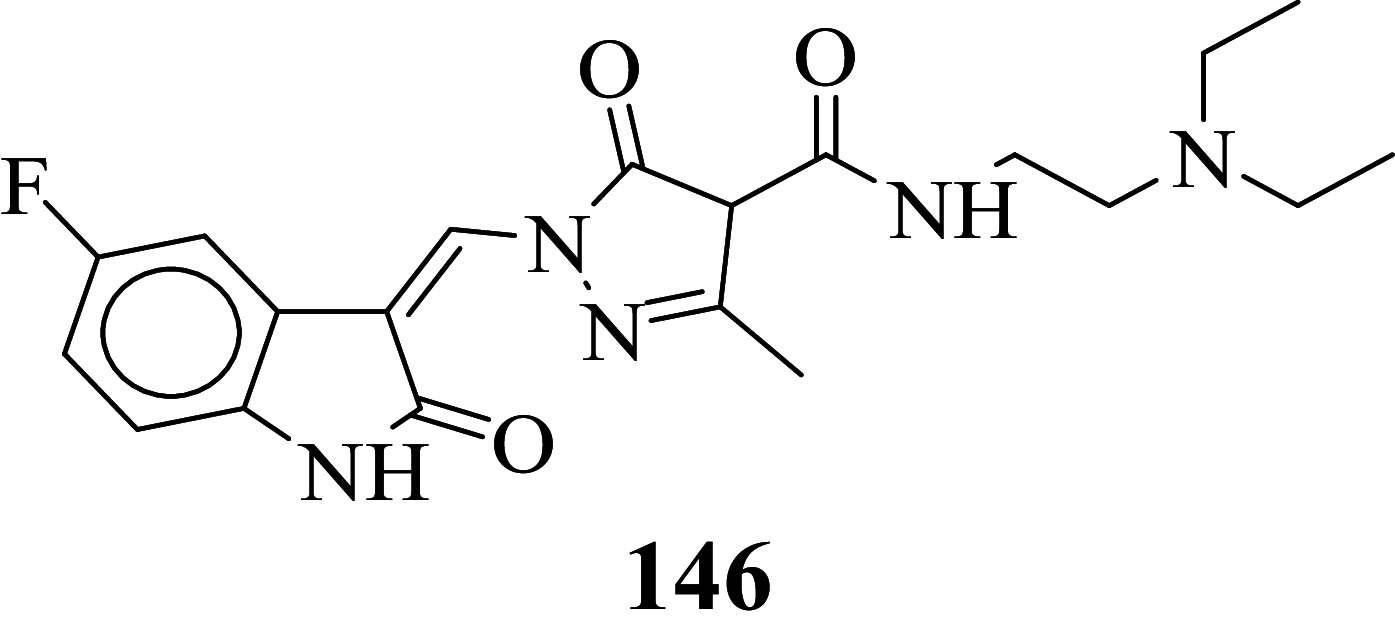
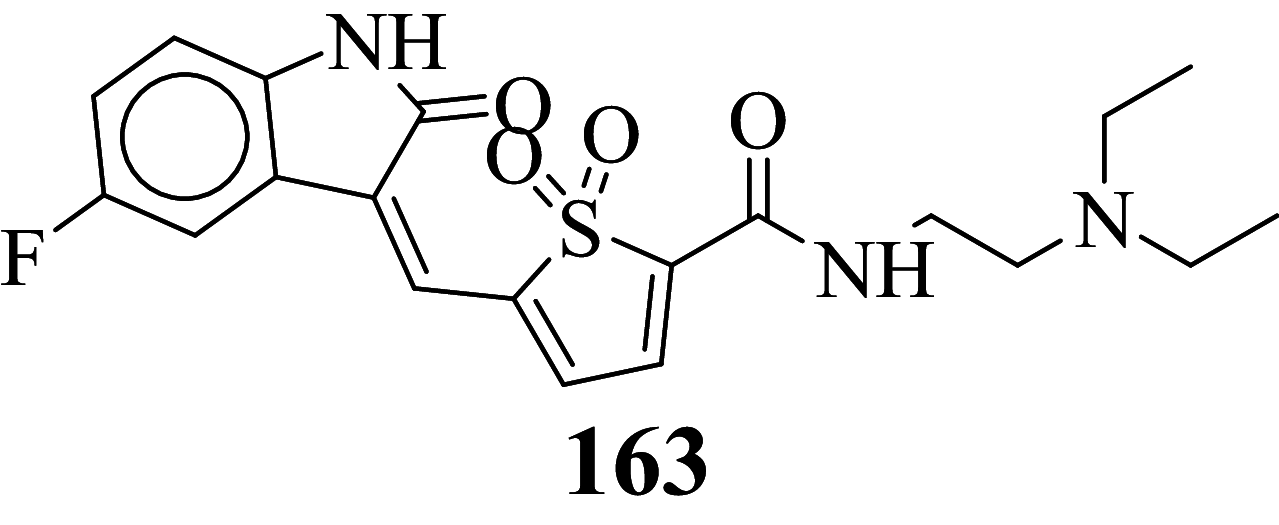
Among the screened Sunitinib analogs, 159 analogs showed a higher binding strength compared to Sunitinib (− 5.58 kcal/mol). Among these analogues, 9 analogues, namely: 138, 146, 114, 121, 163, 128, 344, 253 and 432 were showing the highest binding potency (− 10.579, − 10.342, − 10.267, − 10.248, − 10.212, − 10.086, − 10.051, − 9.979, and − 9.935 kcal/mol) than Sunitinib (− 5.58 kcal/mol). Comparing the docking result and MM− GBSA binding energy, we found that the compounds 163 and 432 showed the highest MM-GBSA binding energy and also a good docking score than Sunitinib (− 5.58 kcal/mol). Further, we have subjected the ligands to DFT analysis for a more specific result. The docking interaction showed that Sunitinib forms pi-cation stacking with Phe1047 as shown in Fig. 3F.
Fig. 3.

Binding interaction analysis of A 2D interaction of compound 163, B 3D interaction compound 163, C 2D interaction of compound 432, D 3D interaction of compound 432, E 2D interaction of Sunitinib, F 3D interaction of Sunitinib with VEGFR-2 binding enzyme (PDB ID: 4AGD) domain
The top docked score analogs compound 163 (Fig. 3A 2D, B 3D), and compound 432 (Fig. 3C 2D, B 3D) were shown similar binding approaches.
MMGBSA binding free energy analysis
The potent protein–ligand composites including Sunitinib for assessing the affinity by ligands to the receptor of the desired protein, the analysis was carried out. In comparison to the Glide score result obtained from MMGBSA binding free energy (∆GBind) which were more efficient for protein–ligand interactions.
