

Abstract
The blood-brain barrier (BBB) hinders therapeutic delivery to the central nervous system (CNS), thereby impeding the development of therapies for brain injury and disease. Receptor-mediated transcytosis (RMT) systems are a promising way to shuttle a targeted therapeutic into the brain. Here, we developed and evaluated an RMT antibody-targeted liposomal system. A previously identified antibody, scFv46.1, that binds to the human and murine BBB and can pass through the murine BBB by transcytosis after intravenous injection was used to decorate the surface of liposomes. Using an in vitro BBB model, we demonstrated the cellular uptake of scFv46.1-modified liposomes (46.1-Lipo). Next, the biodistribution and brain uptake capacity of 46.1-targeted liposomes were assessed after intravenous administration. Our results showed that 46.1-Lipo can lead to increased brain accumulation through targeting of the brain vasculature. Initial rate pharmacokinetic experiments and biodistribution analyses indicated that 46.1-Lipo loaded with pralidoxime exhibited a 10-fold increase in brain accumulation compared with a mock-targeted liposomal group, and this increased accumulation was brain-specific. These studies indicate the potential of this 46.1-Lipo system as a synthetic vehicle for the targeted transport of therapeutic molecules into the CNS.
Keywords: antibody, brain drug delivery, blood-brain barrier, liposome
INTRODUCTION
One of the major challenges in brain drug development and therapy is that drug delivery to the central nervous system (CNS) is limited by the blood-brain barrier (BBB), which separates the bloodstream from the brain parenchyma (1). The BBB possesses low and selective permeability to therapeutic molecules as a result of (i) tight junctions between brain microvascular endothelial cells that essentially eliminate passive diffusion pathways, (ii) very low levels of pinocytosis and (iii) efflux transporters that perform active efflux of the therapeutic molecules back to the blood vessel lumen (2). Thus, identifying improved methods for crossing the BBB has been a longstanding goal (3, 4). Several strategies have been employed to enhance drug delivery to the brain, with systems that co-opt the transcytosis capacity of receptor-mediated transcytosis (RMT) receptors expressed at the brain microvascular endothelial cell (BMEC) surface exhibiting particular promise (5, 6). For example, transferrin (7), insulin (8) and low-density lipoprotein (9) receptors have been used as target RMT receptors to deliver therapeutic cargo across BMECs and into the brain in both pre-clinical settings and, more recently, in clinical trials (10). However, these targeting systems still face challenges related to off-target effects resulting from non-specificity and inefficient brain uptake. These issues have motivated work to identify new RMT targets and targeting systems (6).
We recently reported the identification of a set of new RMT-targeting antibodies that bind the human and murine BBB in brain tissue sections and target the mouse BBB. One promising lead antibody, scFv46.1, was found to pass through the murine BBB by transcytosis, exhibiting substantial concentrations in the brain after intravenous administration (11). The present work was aimed at testing the potential of scFv46.1-modified liposomes as a drug delivery platform. Liposomes have been well studied as drug carriers due to their biocompatible and biodegradable compositions and flexibility in drug loading and release (12). We hypothesized that the surface modification of liposomes with scFv46.1 could enhance brain accumulation of encapsulated drugs and improve pharmacokinetic profiles compared to untargeted liposomes (Figure 1). Thus, we explored the potential of scFv46.1-modified liposomes (46.1-Lipo) for the delivery of pralidoxime (2-PAM), a model drug that exhibits low brain permeability (13) and that is rapidly cleared when delivered intravenously (14). 2-PAM was encapsulated into liposomes that were subsequently decorated with scFv46.1, and the resulting 46.1-Lipo formulations promoted internalization into BMEC-like cells. Mice treated with 46.1-Lipo had significantly improved brain accumulation of 2-PAM compared to control liposomes that were not decorated with scFv46.1, leading to a significant and selective increase in 2-PAM uptake in brain after intravenous injection into mice. These studies further demonstrate the potential of this new BBB targeting ligand for the transport of therapeutic molecules into the brain.
Figure 1. Schematic of scFv-liposome preparation and expression, functionalization, and activity of scFv.
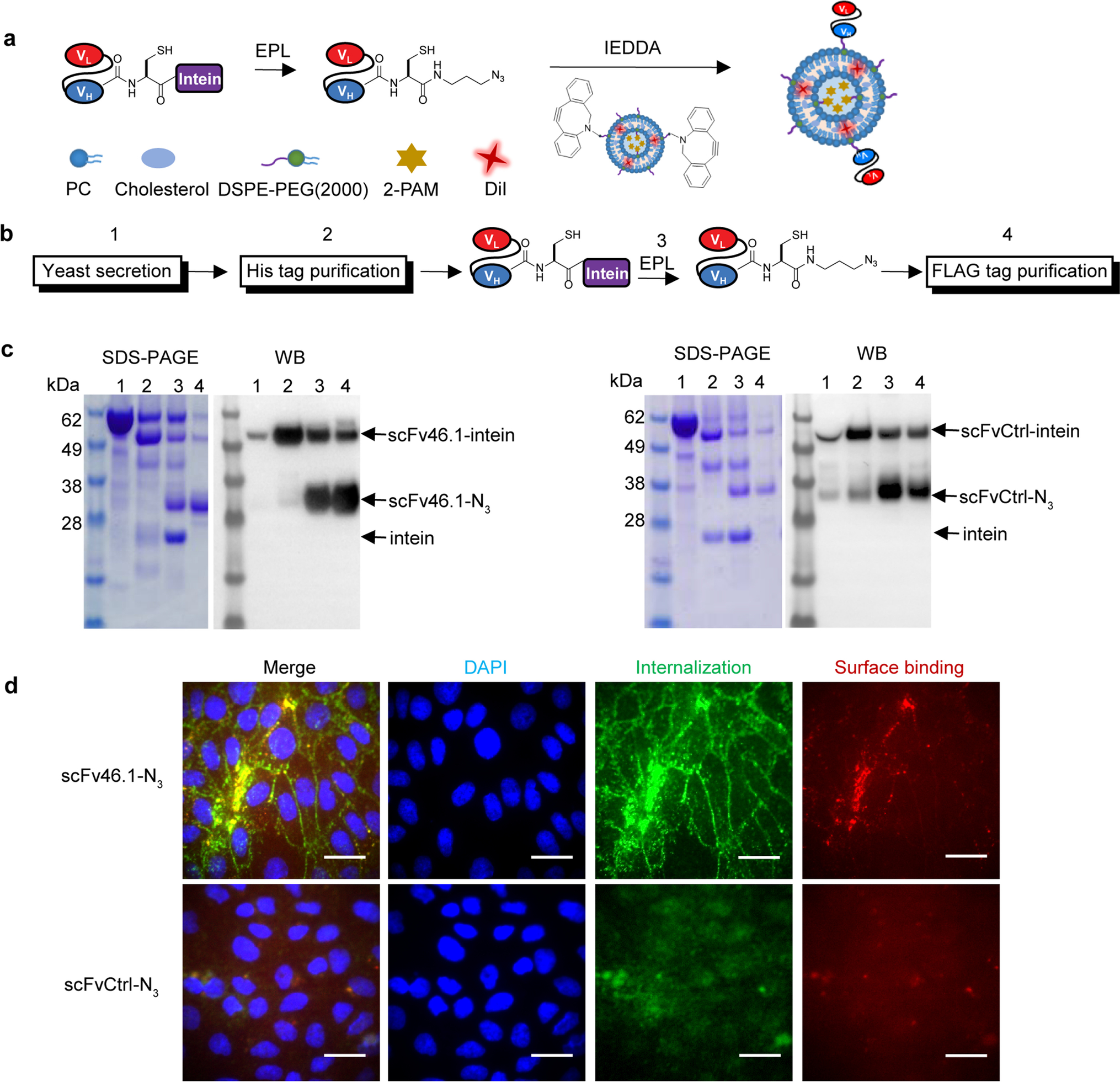
(a) Schematic of conjugation of scFv-N3 with DBCO groups on liposomes by coupling expressed protein ligation (EPL) and inverse electron-demand Diels-Alder (IEDDA) cycloaddition.
(b) Schematic of scFv production, purification, and functionalization strategy. scFv-intein fusion proteins were produced as secreted proteins from yeast (step 1), then enriched by His tag resin via the C-terminal His6 epitope tag (step 2). For intein-mediated protein release and EPL functionalization, MESNA reacts to release the scFv and append a carboxy-terminal thioester, which is then reacted with cysteine azide to covalently link the scFv with an azide (-N3) group (step 3). Finally, scFv-N3 was purified by anti-FLAG affinity resin via the N-terminal FLAG epitope tag (step 4).
(c) Products from each of the four steps depicted in panel (a) were resolved and analyzed by SDS-PAGE followed by Coomassie staining and western blotting against the FLAG epitope tag. The arrows indicate uncleaved scFv-intein, the desired scFv-N3 product, and the released intein.
