

Abstract
T cells modified to express a chimeric-antigen receptor (CAR)targeting CD19 can induce potent and sustained responses in children with relapsed/refractory acute lymphoblastic leukemia (ALL). The durability of remission is related to the length of time the CAR T cells persist. Efforts to understand differences in persistence have focused on the CAR construct, in particular the co-stimulatory signaling module of the chimeric receptor. We previously reported a robust intent-to-treat product manufacturing success rate and remission induction rate in children and young adults with recurrent/refractory B-ALL using the SCRI-CAR19v1 product, a 2nd generation CD19-specific CAR with 4–1BB costimulation co-expressed with the EGFRt cell surface tag (NCT02028455). Following completion of the phase 1 study, two changes to CAR T–cell manufacturing were introduced: switching the T-cell activation reagent and omitting mid-culture EGFRt immunomagnetic selection. We tested the modified manufacturing process and resulting product, designated SCRI-CAR19v2, in a cohort of 21 subjects on the phase 2 arm of the trial. Here, we describe the unanticipated enhancement in product performance resulting in prolonged persistence and B-cell aplasia, and improved leukemia-free survival with SCRI-CAR19v2 as compared to SCRI-CAR19v1.
Keywords: CAR T cell, CD19, T cell persistence, ALL, chimeric antigen receptor
INTRODUCTION
Adoptive cellular therapy has emerged as a highly efficacious approach to improve outcomes for patients with B-cell malignancies [1, 2]. Autologous T cells engineered to express a chimeric-antigen receptor (CAR) with CD19 or CD22 specificity have shown impressive efficacy in early-phase clinical trials for relapsed/refractory B-cell acute lymphoblastic leukemia (B-ALL) and B-cell lymphomas [3–6]. Production of the CAR T cells involves the collection of autologous patient-derived T cells that are activated, genetically modified by viral transduction, and propagated in recombinant cytokines. The impact of each manufacturing step on subsequent clinical therapeutic index is not yet clear and randomized trials to systematically assess these variables are yet to be realized.
Early loss of persistence of CD19-specific CAR T cells increases the risk of CD19+ relapse in ALL [7]. The study of what impacts long-term CAR T–cell persistence amongst various CAR T–cell products has focused predominantly on the CAR construct itself, including the extracellular scFv portion of the receptor as well as intracellular costimulatory signaling domains [8]. FMC63 is the most widely used CD19-targeting scFv and is murine in origin. In select patients, T-cell mediated rejection responses have been reported against FMC63 scFv epitopes, leading to attenuated in vivo persistence following an initial dose of CAR T cells and failure of subsequent doses to engraft [9]. The two most commonly used costimulatory moieties are 4–1BB and CD28. Costimulation with 4–1BB promotes cell survival and retention of function that favors long-term persistence of CAR T cells in vivo, and this has been associated with remission durability [10]. CD28 costimulation, in contrast, supports rapid CAR T–cell expansion and accentuated effector outputs, followed by early loss of CAR T–cell persistence [11]. In pediatric B-ALL trials, the duration of remission appears longer in patients receiving 4–1BB costimulated CAR T–cell products compared with CD28 costimulated CAR T–cell products [3, 5, 6], although there are no published direct comparisons.
We previously reported a 93% MRD-negative complete remission (MRD-CR) of infused subjects in heavily pre-treated B-ALL subjects during the phase 1 portion of our phase1/2 study, Pediatric and young adult Leukemia Adoptive Therapy (PLAT)-02 (NCT02028455) [5]. In the phase 1 portion of the trial, 43 children and young adults with relapsed or refractory B-ALL were infused with SCRI-CAR19v1, which uses a 2nd generation CAR construct targeting CD19 through FMC63 scFv, encoded with 4–1BB costimulation, and formulated as a defined CD4/CD8 composition product with limited effector differentiation [7]. In addition to CAR expression, the lentiviral vector also encodes a truncated epidermal growth factor receptor (EGFR) tag (EGFRt), which facilitates immunomagnetic enrichment of CAR-expressing T cells during manufacturing and tracking of cells by flow cytometric detection in patient specimens following infusion. The manufacturing platform for SCRI-CAR19v1 included a CD4 and CD8 magnetic bead selection from apheresis starting material, enabling separate activation of purified CD4+ and CD8+ T cells with the CTS Dynabead CD3/CD28 bead reagent (Figure 1) followed by propagation in recombinant cytokine mixtures (CD4 culture: rhuIL7/rhIL15; CD8 culture rhuIL2/rhuIL15). Mid-culture, transduced T cells underwent immunomagnetic positive selection for the EGFRt-expressing cells, creating a near-uniform population of CAR-expressing cells at the time of infusion [5].
Figure 1. Process Development of SCRI-CAR19v2.
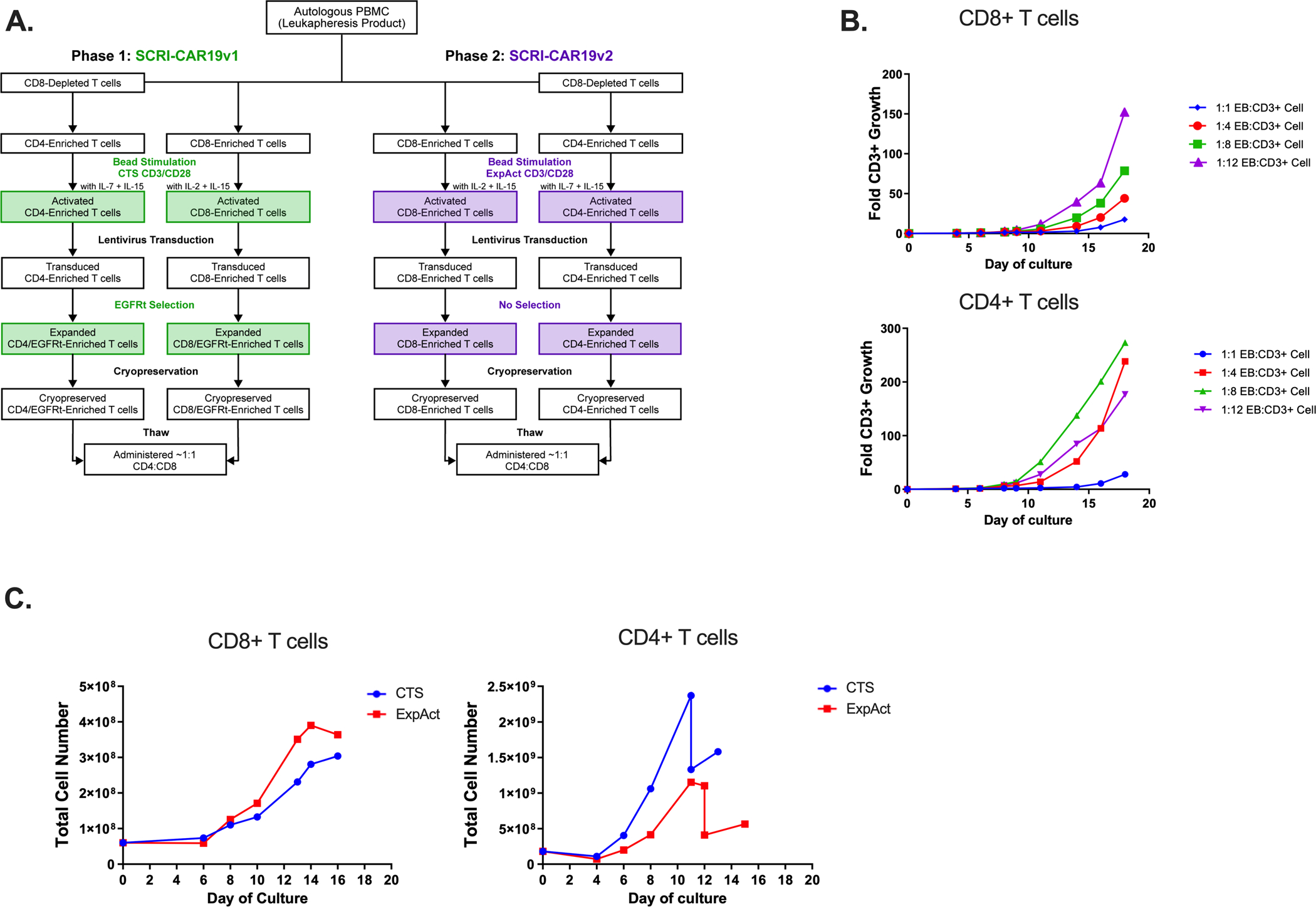
(A) Manufacturing schemas of SCRI-CAR19v1 and SCRI-CAR19v2. (B) Comparison of normal donor–derived cell product growth rates at small scale with varying ExpAct (EB) to CD3+ T-cell ratios for both CD8+ (top graph) and CD4+ T-cell (bottom graph) cultures (n=1). (C) Large scale experiments using a single normal donor comparing growth of CD4+ and CD8+ T cells manufactured with CTS beads versus ExpAct using the SCRI-CAR19v1 platform. Cell counts analyzed periodically throughout cell culture on indicated days post initiation. The blue line indicates use of CTS beads to CD8+ T cells at 3:1 ratio and the red line indicates ExpAct to CD8+ T cells at a 1:8 ratio. For CD4+ T-cell cultures, the blue line indicates CTS beads to T cells at a 3:1 ratio and the red line indicates ExpAct to T cells at a 1:4 ratio.