The analysis was done based on the primary energy components, such as Coulomb or electrostatic interaction energy (∆GBind Coulomb), lipophilic interaction energy (∆GBind Lipo), generalized Born electrostatic solvation energy (∆GBind Solv-GB), and van der Waals interaction energy (∆GBind vdW) altogether contribute to the analysis of MM-GBSA-based relative binding affinity are mentioned in Table 2 and are shown in (Supplementary Table S2).
Table 2.
Binding free energy components for the protein–ligand complexes calculated by MM-GBSA analysis
| Compound code | MMGBSA (kcal/mol) | Prime energy | ||||
|---|---|---|---|---|---|---|
| ∆G bind | ∆G Coulomb | ∆G Lipo | ∆G Solv_GB | ∆G vdW | ||
| 163 | − 71.35 | 30.36 | − 22.15 | − 24.28 | − 60.16 | − 11,923.3 |
| 432 | − 70.96 | 31.49 | − 20.58 | − 31.17 | − 54.11 | − 11,931.7 |
| 436 | − 68.44 | 27.35 | − 16.95 | − 28.75 | − 51.1 | − 11,930.5 |
| 253 | − 67.85 | 26 | − 17 | − 26.62 | − 51.54 | − 11,933.8 |
| 97 | − 67.21 | 38.44 | − 23.21 | − 29.63 | − 55.31 | − 11,957.5 |
| 272 | − 65.64 | 22.57 | − 21.48 | − 25.58 | − 42.75 | − 11,947.2 |
| 325 | − 64.2 | 20.33 | − 24.05 | − 18.04 | − 48.02 | − 11,908.3 |
| 61 | − 63.19 | − 66.11 | − 20.61 | 71.76 | − 50.69 | − 11,972 |
| 275 | − 62.82 | 41.68 | − 21.59 | − 31.19 | − 57.29 | − 11,962.8 |
| 175 | − 62.72 | − 26.04 | − 20.2 | 30.91 | − 49.61 | − 11,990.6 |
| 110 | − 62.27 | 30.78 | − 21.48 | − 24.44 | − 49.13 | − 11,907.4 |
| Sunitinib | − 35.08 | 40.79 | − 20.47 | − 21.16 | − 39.65 | − 11,913.1 |
∆GBind binding free energy, ∆GCoulomb Coulomb or electrostatic interaction energy, ∆GLipo lipophilic interaction energy, ∆GSolv_GB generalized Born electrostatic solvation energy, ∆GvdW Van der Waals interaction energy
Out of 278 compounds, 140 compounds exhibited higher binding free energies in comparison to standard compound Sunitinib (∆GBind = − 35.08 kcal/mol). Out of these 140 compounds, compound no. 163 and 432 (∆GBind = − 71.35, − 70.96 kcal/mol respectively) were found to have relatively higher binding energy.
