

Abstract
Background
Prostate cancer patients experience heterogeneous outcomes after radical prostatectomy. Genomic studies including The Cancer Genome Atlas (TCGA) have reported molecular signatures of prostate cancer, but few studies have assessed the prognostic effects of DNA methylation profiles.
Methods
We conducted the largest methylome subtyping analysis for primary prostate tumors to date, using methylome data from three patient populations: TCGA, a prostate cancer cohort study conducted at the Fred Hutchinson Cancer Research Center (FH), and the Canadian International Cancer Genome Consortium (ICGC) cohort. Four subtypes were detected in the TCGA dataset, then independently assigned to FH and ICGC cohort data. The identified methylation subtypes were assessed for association with cancer prognosis in the above three patient populations.
Results
Using a set of hypermethylated CpG sites, four methylation subtypes were identified in TCGA. Compared to Subtype 1, Subtype 4 had a hazard ratio (HR) of 2.09 (p=0.029) for biochemical recurrence (BCR) in TCGA patients. HRs of 2.76 (p=0.002) for recurrence and 9.73 (p=0.002) for metastatic-lethal (metastasis or prostate cancer-specific death) outcomes were observed in the FH cohort. A similar pattern of association was noted in the Canadian ICGC cohort, though HRs were not statistically significant.
Conclusions
A hypermethylated subtype was associated with an increased hazard of recurrence and mortality in three studies with prostate tumor methylome data. Further molecular work is needed to understand the effect of methylation subtypes on cancer prognosis.
Impact
This study identified a DNA methylation subtype that was associated with worse prostate cancer prognosis after radical prostatectomy.
Keywords: DNA methylation, molecular subtypes, prostate cancer, prognostic markers
Introduction
Prostate cancer accounts for over a quarter of all incident cancer diagnoses in U.S. men and ranks second as a cause of cancer-related deaths (1). The majority of prostate cancer patients present with localized disease at diagnosis. Some have indolent disease that can be cured or safely observed under active surveillance, while others may have highly variable clinical outcomes after radiotherapy or surgery – a portion of these will eventually relapse, with some developing metastasis that ultimately leads to prostate-cancer specific death. A critical unmet clinical need is to identify aggressive prostate cancers that confer a high risk for relapse and metastasis after primary treatment.
Existing risk stratification systems using clinical factors (Gleason score, prostate-specific antigen (PSA) levels, and TNM stage) have not been able to completely predict adverse outcomes (2–4). Over the past decade both genetic and genomic efforts have characterized molecular events contributing to prostate cancer initiation and progression (5–9). Somatic mutations in prostate cancer are less common than in other adult solid tumors (5). A varying degree of copy number aberrations has been observed, with higher burdens of aberrations and instability in aggressive cancers (6,10–12). The most common molecular alteration is the TMPRSS2-ERG gene fusion (13), though its clinical implication is not clear.
In addition to genetic alterations, epigenetic alterations play a crucial role in prostate cancer development. To date, most studies of DNA methylation and prostate cancer progression have been limited to candidate genes in relation to biochemical recurrence (14–19). More recently, genome-wide methylation studies for discovering prognostic markers have been conducted by arrays or pyrosequencing methods (20,21). Among recent studies, TCGA confirmed substantial heterogeneity in DNA methylation and detected widespread changes in hyper- or hypo-methylation in 333 primary prostate tumors when compared to adjacent benign tissue obtained from radical prostatectomies (5,22,23). Using a set of hypermethylated CpG sites, TCGA defined four distinct epigenetic groups, one of which was exclusively associated with ERG fusion positive tumors. The clinical implications of these epigenetic subgroups have yet to be determined, in part due to the short follow-up for patients in TCGA not yielding an adequate number of adverse outcomes. This limitation also applies to characterizing the clinical significance of other molecular subtypes (24), since a disease with a long natural history like prostate cancer generally requires a long follow-up period to observe recurrence and mortality endpoints.
To delineate epigenetic subtypes and determine their clinical significance, we analyzed methylome data from three large primary prostate cancer patient populations who underwent radical prostatectomy (RP), all with clinical information for evaluating outcomes. We performed unsupervised clustering in one of the largest fresh-frozen prostate cancer methylome datasets currently available - the TCGA-PRAD, which includes ~500 samples; and we characterized the risk of recurrence associated with the derived subtypes using the recently curated TCGA clinical data (25). We then applied the classification rule to the other two tumor datasets and tested the resulting subtype associations with clinical factors, recurrence, and mortality.
Materials and Methods
Study participants and data collection
This study includes patients from three cohorts who had localized prostate cancer and treated with RP. CONSORT diagrams for these three cohorts are shown in Supplementary Figure 1.
TCGA-PRAD
The Cancer Genome Atlas Prostate Adenocarcinoma (TCGA-PRAD) dataset includes matched primary tumor and normal (adjacent) specimens from prostate cancer patients who underwent RP as primary therapy (5). All patients provided written informed consent and specimens were approved for collection and distribution by local Institutional Review boards (5). DNA methylation data generated using the Infinium HumanMethylation450 BeadChip (Illumina, San Diego, CA, USA) and RNA-seq data from freshly frozen tumor tissue were downloaded using the TCGAbiolinks R package (v3.1.4) (26); and the HTSeq count gene expression data was log2(count +1) transformed. Tissue images were centrally reviewed by genitourinary pathologists to determine Gleason score. For clinical data, we used the TCGA Pan-Cancer Clinical Data Resource, which includes a progression-free interval (PFI) outcome (27). Prognosis outcomes after RP include non-recurrence (n=387), recurrence only (n=80), and the metastatic-lethal event, that a patient either had metastasis or died from prostate cancer (n=8). There were 475 tumors from patients of Caucasian, African American, and Asian ancestry that had methylation and clinical information available for this analysis.
Fred Hutch cohort
The Fred Hutch (FH) cohort has been previously described in detail (26,27). Briefly, it includes patients with newly diagnosed prostate cancer in 1993–1996 and 2002–2005 who enrolled in population-based studies in King County, WA, and who were identified using the Seattle-Puget Sound Surveillance, Epidemiology, and End Results (SEER) cancer registry. Our study was restricted to 458 men who had RP as the primary therapy for clinically localized adenocarcinoma of the prostate and had genome-wide methylation data and clinical data available. Gleason grade, diagnostic PSA and pathologic tumor stage were collected and centrally coded by Puget Sound SEER (20,30). Pathological stage data were used to define two stage groups: localized stage (pT2, N0/NX, M0); regional stage (pT3-pT4 and/or N1, M0). Time to recurrence or censoring was determined using data from follow-up surveys and review of medical records (n=445). There were 13 patients who did not complete surveys or died of prostate cancer (n=13) prior to mailing of the surveys. Their recurrence time was imputed by a linear regression model including curative therapy and Gleason score and the model was trained to account for the fact that BCR occurs before metastasis or death from prostate cancer. Prognosis outcomes after RP include non-recurrence (n=315), recurrence only (n=114), and metastatic-lethal events (n=29). DNA methylation data were measured using the Infinium HumanMethylation450 BeadChip (Illumina, San Diego, CA, USA) (29) on tumor samples from FFPE blocks and have been deposited to dbGaP (Accession: phs001921.v1.p1). More information about clinical and genomics data collection and quality control procedures have been described previously (20,30). The Fred Hutchinson Cancer Research Center Institutional Review Board approved the study, and all participants provided written informed consent statements.