(d) Activity of scFvs after modification. iPSC-derived BMEC-like cells were incubated with pre-dimerized scFv-N3 (see Methods) for 30 min at 4 °C and subsequently at 37 °C for 45 min. On ice, the membrane bound fraction (red) was labeled with anti-FLAG antibody and followed by anti-mouse AlexaFluor555 secondary antibody. After fixation and permeabilization, the internalized scFv (green) was labeled with anti-FLAG antibody followed by anti-mouse AlexaFluor488 antibody. Nuclei are stained with DAPI (blue). Images were acquired on Zeiss Axio Imager Z2 Upright fluorescence microscope. Scale bar: 20 μm.
MATERIALS AND METHODS
Chemicals and Materials
2-PAM chloride (purity > 97%) and 3H-2-PAM were purchased from Sigma-Aldrich Co. Ltd. (St. Louis, MO, USA) and American Radiolabeled Chemicals, Inc. (St. Louis, MO, USA) respectively. All lipids including phosphatidylcholine (PC), cholesterol, 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (ammonium salt) (DSPE-PEG2000), and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[dibenzocyclooctyl(polyethylene glycol)-2000] (DSPE-PEG2000-DBCO) were purchased from Avanti Polar Lipids, Inc. (Alabaster, AL, USA). 1,1’-Dioctadecyl-3,3,3’,3’-tetramethylindocarbocyanine perchlorate (DiI) was purchased from Thermo Fisher Scientific (Waltham, MA, USA). An extruder (LIPEX® 10 mL extruder) was purchased from Evonik Canada Inc. (Burnaby, B.C., Canada). 2-Mercaptoethanesulfonic acid (MESNA) and Triton-X were purchased from Sigma-Aldrich Co. Ltd. (St. Louis, MO, USA). Cystein-azide was purchased from AnaSpec (Fremont, CA, USA). Anti-FLAG antibody and anti-mouse horseradish peroxidase (HRP)-conjugated antibodies were purchased from Sigma-Aldrich Co. Ltd. (St. Louis, MO, USA). Anti-mouse AlexaFluor488, anti-mouse AlexaFluor555 and anti-mouse AlexaFluor647 were obtained from Invitrogen (Carlsbad, CA, USA). Anti-FLAG AlexaFluor647 antibody was purchased from BioLegend (San Diego, CA, USA). All other chemicals were of analytical reagent or higher-purity grades and were used without further purification unless otherwise noted.
Cell Lines and Animals
The human-induced pluripotent stem cell (iPSC)-derived BMEC-like cell differentiation was performed as previously described using the IMR90-C4 iPSC line (15, 16). At day 8 of differentiation, cells were plated on collagen/fibronectin pre-coated plates or Lab Tek II chamber slides. iPSC-derived BMEC-like cell assays were performed on day 10. bEnd.3 cells [American Type Culture Collection (ATCC CRL-2299)] were cultured in Dulbecco’s modified Eagle’s medium (DMEM):F12 (1:1) supplemented with 10% fetal bovine serum (FBS, Invitrogen, Carlsbad, CA). The rat brain endothelial cell line RBE4 was a kind gift from Dr. Françoise Roux (17). RBE4 cells were grown in 45% alpha minimum essential medium, 45% Ham’s F10 medium and 10% heat inactivated FBS supplemented with 100 mg/mL streptomycin, 100 units/mL penicillin G (Invitrogen, Carlsbad, CA), 0.3 mg/mL geneticin (Fisher Scientific, Pittsburgh, PA) and 1 mg/L basic fibroblast growth factor (Roche Diagnostics, Indianapolis, IN). Both bEnd.3 cells and RBE4 cells were grown in an incubator at 37 °C with 5% CO2 and passaged every 2–3 days.
All animal studies were approved by IACUC at the University of Wisconsin-Madison. Mice C57BL6 (5–6 weeks old) were obtained from Envigo (Indianapolis, IN, USA) and were maintained at 20 °C. Free access to food and water was provided, and the mice were allowed to acclimate to their environment for at least 3 days before the experiments.
ScFv Production and Modification
ScFv46.1 and negative control scFvCtrl (an anti-fluorescein scFv4420 described previously (18)) were subcloned to engineered intein-fused pRS316-FLAG-202–08 vector for protein secretion (18). Yeast secretion strain YVH10 (19) was transformed using the LiAc/ssDNA/PEG method. Transfected YVH10 cells were selected on leucine and uracil deficient SD-2XSCAA + Trp agar plates (20 g/L dextrose, 6.7 g/L yeast nitrogen base, 10.19 g/L Na2HPO4·7H2O, 8.56 g/L NaH2HPO4·H2O, 15 g/L agar 190 mg/L Arg, 108 mg/L Met, 52 mg/L Tyr, 290 mg/L Ile, 440 mg/L Lys, 200 mg/L Phe, 1260 mg/L Glu, 400 mg/L Asp, 480 mg/L Val, 220 mg/L Thr, 130 mg/L Gly, and 40 mg/L Trp, lacking leucine and uracil) as previously described (18) and grown in SD-2XSCAA + Trp liquid medium at 30 °C, 260 rpm overnight. The following day, cultures were reset to OD600 = 0.1 and grown for 72 h at 30 °C at 260 rpm. Yeast were induced by replacing the media with an equivalent volume of SG-2XSCAA + Trp, which is identical to SD-2XSCAA medium except dextrose is substituted for galactose and contains 0.1% w/v bovine serum albumin (BSA), and culturing for 72 h at 20°C and 260 rpm. Where noted, after induction yeast were refed at 48 h with 0.1X of respective culture volume of a 10X solution of yeast nitrogen base and amino acids (67 g/L yeast nitrogenous base, 1900 mg/L Arg, 1080 mg/L Met, 520 mg/L Tyr, 2900 mg/L Ile, 4400 mg/L Lys, 2000 mg/L Phe, 12600 mg/L Glu, 4000 mg/L Asp, 4800 mg/L Val, 2200 mg/L Thr, 1300 mg/L Gly, and 400 mg/L Trp, lacking leucine and uracil). After 72 h of growth in SG-2XSCAA + Trp at 20°C, the yeast supernatant containing the secreted proteins was collected by centrifugation, filtered through 0.22 μm PES membranes, and dialyzed against PBS. ScFv-intein was then purified by Ni-NTA Superflow resin (Qiagen, Hilden, Germany) according to the manufacturer’s protocol.
For the EPL modification, 1 M MESNA and cysteine-azide was added to purified scFv-intein to a final concentration of 100 mM and 5 mM, respectively, and the reaction was allowed to proceed for 20 h at room temperature (20). The reaction mixture was dialyzed against PBS and the scFv-N3 was purified using an anti-FLAG resin (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s protocol.
All of the proteins were individually mixed with SDS-containing sample buffer with reducing reagent and boiled for 10 min prior to loading onto a 4–12% Bis-Tris gel (Thermo Fisher Scientific, Waltham, MA, USA). Gels were stained with Coomassie blue or transferred to a nitrocellulose membrane for Western blotting. Membranes were blocked with 5% fat free dry milk in TBS with 0.1% (v/v) Tween 20, probed with primary antibody anti-FLAG antibody (1:2000 dilution, Sigma F1804) followed by anti-mouse HRP-conjugated antibodies (1:2000 dilution).
Preparation of Liposomes
PC, cholesterol, DSPE-PEG2000, and DSPE-PEG2000-DBCO were dissolved at a mass ratio of 50:8:8:3 in a 1:1 mixture of methanol and chloroform and then dehydrated as previously described to form a film layer (21). Films were then rehydrated in 155 mM (NH4)2SO4 (pH 5.5) via vortexing and heating to >65°C. After sonication for 1 h, the hydrated lipids were extruded through double stacked 100 nm filters (Waterman Filters, Worthington, OH, USA) to obtain liposomes.
For drug loading, liposomes were buffer-exchanged into 123 mM sodium citrate using PD-10 desalting columns (GE Healthcare, Chicago, IL, USA) and immediately mixed with 2-PAM or 3H-2-PAM to facilitate post-formation loading. After incubation at 65 °C for 1 h, liposomes were stored at 4 °C overnight. The next day, free drug was removed using PD-10 desalting columns.
To facilitate scFv decoration (scFv-Lipo), scFv-N3 (scFv46.1-N3 or scFvCtrl-N3) was incubated with DBCO-containing liposomes overnight at 30 °C with shaking. Excess protein was removed by 100 kDa molecular weight cut-off (MWCO) centrifugal filter units (MilliporeSigma, Burlington, MA, USA). The liposomes were then washed with PBS three times. For liposomes labeled with DiI, DiI (at a mass ratio of DiI to PC of 1000:1) was added to the lipid mixture as a fluorescent marker when forming the lipid film. For liposomes labeled with IR800, IR800-N3 was added to scFv-Lipo for another 90 min and the excess IR800-N3 was removed by 10 kDa MWCO centrifugal filter units.