Molecular dynamics simulation study
A molecular modeling study was performed on the crystal structure of VEGFR-2. The study was carried out for optimization of the interaction between the lead compound and target receptor to obtain stable confirmation as mentioned in Table 1. Compounds 163 exhibited relatively higher docking value amongst all along with highest binding free energy (∆GBind = − 71.35 kcal/mol) Hence, co-crystallized for a complex of VEGFR-2 and compound 163 were considered for further studies of molecular dynamics simulation of 100 ns using simple point charge (SPC) water mode. RMSF (root mean square fluctuation) and protein–ligand composition were investigated for MD stimulation trajectories to assess thermodynamics conformational stability during the 100 ns period (Panwar and Singh 2020; Guillemont et al. 2011; Bhowmick et al. 2020).
In the dynamic condition, the MD simulation trajectory's RMSD investigation implies the stability of protein skeleton at the time of binding at specific ligand provides detailed insights into its structural conformation during the MD simulation. During MD simulation, the lower the RMSD value, implies high stability of the protein–ligand complex. A graphical result of RMSD of compound 163 is depicted in Fig. 4. Primarily, the graph line indicated an elevating tendency between 0 to 20 ns along with RMSD value ranging from 0.8 to 3.2 Å, further little stability from 20 to 30 ns. After 30 ns till 65 ns the graph line indicated minute fluctuation, after 65 ns, the promising results were observed, and then the graph line was stable till 100 ns. The overall analysis of RMSD concludes that fluctuations in a graph during 100 ns simulation occurred fall in a standard range of RMSD. RMSD value of ligand between 1 and 3 Å implies lead compound is bound tightly within the cavity of the VEGFR-2 enzyme binding pocket. The RMSF results conclude that protein residues are mobile and flexible during the entire simulation process. Relatively higher RMSF ranges indicate more flexibility, however, the relatively lower value of RMSF indicates the system is stable.
Fig. 4.

Time-dependent protein–ligand RMSD plot (Angstrom) of the compound 163 with VEGFR-2 enzyme binding pocket
163-complex with the VEGFR-2 enzyme showed little bit fluctuations below 2 Å at Glu917, Cys919, and Asn923 residues which is perfectly acceptable Fig. 5.
Fig. 5.

Protein RMSF for the trajectory of 163-complex with VEGFR-2 enzyme. Alpha helical and beta-strand regions of the protein are highlighted in red and blue background. Protein residues that interact with compound 163 are marked with green-colored vertical bars
The present peaks indicate a specific domain of the protein which fluctuates much during the simulation. The secondary structure elements i.e. region containing (beta-strand regions) highlighted in the red and blue background appear to be stable as expected in the complex. Appeared fluctuations are prominently at the tails region (N- and C-terminal) and the unstructured part (loop regions) of the protein. However, the complete protein appears in stable fluctuations in the composites.
Protein–ligand interaction analysis
The interaction of protein–ligand composite was regulated during the simulations. Residue Glu917 and Cys919 play a key factor in hydrogen bonding to compound 163, Phe1047 is only a single residue from Pi–Pi stacking with compound 163. The hydrophobic interaction between protein–ligand take place through Val916, Cys919, and Phe1047 (Fig. 6).
Fig. 6.