Canadian ICGC cohort
The Canadian ICGC cohort includes intermediate-risk patients with pathologically confirmed localized prostate cancer as described previously (23). All patients (n=236) were defined as N0M0 and treated with RP. Fresh-frozen RP samples were stored at the University Health Network (UHN) Pathology BioBank and the Centre Hospitalier Universitaire de Qubec-Universit Laval (CHUQ) Genito-Urinary BioBank prior to analysis. All participants provided written informed consent statements, and tumor tissues were utilized based on UHN Research Ethics Board-approved study protocols (UHN 06–0822-CE, UHN 11–0024-CE and CHUQ 2012–913:H12–03–192). Prognosis outcomes after RP include non-recurrence (n=171), recurrence only (n=42), and metastatic-lethal events (n=23). Gleason scores were evaluated by two genitourinary pathologists using hematoxylin and eosin-stained slides (23). Serum PSA levels were measured at the time of diagnosis.
DNA methylation assays and pre-processing
Level 3 TCGA-PRAD data contain β-values, where the β-value is the ratio of the methylated signal over the total signal. Probes that had a common SNP within 10 bp of the interrogated CpG site, were located within 15bp of a repetitive element, aligned to multiple sites on the human genome, or had detection p-values greater than 0.05 for a specific data point were masked (5).
Methylome data from the FH cohort were profiled using the Infinium HumanMethylation450 BeadChip (Illumina, San Diego, CA, USA). We excluded four low quality samples that clustered together with low signal intensities. None of the samples was found to have at least 5% of probes with a detection p-value greater than 0.05. We also excluded non-CpG sites and CpG sites that had a detection p-value greater than 0.05 in at least 10% of samples, CpG sites in any SNP or within 10 base pairs of a SNP with a minor allele frequency greater than 1% in any 1000 Genomes population, or CpG sites classified as cross-reactive probes. We then performed background correction and applied Noob normalization method (31) on the methylation data.
Global DNA methylation for the Canadian ICGC cohort was evaluated using Illumina Infinium HumanMethylation450 BeadChip kits as described previously (23). The raw methylation microarray data were preprocessed using the dasen method from the R package wateRmelon (32). Methylation intensities were filtered based on the detectability over noise level. Probe positions and chromosome locations were annotated using the IlluminaHumanMethylation450kanno.ilmn12.hg19 package (version 0.6.0). All methylation data processing was performed using the R language (version 4.0.5) (33).
Tumor clustering
The TCGA tumors were clustered based on the following criteria for selecting informative hypermethylated CpG sites: 1) having low mean methylation in adjacent benign samples <0.1; 2) uncorrelated with ABSOLUTE (34) tumor purity estimates based on copy number changes; 3) having β-values greater than 0.3 in at least 5% of tumors, a criterion commonly used to detect reliable methylation sites and guard against measurement errors in methylation array; 4) among the top 5,000 most variable probes in tumors; 5) showing differential variability between ~500 primary prostate tumors and 50 adjacent benign prostate samples by the Levene test (with a family-wise error rate <0.05 by Bonferroni correction); and 6) mean methylation <0.1 in adjacent benign samples. These criteria were motivated by the TCGA clustering analysis (5), but made more stringent by requiring lower methylation in adjacent benign samples and significantly increased variability in tumor samples. The final set contained 1306 CpG sites (Supplementary Table 1). Hierarchical clustering was performed using the function eclust in the R package factoextra (v1.0.7) (35) based on these CpGs with dichotomized β-values (> 0.3 vs. ≤ 0.3). Binary distance metric and the agglomeration method of “ward.D2” (36) were used when performing hierarchical clustering. In the FH and Canadian ICGC studies, dichotomized β-values for the same set of probes were used.
Statistical analysis
CpGs with different variability between tumor samples and benign samples were detected by the Levene method implemented in R package Lawstat (v3.4) (37). Hierarchical clustering of TCGA samples was performed by the R package factoextra (v1.0.7) (35). The heatmap of TCGA methylation data was visualized using the R package ComplexHeatmap (v2.6.2) (38). The two-dimensional t-distributed stochastic neighbor embedding (tsne) plot was visualized using the R package Rtsne (v0.15) (39). Assignment of validation samples (FH and ICGC) to the subtypes was determined by the k-Nearest Neighbor Classification (KNN) method, in which k=21 was determined by the square root of the number of complete cases, which is rounded to the nearest odd integer (40). The binary distance metric was used for cluster assignment. In survival analysis, deaths due to other causes than prostate cancer have been accounted for using the cumulative incidence function plots and cause-specific hazard regression (41,42) . Cause-specific Cox proportional hazards regression was used to compute hazard ratios (HRs), 95% confidence intervals (CIs), and p-values. Cumulative incidence functions (CIFs) were plotted to evaluate associations between subtypes and prostate cancer recurrence or metastatic-lethal events, as implemented in the R package cmprsk (v2.2–10) (43). Pathway analysis was done by the gometh function of the R package missMethyl (v1.24.0) (44). Logistic regression models were used to construct the prediction models with predictors including age, race, Gleason score, methylation subtypes, diagnostic PSA level, and tumor stage. The area under curve measure (AUC) of the receiver operating characteristic (ROC) performance measure was computed for prediction models. The ROC analysis was carried out using the R package pROC (v1.17.0.1) (45).
Results
Characteristics of primary cancer patients in the three studies
Table 1 shows the demographic and clinical characteristics of the three primary prostate cancer datasets analyzed for methylation subtypes. Prostate cancer patients from TCGA were older and had more aggressive disease with a higher PSA, a higher tumor grade and a higher T category when compared to FH, and Canadian ICGC studies. On the other hand, the ICGC cohort has fewer patients with Gleason ≤6 and fewer patients with PSA≥20.
Table 1.
Demographic and clinical characteristics of prostate cancer patients in the three studies.
| TCGA (n= 475) |
Fred Hutch (n= 458) |
Canada ICGC (n= 236) |
p-value | ||
|---|---|---|---|---|---|
| Age | Median (25%, 75%) | 62 (57, 67) | 59 (53, 63) | 62 (59, 67) | 4.82×10−16 |
| Race | Caucasian | 405 (85%) | 430 (94%) | 226 (95%) | |
| African American | 58 (12%) | 28 (6%) | 6 (3%) | 3.66×10−7 | |
| Asian | 12 (3%) | 0 (0%) | 4 (2%) | ||
| Gleason Score | <= 6 | 41 (9%) | 217 (47%) | 21 (9%) | |
| 7 (3+4) | 146 (31%) | 166 (36%) | 137 (58%) | <1.0×10−16 | |
| 7 (4+3) | 97 (20%) | 41 (9%) | 58 (25%) | ||
| 8–10 | 191 (40%) | 34 (7%) | 17 (7%) | ||
| Missing | 0 (0%) | 0 | 3 (1%) | ||
| PSA (ng/mL) | <4 | 50 (11%) | 70 (15%) | 24 (10%) | |
| 4–10 | 264 (56%) | 274 (60%) | 155 (66%) | 2.84×10−8 | |
| 10–20 | 93 (20%) | 60 (13%) | 56 (24%) | ||
| ≥20 | 53 (11%) | 26 (6%) | 1 (0.4%) | ||
| Missing | 15 (3%) | 28 (6%) | 0 (0%) | ||
| Stage | Local/T2 | 178 (37%) | 312 (68%) | 114 (48%) | |
| Regional/T3 | 211 (44%) | 146 (32%) | 102 (43%) | <1.0×10−16 | |
| T4/N1 | 80 (17%) | 0 (0%) | 0 (0%) | ||
| Missing | 6 (1%) | 0 (0%) | 20 (8%) | ||
| Subtype | 1 | 137 (29%) | 123 (27%) | 21 (9%) | |
| 2 | 143 (30%) | 210 (46%) | 77 (33%) | <1.0×10−16 | |
| 3 | 109 (23%) | 97 (21%) | 97 (41%) | ||
| 4 | 86 (18%) | 28 (6%) | 41 (17%) |
Clustering TCGA primary prostate tumor samples by cancer-specific methylation
CpGs with increased variability in TCGA primary tumors relative to adjacent benign samples were selected for clustering analysis. Among 216,605 CpGs passing quality control filters, the volcano plot shows that over 95% of the differential variability CpGs are hypervariable, and 20,446 CpGs show statistically significant increased variability (family wise error rate <0.05 for Levene test, red dots in Figure 1A). Aforementioned filters were applied to identify cancer-specific methylation and to limit the impact of tumor purity on clustering. The final set of 1,306 CpGs (Supplementary Table 1) was used in hierarchical clustering. The vast majority of these CpGs do not fall into copy number alteration (CNA) regions previously reported for the prostate cancer genome (6,11,12), therefore the subtypes defined by these CpGs are unlikely to reflect prostate cancer genomic instability due to copy number changes. Four subtypes were identified using the selected 1,306 CpGs (Figure 1b); mean beta values were 0.128, 0.127, 0.277, 0.249, respectively, from Subtype 1 to Subtype 4. The main differences are between Subtype 1–2 versus 3–4. Subtypes 1 and 2 had relatively smaller methylation differences with a subset of CpGs showing differences between these groups (Supplementary Table 1). A similar pattern was seen for Subtypes 3 and 4. In Figure 1c, the two-dimensional tSNE plot shows that samples in Subtype 1 are surrounded by the other three subtypes; there is a clear separation of Subtypes 3 and 4 relative to Subtypes 1 and 2, with the latter two subtypes slightly overlapping. The heatmaps of assigned subtypes for FH cohort and Canadian ICGC cohort are shown in Supplementary Figure 2.
