

Abstract
Purpose:
The immunosuppressive tumor microenvironment present in the majority of diffuse glioma limits therapeutic response to immunotherapy. As the determinants of the glioma-associated immune response are relatively poorly understood, the study of glioma with more robust tumor-associated immune responses may be particularly useful to identify novel immunomodulatory factors that can promote T cell effector function in glioma.
Experimental Design:
We used multiplex immune-profiling, proteomic profiling, and gene expression analysis to define the tumor-associated immune response in two molecular subtypes of glioma and identify factors that may modulate this response. We then used patient-derived glioma cultures and an immunocompetent murine model for malignant glioma to analyze the ability of tumor-intrinsic factors to promote a CD8+ T cell response.
Results:
As compared with IDH-mutant astrocytoma, MAPK-activated pleomorphic xanthoastrocytoma (PXA) harbored increased numbers of activated cytotoxic CD8+ T cells and Iba1+ microglia/macrophages, increased MHC class I expression, enrichment of genes associated with antigen presentation and processing, and increased tumor cell secretion of the chemokine CXCL14. CXCL14 promoted activated CD8+ T cell chemotaxis in vitro, recruited tumor-infiltrating CD8+ T cells in vivo, and prolonged overall survival in a cytotoxic T cell-dependent manner. The immunomodulatory molecule B7-H3 was also highly expressed in PXA.
Conclusions:
We identify the MAPK-activated lower grade astrocytoma PXA, as having an immune-rich tumor microenvironment and suggest this tumor may be particularly vulnerable to immunotherapeutic modulation. We also identify CXCL14 as an important determinant of the glioma-associated immune microenvironment, sufficient to promote an anti-tumor CD8+ T cell response.
Keywords: Glioma immune microenvironment, tumor infiltrating lymphocytes, macrophage/microglia, CXCL14, MHC class I
INTRODUCTION
Diffuse glioma are characterized as lymphocyte depleted and studies suggest they share an immunosuppressive tumor microenvironment characterized by T cell dysfunction and abundant immunosuppressive myeloid cells (1–3). Yet, not all glioma share this immunophenotype and even within diffuse glioma, the immune microenvironment can vary (4–6). A mechanistic understanding of the determinants of the glioma immune microenvironment across distinct glioma types is critical to develop improved immunotherapeutic strategies for brain tumor patients and identify potential biomarkers for optimal therapeutic stratification.
The glioma immune microenvironment is shaped at multiple levels by tumor cell-extrinsic and tumor cell-intrinsic factors (7). Compared to other peripheral organs, the brain is often considered immunologically ‘distinct’ (8) and certain aspects of the immune microenvironment, including myeloid cell populations, may contribute to an immunosuppressive microenvironment (2,7,9,10). Supporting the important role for tumor-cell intrinsic factors, tumors driven by different epigenetic or genetic alterations can exhibit quantitative and qualitative differences in their tumor-associated immune response (9,11). In pediatric high-grade glioma, mutations in BRAF and NF1 have been associated with increased CD8+ tumor-infiltrating lymphocytes (6). Even within IDH-wildtype GBM, there are differences in the brain tumor immune microenvironment across tumors (4,12). In IDH-mutant astrocytoma several mechanisms may contribute to tumor immune-evasion, including reduced tumor cell expression of interferon-gamma-inducible chemokines, including CXC-chemokine ligand 10 (CXCL10) (1).
To date there has been limited investigation of the determinants of the immune microenvironment in diverse glioma subtypes. We hypothesize that mechanisms supporting a glioma-associated CD8+ T cell response may be relevant across brain tumor subtypes. Thus, in the present study, we identify a MAPK-activated lower grade astrocytoma, pleomorphic xanthoastrocytoma (PXA), as a glioma subtype with a more robust CD8+ T cell response and we use multiplex immune-profiling, spatial profiling, and gene expression analysis to elucidate the tumor immune microenvironment. Then, using human tumor tissues, patient-derived cell lines, and an immunocompetent murine model for glioma, we investigate the tumor-intrinsic determinants of this robust glioma-associated CD8+ T cell response and demonstrate the importance of tumor secreted factors.
MATERIALS and METHODS
Patient Samples and inclusion criteria.
A total of 89 WHO 2016 grade II-IV astrocytoma cases, of which 27 were pleomorphic xanthoastrocytoma (PXA) (mean age 28.7 y), 32 were IDH-mutant astrocytoma (mean age 39.4 y), and 30 were IDH-wildtype glioblastoma (GBM) (mean age 55.1 y), were identified from records in the UCSF Brain Tumor Center Biorepository and the Division of Neuropathology, Department of Pathology, at UCSF. Pleomorphic xanthoastrocytoma (PXA) is genetically characterized by the combination of CDKN2A biallelic inactivation and oncogenic RAF kinase signaling, most commonly the activating BRAF p.V600E (c.1799T>A) mutation (13,14). Less common MAPK alterations identified, include BRAF insertion/deletion mutations (15), fusions involving BRAF or RAF1 (16) and mutation of NF1(13,17). Molecular classification was performed using the UCSF500 targeted next-generation sequencing panel and/or immunostaining for IDH1 R132H, mutant protein, p16, BRAF V600E mutant protein , ATRX, and p53 (18). The samples included in each analysis are specified in the text and figure legend (for additional details please see Supplemental Methods). Non-neoplastic adult brain samples were used as a control. The assays performed on each case are summarized in Supplemental Table S1. The ethics approval number for the use of de-identified human biospecimens is 10-01318. These studies were in accordance with the ethical standards of the institutional research committee and with the 1964 Helsinki declaration and its later amendments.
Multiplex immunofluorescence and immunostaining.
Protein expression, including assessment of tumor-associated immune infiltration, was evaluated using multiplex immunofluorescence (mIFA; for most assays) or immunohistochemistry (for select mouse studies) on FFPE slides using a Discovery XT autostainer (Ventana Medical Systems) with appropriate controls. Cases analyzed by mIFA are listed in Supplemental Table S1, methods described in Supplemental Methods, and all antibodies detailed in Supplemental Table S2.
Image acquisition and analysis.
5X5 tiled images of immunostained slides were acquired at 200x magnification using a Zeiss Spinning Disc confocal & TIRF (Zeiss). For digital quantification FIJI software was used (Supplemental Methods).
Digital spatial profiling of protein expression.
46 immunologically relevant proteins (plus 3 normalization controls and 3 negative isotype controls, Supplemental Table S3) were quantified using the NanoString Inc. GeoMX platform (19) (Supplemental Methods). Slides were also stained with fluorescently labeled antibodies directed against CD3 (Cyan,), Iba1 (red), CD34 (yellow) and DAP1 nuclear staining (blue) which served as visualization markers. ROIs was selected as geometric shapes in T-cell abundant regions.
Nanostring PanCancer Immune Profiling.
Information on cases selected for Nanostring profiling and more detailed Methods are detailed in Supplemental Table S1 and Supplemental Methods. The multiplex gene expression analysis using nCounter® PanCancer immune profiling panel (n = 730 genes involved in the immune response and 40 housekeeping genes, NanoString Technologies™, Seattle, Washington, USA) was performed at the CAT facility, UCSF. Raw counts expression was analyzed using the nSolver™ Analysis Software (NanoString Technologies™ RRID:SCR_02171). The R package “limma” (version 3.29.0) was used for differential gene expression analysis. Multiple comparison testing using the Benjamini-Hochberg method was used to control the false discovery rate (FDR). Genes with FDR less than 0.1 were reported. Two-by two comparisons were performed and differentially expressed genes (DEGs) were selected using expression levels p-value ≤0.05.
Analysis of RNA-sequencing data from Children’s Brain Tumor Network.
Mutational calls and normalized RNA sequencing data from pediatric low-grade (WHO grade I-II) and high-grade astrocytoma (WHO grade III-IV), from the Children’s Brain Tumor Network (CBTN) was downloaded via the Gabriella Miller Kids First Data Resource Portal. Data from a total of 314 tumors were divided into three cohorts based on molecular features: RAF altered astrocytoma (n=38), defined by mutations or fusions involving RAF1 or BRAF; RAF wildtype astrocytoma (n=164), as defined by an absence of RAF1/BRAF alterations; and tumors most likely to be WHO grade I pilocytic astrocytoma (20), as defined by the presence of KIAA1549-BRAF fusion, FAM131-BRAF fusion, or RNF130-BRAF fusions (n=112). T-effector (T-eff) score was calculated as the geometric mean of the TPM (Transcripts Per Kilobase Million) of GZMA, GZMB, PRF1, IFN-γ, EOMES, and CD8A (21). The Cytolytic score (CYT) was calculated in a similar manner using the geometric mean of the TPM of GZMA and PRF1 as previously described (22). Data analysis and visualization were performed with R version 4.0.0.
RNAscope.
FFPE sections were evaluated by RNAscope chromogenic in situ hybridization (CISH) assay for the expression of CXCL14 using Advanced Cell Diagnostics (ACD, RRID:SCR_012481) probes (Newark, CA) specific for CXCL14 (425299-C2). The RNA Probe PPIB (313909) and dapB (312039) were used as positive and negative control probes, respectively.
Cell Culture, condition media and treatments.