In the phase 1 portion of PLAT-02, there was a median duration of functional persistence of 3 months [5]. We identified patient-specific attributes, as well as product features, that correlated with the duration of persistence of CAR T cells. Subjects who had a CD19 antigen load (malignant and normal B cells) of >15% in the bone marrow prior to lymphodepletion exhibited superior in vivo persistence as compared with subjects having limited CD19+ B cells and leukemic cells capable of driving in vivo restimulation and proliferation [5]. Consistent with trials in adults [9], there was also a trend towards increased persistence in those subjects who received lymphodepletion with the combination of fludarabine and cyclophosphamide (Flu/Cy) as compared to those who received one or the other preparative agent. Subsequently, we identified phenotypic and functional characteristics of the final CD8+ CAR T–cell products that correlated with engraftment persistence and ongoing B-cell aplasia beyond 6 months, including high TNFα production and low TIM-3 expression [7]. We did not identify specific features of CD4+ CAR T–cell product phenotype or apheresis product CD4+ T–cell repertoire associated with enhanced persistence.
Following completion of the phase 1 portion of PLAT-02, we implemented two changes to the process by which the CAR T–cell products were manufactured for phase 2 product manufacturing (referred to as SCRI-CAR19v2). Due to restricted access at the time of the clinical trial conduct, the CTS Dynabead CD3/CD28 stimulation reagent was replaced with the ExpAct reagent (Figure 1). Second, the mid-culture EGFRt enrichment step was discontinued to avoid an observed proclivity of positive selection using biotinylated cetuximab and anti-biotin microbeads to co-enrich nonviable cells. Upon initiation of phase 2 cohort infusions with SCRI-CAR19v2, a change in product behavior was observed with earlier onset of cytokine release syndrome (CRS) and an increased portion of subjects experiencing prolonged B-cell aplasia. We hypothesized that the activation reagent used to initiate manufacturing cultures imparts product attributes that affect subsequent product performance in treated subjects. Here, we report on the superior clinical performance of SCRI-CAR19v2 in 21 treated subjects and corresponding product attributes in comparison to SCRI-CAR19v1.
METHODS
Study design and participants
This phase 2 study for relapsed or refractory CD19+ B-ALL in children and young adults was conducted in accordance with US Food and Drug Administration and International Conference on Harmonization Guidelines for Good Clinical Practice, the Declaration of Helsinki, and was approved by the Seattle Children’s and Children’s Hospital of Los Angeles institutional review boards prior to patient enrollment. All patients or their guardians provided written informed consent. Enrollment criteria included: age ≥12 months and <27 years and weight ≥10 kg. Patients with no prior history of allogeneic hematopoietic cell transplantation (HCT) were required to have one of the following characteristics: second or later marrow relapse, with or without extramedullary disease; first marrow relapse at the end of the first month of reinduction, with the marrow having ≥0.01% blasts by multiparameter flow cytometry, with or without extramedullary disease; primary refractory disease, defined as having M2 (5–<25% morphologic disease) or M3 (≥ 25% morphologic disease) marrow after ≥2 separate induction regimens; or an indication for HCT but ineligibility for the procedure. Patients who had undergone allogeneic HCT were required to have a confirmed CD19+ leukemia recurrence, defined as ≥0.01% disease, and were required to be free from active graft-versus-host disease (GVHD) and have ended immunosuppressive therapy ≥4 weeks before enrollment. Patients with central nervous system (CNS) leukemic involvement were eligible for the study provided they were asymptomatic. Patients with significant neurologic deterioration were not eligible until alternate therapies resulted in neurologic stabilization and return to baseline status. Patients had a Lansky performance status score of ≥50 or a Karnofsky score of ≥50 for those age ≥16 years. Patients were required to have a life expectancy of ≥8 weeks and to have recovered from the acute toxic effects of prior chemotherapy, immunotherapy or radiotherapy. Patients were required to have adequate organ function and an absolute lymphocyte count of ≥100 cells per μL.
Clinical lentiviral vector and cell product manufacture
The clinical lentiviral vector and SCRI-CAR19v1 manufacturing process have been previously described [5]. The manufacturing process for SCRI-CAR19v2 is described here and mirrored that of SCRI-CAR19v1 with modifications of the stimulation bead reagent, ratio of bead to T cell, and lack of mid-culture enrichment. In brief, after CD4+ and CD8+ T-cell isolation from patient apheresis product by immunomagnetic separation using the CliniMACS device (Miltenyi Biotec) in positive-selection mode, T-cell cultures for expansion were initiated separately for CD4+ T cells and CD8+ T cells. Cells were placed with X-Vivo15 (Lonza) + 10% defined, irradiated, heat-inactivated fetal bovine serum (Cytiva). Cytokine concentrations were 50U/mL IL-2 (Prometheus Laboratories) and 0.5ng/mL IL15 for CD8+ T-cell cultures, and 5ng/mL IL7 (CellGenix) and 0.5ng/mL IL15 (CellGenix) for CD4+ T-cell cultures. Process development work using healthy donor T cells evaluated a variety of bead to T cell ratios for ExpAct beads (Miltenyi Biotec, cat. #200-070-412) (range of 1:1 to 1:12) and MACS TransAct beads (Miltenyi Biotec, cat #130-020-008) (CD3 reagent 100–400 ng/mL, CD28 reagent 200–800 ng/mL). Clinical products were generated with 180 × 106 CD4+ and CD8+ selected T cells, separately stimulated with GMP ExpAct beads instead of Dynabeads CD3/CD28 CTS (Thermo Fisher) at a 1:8 (bead to T cell) ratio for CD8+ T-cell cultures and 1:4 ratio for CD4+ T-cell cultures and transduced via spinoculation, as previously described [5].The CTS bead ratio was 3:1 for both the CD8+ and CD4+ T-cell cultures. During expansion, the virus is diluted with additional media as per above, however, there is no complete washout of the virus. Similar to the initial expansion methods, approximately 10 days after lentiviral transduction, CD3/CD28 ExpAct beads were removed using the Dynal DynaMag CTS magnet. After bead removal, SCRI-CAR19v2 cultures did not receive immunomagnetic positive selection for EGFRt-expressing cells. Following culture, T cells were cryopreserved and stored until the time of CAR T–cell infusion.