Figure 1.
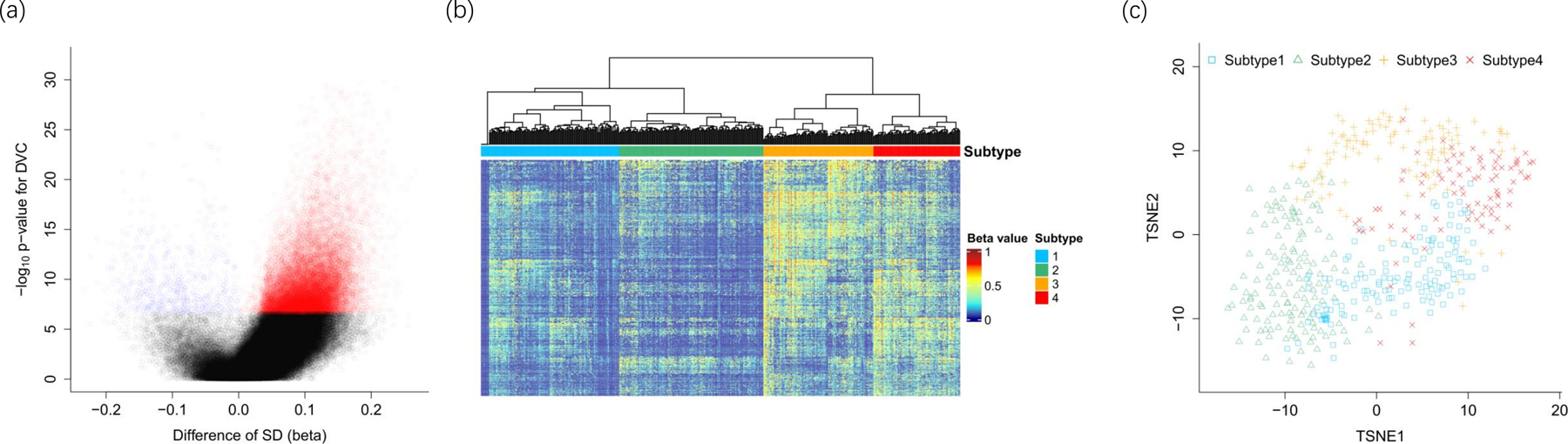
TCGA-PRAD DNA methylation. (a) Volcano plot for testing differentially variable CpGs between cancers and normal samples. (b) DNA methylation subtypes. (c) TSNE plot for tumor DNA methylation subtypes
Table 2 shows the top 20 CpGs that were associated with the 4 Subtypes using the ANOVA test. Nearly all of them were linked to prostate cancer development in the literature, including ESR1 – a gene encoding estrogen receptor 1. Most of them had low methylation in Subtypes 1 and 2, but hypermethylation in Subtypes 3 and 4, consistent with the hierarchical clustering process. Notably, pathway analysis of the 1,306 CpGs showed that the leading enriched pathway is “neuroactive ligand-receptor interaction”, which was previous identified by gene expression data in prostate cancer (46).
Table 2.
Top 20 CpGs associated with the four methylation subtypes.
| CpG | CHR | Position | Gene group | Gene | Subtype1 mean |
Subtype2 mean |
Subtype3 mean |
Subtype4 mean |
p-value* |
|---|---|---|---|---|---|---|---|---|---|
| cg26415547 | 12 | 66583048 | 1stExon|5’UTR | IRAK3 | 0.12 | 0.08 | 0.34 | 0.34 | 2.58×10−45 |
| cg13009111 | 11 | 71350975 | KRTAP5–11|FAM86C | 0.17 | 0.1 | 0.3 | 0.35 | 6.75×10−41 | |
| cg21884421 | 15 | 65648103 | Body | IGDCC3 | 0.13 | 0.16 | 0.48 | 0.32 | 1.59×10−38 |
| cg07462540 | 7 | 8473279 | TSS1500 | NXPH1 | 0.12 | 0.11 | 0.24 | 0.35 | 2.07×10−36 |
| cg20750832 | 5 | 170742345 | TLX3 |NPM1 | 0.21 | 0.22 | 0.49 | 0.49 | 2.88×10−36 | |
| cg16332256 | 4 | 9534388 | DEFB131/MIR548I2 | 0.14 | 0.11 | 0.27 | 0.42 | 4.83×10−36 | |
| cg02145932 | 20 | 13200969 | TSS1500 | ISM1 | 0.12 | 0.05 | 0.3 | 0.36 | 1.10×10−35 |
| cg15720669 | 3 | 138666040 | TSS200 | FOXL2|C3orf72 | 0.09 | 0.13 | 0.37 | 0.3 | 1.56×10−35 |
| cg12072560 | 18 | 12254173 | TSS1500|TSS200 | CIDEA | 0.13 | 0.11 | 0.38 | 0.32 | 9.4×10−35 |
| cg17802942 | 3 | 138666050 | TSS200 | FOXL2|C3orf72 | 0.08 | 0.13 | 0.36 | 0.28 | 1.61×10−34 |
| cg00114029 | 1 | 35351407 | Body | DLGAP3 | 0.14 | 0.13 | 0.31 | 0.37 | 1.19×10−33 |
| cg18932798 | 10 | 105037503 | 1stExon | INA | 0.12 | 0.06 | 0.28 | 0.37 | 1.61×10−32 |
| cg15980539 | 6 | 152128865 | 5’UTR|1stExon | ESR1 | 0.12 | 0.11 | 0.29 | 0.32 | 1.64×10−32 |
| cg11120927 | 2 | 239072674 | KLHL30/ILKAP | 0.1 | 0.19 | 0.42 | 0.33 | 1.82×10−32 | |
| cg15041550 | 6 | 39016590 | 1stExon|5’UTR | GLP1R | 0.16 | 0.16 | 0.41 | 0.33 | 2.95×10−32 |
| cg12664209 | 20 | 13200954 | TSS1500 | ISM1 | 0.21 | 0.07 | 0.42 | 0.49 | 7.97×10−32 |
| cg11850773 | 18 | 904963 | 5’UTR|TSS1500|1stExon | ADCYAP1 | 0.13 | 0.17 | 0.24 | 0.32 | 2.23×10−31 |
| cg00851770 | 18 | 12254175 | TSS1500|TSS200 | CIDEA | 0.09 | 0.08 | 0.29 | 0.24 | 2.30×10−31 |
| cg27109129 | 4 | 74864165 | Body | CXCL5 | 0.23 | 0.28 | 0.63 | 0.5 | 3.20×10−31 |
| cg03382304 | 21 | 27012176 | 1stExon | JAM2 | 0.13 | 0.07 | 0.21 | 0.29 | 3.22×10−31 |
p-value is computed using ANOVA for association with 4 Subtypes.