Patient-derived PXA cell lines, PXA1 (SF12183) and PXA2 (SF12570), were established from biopsy tissue from recurrent anaplastic PXA, WHO grade III, and maintained under adherent conditions in DMEM media. DBTRG-05MG (RRID: CVCL_1169) and the immortalized human fetal astrocytes line SVG-p12 (RRID:CVCL_3797) were purchased from American Type Culture Collection (ATCC). IDH Mt cells are immortalized normal human astrocytes expressing E6/E7 oncoprotein, catalytic subunit of telomerase, and heterozygous IDH1 R132H mutation (23). Tumor prone murine progenitor cells (mTPC) containing EGFRvIII were generated in the laboratory and cultured under non-adherent conditions (24). All the established cell lines and primary cell line used were cultured for less than 30 and 7 passages, respectively, were maintained at 37°C in a humidified 5% CO2 incubator (Supplemental Table S4), authenticated by analysis of short tandem repeats (STR), and confirmed mycoplasma negative by PCR twice per year. Glioma cell condition media (CM) was isolated on day 3 when cells were 80–90% confluent and centrifuged at 1000 rpm at 4°C for 4 minutes to remove debris (Supplemental Methods). Cells were treated for 24 hours with recombinant human CXCL14 (Peprotech, Catalog #300-50) or IFN gamma (Peprotech, Catalog #500-M90). Inhibitor AMD3100 was used at 10μM (Millipore Sigma, Catalog #A5602) for 15 minutes. siRNA knockdown of CXCL14 was performed using siRNA ON-TARGETplus siRNA SMARTpools targeting CXCL14 (siCXCL14, #SO2860488G) or control siScramble (siScr, #D-001810-10-05) (Dharmacon, Lafayette, CO). Cells were analyzed 48 hours from initial transfection. Primer sequences used for qRT-PCR are listed in Supplemental Table S5.
Flow cytometry and ELISA.
Single cell suspensions were incubated with Fc Block (BD Bioscience), stained with antibodies (Supplemental Table S2) according to standard protocols, and run on Attune instrument (BD Biosciences). Samples were evaluated with appropriate isotype controls and analyzed using FlowJo software (TreeStar). The CXCL14 ELISA Kit (R&D Systems; Catalog Number# DY866) was used according to the manufacturer’s protocol and absorbance obtained at 450nm (Epoch Spectrophotometer, BioTek). A standard curve was generated using recombinant ligand and CXCL14 concentration was interpolated.
Isolation of human CD8+T cells and in vitro transmigration assay.
Human CD8+ T cells were isolated using EasySep™ direct human CD8+ T cell isolation kit (Stem Cells; Catalog #19663) and cultured in ImmunoCult ™XF T Cell Expansion Medium (Stem Cells; Catalog #10981), Human Recombinant IL-2 (Stem cells, Catalog # 78220) and ImmunoCult™ Human CD3/CD28 T Cell Activator (Stem cell, Catalog #10970). Cells were incubated at 37°C and 5% CO2 for 3 days before plating in the top of Transwell inserts (5.0-μm pore size; Costar) (Supplemental Methods).
Generation of cell lines transduced with CXCL14.
Murine tumor-prone neural progenitor cells containing EGFRvIII (mTPCs) were transduced with either Lenti ORF particles, CXCL14-GFP-tagged (mTPCs-CXCL14) (Origene technology, Catalog #MR222808L4V) or with pLenti-C-mGFP-P2A (mTPCs-Control) (Origene Technology, Catalog #PS100093V) according to the manufacture’s protocol. The transduced cells were expanded, cultured as described previously using minimal essential media under non-adherent conditions, and GFP-positive cells were selected by cell sorting (24).
Intracranial injection of transduced mTPCs cells.
All experiments were performed in compliance with institutional guidelines and regulations following approval from the appropriate institutional review board (UCSF Office of Research Institutional Animal Care and Use Committee (IACUC), #AN185514-01). FVB mice (male, 4 weeks, weight ranged from 20-21gm) were obtained from Charles River (Charles River Laboratories, Wilmington, MA, RRID: IMSR_CRL:207) and nude mice (Foxn1nu, female, 4 weeks, weight ranged from 19-20gm) were obtained from the Jackson Laboratory (The Jackson Laboratory, Maine, RRID: IMSR_JAX:007850) and were maintained in accordance with the IACUC guidelines. Intracranial allografts were performed as described previously (24) (Supplemental Methods).
CD8 T immune cell depletion in vivo.
Mice implanted with CXCL14-mTPC received intraperitoneal administration of anti-CD8α (clone GK 2.43) or control isotype anti-IgG (mIgG2b, clone LTF-2) beginning 4 days prior to tumor cell implantation and continuing twice a week until day 60. In parallel, mice without implanted tumor were treated with either anti-CD8α or anti-IgG and splenocytes were isolated on days 2, 23 and 44 post-implantation to verify CD8α cell depletion by flow cytometry (Supplemental Tables S2, S6).
Statistical analyses.
For in vivo studies 5-10 mice were used per treatment group. Data analyzed using GraphPad Prism 9.00 software. Data evaluated by one-way ANOVA, 2-sided, unpaired Mann-Whitney U test, and Student’s t test as stated in Figure Legends. Survival graphs were calculated using Kaplan Meier analysis. A p value < 0.05 was defined as statistically significant.
Data Availability Statement.
The human sequence data generated in this study are not publicly available due to patient privacy requirements but are available upon reasonable request from the corresponding author. Other data generated in this study are available within the article and its supplementary data files. Some of the data analyzed in this study were obtained from Children’s Brain Tumor Network (CBTN) and was downloaded via the Gabriella Miller Kids First Data Resource Portal (https://portal.kidsfirstdrc.org/login).
RESULTS
Quantitative differences in the tumor-associated immune response in molecularly distinct astrocytoma subsets.
Tumor-infiltrating lymphocytes (TILs) play an important role in the tumor microenvironment by influencing the growth and progression of cancer cells (25). Given previous reports suggesting that some RAF-altered glioma, predicted to be MAPK-activated, exhibit an increased CD8+ T cell response (6), we investigated CD3+ TILs in three molecular glioma subsets with astrocytic morphology, as follows: (i) pleomorphic xanthoastrocytoma, characterized by RAF alterations, most commonly BRAF p.V600E mutation, and concomitant CDKN2A homozygous deletion, WHO grade II and III (PXA, n=22); (ii) IDH-mutant astrocytoma WHO grade II and III (IDH-mt, n=29); and (iii) glioblastoma, IDH-wildtype, WHO grade IV (GBM, n=30) (Figure 1). PXAs harbored a significantly higher mean percentage of CD3+ T cells (8.1%) as compared with IDH-mt astrocytoma (3.9%, p<0.01) and IDH-wt GBM (2.1%, p<0.0001) (Figure 1A). In PXA, 23.7% of CD3+ T cells were cytotoxic T cells (dual CD8+CD3+), a 12-fold increase relative to IDH mt astrocytoma (p<0.05; Figure 1B and 1D, Supplemental Figure S1A). In contrast, few CD3+ T cells were regulatory T cells (dual FOXP3+CD3+) (mean 1.7% vs. 28.9% in IDH-mt astrocytoma, p<0.05; Figure 1C, Supplemental Figure S1B). In PXA, CD3+ T cells were located in perivascular regions as well as diffusely throughout the tumor (Figure 1E). Cytotoxic activity of T cells requires an activated effector T cell state, including expression of granzymes, including granzyme B (GZMB), a serine protease secreted by activated cytotoxic effector T cells. In PXA, 22.1.% of CD8+ T cells co-expressed GZMB while in IDH-mt astrocytoma less than 1% of CD8+ T cells co-expressed GZMB (p<0.05, Figure 1F–G).
Figure 1. Quantitative analysis of immune infiltrates in molecularly distinct subsets of astrocytoma.

(A) CD3+ T-cells as a percentage of total cells in PXA (n=22), IDH-mutant (IDH-mt, n=29) and glioblastoma, IDH-wildtype (GBM, n=30). (B,C) Dot plot showing CD3+ T cells double-positive for CD8 (B) or FOXP3 (C) as a percentage of total CD3+ cells in PXA (n=22) and IDH-mt (n=27). (D) Representative images of dual CD3 (green) and CD8 (violet) immunostaining in PXA. Dotted lines denote magnified region at lower right. Arrowhead highlights CD3+CD8+ T cell. (E) Representative images demonstrating regions of PXA with predominant perivascular (arrows) and parenchymal (arrowheads) CD3+ T cells (brown) and associated Iba1+ microglia/macrophages (purple). (F) Dot plot showing granzyme B (GZMB)+CD8+ cells as a percentage of total CD8+ cells in PXA (n=22) and IDH-mt (n=5). (G) Representative images demonstrating granzyme B+ cells (yellow), CD8+ T cells (violet), and dual positive cells (white) in PXA. Arrowhead highlights GZMB+CD8+ T cell. (H-I) T effector cell score (H) and cytolytic cell score (I) from gene expression analysis of pediatric low-grade and high-grade astrocytoma comparing RAF altered astrocytoma (n=38) and RAF wildtype astrocytoma (n=164). Data from CBTN downloaded via Kids First Data Resource Portal. (J-K) Dot plot showing Iba1+ and CD163+ cells as a percentage of total cells in PXA (n=23) and IDH-mt (n=27). (L) Representative images of multiplex immunostaining showing Iba1+ (green), CD163+ (violet), and dual positive (white), cells in PXA and IDH-mt. Arrowhead denotes Iba1+CD163+ cells. In (A-C), (F), and (H-K) each dot represents a value from a single patient, black lines represent the mean, and error bars indicate the SD. Nuclei are stained with DAPI (blue). Significance evaluated by one-way ANOVA and 2-sided, unpaired Mann-Whitney U test. *p<0.05, **p < 0.01, ***p < 0.001, n.s. indicates p > 0.05. Scale bar 20 μm.
Alterations in RAS/RAF/MAPK pathway signaling are relatively common in pediatric and young adult low grade glioma (26). Given the relatively high cytotoxic T effector TIL population in PXA, we investigated whether this might be a broader phenotype across RAF altered glioma. Using available genomic and transcriptomic data from 314 pediatric low- and high-grade astrocytoma, WHO grade I-IV, from the Children’s Brain Tumor Network (CBTN) we identified three cohorts of tumors (defined in Methods): RAF altered astrocytoma (n=38), RAF wildtype astrocytoma (n=164), and pilocytic astrocytoma (n=112). RAF altered astrocytoma had an increased T cell score (p<0.001) and an increased cytolytic T cell score (p<0.0001) relative to RAF wildtype astrocytoma (Figure 1H–I) and pilocytic astrocytoma (Supplemental Figure S1C–D).