Evaluations
The primary end points of this study were to evaluate feasibility, efficacy and toxicity. Feasibility was evaluated by the ability to generate a therapeutic product after 2 attempts using a single apheresis product as starting material. Toxicity was evaluated relative to a baseline assessment conducted at some point during the 24 hours before T-cell infusion. The primary efficacy end point was the MRD-negative rate by bone marrow aspirate by day 63; assessments were also conducted on day 21 post-infusion. Responses were graded per standard ALL criteria [12]. Secondary end points included persistence of transferred T cells (defined as the detection of transferred T cells in the peripheral blood and bone marrow), disease response and anti-CD19 activity response (defined as a measurable effect of disease reduction and absence of CD19 cells).
Flow staining
Persistence of transferred T cells
Peripheral blood, bone marrow aspirate and whole cerebrospinal fluid were stained as previously described [7] or as follows. Peripheral blood and bone marrow aspirates were treated to lyse red blood cell (Invitrogen Cat# 00-4333-57) prior to persistence tracking via flow staining. Isolated white blood cells and whole cerebrospinal fluid were stained with a viability dye (BD Cat# 564407) and monoclonal antibodies specific for the following human markers: CD3 (BD Cat# 652356), CD4 (BD Cat# 562658), CD8 (BD Cat# 560662), CD19 (BD Cat# 340720), and CD36 (BD Cat# 555454). CAR expression on the T cells was quantified using Cetuximab (Creative Diagnostics Cat# TAB-003) custom-conjugated to allophycocyanin by BD Biosciences to detect the transduction marker, truncated EGFR.
Final Products
Immunophenotyping of surface markers on final products was performed using standard staining and flow cytometry techniques with combinations of fluorophore-conjugated monoclonal antibodies specific for the following human markers: CD3 (BD Biosciences Cat# 562426, RRID:AB_11152082), CD4 (BD Biosciences Cat# 562658, RRID:AB_2744420), CD8a (BD Biosciences Cat# 560662, RRID:AB_1727513), CD14 (BD Biosciences Cat# 555397, RRID:AB_395798), CD27 (BD Biosciences Cat# 564301, RRID:AB_2744350), CD39 (BioLegend Cat# 328212, RRID:AB_2099950) CD45RA (BD Biosciences Cat# 563870, RRID:AB_2738459), CD45RO (BD Biosciences Cat# 564291, RRID:AB_2744410), CD95 (BD Biosciences Cat# 561633, RRID:AB_10894384), CCR7 (BD Biosciences Cat# 552176, RRID:AB_394354), PD-1 (BD Biosciences Cat# 565299, RRID:AB_2739167), LAG-3 (BD Biosciences Cat# 565616, RRID:AB_2571727), and TIM-3 (BioLegend Cat# 345006, RRID:AB_2116576). Cells were also stained with a live/dead viability dye (BD Cat# 564407) and the custom-conjugated Cetuximab described above in order to assess CAR expression.
Intracellular cytokine staining was performed on final products post-cryopreservation. Samples were thawed and allowed to rest at 37°C in a 5% CO2 incubator for 6 hours in R10 media [RPMI 1640 (Gibco Cat# 22400–089), 10% FBS (VWR Cat#97068–085), 1% L- Glutamine (Gibco Cat#25030-081)]. Following rest, associated T-cell subsets were mixed at a 1:1 ratio of EGFRt+ CD4:CD8 for stimulation. Staphylococcal enterotoxin B (1ug/mL, Toxin Technology Cat#BT202) or Cell Stim Cocktail (Invitrogen Cat#00-4970-93) was added to mixed CD4/CD8 final product cells for universal T-cell stimulation as a positive stimulation control. K562 parental cells or CD19-expressing K562 cells were added at 1:1 ratio of CAR T cell:Target to determine antigen-specific response. Anti-human CD107a (BD Cat#555801) was added to stimulation cultures immediately following the addition of antigen. Stimulation cultures were incubated for 1 hour at 37°C post-initiation before a cocktail of 10.6uM Brefeldin A and 2uM Monensin (Invitrogen Cat#00-4980-03) was added to prevent protein transport out of the cell and acidification of the lysosomes. Culture was continued for an additional 17 hours followed by flow cytometry staining. Surface staining was performed to define effector cells using fluorophore-conjugated anti-human monoclonal antibodies targeting CD3, CD4, CD8α, Cetuximab, CD14 and viability dye as described above. Cell fixation/permeabilization buffers (BD Cat#554714) were used according to manufacturer recommendations and intracellular cytokines were detected using monoclonal antibodies specific for human IL2 (BD Cat#560707), IFNγ (BD Cat#563731) and TNFα (BD Cat#563996). Degranulation activity was determined by the presence of the fluorochrome-conjugated CD107a antibody taken up by cells during the stimulation culture. Antigen-specific response was determined by subtracting cytokine response detected in the K562 parental cell line stimulation from the K562-CD19 treated samples.
Acquisition and Analysis
Stained cells were acquired on an LSRFortessa (BD Biosciences), and flow cytometric analysis was performed using FlowJo software Version 10 (BD Biosciences). CAR T cells were defined as singlets/lymphocytes/viableCD36–/CD3+/EGFRt+ (persistence tracking) or singlets/lymphocytes/viableCD14–/CD3+/EGFRt+/CD4+ or CD8+ (Final Product analysis), while CD19 antigen in persistence tracking was defined as singlets/lymphocytes/viableCD36–/CD3–/CD19+ (Supplementary Figure S1). Multi-activation marker and poly-functional cytokine profiles were analyzed via Boolean gating and SPICE analysis software version 6 per standard use [13].
Luminex cytokine profiling
CAR T cells were stimulated as isolated CD4+ or CD8+ final products according the procedures described above prior to intracellular cytokine staining, with the exclusion of CD107a and Protein Transport Inhibitor in the stimulation culture. Following the 18h incubation at 37°C, cultures were pelleted by centrifugation and the supernatant was removed for cryopreservation at −80°C. Batched supernatant was thawed and analyzed according to manufacturer directions for the 25-plex Human Th17 Luminex Kit (Millipore Cat#HTH17MAG-14K). Antigen-specific response was determined by subtracting cytokine response detected for the K562 parental cell line stimulation from the K562-CD19 treated samples. Hierarchical clustering and principle component analyses (PCA) of SCRI-CAR19v1 and SCRI-CAR19v2 product Luminex data was performed using Log10 values of cytokine production. Hierarchical clustering analyses used the “Ward.D” algorithm with Euclidean distance matrix. PCA were conducted using covariance matrix.