Tables 3 and 4 show the characteristics of the four subtypes in TCGA patients and FH patients. Consistently in both study cohorts, Subtypes 3 and 4 are more likely to include older men, men having higher Gleason scores, higher PSA levels, and presenting with later-stage disease at diagnosis (Tables 3 and 4). Consistent with the previous paper clustering TCGA-PRAD samples (5), there is a cluster with a substantially higher proportion of patients with the TMPRSS2-ERG fusion transcript (Subtype 2, 88%). Subtype 3 also has a higher portion of patients with the fusion transcript than Subtypes 1 and 4, with Subtype 4 having the lowest frequency of the fusion transcript (6% in TCGA and 11% in FH).
Table 3.
Distribution of relevant demographic and clinical characteristics by subtypes in the TCGA-PRAD cohort.
| Subtype 1 | Subtype 2 | Subtype 3 | Subtype 4 | p-value | ||
|---|---|---|---|---|---|---|
| Age | Median (25%, 75%) | 60 (56, 66) | 60 (55, 65) | 64 (59, 68) | 64 (58, 67) | 1.02×10−5 |
| Race | ||||||
| Caucasian | 112 (82%) | 131 (92%) | 86 (79%) | 76 (88%) | ||
| African American | 19 (14%) | 9 (6%) | 21 (19%) | 9 (10%) | 0.037 | |
| Asian | 6 (4%) | 3 (2%) | 2 (2%) | 1 (1%) | ||
| Gleason Score | ||||||
| <= 6 | 19 (14%) | 15 (10%) | 3 (3%) | 4 (5%) | ||
| 7 (3+4) | 49 (36%) | 56 (39%) | 21 (19%) | 20 (23%) | 8.32×10−5 | |
| 7 (4+3) | 23 (17%) | 23 (16%) | 28 (26%) | 23 (27%) | ||
| 8–10 | 46 (34%) | 49 (34%) | 57 (52%) | 39 (45%) | ||
| PSA (ng/mL) at diagnosis | ||||||
| <4 | 18 (13%) | 19 (13%) | 6 (6%) | 7 (8%) | ||
| 4–10 | 76 (55%) | 88 (62%) | 55 (50%) | 45 (52%) | ||
| 10–20 | 31 (23%) | 25 (17%) | 23 (21%) | 14 (16%) | 7.38×10−3 | |
| ≥20 | 11 (8%) | 7 (5%) | 19 (17%) | 16 (19%) | ||
| Missing | 1 (1%) | 4 (3%) | 6 (6%) | 4 (5%) | ||
| Stage | ||||||
| Local/T2 | 60 (44%) | 60 (42%) | 29 (27%) | 29 (34%) | ||
| Regional/T3 | 54 (39%) | 59 (41%) | 62 (57%) | 36 (42%) | 0.024 | |
| T4/N1 | 21 (15%) | 22 (15%) | 16 (15%) | 21 (24%) | ||
| Missing | 2 (1%) | 2 (1%) | 2 (2%) | 0 (0%) | ||
| TMPRSS2-ERG gene fusion | ||||||
| No fusion | 119 (87%) | 17 (12%) | 75 (69%) | 81 (94%) | 5.9×10−49 | |
| Fusion | 18 (13%) | 126 (88%) | 34 (31%) | 5 (6%) | ||
Table 4.
Distribution of relevant demographic and clinical characteristics by subtypes in the FH cohort.
| Subtype 1 | Subtype 2 | Subtype 3 | Subtype 4 | p-value | ||
|---|---|---|---|---|---|---|
| Age | Median (25%, 75%) | 60 (53, 64) | 57 (51, 61) | 61 (56, 64) | 60 (56, 65) | 6.28×10−6 |
| Race | ||||||
| Caucasian | 106 (86%) | 203 (97%) | 93 (96%) | 28 (100%) | 1.36×10−3 | |
| African American | 17 (14%) | 7 (3%) | 4 (4%) | 0 (0%) | ||
| Gleason Score | ||||||
| <= 6 | 67 (54%) | 110 (52%) | 34 (35%) | 6 (21%) | ||
| 7 (3+4) | 42 (34%) | 75 (36%) | 35 (36%) | 14 (51%) | 1.08×10−3 | |
| 7 (4+3) | 8 (7%) | 13 (6%) | 16 (16%) | 4 (14%) | ||
| 8–10 | 6 (5%) | 12 (6%) | 12 (12%) | 4 (14%) | ||
| PSA (ng/mL) at diagnosis | ||||||
| <4 | 18 (15%) | 36 (17%) | 14 (14%) | 2 (7%) | ||
| 4–10 | 79 (64%) | 132 (63%) | 47 (48%) | 16 (57%) | ||
| 10–20 | 14 (11%) | 20 (10%) | 20 (21%) | 6 (21%) | 0.031 | |
| ≥20 | 3 (2%) | 11 (5%) | 9 (9%) | 3 (11%) | ||
| Missing | 9 (7%) | 11 (5%) | 7 (7%) | 1 (4%) | ||
| Stage | ||||||
| Local/T2 | 94 (76%) | 141 (67%) | 63 (65%) | 14 (50%) | 0.034 | |
| Regional/T3 | 29 (24%) | 69 (33%) | 34 (35%) | 14 (50%) | ||
| TMPRSS2-ERG gene fusion | ||||||
| No fusion | 95 (77%) | 27 (13%) | 53 (55%) | 23 (82%) | ||
| Fusion | 19 (15%) | 172 (82%) | 41 (42%) | 3 (11%) | 1.8×10−35 | |
| Missing | 9 (7%) | 11 (5%) | 3 (3%) | 2 (7%) | ||
Methylation subtypes predict recurrence and metastasis outcomes
The association of the four methylation subtypes with cancer recurrence was assessed using cause-specific proportional hazards models (Table 5 and Supplementary Table 2). Patients with metastasis or prostate cancer death were also compared to patients without recurrence whenever such data were available. A base (minimally adjusted) model was first assessed with age and race to predict BCR; Gleason score was additionally adjusted for to evaluate whether the subtypes were an independent prognostic factor (Supplementary Table 2). In TCGA data, Subtypes 2–4 showed increasing hazards for cancer progression relative to Subtype 1, reaching a hazard ratio of 2.09 (p-value=0.029) for Subtype 4 in the base model (Table5). Adjusting for Gleason score, stage and PSA increased p-values for the associations, to a different degree for the four subtypes in TCGA, e.g., the p-value for Subtype 4 increased to 0.28 (HR=1.47). The association between methylation subtypes and the metastatic-lethal outcome was not assessed because there is a limited number of TCGA patients developed metastatic-lethal event (n=8).
Table 5.