Characterization of Liposomes
2-PAM loading capacity and encapsulation efficiency
2-PAM drug-loading (DL) and encapsulation efficiency (EE) were quantified by separating 2-PAM from lipids and proteins and measuring 2-PAM absorbance at 294 nm compared to a standard curve. The liposomes were treated with methanol first and then lyophilized. The separation was accomplished by adding ddH2O to lyophilized liposomes and centrifuging through 10 kDa MWCO centrifugal filter units (MilliporeSigma, Burlington, MA, USA) at 14000 g for 10 min. Free 2-PAM in the aqueous phase in the filtration tube was quantified, and DL and EE were calculated with the following equations: DL = (mass of loaded drug in liposomes) / (mass of total lipids) × 100%; EE = (mass of loaded drug in liposomes) / (mass of initially added drug) × 100%.
Quantitative determination of scFvs bound to liposomes
scFvs covalently bound to the liposome surface were quantified using a BCA assay in non-2-PAM-loaded liposomes using the manufacturer’s protocol (Pierce). Liposome concentration was measured by Nanoparticle tracking analysis (NTA; Malvern Panalytical, UK). Then, the numbers of scFvs per liposome were quantitatively determined by a combination of BCA assay and Nanoparticle tracking analysis.
Where NA is Avogadro’s number; Cprotein is protein concentration measured by BCA assay; MWprotein is molecular weight of the scFv; Nliposomes is concentration of liposomes measured by Nanoparticle tracking analysis.
Size distribution and zeta potential analysis
Dynamic light scattering (DLS; Malvern Zetasizer, Malvern Panalytical, UK) was used to measure the hydrodynamic diameter, size distribution and zeta potential of liposomes at a 173° detection angle with a sample concentration at 0.07 mg/mL. The stability of the liposomal formulation was evaluated by measuring the size distribution after storing for 30 days at 4°C in physiological saline.
Characterization of in vitro 2-PAM Release Profile
Experiments for the in vitro release of 2-PAM were performed at 37 °C in PBS (pH 7.4) with 10% FBS with constant stirring. In brief, 1 mL of 2-PAM@scFv-Lipo solution was placed in a dialysis bag (MWCO 8000), which was then immersed in 49 mL of buffer solution incubated at 37 °C with continuous stirring. At desired time intervals, 1 mL of the released medium as withdrawn for absorbance analysis (at 294 nm) and 1 mL of the corresponding fresh buffer was added to keep a constant volume. All of the release experiments were carried out in three independent liposomal sample preparations.
In vitro Cytotoxicity Assay
Cytotoxicity assays were performed by using Cell Counting Kit-8 (CCK-8, Sigma-Aldrich Co. Ltd.; St. Louis, MO, USA) to evaluate the influence of 2-PAM-loaded liposomes on the viability of bEnd.3 and RBE4 cells. Both bEnd.3 and RBE4 cells were seeded onto 96-well plates at a seeding density of 1 × 104 cells per well. After the cells grew to about 90% confluence, the medium was replaced by fresh medium containing 2-PAM-loaded 46.1-Lipo at different 2-PAM concentrations ranging from 0–40 μg/mL. The plates were incubated for another 24 h. CCK-8 was added to the plate according to the manufacturer’s protocol. After incubation for 1 h at 37 °C, absorbance at 450 nm of each well was measured using a microplate reader. The absorbance values were normalized to the wells in which cells were not treated with samples. The cell viability was calculated by the equation: Absample/Abcontrol × 100%, in which Absample and Abcontrol are the absorbance values of the testing well (in the presence of samples) and the control well (in the absence of samples), respectively. Data are presented as an average of six independent wells of cells with standard deviations.
Cellular Uptake Study
iPSC-derived BMEC-like cells were differentiated according to a previous protocol (16) and subcultured at day 8 onto collagen/fibronectin-coated plates or Lab Tek II chamber slides. On day 10 when transendothelial electrical resistance reached > 2000 Ω×cm2, iPSC-derived BMEC-like cells were washed once with PBS and incubated with blocking buffer PBSG (10% goat serum in PBS) for 30 min on ice. scFv-intein, scFv-N3, or scFv-Lipo were added to cells and incubated for an additional 30 min on ice to allow binding. The chamber slides were then transferred to 37 °C for 45 min to allow internalization. Afterwards, cells were washed with cold PBS and incubated with anti-FLAG antibody (1:500 in PBSG) for 30 min on ice to label the membrane-bound fraction of scFv. Cells were washed three times more on ice and incubated with anti-mouse AlexaFluor555 or anti-mouse AlexaFluor647 (1:1000 in PBSG) on ice for another 30 min. Then cells were washed with cold PBS for three times and fixed with 4% PFA on ice for 10 min and permeabilized with 0.2% Triton-X for 2 min. At this stage, cells were additionally incubated with anti-FLAG antibody (1:500 in PBSG) for 30 min at room temperature. Cells were then washed with PBS three times and incubated with anti-mouse AlexaFluor 488 (1:1000 in PBSG) for 30 min at room temperature. Finally, cells were washed and mounted with ProLong Gold antifade reagent with DAPI (Invitrogen, Waltham, MA). Images were acquired on Zeiss Axio Imager Z2 Upright microscope or Nikon A1RS HD Confocal Microscope.
In vivo Targeting Efficacy in Brain Tissue
2-PAM@46.1-Lipo and 2-PAM@Ctrl-Lipo were intravenously administered through retro-orbital injection to anesthetized C57BL6 mice in a dose of 5 mg/kg scFv. Mice were anesthetized by intraperitoneal injection of ketamine (100 mg/kg) and xylazine (10 mg/kg). After 1 h, mice were anesthetized and whole-body perfusion was performed at a rate 5 mL/min for 5 min with a physiological solution, supplemented with 100 U/mL heparin, 4 μg/mL fluorescently labeled lectin (LEL Dylight488, Vector laboratories, Burlingame, CA, USA) and 0.1% BSA, followed by additional 5 min of perfusion with 4% PFA. Organs such as brain, heart, lung, spleen, liver, and kidney were collected and snap frozen in liquid nitrogen and stored at −80°C. Whole brain images were acquired using a LI-COR scanner Odyssey (LI-COR Biosciences, Lincoln, NE, USA) for IR800 signal. Sections 8 μm thick were made using a Thermo Scientific CryoStat NX70. Before immunolabeling, sections were air dried for 1 h, permeabilized with 0.2% Triton-X for 30 min, and blocked with PBSG for 30 min at room temperature. To visualize bound scFv, sections were incubated with anti-FLAG AlexaFluor647 antibody and diluted 1:500 in PBSG with 0.2% Triton-X overnight at 4°C. Washing steps were performed using 0.2% Triton-X in PBS. Sections were mounted with ProLong Gold antifade reagent with DAPI and analyzed on Zeiss Axio Imager Z2 Upright microscope.
Pharmacokinetic Studies of 3H-2-PAM@scFv-Lipo in Plasma and Brain Tissue
3H-2-PAM@46.1-Lipo and 3H-2-PAM@Ctrl-Lipo were intravenously administered through retro-orbital injection to anesthetized C57BL6 mice in a dose of 5 mg/kg scFvs. Mice were anesthetized by intraperitoneal injection of ketamine (100 mg/kg) and xylazine (10 mg/kg). An aliquot of orbital vein blood was collected at 0.5, 2, 15, 30, 60, 120 and 240 min after injection. The blood was centrifuged for collection of plasma. At 4 h after injection, mice were anesthetized and whole-body, transcardiac perfusion was performed at a rate 5 mL/min for 5 min with a physiological solution. Brain, heart, lung, liver, spleen, and kidney were removed and weighed for each mouse. Plasma and tissue samples were prepared according to the scintillation analysis protocol provided by Perkin Elmer (Waltham, MA, USA). In brief, blood sample (5–10 μL) or tissue sample (0.1–0.4 g) was mixed with 2 mL SOLVABLE (Perkin Elmer, Waltham, MA, USA) with swirling in 60 °C oven for at least 2 h. After cooling to room temperature, the sample was decolorized by adding 200 μL of 30% hydrogen peroxide and heated at 60 °C for another 30 min. Liquid scintillation cocktail (10 mL; Perkin Elmer) was added to the samples and the mixture was vortexed before being quenched for 1 h in the dark. The samples were then measured using a Tri-Carb 2100TR liquid scintillation counter. Single time point initial rate pharmacokinetic parameters were calculated as previously described (22) and as denoted below:
Where Cplasma(t) is the detected 3H-2-PAM radioactivity (DPM) per microliter of plasma at each blood sampling time (t) (DPM/μL); DPMinjection is the total injected 3H-2-PAM radioactivity; DPMorgans is the detected 3H-2-PAM radioactivity in different organ tissues such as heart, liver, lung, spleen, kidneys; Morgans is the mass weight of the organs; Since the animals were perfused, the (DPMorgans/g organs)/Cplasma at 240 min is a measure of (Vd - V0) (μL/g), the volume of distribution of the 3H-2-PAM in organ tissue in excess of the organ plasma volume (V0) at the final sampling time; Cplasma at 240 min is the terminal detected 3H-2-PAM radioactivity per microliter of plasma after 240 min injection; AUC0–240 min is the area under the plasma concentration curve (DPM*t/μL) and PS is the permeability surface area product (μL/min/g).