Simulation interaction diagram, A 2D binding interaction of compound 163 with VEGFR-2 enzyme binding pocket. B Bar diagram
To demonstrate the stability of ligand at the binding site of protein, five molecular properties were studied during the simulation of 100 ns as shown in Fig. 7. (1) Ligand RMSD: root mean square deviation of a ligand for the reference conformation (typically the first frame is used as the reference and it is regarded as time t = 0); (2) radius of gyration (rGyr): it is used to measure the 'extendedness' of a ligand and is equivalent to its principle moment of inertia; (3) molecular surface area (MolSA): molecular surface was calculated with 1.4 Å probe radius; (4) solvent accessible surface area (SASA): molecular surface area was accessible by a water molecule; (5) polar surface area (PSA): solvent accessible surface area in a molecule (Jordaan et al. 2020; Jin et al. 2020). As shown in Fig. 7 the RMSD value of compound 163 was below 2 Å; the radius of gyration (rGyr) value for the compound 163 was 4.8–5.4 Å from 0 to 10 ns, and radius value was 4.9–5.3 Å after 20 ns. The constant value indicated that steady behavior and MolSA (360–380 Å2), SASA (160–240 Å2), and PSA (150–180 Å2) plot suggested the stability of compound during the simulation process.
Fig.7.

Ligand properties during 100 ns simulations for compound 163. A Ligand RMSD (root mean square deviation), B radius of gyration (rGyr), C molecular surface area (MolSA), D solvent accessible surface area (SASA), and E polar surface area (PSA)
Density functional theory analysis
Frontier molecular orbital of compounds specifies a key factor for interactions associated with charge transfer at the binding pocket of the VEGFR-2 enzyme. The virtually screened compound 163 exhibiting good binding energy lower than compound 432 and Sunitinib was studied to recognize the electronic energy state for better charge-transfer interaction. The higher HOMO value denotes a molecule with a good electron donor, whereas a lower value implies a weak electron acceptor. Furthermore, a smaller energy gap between the LUMO and HOMO energies has a considerable influence on the intermolecular charge transfer and bioactivity of molecules. Thus, the wide energy gap observed in the hit molecule negatively affects the electron to move from the HOMO to the LUMO, which subsequently leads to a weak affinity of the inhibitor for the VEGFR-2 enzyme (Amala et al 2019).
The HOMO, LUMO, and their energy gap (∆E) and molecular electrostatic potential surface (MESP) of three prime hits are depicted in Table 3. For compounds 163, 432, and Sunitinib, for which HOMO's results ranges in − 0.160346 to − 0.274559 eV and LUMO's in ranges − 0.075863 to − 0.148101 eV and difference in energy gap was 0.084483, 0.132755, and 0.126458 eV respectively.
Table 3.
Single point energy (Jaguar) output value of frontier orbital energies
| Compounds | HOMO (eV) | LUMO (eV) | Energy gap/∆E (eV) | MESP (kcal/mol) |
|---|---|---|---|---|
| 163 | − 0.160346 | − 0.075863 | 0.084483 | − 83.459 to 72.3565 |
| 432 | − 0.286260 | − 0.153505 | 0.132755 | − 9.0709 to 144.1137 |
| Sunitinib | − 0.274559 | − 0.148101 | 0.126458 | − 5.2164 to 126.7861 |
The result graphical representations of DFT (density functional theory) are depicted in Fig. 8A–F. The area highlighted in red and blue parts postulate the cloud density of frontier orbital at HOMO or LUMO states. Representation of negative values of HOMO–LUMO for three candidates exhibited greater stability and forms stable ligand–protein composites. Comparing the ∆E values of compound 163 with Sunitinib (∆E = 0.126458), the energy gap ∆E value of compound 163 (∆E = 0.084483 eV) is lower which may be considered as a potential binding with the VEGFR-2 enzyme. The molecular electrostatic potential surface (MESP) provides concise positive and negative charges, also identifies the reactive sites for electrophilic and nucleophile attack in a compound for protein binding interaction with ligand. The different charges are denoted in different colors. The red color indicates negative potential (negative regions) exhibiting proton alluring property whereas the blue region depicted positive potential (positive region) having proton revolting properties. The green region represents zero potential. A region which is negative potential contributes to the formation of a hydrogen bond with the protein. In compound 163 (Fig. 8B) highest negative potential was detected at thiophene-1, 1-dioxide while in compound 432, the negative potential was detected within tetrahydro-thiophene scaffold (Fig. 8D) and can be liable for interaction with the protein via hydrogen bonding (Murray and Politzer 2011; Ganesan et al. 2020).