Associations between methylation subtypes and biochemical recurrence (BCR) or metastatic-lethal events. Cause-specific Cox proportional hazards regression models are used. In the base models, BCR is adjusted for age and race; the metastatic-lethal events models are adjusted for age. In the full models, BCR is adjusted for age, race, Gleason score, PSA, and tumor stage; metastatic-lethal events are adjusted for age, Gleason score, PSA, and tumor stage. (a) TCGA-PRAD, results of metastatic-lethal events are not shown due to the limited sample size. (b) FH, (c) Canadian ICGC.
| a) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Base model |
Full model |
||||||||||
| No. patients (No. events) | HR (95% CI) | p-value | Global p-valuea |
No. patients (No. events) | HR (95% CI) | p-value | Global p-valuea |
||||
| BCR | |||||||||||
| Subtype 1 | 137(16) | REF | 134(16) | REF | |||||||
| Subtype 2 | 143(28) | 1.59 (0.86–2.95) | 0.14 | 137(27) | 1.55 (0.83–2.91) | 0.17 | |||||
| Subtype 3 | 109(23) | 1.89 (0.98–3.62) | 0.057 | 0.07 | 101(22) | 1.40 (0.72–2.74) | 0.32 | 0.25 | |||
| Subtype 4 | 86(21) | 2.09 (1.08–4.03) | 0.029 | 82(18) | 1.47 (0.73–2.98) | 0.28 | |||||
|
| |||||||||||
| b) | |||||||||||
|
| |||||||||||
| Base model |
Full model |
||||||||||
| No. patients (No. events) | HR (95% CI) | p-value | Global p-valuea |
No. patients (No. events) | HR (95% CI) | p-value | Global p-valuea |
||||
|
|
|||||||||||
| BCR | |||||||||||
| Subtype 1 | 123(26) | REF | 114(25) | REF | |||||||
| Subtype 2 | 210(65) | 1.44 (0.91–2.29) | 0.12 | 199(59) | 1.14 (0.71–1.86) | 0.57 | |||||
| Subtype 3 | 97(38) | 1.80 (1.09–2.99) | 0.03 | 0.007 | 90(37) | 1.09 (0.64–1.86) | 0.75 | 0.36 | |||
| Subtype 4 | 28(14) | 2.76 (1.43–5.31) | 0.002 | 27(14) | 1.41 (0.70–2.82) | 0.33 | |||||
| Metastatic-lethal | |||||||||||
| Subtype 1 | 100(3) | REF | 91(2) | REF | |||||||
| Subtype 2 | 156(11) | 2.19 (0.60–7.94) | 0.23 | 149(9) | 2.06 (0.43–9.80) | 0.36 | |||||
| Subtype 3 | 69(10) | 4.75 (1.30–17.33) | 0.02 | 0.004 | 63(10) | 3.35 (0.69–16.38) | 0.14 | 0.12 | |||
| Subtype 4 | 19(5) | 9.73 (2.32–40.75) | 0.002 | 18(5) | 5.90 (1.06–32.92) | 0.043 | |||||
|
| |||||||||||
| c) | |||||||||||
|
| |||||||||||
| Base model |
Full model |
||||||||||
| No. patients (No. events) | HR (95% CI) | p-value | Global p-valuea |
No. patients (No. events) | HR (95% CI) | p-value | Global p-valuea |
||||
|
|
|||||||||||
| BCR | |||||||||||
| Subtype 1 | 21(4) | REF | 21(4) | REF | |||||||
| Subtype 2 | 77(19) | 1.60 (0.54–4.73) | 0.39 | 73(16) | 0.84 (0.27–2.63) | 0.77 | |||||
| Subtype 3 | 97(34) | 1.93 (0.69–5.34) | 0.18 | 0.19 | 86(28) | 1.15 (0.39–3.40) | 0.81 | 0.81 | |||
| Subtype 4 | 41(8) | 1.06 (0.32–3.53) | 0.93 | 33(4) | 0.47 (0.12–1.90) | 0.29 | |||||
| Metastatic-lethal | |||||||||||
| Subtype 1 | 18(1) | REF | 18(1) | REF | |||||||
| Subtype 2 | 65(7) | 2.67 (0.32–22.23) | 0.36 | 63(6) | 1.50 (0.16–14.14) | 0.72 | |||||
| Subtype 3 | 76(13) | 3.30 (0.43–25.49) | 0.25 | 0.25 | 68(10) | 1.66 (0.20–14.02) | 0.64 | 0.59 | |||
| Subtype 4 | 35(2) | 1.46 (0.13–16.41) | 0.76 | 30(1) | 0.59 (0.04–10.08) | 0.72 | |||||
Global p-value is for testing association of all 4 subtypes.
Using the KNN method, samples from independent datasets (FH and Canadian ICGC) were assigned to the four subtypes using the classification rule developed in TCGA, and were then evaluated in relation to cancer recurrence or metastatic/death events. Due to quality control criteria, a small number of CpGs among the 1,306 CpGs used for clustering were missing in the FH cohort (90 missing CpGs) and the Canadian ICGC cohort (14 missing CpGs), and were therefore omitted when computing distances. The fractions of the four subtypes were similar in TCGA and FH (Table 1), though FH has fewer Subtype 4 samples; there were proportionally less Subtype 1 and more Subtype 3 samples in the Canadian ICGC cohort, which may reflect its intermediate-risk patient population. Consistent with the TCGA associations, FH patients in Subtypes 2–4 showed increasing HRs (Table 5), reaching 2.76 for Subtype 4 (p-value=0.002) in the base model. The association with metastatic-lethal prostate cancer as compared to no recurrence was even more pronounced, with patients in Subtype 4 showing a HR=9.73 in the base model (p-value=0.002). Adjusting for Gleason score, PSA, and tumor stage diminished the significance, yet the association of Subtype 4 with metastatic-lethal prostate cancer remained significant (p-value=0.043, HR=5.90). The BCR association results in the Canada ICGC cohort are not significant for either of the subtypes, nor the associations with metastasis/death events. In Supplementary Table 2, the associations between subtypes and prognosis were also assessed when adjusting for age and Gleason score, Subtype 4 remained significantly associated with metastatic-lethal prostate cancer in the FH cohort. The global association of 4 subtypes with prognosis outcomes are statistically significant for the FH cohort. A similar trend of hazard ratios was observed for stratified analysis in the three risk groups, though with less significance (Supplementary Table 3).
Figure 2 shows the cumulative incidence function plots for the four subtypes in TCGA and FH, accounting for deaths due to other reasons and separated by BCR events and metastasis/death events. From Subtype 1 to Subtype 4, the gradual increase of hazards for cancer recurrence and for metastasis/death events was more evidently seen with statistical significance in the FH cohort than in TCGA patients, due to a larger number of events in the FH cohort. Adding the 4 subtypes to a logistic regression model with age, Gleason score, PSA, and stage for predicting metastatic-lethal prostate cancer increased the AUC for ROC from 0.8 to 0.831, though the increment is borderline statistically significant (p=0.12, Supplementary Figure 3). Similarly, adding the 4 subtypes to a logistic regression model with age, Gleason score, PSA, and stage for predicting BCR outcomes increased the AUC for ROC from 0.721 to 0.733, with a p-value of 0.12. This is largely due to the observation that among the 4 subtypes, subtype 4 is the one that is significantly associated with prognosis, less so for subtype 2 and 3.
Figure 2.