Statistical Analyses
Detailed replication strategy is described in figure legends. The comparison of means from two experimental groups was performed with a two-tailed unpaired Student’s t-test; The comparison of means from three or more groups was performed by one-way analysis of variance (ANOVA) followed by Tukey’s honest significant difference (HSD) post-hoc test, p values < 0.05 were considered statistically significant.
RESULTS
ScFv Production and Chemical Functionalization
The targeting antibody was used in single-chain antibody form (scFv) and both scFv46.1 and a negative control scFv, scFvCtrl, were fused to the N-terminus of an engineered intein (18) to allow for intein-mediated protein release and conjugation to chemical moieties suitable for liposomal attachment via an inverse electron-demand Diels-Alder (IEDDA) reaction (Figure 1a). Yeast were used to produce both scFv46.1 and scFvCtrl as scFv-intein fusion constructs, and secreted proteins contained N-terminal FLAG and C-terminal c-myc and His6 tags to allow for affinity purification and detection (Figure 1b). As reflected in Figure 1c, scFv-intein fusions migrated at the expected molecular size of approximately 55 kDa (Figure 1c, Lane 2). Upon reaction with the sulfur nucleophile MESNA, the scFvs were largely cleaved from the intein, which undergoes an N- to S-acyl shift at its N-terminal cysteine, forming a thioester intermediate susceptible to nucleophilic attack (Figure 1b, step 3). This thioester was subsequently reacted with a cysteine-azide to append the azide (N3) group at the C-terminus of the scFv to yield the scFv-N3 with a molecular weight around 30 kDa (Figure 1c, Lane 3). Finally, the modified scFv-N3 was purified from the reaction mixture using an anti-FLAG resin, and scFv-N3 proteins were confirmed using SDS-PAGE and western blot (Figure 1c, Lane 4).
Confirmation of scFv-N3 Activity
To confirm the activity of the scFv-N3 after modification, antibodies were incubated with iPSC-derived BMEC-like cells, which were the original BMEC cells used to identify scFv-46.1 (11), on ice to allow binding to cell surface antigens (Figure 1d, red). Cells were then incubated at 37 °C to promote endocytosis (Figure 1d, green). As depicted in Figure 1d, scFv46.1-N3 retains its capacity to bind and to endocytose in iPSC-derived BMEC-like cells (Figure 1d). As previously described (11), the internalized scFv46.1-N3 localizes to areas proximal to the cell-cell tight junctions upon uptake (Figure 1d). These results indicated that scFv46.1 retained its expected transport capacity after chemical functionalization.
Preparation and Characterization of 2-PAM-loaded Liposomes
A schematic diagram of the construction of the 2-PAM-loaded scFv-Lipo is shown in Figure 1. Pegylated liposomes were synthesized using a composition of PC, cholesterol, DSPE-PEG2000, and DSPE-PEG2000-DBCO at a mass ratio of 50:8:8:3 (21). The addition of the strained cyclooctyne (DBCO) chemical moieties allowed for the covalent linkage of the scFv-N3 to the liposome surface through an inverse electron-demand Diels-Alder (IEDDA) reaction. Specific scFv attachment was confirmed by significant increases in protein concentrations in liposomal formulations possessing the scFv-N3 compared with those scFvs lacking a chemically reactive carboxy-terminal group or a non-azido modified albumin control for protein adsorption (Figure S1b), although scFv adsorption to the liposomes was also observed. The average number of scFvs per liposome was estimated at 70 copies (see Methods for details). The zeta potential of the liposomes was measured and liposomes possessed a net negative charge (Table S1), and no significant difference between 46.1-Lipo and Ctrl-Lipo was observed (p > 0.05 by two-tailed unpaired Student’s t test).
Next, the physical characterization of liposomes before and after 2-PAM loading and scFv modification was performed (Table 1). The unloaded, non-conjugated liposomes had an average hydrodynamic diameter of 86.00 ± 0.60 nm with polydispersity index (PDI) of 0.133 ± 0.007 by DLS (Figure S1a and Table 1) and the liposomal size and PDI were 89.52 ± 0.69 nm and 0.049 ± 0.028, respectively, after 2-PAM loading (2-PAM@liposomes). The 2-PAM drug-loading (DL) and encapsulation efficiency (EE) were 4.7 ± 0.37% and 33.9 ± 3.2%, respectively. After modification with scFvs, both 2-PAM@46.1-Lipo and 2-PAM@Ctrl-Lipo showed similar size distributions of 88.16 ± 0.14 nm and 88.14 ± 1.93 nm, respectively, with narrow PDIs of 0.084 ± 0.011 and 0.096 ± 0.042 (Figure 2a). We also note that the LC and EE of 46.1-Lipo were statistically indistinguishable from those for the Ctrl-Lipo (Table 1). Finally, stability of the liposomal formulation was evaluated after storing for 30 days at 4°C in physiological saline. Neither the 2-PAM@46.1-Lipo nor 2-PAM@Ctrl-Lipo preparations exhibited an increase in average size as a result of storage (Table 1).
Table 1.
Liposome characterization
| Sample | Size (nm) | PDI | Drug-Loading (%) | Encapsulation Efficiency (%) |
|---|---|---|---|---|
| Lipo | 86.0 ± 0.6 | 0.133 ± 0.007 | - | - |
| 2-PAM@Lipo | 89.5 ± 0.6a | 0.062 ±0.023b | 4.7 ± 0.4% | 33.9 ± 3.2% |
| 2-PAM@46.1-Lipo | 88.2 ± 0.1 | 0.084 ± 0.011 | 4.4 ± 0.4% | 30.6 ± 3.3% |
| 2-PAM@Ctrl-Lipo | 88.1 ± 1.9 | 0.096 ± 0.042 | 4.6 ± 0.2% | 31.7 ± 2.1% |
| 2-PAM@46.1-Lipo (30 days) | 93.6 ± 3.2 nmc | 0.194 ± 0.069 | - | - |
| 2-PAM@Ctrl-Lipo (30 days) | 91.6 ± 0.3 nmd | 0.073 ± 0.017 | - | - |
Size for Lipo versus 2-PAM@Lipo, p<0.05
PDI for Lipo versus 2-PAM@Lipo, p<0.05
Size for 2-PAM@46.1-Lipo (30days) versus 2-PAM@46.1-Lipo, no change, p>0.05
Size for 2-PAM@Ctrl-Lipo (30days) versus 2-PAM@Ctrl-Lipo, no change, p>0.05
All other comparisons: p > 0.05, no significant differences among groups from 2-PAM@Lipo, 2-PAM@46.1-Lipo and 2-PAM@Ctrl-Lipo for hydrodynamic size, PDI, DL and EE by one-way ANOVA followed by Tukey’s HSD test.
Figure 2. Characterization and confirmation of the binding activity of scFv-Lipo.
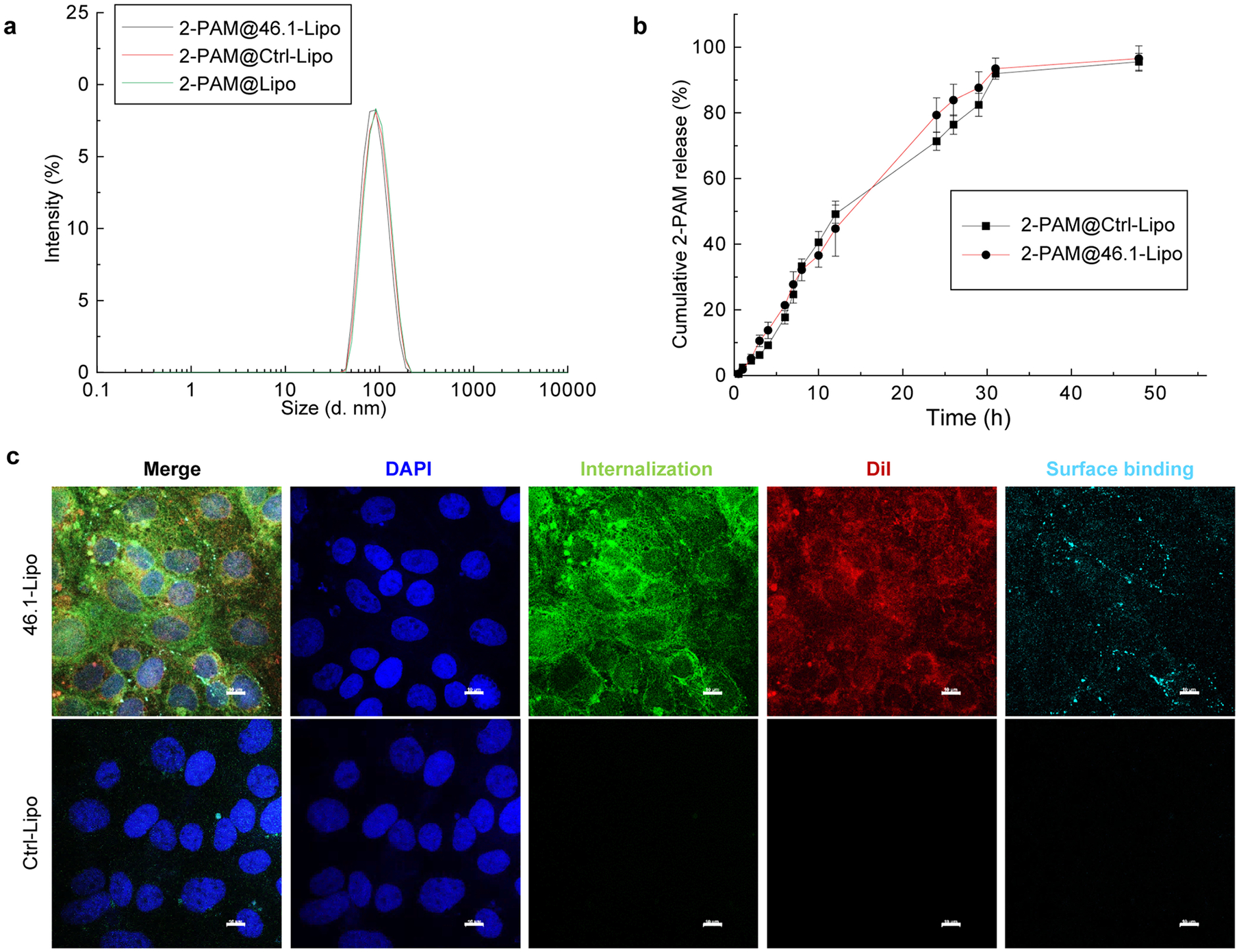
(a) Hydrodynamic size distribution of 2-PAM loaded liposomes as determined using dynamic light scattering. Hydrodynamic size and drug-loading data are presented in Table 1. Reported are means ± S.D., n=3 independent sample preparations.