Myeloid cells help regulate the tumor-associated immune response in glioma (9,10). In PXA (n=23), there were increased numbers of tumor-associated microglial/macrophages (TAMs), as defined by Iba1 expression, relative to IDH-mt astrocytoma (n=27) (mean 32.2% vs. 23.3%, respectively; p<0.001) (Figure 1J–L). TAMs can exhibit different activation phenotypes. The proportion of CD204 (macrophage scavenger receptor 1) expressing cells was comparable in PXA and IDH-mt astrocytoma (15.9% vs 15.4%, respectively; p=0.9, ns; Supplemental Figure S1E). In contrast, the proportion of CD163 cells was increased in PXA (3.7% vs 1.4%, respectively; p<0.05), although the total population of CD163 cells was low in both tumor types (Figure 1K–L). Taken together, these data suggest that PXA have a more robust cytotoxic TIL response and TAM response as compared to IDH-mt astrocytoma.
High-plex proteomic profiling of the immune response in PXA and identification of potential therapeutic targets.
To better characterize the tumor-associated immune response in PXA, we performed high-plex spatial profiling of 46 immune-response related proteins in a cohort of PXA (n=9) and IDH-mt astrocytoma (n=8). Analysis of CD3+ T cell rich regions revealed both quantitative and qualitative differences in the tumor-associated immune response (Figure 2A). PXA demonstrated an increase in overall CD45+ immune cells, CD3+ T cells, CD8+ T cells, HLA-DR antigen-presenting cells, and CD20+ B cells (p<0.05 for all). In contrast, CD56+ NK cells were not significant but trended lower in PXA and CD11c+ myeloid cells also did not demonstrate a difference (Figure 2B).
Figure 2. High-plex spatial proteomic profiling of immune response in PXA and IDH-mutant astrocytoma.
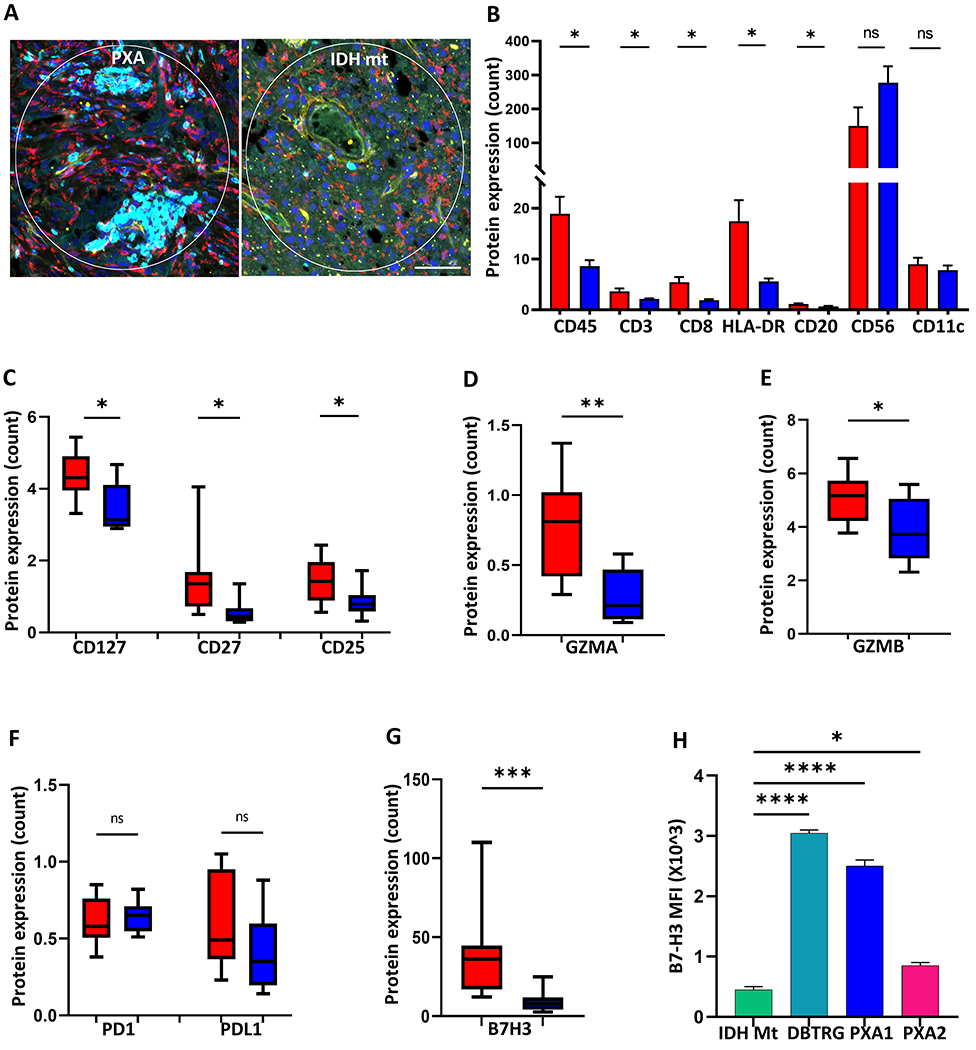
(A) Representative images of PXA (left) and IDH-mutant astrocytoma (IDH-mt, right) analyzed by digital spatial profiling. CD3+ T cell rich regions of interest were identified by multiplex immunostaining for CD3 (cyan), Iba1 (red), CD34 (yellow), and nuclei (blue) in PXA (n=9) and IDH-mt (n=8) (B) Bar graph showing mean immune cell protein marker expression level for PXA (red) and IDH-mt (blue). (C-E) Box plots showing protein expression of T lymphocyte co-stimulatory (CD127, CD27, and CD25) and activation (GZMA, GZMB) molecules in PXA (red) and IDH-mt (blue). (F-G) Box plots showing protein expression of immunomodulatory molecules in PXA (red) and IDH-mt (blue). (H) Cell surface expression of B7-H3 on patient derived PXA cell lines (PXA1, PXA2), DBTRG, and NHA-IDH1 mutant cell line (IDH Mt) as determined by flow cytometry expressed as mean fluorescent intensity (MFI) (n=3 replicates per line). Data shown are Mean ± SEM or box plots denoting 25%, median, 75%, and whiskers represent the minimum and maximum. All experiments analyzed by two-sided, unpaired Mann-Whitney U test.*p<0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. n.s. indicates p > 0.05. Scale bar: 20 μm.
T-lymphocyte activation and immune function are regulated by co-stimulatory molecules such as CD25, CD27 and CD127. CD27 is required for the T cell expansion and contributes to the accumulation of effector T cells, CD25 (IL-2) promotes the differentiation of T cells into effector cells and is transiently up-regulated upon activation of T-effectors, and CD127 helps mediate T cell homeostasis (27). Each of these co-stimulatory molecules was significantly increased in PXA relative to IDH-mt astrocytoma (p<0.05, Figure 2C). In addition, cytotoxic T cell activation markers, granzyme A (GZMA) and granzyme B (GZMB), were increased in PXA relative to IDH-mt astrocytoma (p<0.01 and 0.05, respectively, Figure 2D–E), providing further support to the activated T cell phenotype.
Immune checkpoints are also crucial for controlling effector T cell function. Increased expression of inhibitory immune checkpoints is common across diverse tumor types, yet the exact mechanism of immune evasion appears to differ. Neither programmed death 1 (PD-1) nor programmed death ligand 1 (PD-L1) expression levels were significantly different in PXA and IDH-mt astrocytoma (Figure 2F). However, PXA had robust expression of B7 Homolog 3 (B7-H3), and this was significantly higher than in IDH-mt astrocytoma (Figure 2G). Protein expression of CTLA-4 and OX40L were also increased and expression of GITR was slightly decreased in PXA relative to IDH-mt astrocytoma (Supplemental Figure S2A). The expression of B7-H3 on the cell surface is critical for its immunomodulatory functions and for it to be targeted therapeutically. Therefore, we investigated the cell surface expression of B7-H3 protein by flow cytometry in three RAF-altered glioma lines, two primary patient-derived PXA cell lines, PXA1 and PXA2, and DBTRG (Supplemental Table S4). All three lines had robust cell surface B7-H3 expression relative to the IDH-mutant expressing normal human astrocytes (NHA), (IDH Mt) (Figure 2H). These data suggest PXA has a relatively immune-rich microenvironment and identify B7H3 as a potential immunotherapeutic target in PXA.
Enrichment of immune-related genes including antigen processing and presentation in PXA.
To identify factors that may promote an immune-rich microenvironment in PXA, we performed transcriptional analysis of immunologically relevant genes in PXA (n=21), IDH-mt astrocytoma (n=6), and non-neoplastic brain (NB) (n=3) using Nanostring. Differential gene expression analysis identified 151 differentially expressed (DE) genes between PXA and IDH-mt astrocytoma (p<0.05) the majority of which (70%;105/151) were upregulated in PXA consistent with a more immune-rich microenvironment. The list of the top 20 upregulated gene in PXA is shown in Supplemental Table S7. Indeed, several inflammatory immune response pathways were enriched based on Gene Ontology (GO) enrichment analysis with the antigen processing and presentation pathway as most significantly enriched (Figure 3A).
Figure 3. Enrichment of immune-related genes including antigen processing and presentation in PXA.