Incucyte Assay
T-cell products were plated in triplicate at a 50:50 CD4/CD8 ratio with mcherry-positive, CD19-expressing K562 tumor cells. CAR products and tumor cells were plated at a 1:1 effector:target ratio, with a seeding density of 104 EGFR+ CAR product cells per well with 104 tumor cells per well in 96-well plates. Four images were taken per well every 2 hours on a Sartorius Incucyte Live Cell Analysis system plate imager, in both the phase and red channel with a 20x objective lens. Tumor re-challenges of 104 mcherry-CD19 K562 cells were added to each well 72 and 144 hours after plating, and plates were imaged for 11 days in total. Tumor killing was measured by the decrease in the red relative total integrated intensity, normalized to timepoint 0 for each well. Raw Incucyte data was analyzed using an Incucyte basic analyzer program with a fluorescence threshold of 3.0 red corrected units (RCU), an object area range of 75 μm2 – 1000 μm2, the edge split tool turned on with an edge sensitivity of 5. The normalized red relative total integrated intensity per well was subsequently exported into GraphPad Prism to create figures
Supernatant samples were taken from the plates 18h after each tumor challenge and stored at −80°C until assay completion. Supernatants were analyzed for cytokine content using a MesoScale V-Plex Proinflammatory Panel 1 Human Kit (MesoScale Discovery, Cat# K151A9H-4), detecting IFNγ, IL-2, and TNFα by electrochemiluminescence immunoassay on a MESO QuickPlex SQ 120 imager. Raw cytokine release data was exported into GraphPad Prism to create figures.
Statistical analysis
Baseline characteristics and disease outcomes were compared between 43 subjects infused with SCRI-CAR19v1 and 21 subjects infused with SCRI-CAR19v2. Quartiles are presented for continuous variables, whereas counts and percentages are presented for categorical variables. Table 1 summarizes subject baseline characteristics captured pre-lymphodepletion including age, sex, disease status, CNS status, history and number of prior allogeneic stem cell transplants, time since most recent stem cell transplant, disease burden, CD19 antigen burden, absolute lymphocyte count and absolute blood cell count. P-values are presented summarizing differences between cohorts; generated using the Wilcoxon rank sums test for continuous variables, the Fisher Exact test for categorical variables and the logrank test for time to event variables. Table S1 summarizes, by pre-infusion subgroup, point estimates and 95% confidence intervals for the percentage of subjects who achieved a best post-infusion disease response of MRD-CR. Also presented are the estimated differences and 95% confidence intervals in MRD-CR proportions between cohorts. Confidence intervals and p-values were calculated using the Clopper-Pearson exact method to account for small samples. The number and percentage of SCRI-CAR19v2 subjects experiencing at least one adverse event are summarized by highest toxicity grade. Kaplan-Meier figures are presented comparing overall survival (OS), leukemia-free survival (LFS), event-free survival (EFS) and time to loss of B-cell aplasia (BCA). BCA is <1% of lymphocytes expressing CD19. Comparisons were made between subjects defined by CAR T–cell product. P-values for differences were computed using the logrank test. All survival endpoints are defined as time from CAR T–cell infusion. OS is defined as time to death from any cause. LFS is defined as time to first relapse or death, and only includes subjects who achieved complete remission. Event-free survival is defined as time to first relapse, disease progression or death. Subjects who did not achieve complete remission by day 21 were considered as having an event on day 21, unless they died or progressed earlier. Loss of BCA is defined as time to first detection of B cells >1% of lymphocytes in either the bone marrow or peripheral blood, and only includes subjects who achieved BCA after first CAR T–cell infusion. Death, second CAR T–cell infusion and stem cell transplant are treated as competing risks for loss of BCA if it is known that the subject had ongoing BCA at time of event.
Table 1.
Summary of Subject Characteristics Pre-Infusion
| Characteristic | SCRI-CAR19v2 N (%) or median (Q1, Q3) N=21 |
SCRI-CAR19v1 N (%) or median (Q1, Q3) N=43 |
P Value |
|---|---|---|---|
| Age, yrs | 13 (8, 17) | 12 (6, 17) | 0.8 |
| Female | 7 (33%) | 22 (51%) | 0.2 |
| Disease Status | 0.07 | ||
| Primary refractory | 4 (19%) | 2 (5%) | |
| 1st relapse | 5 (24%) | 16 (37%) | |
| 2nd relapse | 6 (29%) | 20 (47%) | |
| 3rd relapse | 6 (29%) | 5 (12%) | |
| ALC, cells/uL | 840 (545, 1420) | 954 (529, 1664) | 0.6 |
| History of HSCT | 0.3 | ||
| 0 | 11 (52%) | 15 (35%) | |
| 1 | 10 (43%) | 24 (56%) | |
| 2 | 4 (9%) | ||
| Time from most recent HSCT, mo* | 18.7 (8.8, 43.2) | 18.6 (10.6, 34.5) | 1.0 |
| Disease Burden | 0.02 | ||
| M1 | 10 (48%) | 8 (19%) | |
| M2 | 3 (14%) | 6 (14%) | |
| M3 | 7 (33%) | 22 (51%) | |
| Poor quality | 1 (5%) | 0 (0%) | |
| MRD-negative | 0 (0%) | 7 (16%) | |
| CD19 leukemic blasts (%) | 18 (0.05, 66) | 22.5 (0.34, 75.8) | 0.8 |
| CD19 antigen load (%) | 30.2 (5.8, 76.1) | 37.8 (12,79.1) | 0.8 |
| CNS Status | 0.4 | ||
| CNS1 | 18 | 40 | |
| CNS2 | 3 | 3 | |
| CNS3 | 0 | 0 |
CNS, central nervous system – status is pre-lymphodepletion
ALC, absolute lymphocyte count; taken at apheresis
HSCT – hematopoietic stem cell transplant,
calculated using only subjects who received prior HSCT
P value is calculated using Wilcoxon rank sum tests for continuous variables, Fischer exact tests for categorical variables, and log rank test for time to event variables
The area under the curve (AUC) values of the CAR T–cell product engraftment profile over time were computed using the Simpson’s rule. The 95% confidence intervals of these AUC values were estimated using ordinary nonparametric bootstrap resampling for clustered data where 2,000 samples were simulated for SCRI-CAR19v1 and SCRI-CAR19v2 products respectively. Statistical significance for the AUC differences were determined using a one-tailed t-test (P<0.05).
Comparison of phenotype and cytokine profile between SCRI-CAR19v1 and SCRI-CAR19v2 products was performed using a two-tailed Mann-Whitney test (exact P<0.05). Statistical significance for pie charts was determined using the nonparametric partial permutation (Monte Carlo simulation) analysis in SPICE software with 10,000 iterations per test. Comparison of the four participant-matched SCRI-CAR19v2 and SCRI-CAR19v1.5 product flow data was performed using a two-tailed Wilcoxon matched-pairs signed ranks test (exact P<0.05). Comparison of cytokine concentrations in supernatants from the recursive tumor killing assay for participant-matched SCRI-CAR19v2 and SCRI-CAR19v1.5 products was performed using paired t-tests.