(b) Release profiles of 2-PAM@46.1-Lipo at 37 °C. Liposomes containing 2-PAM were incubated at 37 °C in physiological saline plus 10% FBS for the time period indicated. 2-PAM release to the buffer was determined by absorbance as detailed in the Methods. Reported are means ± S.D., n=3 independent drug loading and releasing experiments, p > 0.05, no significant differences between 2-PAM@46.1-Lipo and 2-PAM@Ctrl-Lipo at each time point by two-tailed unpaired Student’s t test.
(c) 46.1-Lipo bind to and are internalized by iPSC-derived BMEC-like cells. iPSC-derived BMEC-like cells were incubated with scFv-Lipo at 4 °C and subsequently at 37 °C for 45 min. The cell membrane was washed with cold buffer and the membrane bound fraction (cyan) was labeled with anti-FLAG antibody (recognizing scFv) for 30 min on ice and then anti-mouse AlexaFluor555 antibody on ice for another 30 min. After fixation and permeabilization the internalized scFv (green) was labeled with anti-FLAG antibody at room temperature for 30 min and then labeled with anti-mouse AlexaFluor488 antibody at room temperature for another 30 min. Liposomal DiI fluorescence (red) is used to visualize both cell surface and internalized liposome. Nuclei are stained with DAPI in blue. Images were acquired on Nikon A1RS HD Confocal Microscope and displayed as single z-slices. Scale bar:10 μm.
The release profile of 2-PAM from loaded liposomes was evaluated in physiological saline supplemented with 10% FBS. As shown in Figure 2b, approximately 45% of the 2-PAM was released during the first 12 h, and complete release of the remaining 2-PAM occurred over an additional 18 h; and there were no significant differences in the release of 2-PAM from 46.1-Lipo and Ctrl-Lipo (Figure 2b). To characterize the potential cytotoxicity of these liposomal systems, cell viability assays were performed on both immortalized rat brain (RBE4) and mouse brain (bEnd.3) endothelial cells. Both RBE4 and bEnd.3 cell viability was greater than 90% at 2-PAM concentrations of up to 40 μg/mL with liposome concentration at around 1 mg/mL after 24 h incubation, indicating the low toxicity of the drug delivery system (Figure S2).
46.1-Lipo Uptake into BMEC-like Cells
To determine whether scFv46.1 could trigger endosomal transport at the BBB after conjugation to a liposome, we characterized the uptake of scFv-Lipo using iPSC-derived BMEC-like cells. For these studies, the fluorophore DiI was also directly incorporated into the liposomes to visualize liposomal trafficking behavior using fluorescence microscopy (see Methods). As with the unconjugated antibody above, 46.1-Lipo and the negative control, Ctrl-Lipo, were incubated with iPSC-derived BMEC-like cells on ice to allow binding of the surface antigens and then the cells were warmed to 37 °C to promote internalization. 46.1-Lipo exhibited a clear capacity to bind (cyan) BMEC-like cells, and the liposomes endocytosed and trafficked (green) to the cell-cell junctions (Figure 2c). By comparison, for cells treated with Ctrl-Lipo, only weak fluorescence signal at the level of background was observed. In these experiments, we also observed Dil fluorescence (red) derived from the liposomes to be substantially co-localized with the antibody-derived fluorescence (green) from scFv46.1, suggesting that the liposomes were internalized and trafficked in a largely intact form.
Biodistribution and Pharmacokinetics of 46.1-Lipo
We next investigated the brain uptake of scFv-Lipo after intravenous administration in mice. For initial experiments, both DiI and IR800 were incorporated into liposomes for ex vivo imaging (see Methods for details). Mice were intravenously injected with an scFv-Lipo dose corresponding to 5 mg/kg of scFv and allowed to circulate for 1 h. The unbound fraction of scFv-Lipo was cleared from the blood vessels by whole body perfusion using a physiological saline solution containing fluorescently labeled lectin to visualize the lumen of the blood vessels and as an indirect measure of BBB integrity. Whole brains were imaged by LI-COR scanning using the liposomal-derived IR800 signal. As shown in Figure 3a and quantified in Figure 3b, mice treated with 46.1-Lipo had a significant, 3.1-fold increase in accumulated brain Lipo-associated fluorescence compared to Ctrl-Lipo-treated mice. Immunohistochemical analysis of brain sections also revealed 46.1-Lipo to be associated with the brain endothelium. As shown in Figure 3c, the fluorescence from both the liposome (DiI in red) and scFv46.1 (visualized using a fluorescent anti-FLAG antibody and pseudocolored in cyan) were often found co-localized with lectin-labeled blood vessels (pseudocolored in green), whereas such occurrences were rare in the Ctrl-Lipo-treated group. These observations are consistent with the in vitro model results above, and indicate that 46.1-Lipo can lead to vascular targeting and brain accumulation in vivo.
Figure 3. Accumulation of 46.1-Lipo in mouse brain.
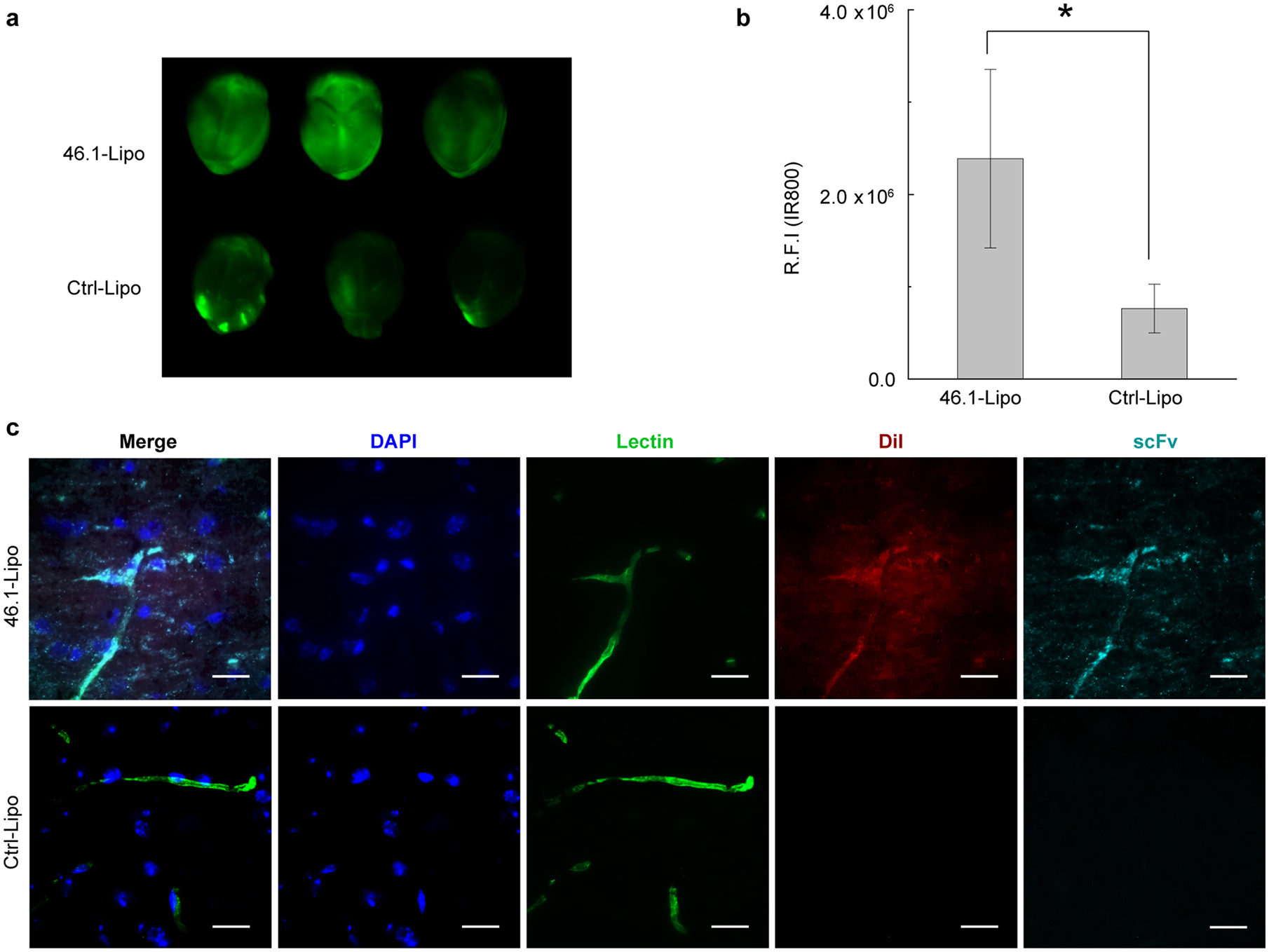
(a) C57BL6 mice were intravenously injected with scFv-Lipo-IR800 at a dose of 5 mg/kg of scFvs (n=3). After one hour of circulation, whole body perfusion was used to remove unbound circulating Lipo. Whole brains were collected and placed on a LI-COR Odyssey scanner and imaged to quantify IR800 labeled 46.1-Lipo or Ctrl-Lipo.