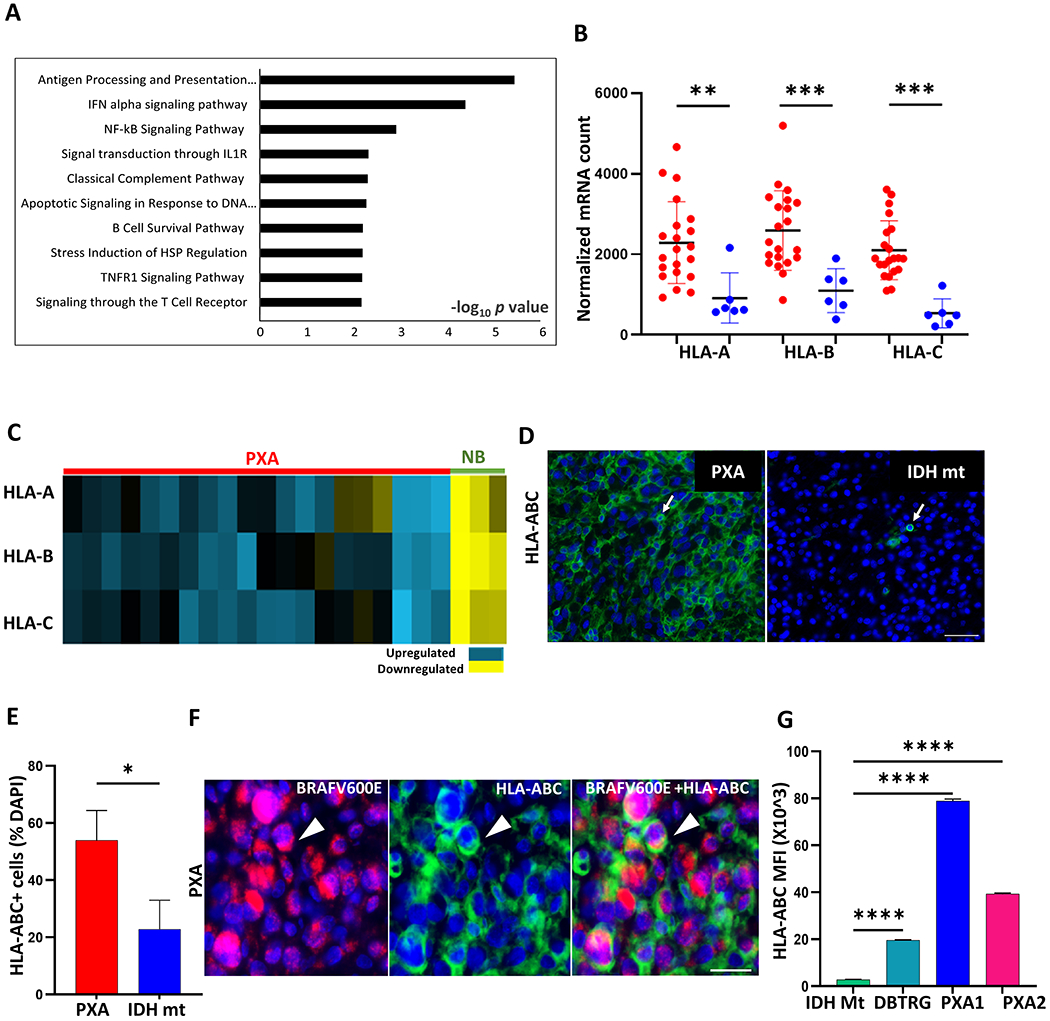
(A) Top 10 enriched biological processes from Gene Ontology (GO) enrichment analysis of differentially expressed genes between PXA and IDH-mt astrocytoma (IDH-mt). (B) Dot plot showing high expression of MHC class I genes (HLA-A, -B and -C) in PXA (red, n=21) compared to IDH-mt (blue, n=6). (C) Heatmap showing upregulated MHC class I genes in PXA (n=21, red) compared with normal brain (NB, n=3, green), p< 0.01 for all, Student’s t-test. Fold change ≤ 0.5 represents down-regulated (yellow) and ≥ 1.5 represents upregulated (blue). (D-E) Representative images and quantification of HLA-ABC (green) expression in PXA (n=9) and IDH-mt (n=7). Arrow highlights HLA-ABC positive cell. (F) Representative image of multiplex immunostaining highlighting HLA-ABC (green) expression on BRAFV600E-mutant tumor cells (violet) denoted by arrowhead in PXA. (G) Surface expression of HLA-ABC assessed by flow cytometry expressed as mean fluorescent intensity (MFI) (n=3 replicates per line) in NHA-IDH1 mutant cells lines (IDH Mt), DBTRG, PXA1, and PXA2. In (B) each dot represents a value from a single patient, black lines represent mean±SD. In (E) and (G) box plots are Mean+SEM. Results are data from at least technical triplicates and are representative of experiments performed in biologic triplicate. Significance obtained using ANOVA, 2-sided (G), unpaired Mann-Whitney U test (E) .*p<0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Nuclei are stained with DAPI (blue). Scale bar: (D) 50 μm and (F) 20 μm.
Major histocompatibility complex (MHC) class I expression is essential for antigen presentation and processing to CD8+ T cells. In PXA, the expression of MHC-I molecules, human leukocyte antigen (HLA)-A, -B and -C, were upregulated relative to IDH-mt astrocytoma (p<0.01, for all; Figure 3B) and non-neoplastic brain (p <0.01, for all; Figure 3C). In dense PXA tumor regions, most cells expressed HLA-ABC protein, mean 57.8 % of total cells, and the percentage was increased relative to IDH-mt astrocytoma, mean 21% (p<0.05; Figure 3D–E). Dual immunostaining demonstrated HLA-ABC expression on BRAF V600E-mutant tumor cells (Figure 3F), and cell surface expression of HLA-ABC was robust in all three RAF-altered glioma lines (Figure 3G). Taken together, these data suggest that an increased antigen processing and presentation pathway, including elevated MHC-I expression, may help promote a robust tumor-associated immune response in PXA.
CXCL14 is upregulated and secreted by tumor cells and patient-derived PXA cell lines.
Chemokines are chemotactic cytokines known to help regulate immune cell trafficking and influence the TIL response (28). Differential gene expression analysis revealed an upregulation of the chemokine (C-X-C motif) ligand 14 (CXCL14) in PXA relative to IDH-mt astrocytoma (Figure 4A; p<0.0001). The mean normalized mRNA count in PXA was 20-fold higher relative to that in IDH-mt astrocytoma (Figure 4B; p<0.001). Comparing CXCL14 expression across pediatric low- and high-grade astrocytoma, WHO grade II-IV, RAF altered astrocytoma had increased CXCL14 expression relative to RAF wildtype astrocytoma (p<0.0001, Figure 4C) and pilocytic astrocytoma (Supplemental Figure S3A).
Figure 4. Increased CXCL14 expression in PXA tumors and patient-derived cell lines.
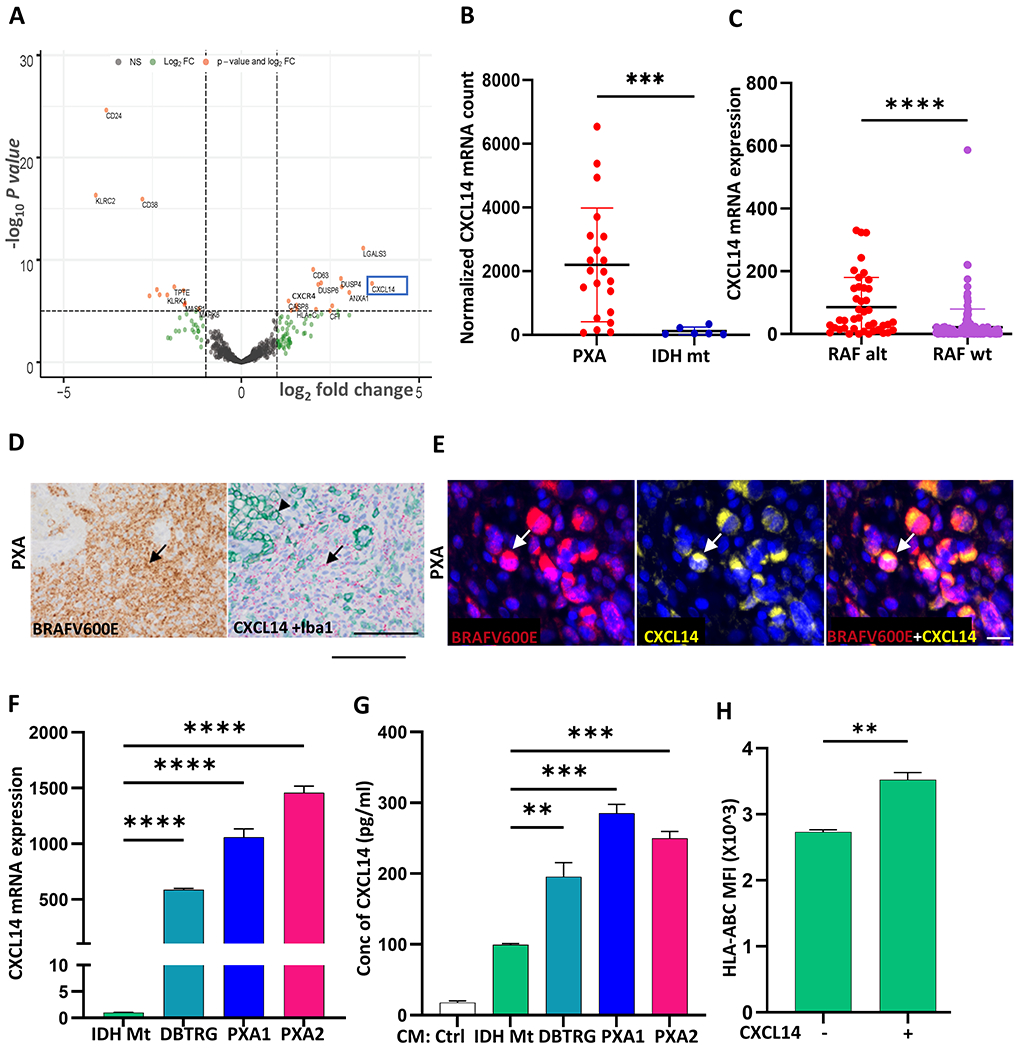
(A) Volcano plot showing differentially expressed genes between PXA and IDH-mt astrocytoma (IDH-mt) highlighting CXCL14 (blue box). (B) Dot plot showing CXCL14 normalized mRNA count in PXA (red, n=21) versus IDH-mt (blue, n=6). (C) Increased expression of CXCL14 in RAF altered astrocytoma (n=38) versus RAF wildtype astrocytoma (n=164) in a cohort of pediatric low-grade and high-grade astrocytoma. Data from CBTN downloaded via Kids First Data Resource Portal. (D) Representative images from serial sections showing BRAF V600E-mutant tumor cells (brown, arrow) corresponding to regions of abundant CXCL14 mRNA expressing cells (red, arrow). Dual immunostaining for Iba1 (green, arrowhead) highlights lack of CXCL14 expression in microglia/macrophages. (E) Representative image of BRAFV600E (red), CXCL14 (yellow), and nuclei (blue) in BRAF V600E-mutant PXA. (F) Expression of CXCL14 in DBTRG and patient-derived PXA lines PXA1 and PXA2 as compared to NHA-IDH1 mutant (IDH Mt) cell line by quantitative RT-PCR. (G) Secreted CXCL14 protein in cell-free conditioned media (CM) from DBTRG, PXA1, PXA2 and NHA-IDH1 mutant (IDH Mt) cell lines measured by ELISA. (H) Flow cytometric analysis of cell surface HLA-ABC expression on IDH Mt after addition of CXCL14 ligand (200ng/ml, 24 hrs). In (B) and (C) each dot represents a value from a single patient, black lines represent the mean ± SD. In (D) red dots correspond to single RNA molecules. In (F-H) data shown in box plots are Mean+SEM. Results are data from at least technical triplicates and are representative of experiments performed in biologic triplicate. Significance obtained using ANOVA, and 2-sided, unpaired Mann-Whitney U test. **p < 0.01, ***p < 0.001, ****p < 0.0001. Scale bar: (D) 100 μm (E) 20 μm.