Fig. 8.

A HOMO, LUMO and B MESP of compound 163, C HOMO, LUMO, and D MESP of compound 432, E HOMO, LUMO and F MESP of Sunitinib
Conclusion
Sunitinib has more effect on the immune function with advanced renal cell carcinoma, and the progression of renal cell carcinoma is related to the immune function. It suggests that targeted drug therapy should be combined with biological immunotherapy, which may be the research direction for the treatment of advanced renal cell carcinoma with minimal toxicity. So, the development of novel antitumor agents is a thrust area of the researcher. Also, the developed antineoplastic agents exhibiting high efficiency and also low toxicity which is prime work for researchers. Total 478 structurally Sunitinib analogs were generated out of which 295 structure exhibits minute cardiotoxicity and hypothyroidism, which are then subject to molecular docking, binding free energy, molecular dynamic simulation, and density functional theory studies. Amongst 295 compounds, 9 analogs namely: compounds 138, 146, 114, 121, 163, 128, 344, 253, and 432 exhibits the highest VEGFR-2 activity. From identified hits, Compounds 163 and 432 exhibit relatively higher binding free energy, and a considerable docking score was selected for atomistic MD simulation and density functional theory analysis. Simulation study concludes that composites are stable at 100 ns. The HOMO LUMO energy difference values can be considered as a hit for further development of VEGFR-2 enzyme inhibitors.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
Authors are thankful to Principal, BVDU Poona College of Pharmacy, Pune and Principal, Vice Principal for conducting collaborative research work R. C. Patel Institute of Pharmaceutical Education and Research, authors are also thankful to Dr. Harun Patel, Division of Computer-Aided Drug Design for contributing for designing and carrying out research work at CADD division at R. C. Patel Institute of Pharmaceutical Education and Research.
Funding
The author(s) received no financial aid or support in any form for this research work.
Declarations
Conflict of interest
Authors have no conflict of interest to declare.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Harun Patel, Email: hpatel_38@yahoo.com.
Deepali Bansode, Email: deepali_mhaske@rediffmail.com.
References
- Amala M, Rajamanikandan S, Prabhu D, Surekha K, Jeyakanthan J. Identification of anti-filarial leads against aspartate semialdehyde dehydrogenase of Wolbachia endosymbiont of Brugia malayi: combined molecular docking and molecular dynamics approaches. J Biomol Struct Dyn. 2019;37(2):394–410. doi: 10.1080/07391102.2018.1427633. [DOI] [PubMed] [Google Scholar]
- Ammar YA, Sh El-Sharief AM, Belal A, Abbas SY, Mohamed YA, Mehany ABM, Ragab A. Design, synthesis, antiproliferative activity, molecular docking and cell cycle analysis of some novel (morpholinosulfonyl) isatins with potential EGFR inhibitory activity. Eur J Med Chem. 2018;5(156):918–932. doi: 10.1016/j.ejmech.2018.06.061. [DOI] [PubMed] [Google Scholar]
- Arora A, Scholar EM. Role of tyrosine kinase inhibitors in cancer therapy. J Pharmacol Exp Ther. 2005;315(3):971–979. doi: 10.1124/jpet.105.084145. [DOI] [PubMed] [Google Scholar]
- Ayllon J, Beuselinck B, Morel A, Barrascout E, Medioni J, Scotte F, Oudard S. Long-term response and postsurgical complete remissions after treatment with sunitinib malate, an oral multitargeted receptor tyrosine kinase inhibitor, in patients with metastatic renal cell carcinoma. Cancer Invest. 2011;29(4):282–285. doi: 10.3109/07357907.2011.568560. [DOI] [PubMed] [Google Scholar]