(b) Quantification of relative IR800 fluorescence intensity in brains of (a). Reported are means ± S.D., n=3 independent mouse brains, * p<0.05 by two-tailed unpaired Student’s t test.
(c) Representative brain section images demonstrating co-localization of 46.1-Lipo (red) with blood vessels (green). scFv-Lipo-DiI were injected into C57BL6 mice at a dose of 5 mg/kg of scFvs (n=3). At one hour post-injection, mice were perfused and brains were collected for sectioning. ScFvs (cyan) were labeled with anti-FLAG AlexaFluor647 antibody, liposomes were visualized by DiI (red), and blood vessels (green) were visualized with DyLight488 lectin that was present in the perfusion buffer. Scale bar: 20 μm.
To evaluate the biodistribution and pharmacokinetics, we next loaded tritium-labeled 2-PAM (3H-2-PAM) into scFv-Lipo. The 3H-2-PAM plasma clearance after a single retro-orbital administration of 3H-2-PAM loaded liposomes is shown in Figure 4. 3H-2-PAM@46.1-Lipo showed a longer half-life in vivo than Ctrl-Lipo. After 4 hours of circulation, mice were whole body perfused and organs were isolated to determine 2-PAM uptake and pharmacokinetic parameters. As these are bulk tissue measurements, the organ accumulation data combine vascular and parenchymal contributions. As shown in Table 2, brain uptake of 3H-2-PAM@46.1-Lipo was approximately 10-fold higher than that for control 3H-2-PAM@Ctrl-Lipo group in terms of %ID/g, and 6.6-fold higher in terms of volume of distribution (Vd-V0) and permeability surface area product (PS). Moreover, while the scFv46.1 targeting moiety substantially impacted brain accumulation, uptake of 46.1-Lipo in peripheral tissues was at or below that for Ctrl-Lipo, indicating a brain-specific targeting property endowed by scFv46.1 in a functionalized liposome context.
Figure 4. Plasma clearance after IV injection of 3H-2-PAM@scFv-Lipo.
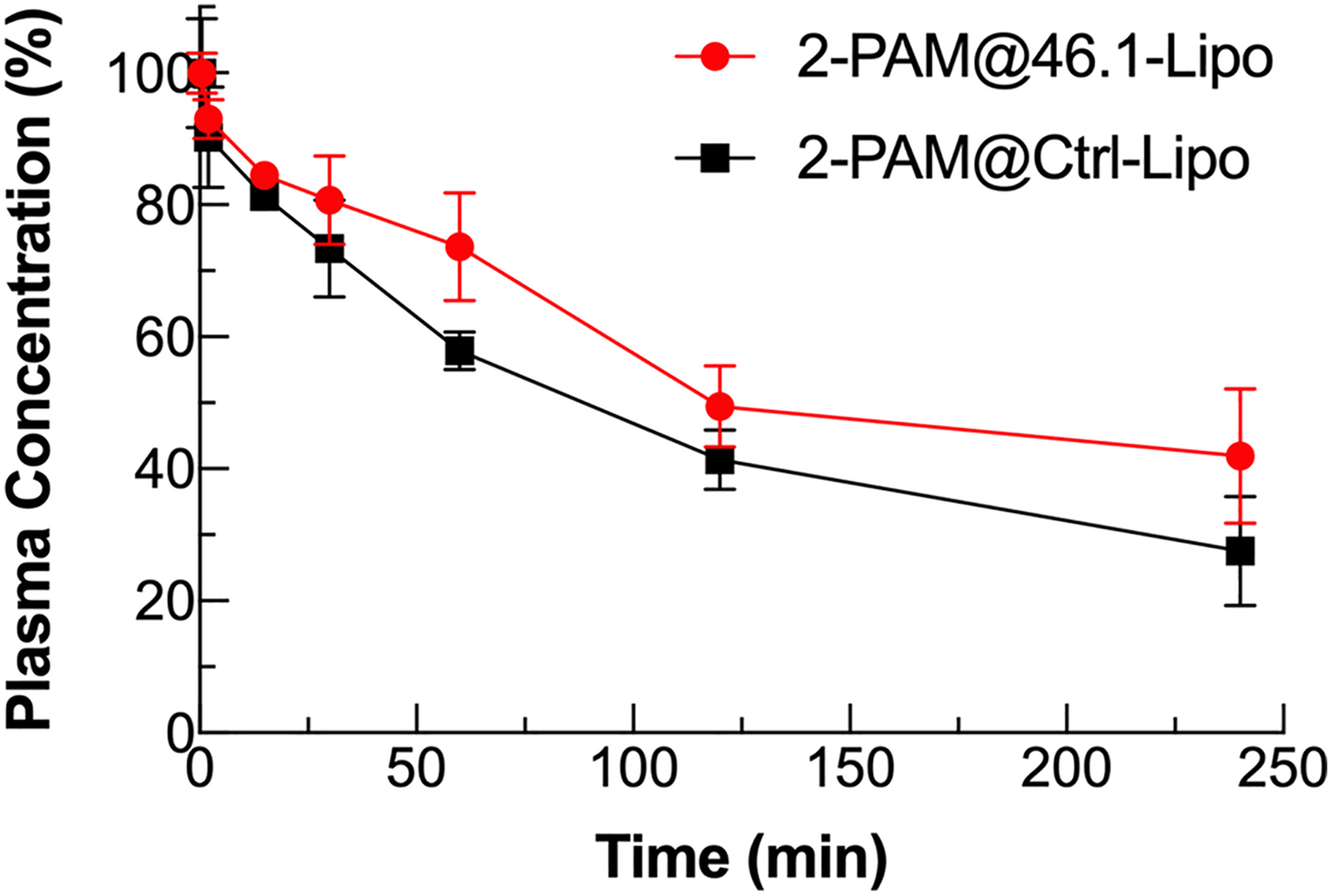
3H-2-PAM@scFv-Lipo was intravenously administered to C57BL6 mice at a dose corresponding to 5 mg/kg of liposomally-conjugated scFvs, and 3.4–4.3 × 107 DPM/kg of 3H-2-PAM (n=3). An aliquot of orbital vein blood sample was collected at 0.5, 2, 15, 30, 60, 120 and 240 min after injection. Plasma amounts of 3H-2-PAM were determined by scintillation counting. The radioactivity at each time point was normalized to that at the 0.5 min time point to allow direct comparison of clearance between the 46.1 and control groups. Reported are means ± S.D., n=3 independent mice.
Table 2.