In glioma, potential cell sources for CXCL14 include both immune cells and tumor cells (29). In BRAF V600E-mutant tumor dense regions, dual RNAscope revealed robust expression of CXCL14 transcript in Iba1-negative regions (Figure 4D). Furthermore, dual immunostaining demonstrated CXCL14 protein expression in BRAF V600E-mutant tumor cells (Figure 4E). In addition, all three RAF-altered glioma lines, PXA1, PXA2, and DBTRG, had high expression of CXCL14 transcript (Figure 4F) and ELISA demonstrated robust secretion of CXCL14 protein in PXA1 (285.5 pg/ml), PXA2 (250 pg/ml) and DBTRG (196 pg/ml) cells. (Figure 4G).
In addition to demonstrated roles in immune cell modulation, CXCL14 has also been implicated in the regulation of MHC class I expression (30). Given the increased MHC-I expression in PXA (Figure 3C), we investigated whether CXCL14 could alter cell surface expression of HLA-ABC. Addition of exogenous CXCL14 increased cell surface HLA-ABC expression on IDH Mt cells relative to control (Figure 4H, Supplemental Figure S3B, p<0.01). These data suggest CXCL14 is upregulated in RAF altered tumor cells and may promote increased cell surface MHC-I.
Secreted CXCL14 promotes activated CD8 T cell chemotaxis in vitro.
Our analysis suggested that PXAs harbor abundant CD8+ cytotoxic T cells (Figure 1B–G). To investigate whether factors secreted from tumor cells may promote chemotaxis of activated CD8+ T cells, we performed transwell migration assays using cell-free conditioned media (CM) from the three RAF-altered glioma lines, PXA1, PXA2, and DBTRG. CM from all three lines induced increased chemotaxis of activated CD8+ T cells relative to media alone (Figure 5A). Given that CXCL14 has been implicated in promoting immune cell recruitment in neoplastic and non-neoplastic settings (30,31), we first demonstrated CXCL14 was sufficient to promote directional migration of activated CD8+T cells in a dose-dependent manner (Figure 5B). Next, we knocked down CXCL14 in DBTRG cells (Figure 5C) and observed a reduction in CM-induced chemotaxis of activated CD8+ T cells to background control levels (Figure 5D).
Figure 5. Secreted factors from tumor cells promote activated CD8 T cell chemotaxis in vitro.
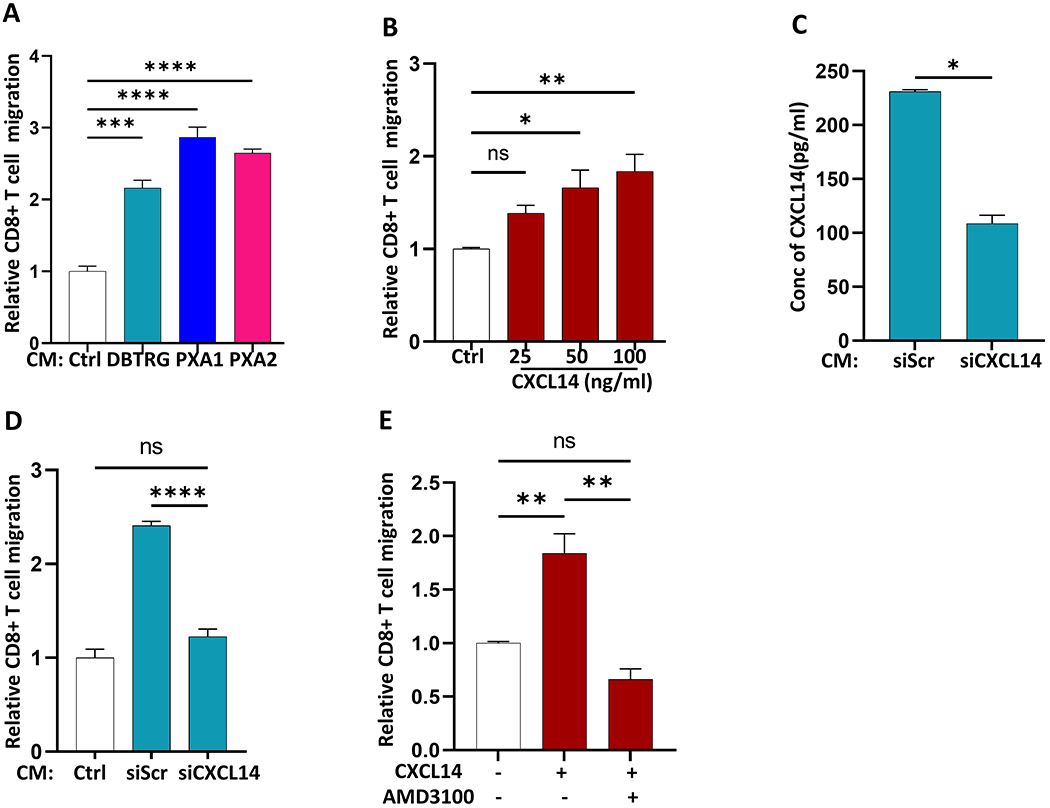
(A) Relative chemotaxis of activated CD8 T cells toward cell free-conditioned media (CM) derived from DBTRG, PXA1 and PXA2 as compared to control media (Ctrl) by transwell migration assay. (B) Chemotaxis of activated CD8+ T cells toward increasing concentration of CXCL14 ligand (25-100ng/ml) by transwell migration assay (C) CXCL14 ELISA of cell-free conditioned media (CM) after transduction of DBTRG with 4 pooled siRNA targeting CXCL14 gene (siCXCL14) versus scrambled siRNA (siScr) (D) Chemotaxis of activated CD8+ T cells toward cell-free conditioned media (CM) from siCXCL14 and siScr transduced DBTRG. (E) CXCL14-induced chemotaxis of activated CD8+ T cells following inhibition of CXCR4 (AMD3100) compared to control. Results are data from at least technical triplicates and are representative of experiments performed in biologic triplicate. Mean+SEM. Significance were obtained using one-way ANOVA and Student’s t test. *p<0.05, **p < 0.01, ****p < 0.0001 and ns indicates p > 0.05.
CXCR4 is one of potentially several receptors that CXCL14 can use or modulate (32,33). In PXA, CXCR4 expression was 5-fold higher relative to IDH-mutant astrocytoma (p<0.001; Supplemental Figure S4A) and activated CD8+ T cells demonstrated robust expression of CXCR4 (Supplemental Figure S4B). Inhibition of CXCR4 with AMD3100 (34) blocked CXCL14-induced chemotaxis of activated CD8+ T cells in vitro (Figure 5E). These data demonstrate that secreted CXCL14 can promote activated CD8+ T cell chemotaxis in a CXCR4-dependent manner, further supporting a potential role for CXCL14 in the tumor-associated immune response.
PXA also demonstrated increased tumor-associated microglia/macrophages relative to IDH-mt astrocytoma (Figure 1J–L). As CXCL14 has previously been implicated in myeloid cell migration (35), we also evaluated the ability of secreted factors from tumor cells, including CXCL14, to influence macrophage chemotaxis. Cell-free conditioned media (CM) from PXA1, PXA2, and DBTRG, promoted chemotaxis of the macrophage cell line MV4-11 in transwell migration assays relative to media alone (Supplemental Figure S4C). We also confirmed that exogenous CXCL14 was sufficient to promote the directional migration of MV4-11 cells in a dose-dependent manner (Supplemental Figure S4D).
Secreted CXCL14 promotes a tumor-associated CD8+ T cell response and prolongs survival.