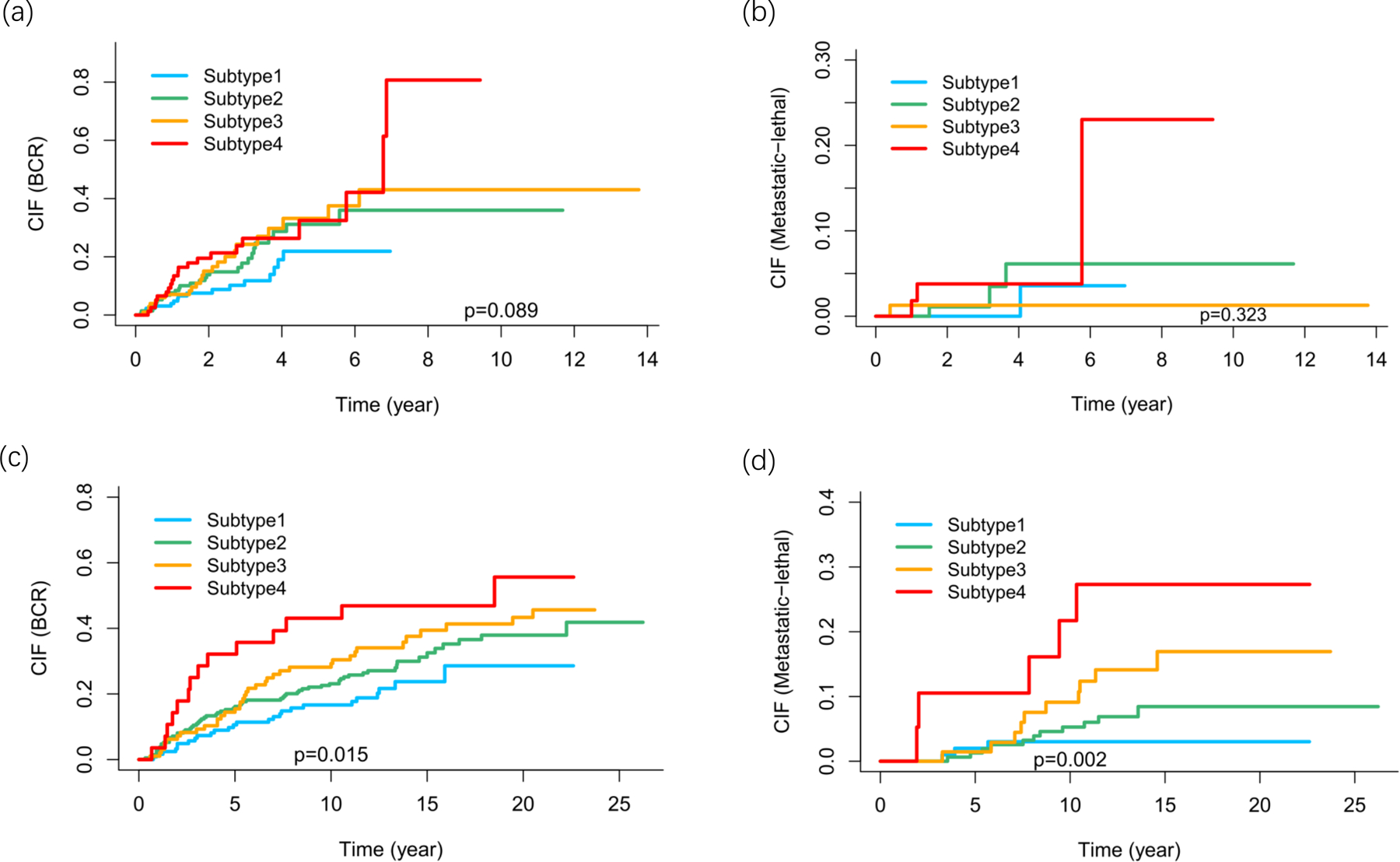
Cumulative incidence functions (CIFs) for developing biochemical recurrence (BCR) or metastatic–lethal events, after accounting for deaths due to other reasons. (a) TCGA, recurrence. (b) TCGA, metastatic-lethal (BCR excluded). (c) FH, recurrence. (d) FH, metastatic-lethal (BCR excluded).
Discussion
Using a set of differentially-variable hypermethylated CpG sites, we have identified four subtypes of primary prostate tumors from patients with localized disease at diagnosis and had been treated with RP, which predict gradually worsening prognosis from Subtype 1 to 4, i.e., increased hazard of biochemical relapse and eventual metastasis and cancer-specific mortality. The subtypes were detected in the TCGA dataset using a clustering algorithm, then the classification rule was independently applied to the FH and Canadian ICGC studies. One of the most striking findings is that, when compared to the first subtype and adjusting for age and race, the fourth subtype had a HR of 2.09 for recurrence (p=0.029) and of 6.05 for metastatic-lethal outcomes (p=0.13) in the TCGA dataset; a HR of 2.76 for recurrence (p=0.002) and a HR of 9.73 for metastatic-lethal outcomes (p=0.002) in the FH dataset. Though these results failed to replicate in the ICGC cohort, the consistency across three studies of an increased risk of recurrence and cancer-specific mortality supports the clinical significance of Subtype 4.
Nearly all previous methylation studies for prostate cancer prognosis were supervised analysis comparing methylation between prognosis groups (14–19,21). To our knowledge, this study is the only analysis that used an unsupervised clustering algorithm to define methylation subtypes first, then identified subtype-prognosis associations. The association between subtype 4 and prognosis was validated in the Fred Hutch cohort. We suspect that the inconsistency in the Canadian ICGC dataset is driven by cohort heterogeneity and the potential batch effects from different methylation experiments. TCGA patients typically presents more aggressive diseases because of the large tumor size TCGA needed to perform multi-omics analyses. The Fred Hutch cohort contains patients with less aggressive disease presentations drawn from a SEER population. The ICGC cohort is an intermediate risk group with fewer patients with Gleason score ≤ 6, and fewer patients with high PSA >20, and so there might be fewer high-risk patients that can be identified to have adverse outcomes (Tables 1 and 5). For example, there were only two metastasis/death events in Subtype 4 in the ICGC cohort. It is also possible that the relatively small proportion of patients assigned to Subtype 1, which was the basis of the comparison, affected our ability to detect a significant association.
In TCGA, Subtypes 3 and 4 had higher Gleason scores, though adjusting for Gleason score did not remove the association between subtypes and outcomes. In the only prospective cohort study (FH), Subtype 4 contained 24 (86%) patients with Gleason score <= 7 (3+4), yet showed a 5 to 10 fold increase in the risk for metastasis and death. Indeed, when the four subtypes were added to a prediction model with age, Gleason score, PSA and tumor stage, the AUC for ROC increased from 0.8 to 0.831 (p-value=0.12) in the FH dataset (Supplementary Figure 3). These results suggest that the methylation subtypes may be an independent predictor of prognosis, likely occurring earlier in the carcinogenesis pathway than before the pathologic indicators at diagnosis such as grade and stage.
An interesting observation is that Subtype 4 had the lowest proportion of tumors with the TMPRSS2-ERG fusion (6% in TCGA and 9% in FH, Tables 3 and 4). Since much higher proportions of the fusion were found in TCGA (31%) and FH (42%), Subtypes 3 and 4 appear to be distinct subgroups with different genomic features. Despite being the most common genomic alteration (~50%) in primary prostate tumors, the clinical implication of TMPRSS2-ERG fusion is not clear (47,48). While there is some evidence that the TMPRSS2-ERG fusion is associated with shorter survival among men managed conservatively or by watchful waiting (49,50), it has not been consistently associated with recurrence or survival among men treated with RP (32,51). Our results add to the existing knowledge of prostate cancer genomics by identifying a high-risk subtype for adverse cancer outcomes that has a low frequency of the TMPRSS2-ERG fusion.
In a similar clinical context, prostate cancer DNA copy number alterations have been shown to predict patient outcome after primary treatment (11). Though DNA methylation may be intertwined with copy number changes, we found no evidence to support that these subtypes are driven by DNA copy number alterations. We examined the overlap of the 1,306 selected CpGs and the most recurrently aberrant regions (95 genes) reported in Table 2 of (11), and we only found four CpGs among the 1,306 CpGs used for clustering, namely cg10722846, cg10795666, cg26559315, and cg26699292, which reside in 4 genes (TRAPPC9, MLNR, LYNX1, WWOX). Similar efforts were made for another two larger sets of CNA regions reported for the prostate cancer genome (6,12): for CNA segments reported in (6), there are 52 CpGs (4%) out of the 1,306 CpGs falling into the reported CNA regions; for CNA segments reported in (12), there are 183 CpGs (14%) falling into the reported CNA regions.