Pharmacokinetic analysis of 3H-2-PAM after IV injection of 3H-2-PAM@scFv-Lipo in mice.
| Organs | Vd-V0 (μL/g) | PS (μL/min/g) | %ID/g | |||
|---|---|---|---|---|---|---|
| 3H-2PAM@46.1-Lipo | 3H-2PAM@Ctrl-Lipo | 3H-2PAM@46.1-Lipo | 3H-2PAM@Ctrl-Lipo | 3H-2PAM@46.1-Lipo | 3H-2PAM@Ctrl-Lipo | |
| brain | 5.39 ± 0.35* | 0.82 ± 1.43 | 0.016 ± 0.0036* | 0.0024 ± 0.0041 | 0.24 ± 0.016* | 0.023 ± 0.040 |
| heart | 32.36 ± 2.58 | 41.86 ± 9.91 | 0.097 ± 0.023 | 0.10 ± 0.045 | 1.43 ± 0.11 | 1.17 ± 0.28 |
| lung | 21.32 ± 1.93 | 31.46 ± 10.60 | 0.063 ± 0.010 | 0.076 ± 0.036 | 0.94 ± 0.086 | 0.88 ± 0.30 |
| liver | 67.60 ± 10.03* | 162.80 ± 11.56 | 0.20 ± 0.039 | 0.39± 0.13 | 2.99 ± 0.44* | 4.55 ± 0.32 |
| spleen | 38.93 ± 4.85* | 108.52 ± 14.24 | 0.12 ± 0.026* | 0.25± 0.040 | 1.72 ± 0.21* | 3.03 ± 0.40 |
| kidney | 27.72 ± 3.40* | 53.63 ± 8.04 | 0.081 ± 0.0093 | 0.12 ± 0.026 | 1.23 ± 0.15 | 1.50 ± 0.22 |
3H-2-PAM@scFv-Lipo was intravenously administered to C57BL6 mice at a dose corresponding to 5 mg/kg of liposomally-conjugated scFvs, and 3.4–4.3 × 107 DPM/kg of 3H-2-PAM (n=3). An aliquot of orbital vein blood sample was collected at 0.5, 2, 15, 30, 60, 120 and 240 min after injection. Plasma amounts of 3H-2-PAM were determined by scintillation counting and used to determine the AUC. After 4 hours of circulation, mice were whole body perfused, organs were isolated, and scintillation counting was used to measure 2-PAM uptake in the whole organ homogenates. Organ uptake parameters were calculated as described in the Materials and Methods. Reported are means ± S.D., n=3 independent mice.
p<0.05 by two-tailed unpaired Student’s t test.
DISCUSSION
In this paper, we developed and verified a new scFv-modified liposomal system that can selectively target molecular cargo to the brain. The modification of liposomes with scFv46.1 selectively increased the brain uptake while uptake by heart, lung, liver, spleen and kidney were not increased compared with the control group, contrasting favorably with other targeted nanoparticulate systems reported in literature that exhibited more widespread biodistribution (7, 23, 24). To make the scFv compatible with liposomal modification, the scFv was first modified with an azido (N3) group in a site-specific manner and reacted with the DBCO groups on the liposomes to yield scFv-targeted liposomes. The decoration of liposomes using scFv-46.1 enabled the binding to iPSC-derived BMEC-like cells in vitro and subsequent internalization and trafficking to cell-cell junctions (Figure 2c). In addition, the colocalization of scFv46.1 and liposomes observed here strongly suggests that the liposomes remain intact during internalization and trafficking in iPSC-derived BMEC-like cells. Generally speaking, there are three pathways that can be followed by internalized scFv-Lipos including recycling back to the apical plasma membrane, degradation in lysosomal compartments, or shuttling to the basolateral plasma membrane (5). The shuttling of 46.1-Lipo to cell junctions is reminiscent of the free 46.1 antibody, which has previously been demonstrated to traffic to the basolateral side of iPSC-derived BMEC-like cells and fully transcytose, yielding parenchymal uptake in vivo (11). The potential of scFv46.1 as a liposomal BBB-targeting ligand was further confirmed in vivo after intravenous administration of scFv-Lipo. After 1 h of circulation, 46.1-Lipo showed enhanced accumulation in brain tissue compared with the control system, and the 46.1-Lipo injected brain section immunostaining studies yielded a clear localization along the brain microvessels.
To benchmark the potential drug delivery capacity of 46.1-Lipo, and as a proof-of-concept, we loaded 2-PAM into liposomes. 2-PAM is an oxime used for the treatment of poisonings with pesticides like paraoxon (13, 25). However, due to its chemical nature and pharmacokinetic profile, 2-PAM is unable to penetrate the BBB to pharmacologically relevant concentrations, which limits its therapeutic effects in the CNS (26). It has been reported that free 2-PAM exhibits an elimination half-life of 24 min in rats after a single intravenous administration (25). To address these issues, a broad range of nanoparticles with different compositions, sizes and surface properties have been explored for 2-PAM delivery. These include human serum albumin (27, 28), PLGA nanoparticles (29), solid lipid nanoparticles (25, 30), mesoporous silica nanoparticles (31), and liposomes (32, 33). As one example, Pashirova et al. loaded 2-PAM into solid lipid nanoparticles and 2-PAM concentrations in brain peaked 45 min after intravenous administration, while free 2-PAM exhibited a rapid clearance (25). Zhang et al. also demonstrated that transferrin receptor (TfR)-targeting aptamer-functionalized liposomes could deliver oximes both in vitro and in vivo (33). However, none of these previously-reported 2-PAM drug delivery systems examined the brain specificity driven by the targeting ligand, and such targeting could be critical for other payloads that possess peripheral toxicity. After intravenous administration of scFv-Lipo, and 4 h of circulation time, the brain distribution of 3H-2-PAM delivered by 46.1-Lipo was 10-fold higher than that of controls (Table 2), indicating a clear impact of BBB targeting on liposomal accumulation in the brain. Although care needs to be taken when comparing to studies using other targeting approaches as a result in experimental differences such as dose or timing endpoint, the amount of drug targeted to the brain for the 46.1-Lipo system (0.24 %ID/g) was in the range of that previously reported for studies of drug loaded liposomes targeted with engineered transferrin receptor antibody (0.01–0.33 %ID/g) (24, 34–36) or β-amyloid25–35 (%ID/g = 0.93%) (37), although a combination of transferrin receptor antibody with cationic peptides could yield higher brain uptake (2.9%ID/g) (38, 39). In addition, the 10-fold increase in brain targeted accumulation of 2-PAM provided by the 46.1-Lipo was similar to that previously reported for β-amyloid25–35 decorated drug loaded liposomes where treated mice had 14.5-fold higher brain doxorubicin distribution than non-modified liposomes (37). Finally, in contrast to the transferrin receptor targeting approaches where uptake in peripheral organs was 2–50 %ID/g (7, 23, 24, 37), the 46.1-Lipo uptake in peripheral organs was at or below control scFv targeted Liposomes, indicating the rather unique brain selective targeting of the 46.1-Lipo system.
CONCLUSION
Taken together, the results presented here validate this new 46.1 scFv-targeted liposomal platform as capable of selective brain targeting and uptake, and holds the promising potential for selective delivery of liposomal payloads to brain. Future work will be needed to validate that this system is capable of delivering pharmacologic amounts of encapsulated drug payload across the BBB.
Supplementary Material
ACKNOWLEGEMENTS
We would like to thank Lance Rodenkirch and the University of Wisconsin Optical Imaging Core for the help with confocal image data collection. The authors acknowledge the use of instrumentation supported by the NSF through the UW-Madison Materials Research Science and Engineering Center (MRSEC; DMR-1720415). We would like to thank Julia V. Georgieva for help with cellular imaging and animal work.
FUNDING STATEMENT
This work was supported by Defense Threat Reduction Agency grant HDTRA1-15-1-0012 and National Institutes of Health grants NS118028 and NS099158. B.D.G. was supported by NIH Biotechnology Training Program grant T32 GM008349 and the National Science Foundation Graduate Research Fellowship Program under grant number 1747503
Footnotes
DECLARATION OF COMPETING INTEREST
E.V.S. is an inventor on a US patent application dealing with the utility of scFv 46.1. The remaining authors declare that they have no conflict of interest.