IDH-mutant diffuse glioma are characterized as lymphocyte depleted (1–3), and median CXCL14 expression is lower than that in normal brain (Supplemental Figure S5A). To investigate whether CXCL14 might be sufficient to promote a CD8+ T cell response in diffuse glioma in vivo, we engineered murine tumor progenitor cells (mTPC) from an immunocompetent murine model of malignant glioma (25) to express CXCL14 (CXCL14-mTPC) (Figure 6). CXCL14-mTPC had increased expression of CXCL14 mRNA (Supplemental Figure S5B) compared to control mTPCs and secreted CXCL14 protein at similar levels to human patient-derived PXA cell lines (282.8 pg/ml) (Figure 6A and Figure 4G). Murine TPCs were then implanted into the striatum of immunocompetent mice and brains were examined at study end point. Histologic and immunophenotypic analysis revealed a striking increase in the CD3+ and CD8+ T cell infiltrates in tumors generated from CXCL14- as compared to control-mTPCs (Figure 6B–C and Supplemental Fig S5C). In contrast, the number of Iba1+ microglia/macrophages was similar (Figure 6D and Supplemental Fig S5D). There was no increased immune infiltrate outside of the tumor region (Supplemental Figure S5E). In vitro, the CXCR4 inhibitor AMD3100 inhibited CXCL14-induced chemotaxis of activated CD8+ T cells (Figure 5E). In vivo, treatment with the CXCR4 inhibitor abrogated the CXCL14-mediated CD8+ T cell response (Supplemental Figure S5F).
Figure 6. Secreted CXCL14 promotes an anti-tumor CD8+ T cell response in vivo.
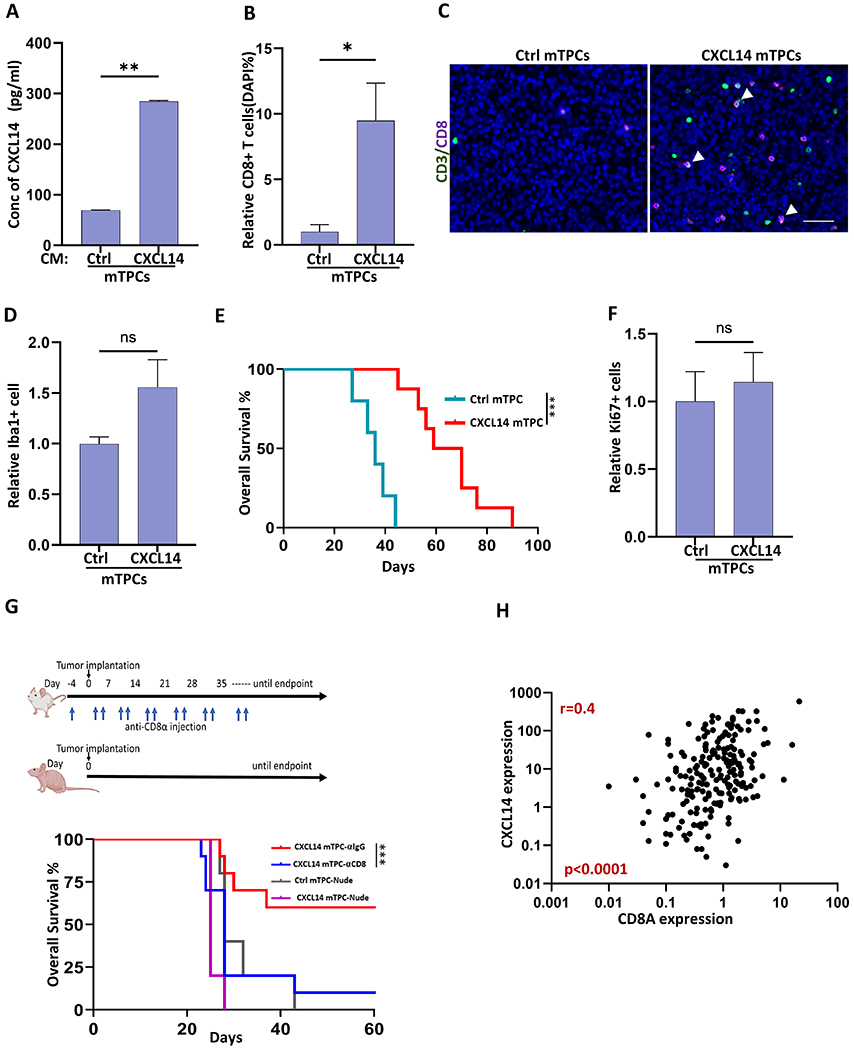
(A) Secreted CXCL14 in cell-free conditioned media (CM) from murine tumor progenitor cells (mTPCs) expressing CXCL14 (CXCL14) or GFP-control (Ctrl). (B) Quantification of CD8+ T cells as percentage of total cells in CXCL14 (n=8 mice) and Ctrl tumors (n=5 mice). (C) Representative images demonstrating CD3+ (green), CD8+ (violet), and dual CD3+CD8+ (white,arrow) T cells in CXCL14 and Ctrl tumors. (D) Quantification of Iba1+ tumor-associated microglia/macrophages per tumor area in CXCL14 (n=7 mice) relative to Ctrl tumors (n=4 mice). (E) Kaplan-Meier survival analysis. Mice with intracerebral allograft of CXCL14 mTPC have prolonged survival (median survival of 64.8 days) relative to Ctrl mTPC (median survival of 35.8 days) (p<0.003, n=8 and n=5, respectively) (F) Quantification of proliferating cells as denoted by Ki-67 per tumor area in CXCL14 (n=7 mice) relative to Ctrl tumors (n=4 mice). (G) Schema for tumor implantation in nude mice (n=5 per group) and FVB mice (n =10 per group) with biweekly anti-CD8α treatment to deplete CD8+ T cells (blue arrows). Corresponding isotype antibody used as control. Nude mice were implanted with either CXCL14 mTPC or Ctrl mTPC and FVB mice were implanted with CXCL14 mTPC. Kaplan-Meier survival analysis (below).(H) Correlation between CXCL14 gene expression and CD8A gene expression in pediatric astrocytoma (CBTN; Spearman’s correlation coefficient, r=0.45, n=202, p<0.0001). Bar graphs represent Mean+SEM. Significance were obtained using Student’s t test. *p<0.05, **p < 0.01, ****p < 0.0001 and ns indicates p > 0.05. Scale bar: 100 μm. Figure 6G (top FVB and nude mouse) was created with BioRender.com.
CXCL14 expression had no influence on tumor cell proliferation in vivo (Figure 6F and Supplemental Figure S5G) or cell growth in vitro, as demonstrated by similar doubling times for CXCL14-mTPC and control mTPC (29.28 h and 27.26 h, respectively; p=0.4). Yet, overall survival was significantly prolonged in mice harboring CXCL14-expressing tumors relative to control (Figure 6E). To evaluate whether T lymphocytes, specifically CD8+ cytotoxic T cells, were required for the survival difference between CXCL14-expressing and control-mTPC two approaches were taken. As demonstrated in Figure 6G, there was no CXCL14-mediated survival advantage in nude mice lacking mature T cells or in otherwise immunocompetent mice with CD8+ T cell depletion (Supplemental Figure S5H). CD8+ T cells were required for the prolonged survival mediated by CXCL14 (anti-CD8, n=10) versus anti-IgG control (n=10, p=0.0035). While median CXCL14 expression is low in several tumor subtypes, variation across tumors is evident. Using gene expression data, CXCL14 expression was correlated with increased CD8A expression in both pediatric and adult astrocytoma (r=0.45, p<0.0001, CBTN n=202, and r=0.60, p<0.0001, TCGA n=310, respectively; Figure 6H and Supplemental Figure S5I). Together these data suggest CXCL14 expression in tumor cells can promote an anti-tumor cytotoxic CD8+ T cell response.
DISCUSSION
Improved immunotherapeutic strategies for brain tumor patients requires a mechanistic understanding of the determinants of the tumor-associated immune response. Using a RAF-driven glioma subtype with a robust immune response, we identify tumor-intrinsic factors that modulate the tumor microenvironment and promote a cytotoxic CD8+ T cell response, including increased expression of MHC Class I and secretion of CXCL14. Highlighting the role for CXCL14 in the tumor microenvironment, increased expression of CXCL14 in an immunocompetent murine model for malignant glioma resulted in an increased tumor-associated immune response, including increased CD8+ T cells, and conferred prolonged survival in a CD8 T cell-dependent manner. Interestingly, CXCL14 expression was correlated with increased CD8 T cell expression in both pediatric and adult astrocytic tumors. Modulation of the tumor microenvironment may be an important immunotherapeutic strategy to promote T cell effector function in diverse brain tumors.
Integrating data from multiplex immune profiling, spatial proteomics, and gene expression analysis we demonstrate a robust immune microenvironment in PXA, including increased cytotoxic CD8+ T cells and tumor-associated microglia/macrophage (TAMs). These data are consistent with several studies suggesting that molecularly distinct brain tumor subtypes harbor unique immune microenvironments (1,4–6,12,36), including those suggesting that some subsets of MAPK-activated brain tumors have a pronounced CD8+ T cell response (6,36). While our study focused on the tumor-associated immune response in PXA, analysis of RNA sequencing data from pediatric astrocytoma suggests a larger subset of RAF altered astrocytic glioma may have an increased cytotoxic TIL response and increased CXCL14 expression. These tumors may be more vulnerable to immunotherapeutic strategies. In support of this, activating MAPK mutations have been associated with GBM response to immunotherapy (37,38). Additional determinants beyond MAPK activation, however, are likely as pilocytic astrocytoma with activating MAPK mutations did not show increased cytolytic T cell score or CXCL14 expression.