- Bhowmick S, Al Faris NA, Al Tamimi JZ, Al Othman ZA, Aldayel TS, Wabaidur SM, Islam MA. Screening and analysis of bioactive food compounds for modulating the CDK2 protein for cell cycle arrest: multi-cheminformatics approaches for anticancer therapeutics. J Mol Struct. 2020 doi: 10.1016/j.molstruc.2020.128316. [DOI] [Google Scholar]
- Bochevarov AD, Harder E, Hughes TF, Greenwood JR, Braden DA, Philipp DM, Rinaldo D, Hall MD, Zhang J, Friesner RA. Jaguar: a high-performance quantum chemistry software program with strengths in life and materials sciences. Int J Quantum Chem. 2013;113(18):2110–2142. doi: 10.1002/qua.24481. [DOI] [Google Scholar]
- Bono P, Rautiola J, Utriainen T, Joensuu H. Hypertension as predictor of sunitinib treatment outcome in metastatic renal cell carcinoma. Acta Oncol. 2011;50(4):569–573. doi: 10.3109/0284186X.2010.543696. [DOI] [PubMed] [Google Scholar]
- Bruce D, Tan PH. Blocking the interaction of vascular endothelial growth factor receptors with their ligands and their effector signaling as a novel therapeutic target for cancer: time for a new look? Expert Opin Investig Drugs. 2011;20(10):1413–1434. doi: 10.1517/13543784.2011.611801. [DOI] [PubMed] [Google Scholar]
- Buda-Nowak A, Kucharz J, Dumnicka P, Kuzniewski M, Herman RM, Zygulska AL, Kusnierz-Cabala B. Sunitinib-induced hypothyroidism predicts progression-free survival in metastatic renal cell carcinoma patients. Med Oncol. 2017;34(4):68. doi: 10.1007/s12032-017-0928-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casalvieri KA, Matheson CJ, Backos DS, Reigan P. Molecular docking of substituted pteridinones and pyrimidines to the ATP-binding site of the N-terminal domain of RSK2 and associated MM/GBSA and molecular field datasets. Data Brief. 2020;29:105347. doi: 10.1016/j.dib.2020.105347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosconati S, Forli S, Perryman AL, Harris R, Goodsell DS, Olson AJ. Virtual screening with AutoDock: theory and practice. Expert Opin Drug Discov. 2010;5(6):597–607. doi: 10.1517/17460441.2010.484460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui W, Zhang ZJ, Hu SQ, Mak SH, Xu DP, Choi CL, Wang YQ, Tsim WK, Lee MY, Rong JH, Han YF. Sunitinib produces neuroprotective effect via inhibiting nitric oxide overproduction. CNS Neurosci Ther. 2014;20(3):244–252. doi: 10.1111/cns.12203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debnath B, Ganguly S. Synthesis, biological evaluation, in silico docking, and virtual ADME studies of 2-[2-Oxo-3-(arylimino) indolin-1-yl]-N-arylacetamides as potent anti-breast cancer agents. Monatsh Chem. 2016;147(3):565–574. doi: 10.1007/s00706-015-1566-9. [DOI] [Google Scholar]
- Desmond Molecular Dynamics System (2017) D. E. Shaw Research, New York, NY, 2018-4. Maestro-Desmond Interoperability Tools, Schrödinger, New York, NY, 2018-4
- Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47(7):1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- Ganesan MS, Raja KK, Murugesan S, Kumar BK, Rajagopal G, Thirunavukkarasu S. Synthesis, biological evaluation, molecular docking, molecular dynamics and DFT studies of quinoline-fluoroproline amide hybrids. J Mol Struct. 2020 doi: 10.1016/j.molstruc.2020.128360. [DOI] [Google Scholar]
- Gingrich DE, Reddy DR, Iqbal MA, Singh J, Aimone LD, Angeles TS, Albom M, Yang S, Ator MA, Meyer SL, Robinson C, Ruggeri BA, Dionne CA, Vaught JL, Mallamo JP, Hudkins RL. A new class of potent vascular endothelial growth factor receptor tyrosine kinase inhibitors: structure-activity relationships for a series of 9-alkoxymethyl-12-(3-hydroxypropyl)indeno[2,1-a]pyrrolo[3,4-c]carbazole-5-ones and the identification of CEP-5214 and its dimethylglycine ester prodrug clinical candidate CEP-7055. J Med Chem. 2003;46(25):5375–5388. doi: 10.1021/jm0301641. [DOI] [PubMed] [Google Scholar]