APPENDIX A. SUPPLEMENTARY DATA
REFERENCES
- 1.Oller-Salvia B, Sánchez-Navarro M, Giralt E, Teixidó M. Blood–brain barrier shuttle peptides: an emerging paradigm for brain delivery. Chem Soc Rev. 2016;45. [DOI] [PubMed] [Google Scholar]
- 2.Abbott NJ, Rönnbäck L, Hansson E. Astrocyte–endothelial interactions at the blood–brain barrier. Nat Rev Neurosci. 2006;7:41–53. [DOI] [PubMed] [Google Scholar]
- 3.Chen Y, Liu L. Modern methods for delivery of drugs across the blood–brain barrier. Adv Drug Deliv Rev. 2012;64:640–65. [DOI] [PubMed] [Google Scholar]
- 4.Furtado D, Björnmalm M, Ayton S, Bush AI, Kempe K, Caruso F. Overcoming the Blood-Brain Barrier: The Role of Nanomaterials in Treating Neurological Diseases. Adv Mater. 2018;30:1801362. [DOI] [PubMed] [Google Scholar]
- 5.Lajoie JM, Shusta EV. Targeting Receptor-Mediated Transport for Delivery of Biologics Across the Blood-Brain Barrier. Annu Rev Pharmacol Toxicol. 2015;55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Terstappen GC, Meyer AH, Bell RD, Zhang W. Strategies for delivering therapeutics across the blood–brain barrier. Nat Rev Drug Discov. 2021. [DOI] [PubMed] [Google Scholar]
- 7.Johnsen KB, Burkhart A, Melander F, Kempen PJ, Vejlebo JB, Siupka P, et al. Targeting transferrin receptors at the blood-brain barrier improves the uptake of immunoliposomes and subsequent cargo transport into the brain parenchyma. Sci Rep. 2017;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boado RJ, Zhang Y, Zhang Y, Pardridge WM. Humanization of anti-human insulin receptor antibody for drug targeting across the human blood–brain barrier. Biotechnol Bioeng. 2007;96:381–91. [DOI] [PubMed] [Google Scholar]
- 9.Axmann M, Sezgin E, Karner A, Novacek J, Brodesser MD, Röhrl C, et al. Receptor-Independent Transfer of Low Density Lipoprotein Cargo to Biomembranes. Nano Lett. 2019;19:2562–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Giugliani R, Giugliani L, de Oliveira Poswar F, Donis KC, Corte AD, Schmidt M, et al. Neurocognitive and somatic stabilization in pediatric patients with severe Mucopolysaccharidosis Type I after 52 weeks of intravenous brain-penetrating insulin receptor antibody-iduronidase fusion protein (valanafusp alpha): an open label phase 1–2 trial. Orphanet J Rare Dis. 2018;13:110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Georgieva JV, Goulatis LI, Stutz CC, Canfield SG, Song HW, Gastfriend BD, et al. Antibody screening using a human iPSC-based blood-brain barrier model identifies antibodies that accumulate in the CNS. FASEB J. 2020;34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Agrawal M, Ajazuddin, Tripathi DK, Saraf S, Saraf S, Antimisiaris SG, et al. Recent advancements in liposomes targeting strategies to cross blood-brain barrier (BBB) for the treatment of Alzheimer’s disease. J Control Release. 2017;260:61–77. [DOI] [PubMed] [Google Scholar]
- 13.Faiz Norrrahim MN, Idayu Abdul Razak MA, Ahmad Shah NA, Kasim H, Wan Yusoff WY, Halim NA, et al. Recent developments on oximes to improve the blood brain barrier penetration for the treatment of organophosphorus poisoning: a review. RSC Adv. 2020;10:4465–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johnson MK, Jacobsen D, Meredith TJ, Eyer P, Heath AJ, Ligtenstein DA, et al. Evaluation of antidotes for poisoning by organophosphorus pesticides. Emerg Med Australas. 2000;12:22–37. [Google Scholar]
- 15.Lippmann ES, Azarin SM, Kay JE, Nessler RA, Wilson HK, Al-Ahmad A, et al. Derivation of blood-brain barrier endothelial cells from human pluripotent stem cells. Nat Biotechnol. 2012;30:783–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stebbins MJ, Wilson HK, Canfield SG, Qian T, Palecek SP, Shusta EV. Differentiation and characterization of human pluripotent stem cell-derived brain microvascular endothelial cells. Methods. 2016;101:93–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roux F, Durieu-Trautmann O, Chaverot N, Claire M, Mailly P, Bourre J-M, et al. Regulation of gamma-glutamyl transpeptidase and alkaline phosphatase activities in immortalized rat brain microvessel endothelial cells. J Cell Physiol. 1994;159:101–13. [DOI] [PubMed] [Google Scholar]
- 18.Marshall CJ, Grosskopf VA, Moehling TJ, Tillotson BJ, Wiepz GJ, Abbott NL, et al. An Evolved Mxe GyrA Intein for Enhanced Production of Fusion Proteins. ACS Chem Biol. 2015;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shusta EV, Raines RT, Plückthun A, Wittrup KD. Increasing the secretory capacity of Saccharomyces cerevisiae for production of single-chain antibody fragments. Nat Biotechnol. 1998;16:773–7. [DOI] [PubMed] [Google Scholar]
- 20.Umlauf BJ, Mix KA, Grosskopf VA, Raines RT, Shusta EV. Site-Specific Antibody Functionalization Using Tetrazine–Styrene Cycloaddition. Bioconjug Chem. 2018;29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Umlauf BJ, Clark PA, Lajoie JM, Georgieva JV, Bremner S, Herrin BR, et al. Identification of variable lymphocyte receptors that can target therapeutics to pathologically exposed brain extracellular matrix. Sci Adv. 2019;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bickel U How to measure drug transport across the blood-brain barrier. NeuroRX. 2005;2:15–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hatakeyama H, Akita H, Maruyama K, Suhara T, Harashima H. Factors governing the in vivo tissue uptake of transferrin-coupled polyethylene glycol liposomes in vivo. Int J Pharm. 2004;281:25–33. [DOI] [PubMed] [Google Scholar]
- 24.Ko YT, Bhattacharya R, Bickel U. Liposome encapsulated polyethylenimine/ODN polyplexes for brain targeting. J Control Release. 2009;133:230–7. [DOI] [PubMed] [Google Scholar]
- 25.Pashirova TN, Zueva IV., Petrov KA, Babaev VM, Lukashenko SS, Rizvanov IK, et al. Nanoparticle-Delivered 2-PAM for Rat Brain Protection against Paraoxon Central Toxicity. ACS Appl Mater Interfaces. 2017;9. [DOI] [PubMed] [Google Scholar]
- 26.Gallagher E, Minn I, Chambers JE, Searson PC. In vitro characterization of pralidoxime transport and acetylcholinesterase reactivation across MDCK cells and stem cell-derived human brain microvascular endothelial cells (BC1-hBMECs). Fluids Barriers CNS. 2016;13:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dadparvar M, Wagner S, Wien S, Worek F, von Briesen H, Kreuter J. Freeze-drying of HI-6-loaded recombinant human serum albumin nanoparticles for improved storage stability. Eur J Pharm Biopharm. 2014;88. [DOI] [PubMed] [Google Scholar]
- 28.Dadparvar M, Wagner S, Wien S, Kufleitner J, Worek F, von Briesen H, et al. HI 6 human serum albumin nanoparticles—Development and transport over an in vitro blood–brain barrier model. Toxicol Lett. 2011;206. [DOI] [PubMed] [Google Scholar]
- 29.Chigumira W, Maposa P, Gadaga LL, Dube A, Tagwireyi D, Maponga CC. Preparation and Evaluation of Pralidoxime-Loaded PLGA Nanoparticles as Potential Carriers of the Drug across the Blood Brain Barrier. J Nanomater. 2015;2015:1–5. [Google Scholar]
- 30.Pashirova TN, Zueva IV, Petrov KA, Lukashenko SS, Nizameev IR, Kulik NV, et al. Mixed cationic liposomes for brain delivery of drugs by the intranasal route: The acetylcholinesterase reactivator 2-PAM as encapsulated drug model. Colloids Surfaces B Biointerfaces. 2018;171. [DOI] [PubMed] [Google Scholar]
- 31.Yang J, Fan L, Wang F, Luo Y, Sui X, Li W, et al. Rapid-releasing of HI-6 via brain-targeted mesoporous silica nanoparticles for nerve agent detoxification. Nanoscale. 2016;8. [DOI] [PubMed] [Google Scholar]
- 32.Orbesteanu Ana-Maria; Cojocaru Victor; Ailiesei Ioana; Mircioiu Constantin; Cinteza LO. Studies on the formulation of nanostructured carriers for increasing the bioavailability of pralidoxime chloride. Stud Univ “Vasile Goldis”, Ser Stiint Vietii. 2014;24:357–61. [Google Scholar]
- 33.Zhang Y, He J, Shen L, Wang T, Yang J, Li Y, et al. Brain-targeted delivery of obidoxime, using aptamer-modified liposomes, for detoxification of organophosphorus compounds. J Control Release. 2021;329:1117–28. [DOI] [PubMed] [Google Scholar]
- 34.De Luca MA, Lai F, Corrias F, Caboni P, Bimpisidis Z, Maccioni E, et al. Lactoferrin- and antitransferrin-modified liposomes for brain targeting of the NK3 receptor agonist senktide: Preparation and in vivo evaluation. Int J Pharm. 2015;479:129–37. [DOI] [PubMed] [Google Scholar]
- 35.Schnyder A, Krähenbühl S, Drewe J, Huwyler J. Targeting of daunomycin using biotinylated immunoliposomes: Pharmacokinetics, tissue distribution and in vitro pharmacological effects. J Drug Target. 2005;13:325–35. [DOI] [PubMed] [Google Scholar]
- 36.Huwyler J, Wu D, Pardridge WM. Brain drug delivery of small molecules using immunoliposomes. Proc Natl Acad Sci. 1996;93:14164–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Z, Guan J, Jiang Z, Yang Y, Liu J, Hua W, et al. Brain-targeted drug delivery by manipulating protein corona functions. Nat Commun. 2019;10:3561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sharma G, Modgil A, Layek B, Arora K, Sun C, Law B, et al. Cell penetrating peptide tethered bi-ligand liposomes for delivery to brain in vivo: Biodistribution and transfection. J Control Release. 2013;167:1–10. [DOI] [PubMed] [Google Scholar]
- 39.Sharma G, Modgil A, Zhong T, Sun C, Singh J. Influence of Short-Chain Cell-Penetrating Peptides on Transport of Doxorubicin Encapsulating Receptor-Targeted Liposomes Across Brain Endothelial Barrier. Pharm Res. 2014;31:1194–209. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.