As a major strength of our analysis, we were able to perform methylation subtyping using three large methylome datasets with well-annotated clinical outcomes, including a prospective cohort of prostate cancer cases recruited from a population-based SEER cancer registry (FH study). We detected the methylation subtypes in TCGA and evaluated them in two other studies, diminishing potential confounding in the initial TCGA discovery set. Another strength of our analysis is that it improves on the initial TCGA clustering (5), since we used a subset of differentially variable CpGs that has significantly lower methylation in matched adjacent benign samples to define the subtypes.
A limitation of our analysis is that the number of patients with metastatic-lethal endpoints, the most clinically relevant outcomes, were limited (8 in TCGA, 29 in FH, 23 in Canada ICGC). Though the effect sizes for Subtype 4 were encouraging, the small number of events requires interpreting these associations with caution. Future analyses of larger cohorts with methylation and survival data will be required to further evaluate the prognostic potential of these methylation cluster-defined prostate cancer subtypes.
Supplementary Material
Acknowledgements:
We thank Suzanne Kolb for her help with maintaining and accessing the datasets.
Funding Details:
James Dai and Xiaoyu Wang were supported by the National Cancer Institute (NCI) grant R01 CA222833. Kristina M. Jordahl was supported by the National Cancer Institute (NCI) Training Grant T32 CA094880. Janet L. Stanford was supported by the National Cancer Institute (NCI) grant K05 CA175147. The data collection for this project was supported by NIH grants R01 CA056678, R01 CA092579, and P50 CA097186, with additional support from the Fred Hutchinson/University of Washington Cancer Consortium (P30 CA015704). Computation was in part supported by NIH grant S10OD028685 to the Fred Hutchinson Cancer Research Center. The contents of this work are solely the responsibility of the authors and do not necessarily represent the official views of the NCI. CCSG: This work was supported by the NIH/NCI under award number P30CA016042 and by an operating grant from the National Cancer Institute Early Detection Research Network (1U01CA214194–01) to PCB. PCB was supported by Prostate Cancer Canada and the Canadian Cancer Society.
Footnotes
Disclosure of Interest: The authors report no conflicts of interest.
References
- 1.Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2021. CA Cancer J Clin 2021;71(1):7–33 doi 10.3322/caac.21654. [DOI] [PubMed] [Google Scholar]
- 2.Cooperberg MR, Broering JM, Carroll PR. Risk assessment for prostate cancer metastasis and mortality at the time of diagnosis. J Natl Cancer Inst 2009;101(12):878–87 doi 10.1093/jnci/djp122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.D’Amico AV, Whittington R, Malkowicz SB, Schultz D, Blank K, Broderick GA, et al. Biochemical outcome after radical prostatectomy, external beam radiation therapy, or interstitial radiation therapy for clinically localized prostate cancer. JAMA 1998;280(11):969–74 doi 10.1001/jama.280.11.969. [DOI] [PubMed] [Google Scholar]
- 4.Yoshida T, Nakayama M, Matsuzaki K, Kobayashi Y, Takeda K, Arai Y, et al. Validation of the Prostate Cancer Risk Index (PRIX): a simple scoring system to predict risk of biochemical relapse after radical prostatectomy for prostate cancer. Jpn J Clin Oncol 2011;41(11):1271–6 doi 10.1093/jjco/hyr139. [DOI] [PubMed] [Google Scholar]
- 5.Cancer Genome Atlas Research Network. The molecular taxonomy of primary prostate cancer. Cell 2015;163(4):1011–25 doi 10.1016/j.cell.2015.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fraser M, Sabelnykova VY, Yamaguchi TN, Heisler LE, Livingstone J, Huang V, et al. Genomic hallmarks of localized, non-indolent prostate cancer. Nature 2017;541(7637):359–64 doi 10.1038/nature20788. [DOI] [PubMed] [Google Scholar]
- 7.Boutros PC, Fraser M, Harding NJ, de Borja R, Trudel D, Lalonde E, et al. Spatial genomic heterogeneity within localized, multifocal prostate cancer. Nat Genet 2015;47(7):736–45 doi 10.1038/ng.3315. [DOI] [PubMed] [Google Scholar]
- 8.Li J, Xu C, Lee HJ, Ren S, Zi X, Zhang Z, et al. A genomic and epigenomic atlas of prostate cancer in Asian populations. Nature 2020;580(7801):93–9 doi 10.1038/s41586-020-2135-x. [DOI] [PubMed] [Google Scholar]
- 9.Banerjee, Punnen S. A review on the role of tissue-based molecular biomarkers for active surveillance. World J Urol 2021. doi 10.1007/s00345-021-03610-y. [DOI] [PubMed] [Google Scholar]
- 10.Hieronymus H, Schultz N, Gopalan A, Carver BS, Chang MT, Xiao Y, et al. Copy number alteration burden predicts prostate cancer relapse. Proc Natl Acad Sci U S A 2014;111(30):11139–44 doi 10.1073/pnas.1411446111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lalonde E, Ishkanian AS, Sykes J, Fraser M, Ross-Adams H, Erho N, et al. Tumour genomic and microenvironmental heterogeneity for integrated prediction of 5-year biochemical recurrence of prostate cancer: a retrospective cohort study. Lancet Oncol 2014;15(13):1521–32 doi 10.1016/S1470-2045(14)71021-6. [DOI] [PubMed] [Google Scholar]
- 12.Espiritu SMG, Liu LY, Rubanova Y, Bhandari V, Holgersen EM, Szyca LM, et al. The evolutionary landscape of localized prostate cancers drives clinical aggression. Cell 2018;173(4):1003–13.e15 doi 10.1016/j.cell.2018.03.029. [DOI] [PubMed] [Google Scholar]
- 13.Tomlins SA, Bjartell A, Chinnaiyan AM, Jenster G, Nam RK, Rubin MA, et al. ETS gene fusions in prostate cancer: from discovery to daily clinical practice. Eur Urol 2009;56(2):275–86 doi 10.1016/j.eururo.2009.04.036. [DOI] [PubMed] [Google Scholar]
- 14.Chao C, Chi M, Preciado M, Black MH. Methylation markers for prostate cancer prognosis: a systematic review. Cancer Causes Control 2013;24(9):1615–41 doi 10.1007/s10552-013-0249-2. [DOI] [PubMed] [Google Scholar]
- 15.Ashour N, Angulo JC, Andrés G, Alelú R, González-Corpas A, Toledo MV, et al. A DNA hypermethylation profile reveals new potential biomarkers for prostate cancer diagnosis and prognosis. Prostate 2014;74(12):1171–82 doi 10.1002/pros.22833. [DOI] [PubMed] [Google Scholar]
- 16.Haldrup C, Mundbjerg K, Vestergaard EM, Lamy P, Wild P, Schulz WA, et al. DNA methylation signatures for prediction of biochemical recurrence after radical prostatectomy of clinically localized prostate cancer. J Clin Oncol 2013;31(26):3250–8 doi 10.1200/JCO.2012.47.1847. [DOI] [PubMed] [Google Scholar]
- 17.Horning AM, Awe JA, Wang CM, Liu J, Lai Z, Wang VY, et al. DNA methylation screening of primary prostate tumors identifies SRD5A2 and CYP11A1 as candidate markers for assessing risk of biochemical recurrence. Prostate 2015;75(15):1790–801 doi 10.1002/pros.23052. [DOI] [PubMed] [Google Scholar]