Cell lines
The human myelogenous leukemia cell line K562 was obtained from the European Collection of Cell Cultures (ECACC Cat# 89121407, RRID:CVCL_0004) through Sigma-Aldrich between 1999 and 2001. Cell line authentication was performed via STR profiling matched to the DMZ database by the University of Arizona Genetics Core on October 16, 2015. The CD19-transgene expressing K562 cell line (K562-CD19) was previously described and obtained from Stanley Riddell in 2012 [14]. Both cell lines tested negative for mycoplasma.
The K562 line expressing both the CD19 ectodomain (CD19t) and mCherry used in the Incucyte assay was generated in house via two sequential lentiviral transductions of the ECACC K562 line. The first transduction utilized a lentiviral vector containing EGFRviii linked to CD19t via a T2A cleavable linker in an epHIV7 backbone. Following transduction, cells were cloned by limiting dilution. The second transduction utilized a lentiviral vector containing mCherry linked to DHFRdm via a cleavable T2A linker in the epHIV7.3 backbone. Transduced cells were subsequently selected through the addition of methotrexate (50 nm) to the culture media. Expression of CD19t and mCherry were characterized via flow cytometry (99.9% CD19t positive, 96% mCherry positive). Cells were passaged approximately twice after thaw prior to use in the Incucyte assay. This cell line tested negative for mycoplasma.
All cell lines were cultured in complete RPMI (Gibco Cat # 22400-089) + 10% FBS (Avantor Seradigm Cat# 97068–085) and 1% L-glutamine (Gibco Cat# 25030-081).
Data Availability
The data generated in this study are available within the article and its supplementary data files or available upon request from the corresponding author.
RESULTS
Qualification of alternate GMP-grade T-cell activation platform for SCRI-CAR19v2 manufacturing
Evaluation was undertaken to identify alternative T-cell activation platforms for the manufacture of separate CD4+ and CD8+ products in order to move forward with clinical manufacturing for phase 2 of NCT02028455. At the time of the process development work, both TransAct and ExpAct reagents were available as GMP grade products. Initial small scale experiments directly comparing growth and viability with the TransAct anti-CD3/CD28 conjugated polymeric nanomatrix showed consistent failure of purified CD4+ T cells to expand as compared to the CTS Dynabead CD3/CD28 bead activation platform (Figure S2). In contrast, ExpAct particles, which are loaded with an anti-CD28 and anti-biotin/biotinylated anti-CD3, if sufficiently diluted, were able to expand both effector CD4+ and CD8+ T cells. Optimization of the particle to T cell ratio using ExpAct was performed independently for both purified CD4+ and CD8+ T-cell expansion, and optimal expansion was achieved using a ratio of 1:8 for CD8+ T cells and 1:4 for CD4+ T cells (Figure 1B). Large scale qualification runs were performed for comparison of growth (Figure 1C) and surface phenotype (Table S2) with the CTS beads and the ExpAct particles. The growth kinetics of the CD8+ and CD4+ T cells were similar, as was the phenotype of expanded subsets, with the exception of a higher proportion of cells expressing CD127 in ExpAct expanded cultures. Based on this process development work, the ExpAct activation platform was deployed in the manufacture of SCRI-CAR19v2 products.
Comparable baseline status of subjects treated with SCRI-CAR19v2 versus SCRI-CAR19v1
Twenty-three subjects with CD19+ relapsed or refractory B-ALL enrolled onto the initial portion of phase 2 of PLAT-02 and 21 of these subjects received SCRI-CAR19v2 cell infusion. Products were successfully manufactured for all 23 subjects; two subjects did not receive CAR T cells due to clinical circumstances.
Clinical baseline attributes of the 21 infused subjects are shown in Table 1 and included a median age of 13 years (range, 8–17 years). There were no major differences in demographics between subject characteristics between those who received SCRI-CAR19v2 versus SCRI-CAR19v1 (Table 1). Ten SCRI-CAR19v2 treated subjects (48%) had a history of at least one prior allogeneic transplant, with a median time of 18.7 months from most recent transplantation before enrollment in PLAT-02; 28 (67%) SCRI-CAR19v1 treated subjects had a comparable history, with a median time of 18.6 months from most recent transplantation before enrollment. The average peripheral blood absolute lymphocyte count (ALC) of subjects at the time of apheresis was similar for those receiving SCRI-CAR19v2—840 cells/μL (range, 545–1420 cells)— and those receiving SCRI-CAR19v1—954 cells/μL (range, 529–1664). Although there were more SCRI-CAR19v2 subjects with an M1 marrow, there were more SCRI-CAR19v1 subjects with no evidence of marrow MRD, resulting in a similar median percent of leukemic cells in subjects receiving each product (18% SCRI-CAR19v2 vs. 22.5% SCRI-CAR19v1; P=0.8). Additionally, there was a similar percentage of subjects that had high antigen burden, defined as >15% CD19+ B-cell lineage and leukemic cells in bone marrow by flow cytometry prior to lymphodepletion (62% SCRI-CAR19v2 vs. 60% SCRI-CAR19v1); this level of antigen burden was previously found to be associated with enhanced engraftment magnitude and longer persistence of CAR T cells in treated subjects on the phase 1 portion of the trial [5]. There was a difference in the lymphodepletion regimens administered. All of the SCRI-CAR19v2 subjects received Flu/Cy for lymphodepletion prior to CAR T–cell infusion, whereas only 14 SCRI-CAR19v1 subjects received Flu/Cy.
CRS and neurotoxicity in SCRI-CAR19v2 and SCRI-CAR19v1 treated subjects
In comparison with SCRI-CAR19v1, subjects dosed with SCRI-CAR19v2 demonstrated similar rates of CRS (76% SCRI-CAR19v2 vs. 93% SCRI-CAR19v1; P=0.17) and neurotoxicity (38% SCRI-CAR19v2 vs. 48% SCRI-CAR19v1; P=0.4) (Table 2). Although the severity of CRS following SCRI-CAR19v2 (19% grade 1, 57% grade 2) was similar to that seen following SCRI-CAR19v1 (39.5% grade 1, 48.8% grade 2, 4.7% grade 3; P=0.5), the median onset of CRS was two days earlier with SCRI-CAR19v2 (5 vs. 7 days, P=0.02). Despite an earlier onset, the duration of CRS was similar (5.1 days V2 vs. 4.8 days V1, P=0.35). The severity of neurotoxicity following SCRI-CAR19v2 was also similar to that seen following SCRI-CAR19v1, with 24% of subjects experiencing severe neurotoxicity (≥ grade 3 neurotoxicity and including grade 2 seizure) compared to 21% of subjects with SCRI-CAR19v1 (P=0.4), although a single event of grade 5 toxicity (cerebral edema) was observed following SCRI-CAR19v2. Similar to CRS, the median onset of neurotoxicity was significantly earlier following SCRI-CAR19v2 (5.5 vs. 9 days, P=0.01) but duration was similar (median 6 vs. 4 days, P=0.41).
Table 2.