Recent studies have highlighted the importance of the tumor secretome in shaping many aspects of the microenvironment in diverse tumor types (39,40). In particular, chemokines produced by tumor cells and immune cells help regulate the tumor-associated immune response (28). In our analysis, despite the relatively robust tumor-associated immune response, many of the cytokines commonly implicated in lymphocyte chemotaxis or as mediators of a pro-inflammatory response demonstrated either no significant differences or reduced expression in PXA relative to IDH-mutant astrocytoma. The increased expression of the CXC motif chemokine 14 (CXCL14, BRAK, or MIP-2G) in PXA was a striking exception. CXCL14 is a chemokine implicated in both homeostatic immune functions, particularly in squamous epithelium, and host-tumor immune interactions (41,42). In several cancers CXCL14 expression is altered, with increased expression reported in tumor cells in papillary thyroid carcinoma (43) and decreased tumor cell expression associated with disease progression in human papillomavirus (HPV)-associated cervical and head and neck carcinoma (30,31). Manipulation of CXCL14 expression in other systems has revealed roles for the chemokine in the recruitment of immune cells, including CD8 cytotoxic T cells, dendritic cells, and natural killer cells (30,44). We demonstrated that CXCL14 is expressed and secreted by tumor cells and promotes chemotaxis of activated CD8+ T cells in a dose-dependent manner. Conversely, CXCL14-knockdown blocked condition media-induced chemotaxis of activated CD8+ T cells. While several factors may promote the immune-rich microenvironment in PXA, our data suggest CXCL14 is an important factor.
The diverse roles reported for CXCL14 in tumor progression may reflect a combination of factors including the cell source of CXCL14, whether the chemokine functions predominantly in an autocrine or paracrine manner, and receptor usage. While CXCL14 is considered an orphan chemokine, some CXCL14-dependent functions may be mediated by CXCR4, the receptor for CXCL12 (32). Recent studies suggest CXCL14 may have both CXCL12-dependent and CXCL12-independent roles in CXCR4 signaling (33). In PXA, both CXCL14 and CXCR4 expression was increased relative to IDH-mutant astrocytoma and inhibition of CXCR4 blocked CXCL14-mediated chemotaxis of activated CD8+ T cells. In our murine model for malignant glioma, inhibition of CXCR4 blocked the CXCL14-dependent increase in CD8+ T cells in vivo. Taken together these data suggest the CXCL14-mediated CD8+ T cell response is mediated largely through CXCR4, although contributions from other receptors cannot be excluded.
In PXA, MHC class I molecules, HLA-ABC, were upregulated. Patient-derived PXA lines also had increased cell surface HLA-ABC expression relative to IDH Mt cells. MHC-I mediated antigen presentation is crucial for CD8 effector T lymphocyte responses and MHC-I expression correlates with the presence of tumor infiltrating CD8+ T cells in several cancers (45). Loss or downregulation of MHC-I is considered a mechanism of T lymphocyte evasion and is associated with tumor progression (46,47). While the precise mechanisms that promote MHC-I expression in PXA are an area of ongoing study, CXCL14 may be a factor, as exogenous CXCL14-induced increased cell surface HLA-ABC expression on IDH Mt cells. A similar increase in MHC-I was observed in human papillomavirus (HPV)-associated head and neck carcinoma upon re-expression of CXCL14 (30). Given the importance of MHC-I-mediated antigen presentation for CD8 effector function, robust expression of MHC-I in PXA may promote the CD8+ cytolytic T cell response.
As CXCL14 has been implicated in regulating brain immune development (48), we hypothesized that tumor cell expression of CXCL14 may promote a more robust tumor-associated immune response. Using an immunocompetent murine model for malignant glioma, CXCL14 expression was sufficient to promote an increased tumor-associated CD8+ T cell response. CXCL14 also conferred prolonged survival in a CD8+ T cell-dependent manner.
In conclusion, our study demonstrates the relatively immune-rich microenvironment of PXA and identifies CXCL14 as an important factor in promoting an anti-tumor cytotoxic T cell response in the brain tumor microenvironment. As recurrence and progression is not uncommon in PXA, immunotherapeutic strategies may be important to consider. In this regard, we identified robust B7-H3 expression in PXA and CAR T cells targeting B7-H3 have been proposed as a potentially promising therapy in subgroups of adult and pediatric glioma patients (49,50). More broadly, our data elucidate a novel mechanism of immune regulation in the brain and suggest CXCL14 modulation may be a potential therapeutic strategy to promote an anti-tumor cytotoxic T cell response.
Supplementary Material
TRANSLATIONAL RELEVANCE.
The majority of diffuse glioma exhibit a lymphocyte-depleted phenotype. Improved immunotherapeutic strategies require elucidation of the determinants of an effective anti-tumor cytotoxic T cell response in brain. By studying a MAPK-activated subset of astrocytic glioma, PXA, we identify determinants of a robust cytotoxic T cell response. Tumor cell expression of chemokine CXCL14 was sufficient to promote cytotoxic T cell chemotaxis, and in an immunocompetent murine model for malignant glioma increase the glioma-associated CD8+ T cell response and promote overall survival. Consistent with CXCL14 driving an anti-tumor cytotoxic T cell response, the prolonged survival conferred by CXCL14 was dependent on CD8+ T cells. These studies elucidate a novel mechanism of immune regulation in the brain and suggest CXCL14 modulation is a potential therapeutic strategy to suppress tumor growth. In addition, they identify a subset of glioma with an immune-rich tumor microenvironment that may be particularly vulnerable to immunotherapeutic modulation.
ACKNOWLEDGEMENTS
This study was supported by UCSF Brain Tumor SPORE Developmental Research Project NIH/NCI 5P50CA097257-18 (A.M.M., J.J.P.), T32 CA151022 (E.M.), NIH/NCI U01 CA168878 (J.J.P.), Panattoni Family Research Program [J.J.P.], and UCSF Loglio collective [J.J.P.]. We also thank the T.J. Martell Foundation, the Gerson and Barbara Bakar Philanthropic Fund, and the Sence Foundation for support. Resources were provided by UCSF Brain Tumor SPORE Biorepository NIH/NCI 5P50CA097257-18 (J.J.P.), UCSF Helen Diller Family Comprehensive Cancer Center Laboratory for Cell Analysis (P30CA082103), and data was provided by The Children’s Brain Tumor Network. We thank Dr. Claudia Petritsch for her scientific input and Sonia Patel for her technical input. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The authors declare that they have no conflicts of interest with the contents of this article.
Footnotes
Conflict of interest statement. The authors have declared that no conflict of interest exists.
REFERENCES
- 1.Kohanbash G, Carrera DA, Shrivastav S, Ahn BJ, Jahan N, Mazor T, et al. Isocitrate dehydrogenase mutations suppress STAT1 and CD8+ T cell accumulation in gliomas. J Clin Invest. 2017. Mar 20;127(4):1425–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Müller S, Kohanbash G, Liu SJ, Alvarado B, Carrera D, Bhaduri A, et al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol. 2017. Dec 20;18(1):234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Woroniecka KI, Rhodin KE, Chongsathidkiet P, Keith KA, Fecci PE. T-cell Dysfunction in Glioblastoma: Applying a New Framework. Clin Cancer Res Off J Am Assoc Cancer Res. 2018. Aug 15;24(16):3792–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang Q, Hu B, Hu X, Kim H, Squatrito M, Scarpace L, et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell. 2017. Jul 10;32(1):42–56.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Petralia F, Tignor N, Reva B, Koptyra M, Chowdhury S, Rykunov D, et al. Integrated Proteogenomic Characterization across Major Histological Types of Pediatric Brain Cancer. Cell. 2020. Dec;183(7):1962–1985.e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mackay A, Burford A, Carvalho D, Izquierdo E, Fazal-Salom J, Taylor KR, et al. Integrated Molecular Meta-Analysis of 1,000 Pediatric High-Grade and Diffuse Intrinsic Pontine Glioma. Cancer Cell. 2017. Oct;32(4):520–537.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sampson JH, Gunn MD, Fecci PE, Ashley DM. Brain immunology and immunotherapy in brain tumours. Nat Rev Cancer. 2020. Jan;20(1):12–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kipnis J, Filiano AJ. The central nervous system: privileged by immune connections. Nat Rev Immunol. 2018. Feb;18(2):83–4. [DOI] [PubMed] [Google Scholar]
- 9.Klemm F, Maas RR, Bowman RL, Kornete M, Soukup K, Nassiri S, et al. Interrogation of the Microenvironmental Landscape in Brain Tumors Reveals Disease-Specific Alterations of Immune Cells. Cell. 2020. Jun;181(7):1643–1660.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wei J, Chen P, Gupta P, Ott M, Zamler D, Kassab C, et al. Immune biology of glioma-associated macrophages and microglia: functional and therapeutic implications. Neuro-Oncol. 2020. Feb 20;22(2):180–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lieberman NAP, DeGolier K, Kovar HM, Davis A, Hoglund V, Stevens J, et al. Characterization of the immune microenvironment of diffuse intrinsic pontine glioma: implications for development of immunotherapy. Neuro-Oncol. 2019. Jan;21(1):83–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dejaegher J, Solie L, Hunin Z, Sciot R, Capper D, Siewert C, et al. DNA methylation based glioblastoma subclassification is related to tumoral T-cell infiltration and patient survival. Neuro-Oncol. 2021. Feb 25;23(2):240–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Phillips JJ, Gong H, Chen K, Joseph NM, van Ziffle J, BC Bastian, et al. The genetic landscape of anaplastic pleomorphic xanthoastrocytoma. Brain Pathol. 2019;29(1):85–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schindler G, Capper D, Meyer J, Janzarik W, Omran H, Herold-Mende C, et al. Analysis of BRAF V600E mutation in 1,320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra-cerebellar pilocytic astrocytoma. Acta Neuropathol (Berl). 2011. Mar;121(3):397–405. [DOI] [PubMed] [Google Scholar]