- Guillemont J, Meyer C, Poncelet A, Bourdrez X, Andries K. Diarylquinolines, synthesis pathways and quantitative structure–activity relationship studies leading to the discovery of TMC207. Future Med Chem. 2011;3(11):1345–1360. doi: 10.4155/fmc.11.79. [DOI] [PubMed] [Google Scholar]
- Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, Banks JL. Glide: new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem. 2004;47(7):1750–1759. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- https://www.rcsb.org/structure/4AGD. Accessed on 18th Apr 2021
- Hui EP, Ma BBY, King AD, Mo F, Chan SL, Kam MKM, Loong HH, Ahuja AT, Zee BCY, Chan ATC. Hemorrhagic complications in a phase II study of sunitinib in patients of nasopharyngeal carcinoma who has previously received high-dose radiation. Ann Oncol. 2011;22(6):1280–1287. doi: 10.1093/annonc/mdq629. [DOI] [PubMed] [Google Scholar]
- Isidori A, Lenzi A, Rizza L, Rota F, Giacinto PD. Role of sunitinib-induced hypothyroidism in oncological patients. Endocrinol Metab Int J. 2017;5(4):00129. doi: 10.1002/cncr.25422. [DOI] [Google Scholar]
- Jiao Q, Bi L, Ren Y, Song S, Wang Q, Wang YS. Advances in studies of tyrosine kinase inhibitors and their acquired resistance. Mol Cancer. 2018;17(1):36. doi: 10.1186/s12943-018-0801-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Z, Wang Y, Yu XF, Tan QQ, Liang SS, Li T, Zhang H, Shaw PC, Wang J, Hu C. Structure-based virtual screening of influenza virus RNA polymerase inhibitors from natural compounds: molecular dynamics simulation and MM-GBSA calculation. Comput Biol Chem. 2020;85:107241. doi: 10.1016/j.compbiolchem.2020.107-241. [DOI] [PubMed] [Google Scholar]
- Jordaan MA, Ebenezer O, Damoyi N, Shapi M. Virtual screening, molecular docking studies and DFT calculations of FDA approved compounds similar to the non-nucleoside reverse transcriptase inhibitor (NNRTI) efavirenz. Heliyon. 2020;6(8):04642. doi: 10.1016/j.heliyon.2020.e04642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kappers MH, van Esch JH, Sluiter W, Sleijfer S, Danser AH, van den Meiracker AH. Hypertension induced by the tyrosine kinase inhibitor sunitinib is associated with increased circulating endothelin-1 levels. Hypertension. 2010;56(4):675–681. doi: 10.1161/HYPERTENSIONAHA.109.149690. [DOI] [PubMed] [Google Scholar]
- Le Tourneau C, Raymond E, Faivre S. Sunitinib: a novel tyrosine kinase inhibitor. A brief review of its therapeutic potential in the treatment of renal carcinoma and gastrointestinal stromal tumors (GIST) Ther Clin Risk Manag. 2007;3(2):341–348. doi: 10.2147/tcrm.2007.3.2.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmoud HK, Farghaly TA, Abdulwahab HG, Al-Qurashi NT, Shaaban MR. Novel 2-indolinone thiazole hybrids as sunitinib analogues: design, synthesis, and potent VEGFR-2 inhibition with potential anti-renal cancer activity. Eur J Med Chem. 2020;15(208):112752. doi: 10.1016/j.ejmech.2020.112752. [DOI] [PubMed] [Google Scholar]
- Murray JS, Politzer P. The electrostatic potential: an overview. Wiley Interdiscipl Rev Comput Mol Sci. 2011;1(2):153–163. doi: 10.1002/wcms.19. [DOI] [Google Scholar]
- Panwar U, Singh SK. Atom-based 3D-QSAR, molecular docking, DFT, and simulation studies of acyl hydrazone, hydrazine, and diazene derivatives as IN-LEDGF/p75 inhibitors. Struct Chem. 2020 doi: 10.1007/s11224-020-01628-3. [DOI] [Google Scholar]
- Patel H, Sonawane Y, Jagtap R, Dhangar K, Thapliyal N, Surana S, Noolvi M, Shaikh MS, Rane RA, Karpoormath R. Structural insight of glitazone for hepato-toxicity: resolving mystery by PASS. Bioorg Med Chem Lett. 2015;25(9):1938–1946. doi: 10.1016/j.bmcl.2015.03.020. [DOI] [PubMed] [Google Scholar]