Count and Proportion of Subjects with CRS and Neurotoxicity Adverse Events Related to CAR T Cell Infusion
| Adverse Events of Interest | Grade 1 | Grade 2 | Grade 3 | Grade 4 | Grade 5 |
|---|---|---|---|---|---|
| Cytokine Release Syndrome | |||||
| SCRI-CAR19v1 (n=43) | 39.5% (17) |
48.8% (21) |
4.7% (2) |
0 | 0 |
| SCRI-CAR19v2 (n=21) | 19% (4) |
57% (12) |
0 | 0 | 0 |
| Neurotoxicity | |||||
| SCRI-CAR19v1 (n=43) | 14% (6) |
16% (7) |
9% (4) |
9% (4) |
0 |
| SCRI-CAR19v2 (n=21) | 0 | 14% (3) |
10% (2) |
10% (2) |
5% (1) |
CAR T–cell persistence and LFS are improved with SCRI-CAR19v2
Eighteen (86%) of the 21 SCRI-CAR19v2 infused subjects obtained a best response of MRD-CR within 21 days following infusion (Table S1), which was comparable to the 93% MRD-CR for SCRI-CAR19v1 (P=0.38). The non-responders included the subject who had grade 5 cerebral edema, a subject with engraftment of CAR T cells without tumor clearance, and a subject with a necrotic marrow that was not evaluable for disease response assessment and who ultimately presented with CD19-negative leukemia documented 63 days post CAR T–cell infusion. We observed 5 leukemic relapses in the 21 subjects infused with SCRI-CAR19v2, 3 were CD19 negative, with 2 of the 3 exhibiting lineage switch to acute myeloid leukemia (AML) with genetic relation to the initial ALL. With a minimum of 1-year follow up (range 12–18 months), the 12-month LFS (Figure 2A) and EFS (Figure 2B) for SCRI-CAR19v2 treated subjects was 88.9% (95% CI, 75.5%–100.0%) and 76.2% (95% CI, 59.9%–96.8%), respectively; compared to 55.0% (95% CI, 41.6%–72.8%) and 51.2% (95% CI, 38.2%–68.5%) for SCRI-CAR19v1 treated subjects (P=0.009 and P=0.043, respectively). CD19-negative relapse accounted for 60% (3/5) of relapses with the SCRI-CAR19v2-treated cohort compared to 40% (11/27) in the SCRI-CAR19v1-treated cohort (P=0.63, Fisher’s exact test). Both of the subjects who recurred with CD19+ relapse after SCRI-CAR19v2 treatment had lost persistence of CAR T cells prior to 4 months. The OS at 12 months for SCRI-CAR19v2-treated subjects was 80.9% (95% CI, 65.8–99.6%) compared to 67.4% (95% CI, 54.8%–83.0%) for SCRI-CAR19v1-treated subjects (P=0.26, Figure 2C).
Figure 2. Durability of anti-leukemic response and CAR T–cell engraftment for subjects treated with SCRI-CAR19v2 versus SCRI-CAR19v1.

Kaplan-Meier curves of (A) Leukemia-Free Survival (LFS) (B) Event-Free Survival (EFS) (C) Overall Survival (OS), and (D) time to loss of B-cell aplasia (BCA), comparing subjects treated with SCRI-CAR19v2 (purple) versus SCRI-CAR19v1 (green). Subjects treated with SCRI-CAR19v2 had significantly better LFS (P=0.0091), EFS (P=0.043), and time to loss of BCA (P=0.019). No significant difference was observed in OS (P=0.26) between groups. P values were calculated via log-rank test with weights=1. (E) Mean absolute CAR T–cell engraftment (cells/μL) values at infusion and 10, 14, 21, and 63 days after T-cell infusion are plotted on the primary y-axis along with standard error of the mean. Mean CAR T–cell composition breakdown, namely %CD4+ and %CD8+ of total EGFRt+ cells, at 10, 14, 21, and 63 days after T-cell infusion are scaled to 100% and plotted on the secondary y-axis. (F) CAR T–cell composition breakdown at long term follow up (LTFU) visits ranging from 3 months (3M) to 54 months (54M) after T-cell infusion are plotted in scattered dots. The colors of the dots denote the corresponding LTFU visit timepoint and the sizes of the dots represent the total CAR T–cell engraftment in %EGFRt+ of CD3+ detected at each visit timepoint. Regions corresponding to more %CD8+ and %CD4+ in the CAR T-cell composition breakdown are highlighted for clarity.
To further investigate the improved LFS following SCRI-CAR19v2, we evaluated the duration of CAR T–cell persistence, as that is a major contributor to enhanced durability of remission. The median duration of functional CAR T–cell persistence, as measured by ongoing BCA, was not reached and 61.1% of subjects had ongoing persistence at 1 year post SCRI-CAR19v2, compared to only 20.6% for SCRI-CAR19v1-treated subjects (P=0.019, Figure 2D). Given that some studies have demonstrated that there is an impact of Flu/Cy lymphodepletion on persistence [5, 9], we performed an additional analysis that restricted the comparison of persistence in SCRI-CAR19v2 subjects to a smaller subset of SCRI-CAR19v1 subjects who received Flu/Cy. This comparison demonstrated that there was still a significant improvement in LFS following SCRI-CAR19v2 compared to SCRI-CAR19v1 (P=0.029, Figure S3A). Although EFS and duration of BCA lost significance, they still trended towards improvement in SCRI-CAR19v2-treated subjects (Figure S3B, D). The loss of significance in EFS and BCA may be related to the small numbers in this comparison. In aggregate, these data suggest that enhanced functional persistence of SCRI-CAR19v2 may be the driver of the corresponding improvements in treated subjects’ LFS and EFS.
In vivo CAR engraftment is predominated by CD8+ CAR T cells
To better assess the mechanism(s) responsible for improved performance of SCRI-CAR19v2, we sought to profile which components of the product exhibited more durable engraftment fitness. We examined both total CAR+ T-cell engraftment over time, as well as, the CD4:CD8 ratio of engrafted CAR T cells in treated subjects (Figure 2 E, F). SCRI-CAR19v1 products had similar short term cumulative engraftment as quantified by the mean area under the curve (AUC) of absolute CAR T-cell engraftment, with 5350.20 cells/μL (95% CI: 2029.69–10634.34), compared to that of SCRI-CAR19v2, with a mean AUC of 2553.60 cells/μL (95% CI: 777.37–4818.11, P=0.3). However, caution should be taken with this analysis as day 10 was the first time point for which engraftment was measured post SCRI-CAR19v2 infusion based on the phase 1 data showing peak engraftment at this time point [5]. The earlier onset of CRS with the SCRI-CAR19v2 product could indicate an earlier expansion and engraftment peak of the SCRI-CAR19v2 product, in which case, the engraftment curves may be underestimating the total engraftment of SCRI-CAR19v2 product over the first two months.
Despite SCRI-CAR19v2 and SCRI-CAR19v1 product infusion at equivalent ratios of CD4:CD8 T cells (~1:1 administration), CD8+ CAR T cells were the predominant population to engraft and persist beyond day 10 regardless of manufacturing process (Figure 2E). For subjects with detectable CAR T cells in peripheral blood beyond 3 months (as measured by %EGFRt of CD3+ T cells via flow cytometry), CAR+ T cells were predominantly CD8+ in the majority of samples (58 out of total 95 samples) (Figure 2F). Together these data show that both SCRI-CAR19v2 and SCRI-CAR19v1 CD8+ CAR T-cell products are the predominant population of persisting CAR T cells in the peripheral blood.
SCRI-CAR19v2 products are phenotypically less activated/exhausted
Given the lack of differences in subject characteristics, we hypothesized that the improved persistence of SCRI-CAR19v2 in vivo may be a result of the change in the manufacturing platform. CD8+ CAR T-cell SCRI-CAR19v2 products were in culture for an average of 12.6 days (range of 8–23), with an average of 9.1-fold expansion (range 0.8–18.4), and CD4+ CAR T–cell SCRI-CAR19v2 products were in culture for an average of 10 days (range 8–21), with an average of 11.4-fold expansion (range 6.5–22.1). In comparison to the SCRI-CAR19v1 platform, when both CD8+ and CD4+ products had shorter manufacturing time (CD8 P=0.0007; CD4 P= <0.0001) and a higher fold expansion (CD8 P=0.007; CD4 P=0.0001) (Table S3).