- 18.Holmes EE, Goltz D, Sailer V, Jung M, Meller S, Uhl B, et al. PITX3 promoter methylation is a prognostic biomarker for biochemical recurrence-free survival in prostate cancer patients after radical prostatectomy. Clin Epigenetics 2016;8:104 doi 10.1186/s13148-016-0270-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ahmad AS, Vasiljević N, Carter P, Berney DM, Møller H, Foster CS, et al. A novel DNA methylation score accurately predicts death from prostate cancer in men with low to intermediate clinical risk factors. Oncotarget 2016;7(44):71833–40 doi 10.18632/oncotarget.12377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhao S, Geybels MS, Leonardson A, Rubicz R, Kolb S, Yan Q, et al. Epigenome-wide tumor DNA methylation profiling identifies novel prognostic biomarkers of metastatic-lethal progression in men diagnosed with clinically localized prostate cancer. Clin Cancer Res 2017;23(1):311–9 doi 10.1158/1078-0432.CCR-16-0549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao S, Leonardson A, Geybels MS, McDaniel AS, Yu M, Kolb S, et al. A five-CpG DNA methylation score to predict metastatic-lethal outcomes in men treated with radical prostatectomy for localized prostate cancer. Prostate 2018. doi 10.1002/pros.23667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Geybels MS, Alumkal JJ, Luedeke M, Rinckleb A, Zhao S, Shui IM, et al. Epigenomic profiling of prostate cancer identifies differentially methylated genes in TMPRSS2:ERG fusion-positive versus fusion-negative tumors. Clin Epigenetics 2015;7:128 doi 10.1186/s13148-015-0161-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Houlahan KE, Shiah YJ, Gusev A, Yuan J, Ahmed M, Shetty A, et al. Genome-wide germline correlates of the epigenetic landscape of prostate cancer. Nat Med 2019;25(10):1615–26 doi 10.1038/s41591-019-0579-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stelloo S, Nevedomskaya E, Kim Y, Schuurman K, Valle-Encinas E, Lobo J, et al. Integrative epigenetic taxonomy of primary prostate cancer. Nat Commun 2018;9(1):4900 doi 10.1038/s41467-018-07270-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD, et al. An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell 2018;173(2):400–16.e11 doi 10.1016/j.cell.2018.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stanford JL, Wicklund KG, McKnight B, Daling JR, Brawer MK. Vasectomy and risk of prostate cancer. Cancer Epidemiol Biomarkers Prev 1999;8(10):881–6. [PubMed] [Google Scholar]
- 27.Agalliu I, Salinas CA, Hansten PD, Ostrander EA, Stanford JL. Statin use and risk of prostate cancer: results from a population-based epidemiologic study. Am J Epidemiol 2008;168(3):250–60 doi 10.1093/aje/kwn141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Little RJA, Rubin DB. Statistical analysis with missing data Wiley; 2019. [Google Scholar]
- 29.Stott-Miller M, Zhao S, Wright JL, Kolb S, Bibikova M, Klotzle B, et al. Validation study of genes with hypermethylated promoter regions associated with prostate cancer recurrence. Cancer Epidemiol Biomarkers Prev 2014;23(7):1331–9 doi 10.1158/1055-9965.EPI-13-1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Geybels MS, Wright JL, Bibikova M, Klotzle B, Fan JB, Zhao S, et al. Epigenetic signature of Gleason score and prostate cancer recurrence after radical prostatectomy. Clin Epigenetics 2016;8:97 doi 10.1186/s13148-016-0260-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fortin JP, Triche TJ, Hansen KD. Preprocessing, normalization and integration of the Illumina HumanMethylationEPIC array with minfi. Bioinformatics 2017;33(4):558–60 doi 10.1093/bioinformatics/btw691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pidsley R, Y Wong CC, Volta M, Lunnon K, Mill J, Schalkwyk LC. A data-driven approach to preprocessing Illumina 450K methylation array data. BMC Genomics 2013;14:293 doi 10.1186/1471-2164-14-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Team RC. R: a language and environment for statistical computing: R Foundation for statistical computing; 2021. [Google Scholar]
- 34.Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol 2012;30(5):413–21 doi 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kassambara A, Mundt F. Factoextra: extract and visualize the results of multivariate data analyses. 1.0.72020 [Google Scholar]
- 36.Murtagh F, Legendre P. Ward’s hierarchical agglomerative clustering method: which algorithms implement Ward’s criterion? Journal of Classification 2014;31(3):274–95. [Google Scholar]
- 37.Gastwirth J, Gel Y, Hui W, Lyubchich V, Miao W, Noguchi K. Lawstat: tools for biostatistics, public policy, and law. 3.42020 [Google Scholar]
- 38.Gu Z, Eils R, Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016;32(18):2847–9 doi 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
- 39.Krijthe J. Rtsne: T-distributed stochastic neighbor embedding using a Barnes-Hut implementation 0.152015 [Google Scholar]
- 40.Jonsson P, Wohlin C. An evaluation of k-nearest neighbour imputation using Likert data 2004; Chicago, IL, USA. p 108–18. [Google Scholar]
- 41.Prentice RL, Kalbfleisch JD, Peterson AV, Flournoy N, Farewell VT, Breslow NE. The analysis of failure times in the presence of competing risks. Biometrics 1978;34(4):541–54. [PubMed] [Google Scholar]
- 42.Dignam JJ, Zhang Q, Kocherginsky M. The use and interpretation of competing risks regression models. Clin Cancer Res 2012;18(8):2301–8 doi 10.1158/1078-0432.CCR-11-2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gray B. cmprsk: Subdistribution analysis of competing risks 2020 [Google Scholar]
- 44.Phipson B, Maksimovic J, Oshlack A. missMethyl: an R package for analyzing data from Illumina’s HumanMethylation450 platform. Bioinformatics 2016;32(2):286–8 doi 10.1093/bioinformatics/btv560. [DOI] [PubMed] [Google Scholar]
- 45.Robin X, Turck N, Hainard A, Tiberti N, Lisacek F, Sanchez JC, et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics 2011;12:77 doi 10.1186/1471-2105-12-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.He Z, Tang F, Lu Z, Huang Y, Lei H, Li Z, et al. Analysis of differentially expressed genes, clinical value and biological pathways in prostate cancer. Am J Transl Res 2018;10(5):1444–56. [PMC free article] [PubMed] [Google Scholar]
- 47.Pettersson A, Graff RE, Bauer SR, Pitt MJ, Lis RT, Stack EC, et al. The TMPRSS2:ERG rearrangement, ERG expression, and prostate cancer outcomes: a cohort study and meta-analysis. Cancer Epidemiol Biomarkers Prev 2012;21(9):1497–509 doi 10.1158/1055-9965.EPI-12-0042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gopalan A, Leversha MA, Satagopan JM, Zhou Q, Al-Ahmadie HA, Fine SW, et al. TMPRSS2-ERG gene fusion is not associated with outcome in patients treated by prostatectomy. Cancer Res 2009;69(4):1400–6 doi 10.1158/0008-5472.CAN-08-2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Demichelis F, Fall K, Perner S, Andrén O, Schmidt F, Setlur SR, et al. TMPRSS2:ERG gene fusion associated with lethal prostate cancer in a watchful waiting cohort. Oncogene 2007;26(31):4596–9 doi 10.1038/sj.onc.1210237. [DOI] [PubMed] [Google Scholar]
- 50.Attard G, Clark J, Ambroisine L, Fisher G, Kovacs G, Flohr P, et al. Duplication of the fusion of TMPRSS2 to ERG sequences identifies fatal human prostate cancer. Oncogene 2008;27(3):253–63 doi 10.1038/sj.onc.1210640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hinoue T, Weisenberger DJ, Lange CP, Shen H, Byun HM, Van Den Berg D, et al. Genome-scale analysis of aberrant DNA methylation in colorectal cancer. Genome Res 2012;22(2):271–82 doi 10.1101/gr.117523.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.