- Patel HM, Shaikh M, Ahmad I, Lokwani D, Surana SJ. BREED based de novo hybridization approach: generating novel T790M/C797S-EGFR tyrosine kinase inhibitors to overcome the problem of mutation and resistance in non-small cell lung cancer (NSCLC) J Biomol Struct Dyn. 2021;39(8):2838–2856. doi: 10.1080/07391102.2020.1754918. [DOI] [PubMed] [Google Scholar]
- Patel HM, Ahmad I, Pawara R, Shaikh M, Surana S. In silico search of triple mutant T790M/C797S allosteric inhibitors to conquer acquired resistance problem in non-small cell lung cancer (NSCLC): a combined approach of structure-based virtual screening and molecular dynamics simulation. J Biomol Struct Dyn. 2021;39(4):1491–1505. doi: 10.1080/07391102.2020.1734092. [DOI] [PubMed] [Google Scholar]
- Peng FW, Liu DK, Zhang QW, Xu YG, Shi L. VEGFR-2 inhibitors and the therapeutic applications thereof: a patent review (2012–2016) Expert Opin Ther Pat. 2017;27(9):987–1004. doi: 10.1080/13543776.2017.1344215. [DOI] [PubMed] [Google Scholar]
- Schrödinger Release (2008) Protein preparation wizard. Epik, Schrödinger, LLC, New York
- Santos IS, Guerra FS, Bernardino LF, Fernandes PD, Hamerski L, Silva BV. A facile synthesis of novel isatinspirooxazine derivatives and potential in vitro anti-proliferative activity. J Braz Chem Soc. 2019;30(1):198. doi: 10.21577/0103-5053.20180153. [DOI] [Google Scholar]
- Shah S, Lee C, Choi H, Gautam J, Jang H, Kim GJ, Lee YJ, Chaudhary CL, Park SW, Nam TG, Kim JA, Jeong BS. 5-Hydroxy-7-azaindolin-2-one, a novel hybrid of pyridinol and sunitinib: design, synthesis and cytotoxicity against cancer cells. Org Biomol Chem. 2016;14(21):4829–4841. doi: 10.1039/c6ob00406g. [DOI] [PubMed] [Google Scholar]
- Simons M. An inside view: VEGF receptor trafficking and signaling. Physiology (bethesda) 2012;27(4):213–222. doi: 10.1152/physiol.00016.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh P, Kaur M, Holzer W. Synthesis and evaluation of indole, pyrazole, chromone and pyrimidine based conjugates for tumor growth inhibitory activities—development of highly efficacious cytotoxic agents. Eur J Med Chem. 2010;45(11):4968–4982. doi: 10.1016/j.ejmech.2010.08.004. [DOI] [PubMed] [Google Scholar]
- Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–249. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- Vijayakumar B, Parasuraman S, Raveendran R, Velmurugan D. Identification of natural inhibitors against angiotensin I converting enzyme for cardiac safety using induced fit docking and MM-GBSA studies. Pharmacogn Mag. 2014;10(Suppl 3):S639–S644. doi: 10.4103/0973-1296.139809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vistoli G, Pedretti A, Testa B. Assessing drug-likeness–what are we missing? Drug Discov Today. 2008;13(7–8):285–294. doi: 10.1016/j.drudis.2007.11.007. [DOI] [PubMed] [Google Scholar]
- Wang F, Wang L, Li Y, Wang N, Wang Y, Cao Q, Gong P, Yang J, Wu C. PAC-1 and its derivative WF-210 inhibit angiogenesis by inhibiting VEGF/VEGFR pathway. Eur J Pharmacol. 2018;821:29–38. doi: 10.1016/j.ejphar.2017.12.035. [DOI] [PubMed] [Google Scholar]
- Zhang L, Zheng Q, Yang Y, Zhou H, Gong X, Zhao S, Fan C. Synthesis and in vivo SAR study of indolin-2-one-based multi-targeted inhibitors as potential anticancer agents. Eur J Med Chem. 2014;82:139–151. doi: 10.1016/j.ejmech.2014.05.051. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Adjei AA. Targeting angiogenesis in cancer therapy: moving beyond vascular endothelial growth factor. Oncologist. 2015;20(6):660–673. doi: 10.1634/theoncologist.2014-0465. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.