To assess polyfunctional cytokine release, we performed intracellular cytokine staining on CD4+ and CD8+ CAR products after an 18-hour co-incubation with a CD19 stimulator cell. Polyfunctional cytokine production differed between SCRI-CAR19v2 and SCRI-CAR19v1 CD8+ products (Figure 3A, P=0.0199). Although there appeared to be a higher frequency of polyfunctional cells within the SCRI-CAR19v1 CD8+ products, the SCRI-CAR19v2 products exhibited significantly higher per cell synthesis of TNFα, IFNγ, and IL2 based on intracellular mean fluorescence intensity (MFI) (Figure 3B, MFI of all cytokines P<0.0001). Analysis of activation/exhaustion marker expression demonstrated that co-expression profiles of TIM-3, LAG-3 and PD-1 were significantly less frequent on SCRI-CAR19v2 CD8+ products as compared to their SCRI-CAR19v1 counterparts (Figure 3C; P<0.0001). Overall, the MFI of expression was also decreased for both TIM-3 and PD-1 on SCRI-CAR19v2 CD8+ products compared to SCRI-CAR19v1 CD8+ products (Figure 3D; TIM-3 MFI P<0.001, PD-1 MFI P<0.05). Finally, in comparison to SCRI-CAR19v1 CD8+ products, SCRI-CAR19v2 CD8+ products had a higher percentage of CD8+ CAR T cells expressing CD45RA and a lower percentage of CD45RO- and CD95-expressing cells (Figure 3E, P<0.05). In aggregate, these data suggest that the quality of activation on the SCRI-CAR19v2 ExpAct platform results in less activated and terminally differentiated CAR T cells in the final product.
Figure 3. Multiparameter flow cytometric analysis of SCRI-CAR19v2 versus SCRI-CAR19v1 CAR T cells.
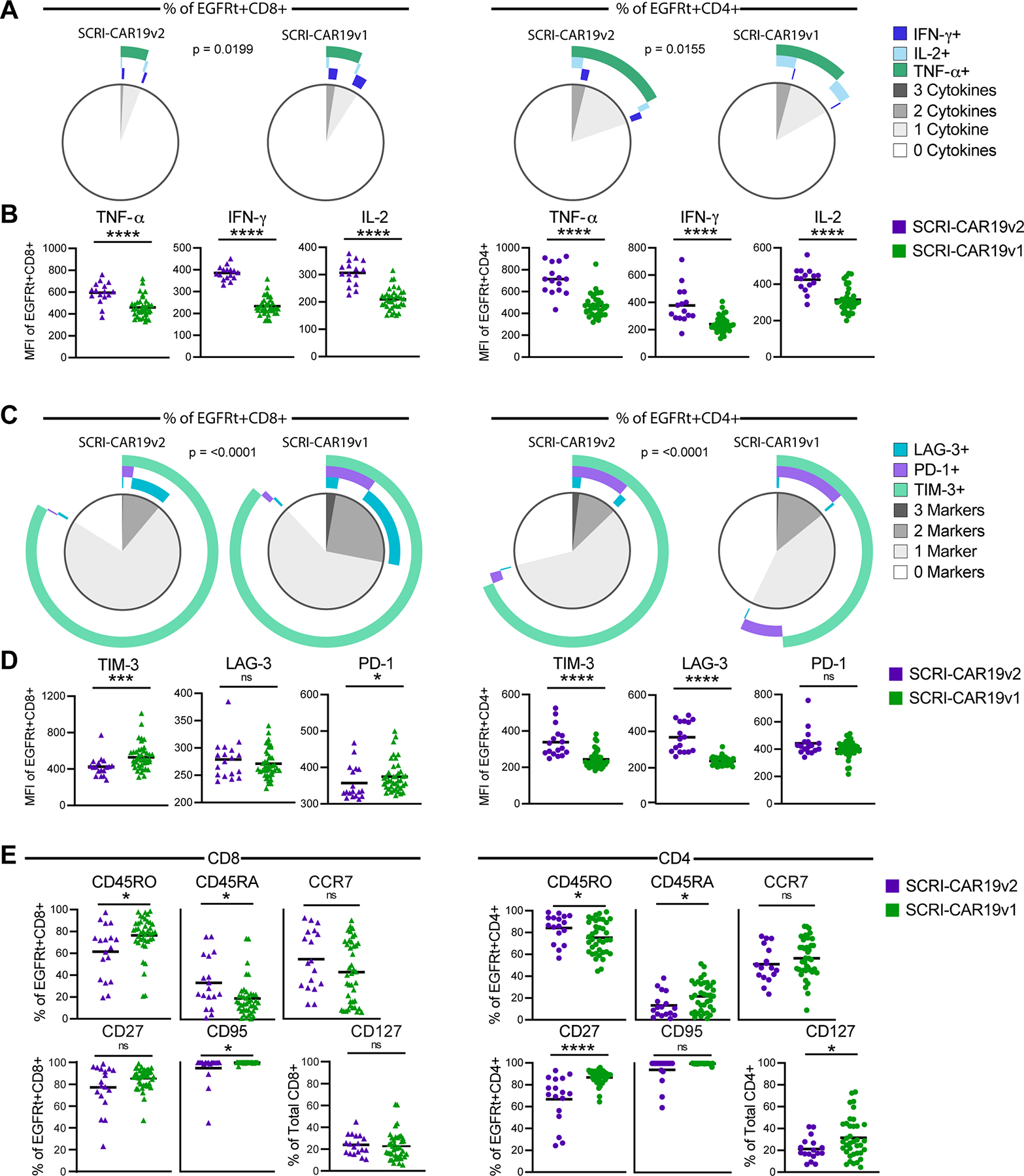
Intracellular cytokine production in response to CD19 stimulation (A-B) and the phenotype of CAR (EGFRt+) T cells in unstimulated SCRI-CAR19v2 versus SCRI-CAR19v1 final products (C-E) was examined by flow cytometry. (A) Following 18h stimulation with CD19 antigen–bearing cells, poly-cytokine profiles of the CD8+EGFRt+ (left) and CD4+EGFRt+ (right) cells of SCRI-CAR19v2 versus SCRI-CAR19v1 products were determined using SPICE analysis software. (B) MFI of the individual cytokines examined in panel A. (C) Comparison of the percent of CD8+EGFRt+ and CD4+EGFRt+ cells in the CD8+ or CD4+ final product material with poly-activation marker expression, visualized via SPICE analysis. (D) MFI of the individual activation markers examined in panel C. (E) Percent of CD8+EGFRt+ and CD4+EGFRt+ cells expressing T-cell differentiation markers in the CD8+ (left) or CD4+ (right) final products. (SCRI-CAR19v2; CD8 n = 16–18 subjects; CD4 n = 15–17 subjects. SCRI-CAR19v1; CD8 n = 37–41 subjects; CD4 n = 37–38 subjects) (****, P ≤ 0.0001; ***, P ≤ 0.001; *, P ≤ 0.05). Significance determined by Mann-Whitney test.