- 15.Pratt D, Camelo-Piragua S, McFadden K, Leung D, Mody R, Chinnaiyan A, et al. BRAF activating mutations involving the β3-αC loop in V600E-negative anaplastic pleomorphic xanthoastrocytoma. Acta Neuropathol Commun. 2018. Dec;6(1):24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Phillips JJ, Gong H, Chen K, Joseph NM, van Ziffle J, Jin L-W, et al. Activating NRF1-BRAF and ATG7-RAF1 fusions in anaplastic pleomorphic xanthoastrocytoma without BRAF p.V600E mutation. Acta Neuropathol (Berl). 2016. Nov;132(5):757–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vaubel R, Zschernack V, Tran QT, Jenkins S, Caron A, Milosevic D, et al. Biology and grading of pleomorphic xanthoastrocytoma—what have we learned about it? Brain Pathol. 2021. Jan;31(1):20–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kline CN, Joseph NM, Grenert JP, van Ziffle J, Talevich E, Onodera C, et al. Targeted next-generation sequencing of pediatric neuro-oncology patients improves diagnosis, identifies pathogenic germline mutations, and directs targeted therapy. Neuro-Oncol. 2017. May;19(5):699–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Amaria RN, Reddy SM, Tawbi HA, Davies MA, Ross MI, Glitza IC, et al. Neoadjuvant Immune Checkpoint Blockade in High-Risk Resectable Melanoma. Nat Med. 2018. Nov;24(11):1649–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Collins VP, Jones DTW, Giannini C. Pilocytic astrocytoma: pathology, molecular mechanisms and markers. Acta Neuropathol (Berl). 2015;129(6):775–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bolen CR, McCord R, Huet S, Frampton GM, Bourgon R, Jardin F, et al. Mutation load and an effector T-cell gene signature may distinguish immunologically distinct and clinically relevant lymphoma subsets. Blood Adv. 2017. Sep 27;1(22):1884–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015. Jan 15;160(1–2):48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chaumeil MM, Larson PEZ, Yoshihara HAI, Danforth OM, Vigneron DB, Nelson SJ, et al. Non-invasive in vivo assessment of IDH1 mutational status in glioma. Nat Commun. 2013;4:2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ohkawa Y, Wade A, Lindberg OR, Chen KY, Tran VM, Brown SJ, et al. Heparan Sulfate Synthesized by Ext1 Regulates Receptor Tyrosine Kinase Signaling and Promotes Resistance to EGFR Inhibitors in GBM. Mol Cancer Res. 2021. Jan;19(1):150–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lohr J, Ratliff T, Huppertz A, Ge Y, Dictus C, Ahmadi R, et al. Effector T-Cell Infiltration Positively Impacts Survival of Glioblastoma Patients and Is Impaired by Tumor-Derived TGF-. Clin Cancer Res. 2011. Jul 1;17(13):4296–308. [DOI] [PubMed] [Google Scholar]
- 26.Ryall S, Zapotocky M, Fukuoka K, Nobre L, Guerreiro Stucklin A, Bennett J, et al. Integrated Molecular and Clinical Analysis of 1,000 Pediatric Low-Grade Gliomas. Cancer Cell. 2020. Apr 13;37(4):569–583.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu W, Putnam AL, Xu-yu Z, Szot GL, Lee MR, Zhu S, et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J Exp Med. 2006. Jul 10;203(7):1701–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nagarsheth N, Wicha MS, Zou W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat Rev Immunol. 2017. Sep;17(9):559–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frederick MJ, Henderson Y, Xu X, Deavers MT, Sahin AA, Wu H, et al. In Vivo Expression of the Novel CXC Chemokine BRAK in Normal and Cancerous Human Tissue. Am J Pathol. 2000. Jun;156(6):1937–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Westrich JA, Vermeer DW, Silva A, Bonney S, Berger JN, Cicchini L, et al. CXCL14 suppresses human papillomavirus-associated head and neck cancer through antigen-specific CD8+ T-cell responses by upregulating MHC-I expression. Oncogene. 2019. Nov;38(46):7166–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cicchini L, Westrich JA, Xu T, Vermeer DW, Berger JN, Clambey ET, et al. Suppression of Antitumor Immune Responses by Human Papillomavirus through Epigenetic Downregulation of CXCL14. mBio. 2016. Jul 6;7(3):e00270–16, /mbio/7/3/e00270-16.atom. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Collins PJ, McCully ML, Martínez-Muñoz L, Santiago C, Wheeldon J, Caucheteux S, et al. Epithelial chemokine CXCL14 synergizes with CXCL12 via allosteric modulation of CXCR4. FASEB J. 2017. Jul;31(7):3084–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tanegashima K, Tsuji K, Suzuki K, Shigenaga A, Otaka A, Hara T. Dimeric peptides of the C-terminal region of CXCL14 function as CXCL12 inhibitors. FEBS Lett. 2013. Nov 29;587(23):3770–5. [DOI] [PubMed] [Google Scholar]
- 34.Fricker SP, Anastassov V, Cox J, Darkes MC, Grujic O, Idzan SR, et al. Characterization of the molecular pharmacology of AMD3100: a specific antagonist of the G-protein coupled chemokine receptor, CXCR4. Biochem Pharmacol. 2006. Aug 28;72(5):588–96. [DOI] [PubMed] [Google Scholar]
- 35.Augsten M, Hägglöf C, Olsson E, Stolz C, Tsagozis P, Levchenko T, et al. CXCL14 is an autocrine growth factor for fibroblasts and acts as a multi-modal stimulator of prostate tumor growth. Proc Natl Acad Sci. 2009. Mar 3;106(9):3414–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tang K, Kurland D, Vasudevaraja V, Serrano J, Delorenzo M, Radmanesh A, et al. Exploring DNA Methylation for Prognosis and Analyzing the Tumor Microenvironment in Pleomorphic Xanthoastrocytoma. J Neuropathol Exp Neurol. 2020. Aug 1;79(8):880–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao J, Chen AX, Gartrell RD, Silverman AM, Aparicio L, Chu T, et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat Med. 2019. Mar;25(3):462–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Arrieta VA, Chen AX, Kane JR, Kang SJ, Kassab C, Dmello C, et al. ERK1/2 phosphorylation predicts survival following anti-PD-1 immunotherapy in recurrent glioblastoma. Nat Cancer. 2021. Dec;2(12):1372–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Georgouli M, Herraiz C, Crosas-Molist E, Fanshawe B, Maiques O, Perdrix A, et al. Regional Activation of Myosin II in Cancer Cells Drives Tumor Progression via a Secretory Cross-Talk with the Immune Microenvironment. Cell. 2019. Feb 7;176(4):757–774.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Venkatesh HS, Johung TB, Caretti V, Noll A, Tang Y, Nagaraja S, et al. Neuronal Activity Promotes Glioma Growth through Neuroligin-3 Secretion. Cell. 2015. May 7;161(4):803–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hromas R, Broxmeyer HE, Kim C, Nakshatri H, Christopherson K, Azam M, et al. Cloning of BRAK, a Novel Divergent CXC Chemokine Preferentially Expressed in Normal versus Malignant Cells. Biochem Biophys Res Commun. 1999. Feb;255(3):703–6. [DOI] [PubMed] [Google Scholar]
- 42.Frederick MJ, Henderson Y, Xu X, Deavers MT, Sahin AA, Wu H, et al. In Vivo Expression of the Novel CXC Chemokine BRAK in Normal and Cancerous Human Tissue. Am J Pathol. 2000. Jun;156(6):1937–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oler G, Camacho CP, Hojaij FC, Michaluart P, Riggins GJ, Cerutti JM. Gene Expression Profiling of Papillary Thyroid Carcinoma Identifies Transcripts Correlated with BRAF Mutational Status and Lymph Node Metastasis. Clin Cancer Res. 2008. Aug 1;14(15):4735–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shurin GV, Ferris R, Tourkova IL, Perez L, Lokshin A, Balkir L, et al. Loss of New Chemokine CXCL14 in Tumor Tissue Is Associated with Low Infiltration by Dendritic Cells (DC), while Restoration of Human CXCL14 Expression in Tumor Cells Causes Attraction of DC Both In Vitro and In Vivo. J Immunol. 2005. May 1;174(9):5490–8. [DOI] [PubMed] [Google Scholar]
- 45.Perea F, Bernal M, Sánchez-Palencia A, Carretero J, Torres C, Bayarri C, et al. The absence of HLA class I expression in non-small cell lung cancer correlates with the tumor tissue structure and the pattern of T cell infiltration: HLA class I expression in NSCLC. Int J Cancer. 2017. Feb 15;140(4):888–99. [DOI] [PubMed] [Google Scholar]
- 46.Cordon-Cardo C, Fuks Z, Drobnjak M, Moreno C, Eisenbach L, Feldman M. Expression of HLA-A,B,C Antigens on Primary and Metastatic Tumor Cell Populations of Human Carcinomas. Cancer Res. 1991. Dec 1;51(23 Part 1):6372–80. [PubMed] [Google Scholar]
- 47.McGranahan N, Rosenthal R, Hiley CT, Rowan AJ, Watkins TBK, Wilson GA, et al. Allele-Specific HLA Loss and Immune Escape in Lung Cancer Evolution. Cell. 2017. Nov 30;171(6):1259–1271.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li Z, Li Y, Jiao J. Neural progenitor cells mediated by H2A.Z.2 regulate microglial development via Cxcl14 in the embryonic brain. Proc Natl Acad Sci. 2019. Nov 26;116(48):24122–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Majzner RG, Theruvath JL, Nellan A, Heitzeneder S, Cui Y, Mount CW, et al. CAR T Cells Targeting B7-H3, a Pan-Cancer Antigen, Demonstrate Potent Preclinical Activity Against Pediatric Solid Tumors and Brain Tumors. Clin Cancer Res. 2019. Apr 15;25(8):2560–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Haydar D, Houke H, Chiang J, Yi Z, Odé Z, Caldwell K, et al. Cell-surface antigen profiling of pediatric brain tumors: B7-H3 is consistently expressed and can be targeted via local or systemic CAR T-cell delivery. Neuro-Oncol. 2021. Jun 1;23(6):999–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The human sequence data generated in this study are not publicly available due to patient privacy requirements but are available upon reasonable request from the corresponding author. Other data generated in this study are available within the article and its supplementary data files. Some of the data analyzed in this study were obtained from Children’s Brain Tumor Network (CBTN) and was downloaded via the Gabriella Miller Kids First Data Resource Portal (https://portal.kidsfirstdrc.org/login).