Although we have not previously identified factors associated with long-term engraftment related to the CD4+ products, we explored similar studies of the CD4+ CAR T–cell products to look for differences between SCRI-CAR19v2 and SCRI-CAR19v1 CD4+ products. Similar to the CD8+ products, polyfunctional cytokine production differed between SCRI-CAR19v2 and SCRI-CAR19v1 CD4+ products (Figure 3A; P=0.0155), and SCRI-CAR19v2 CD4+ products had significantly elevated per cell expression of TNFα, IFNγ, and IL2 in response to CD19 antigen stimulation compared to SCRI-CAR19v1 CD4+ products (Figure 3B; MFI of all cytokines P<0.0001). Co-expression profiles of TIM-3, LAG-3 and PD-1 differed between SCRI-CAR19v2 and SCRI-CAR19v1 CD4+ products (Figure 3C; P<0.0001). However, the difference was the reverse of that observed for the CD8+ products. The SCRI-CAR19v2 CD4+ CAR T cells expressed significantly higher TIM-3 and LAG-3 compared to SCRI-CAR19v2 CD4+ CAR T cells (Figure 3D; MFI P<0.0001). SCRI-CAR19v2 CD4+ products had a higher percentage of CAR T cells expressing CD45RO (P<0.05) and a lower percentage expressing CD45RA and CD27 (P<0.05 and P<0.0001, respectively) (Figure 3E). Finally, SCRI-CAR19v2 CD4+ products also had a significantly lower percentage of CD127+CD4+ T cells similar to the finding during the large scale process development work. To date, the characteristics of CD4+ CAR T–cell products that may enhance or contribute to prolonged functional engraftment of CD8+ CAR T cells have not been identified.
Together, these data demonstrate that the changes in manufacturing platform between SCRI-CAR19v1 and SCRI-CAR19v2 resulted in CAR T–cell products that were phenotypically and functionally distinct. Most notable to us was that the CD8+ CAR T–cell products from SCRI-CAR19v2 were enhanced for attributes previously associated with improved engraftment fitness.
Cytokine profiles differentiate between SCRI-CAR19v2 and SCRI-CAR19v1 products
Polyfunctional cytokine production has been identified as a predictor of outcomes as it relates to both efficacy and toxicity [15, 16]. To further define polyfunctional cytokine differences between SCRI-CAR19v2 and SCRI-CAR19v1 products and identify cytokine signatures associated with prolonged persistence, we performed an unbiased hierarchical clustering analysis of CD8+ and CD4+ products comparing antigen-specific cytokine release by the CAR T cells following CD19 antigen stimulation in vitro. Three clusters emerged for both CD8+ and CD4+ products (designated 1–3), with a majority of SCRI-CAR19v2 products falling into cluster 3 (Figure 4A, orange). SCRI-CAR19v2 CD8+ products produced higher levels of IL2, IL5 and IL13 compared to SCRI-CAR19v1 CD8+ products (Figure 4A–C; IL2 P<0.0001, IL5 P<0.0001, IL13 P<0.001). Of potential biological significance, PCA revealed that increased secretion of IL2 by SCRI-CAR19v2 CD8+ products was in stark contrast to a discrete subset of SCRI-CAR19v1 CD8+ products that did not release detectable IL2 during the stimulation (Figure 4B; cluster 2, blue).
Figure 4. Antigen-specific cytokine secretion by SCRI-CAR19v2 versus SCRI-CAR19v1 CAR T cells.

Following 18h stimulation with CD19-positive or -negative K562 cells, cytokine secretion by CD8+ (left) and CD4+ (right) products was determined by Luminex analysis of the culture supernatant. The antigen-specific cytokine production was determined by subtracting the response to CD19-negative control stimulation from the response to CD19-positive stimulation. (A) Heat map with dendrogram and (B) Principle component analysis (PCA) plot of cytokine secretion by SCRI-CAR19v2 (purple) and SCRI-CAR19v1 (green) CD8+ or CD4+ products. Log10 values of the cytokine production are used in these analyses. Analytes that were not detected in both SCRI-CAR19v2 and SCRI-CAR19v1 products were excluded. Last column of heat maps are pseudo-colored to indicate product designation. In panel A, unbiased hierarchical clustering analyses were carried out using “Ward.D” algorithm with Euclidean distance matrix on both products (rows) and analytes (columns). Data points in the PCA plots are colored with product designation and are grouped by the unbiased hierarchical clustering assignment on products (rows) presented in panel A. PCA analyses were conducted using covariance matrix. (C) Cytokine production (in pg/mL) represented by box plots displaying the median and 25th to 75th percentiles with whiskers extending to the minimum to maximum detected values. Only analytes with significantly different secretion between SCRI-CAR19v2 and SCRI-CAR19v1 products in either the CD8+ or CD4+ CAR T–cell populations are shown. (SCRI-CAR19v2; CD8 n = 14 subjects; CD4 n =17 subjects. SCRI-CAR19v1; CD8 n = 35 subjects; CD4 n = 38 subjects.****, P ≤ 0.0001; ***, P ≤ 0.001; **, P ≤ 0.01; *, P ≤ 0.05) Significance determined by Mann-Whitney test.
PCA analysis of the CD4+ products showed that separation between cluster 3 and clusters 1 and 2 was predominantly driven by elevated secretion of IL17A, IL17F, IL22, and TNFβ, and to a lesser extent TNFα and IL21, by SCRI-CAR19v2 CD4+ products compared to SCRI-CAR19v1 CD4+ products (Figure 4B). These cytokines were all significantly increased in SCRI-CAR19v2 compared to SCRI-CAR19v1 CD4+ products (Figure 4C; IL17A P<0.0001, IL17F P<0.001, IL21 and IL22 P<0.01, TNFα and TNFβ P<0.05). PCA further revealed that secretion of IL4, IL5, IL13 and IL23 separated cluster 2 from 1 and was orthogonal to cluster 3. In addition, SCRI-CAR19v2 CD4+ product secretion of IL4, IL5, IL13 and IL23 was significantly increased compared to secretion by SCRI-CAR19v1 CD4+ products (Figure 4C; IL-4 and IL-5 P<0.0001, IL-13 P<0.001, IL-23 P<0.05)
These data further highlight differences between SCRI-CAR19v1 and SCRI-CAR19v2 products and show that changes during product manufacturing can significantly effect antigen-specific cytokine release by the CAR T cells, which may in turn impact their ability to engraft and persist in patients.
Stimulation reagent drives phenotypic and functional changes in CAR products
Given that there were two manufacturing changes in the platform for SCRI-CAR19v2—the activation bead reagent was changed and the EGFRt enrichment step was discontinued—we wanted to determine the impact of the activation platform change on product phenotype and function independent of any possible effect imparted by eliminating the EGFRt immunomagnetic enrichment step. Additionally, although our prior work did not identify apheresis T-cell repertoire features associated with final product engraftment fitness [17], this additional analysis allowed for comparison of manufacturing strategies without the variability of inter-subject differences in apheresis composition by utilizing the same starting material for both product versions. To do this, we generated participant-matched CD8+ and CD4+ CAR T–cell products for four PLAT-02 subjects using two different manufacturing strategies: 1) the SCRI-CAR19v2 platform and 2) a new platform using the CTS Dynabead CD3/CD28 stimulation reagent used for SCRI-CAR19v1 but without EGFRt enrichment (referred to as SCRI-CAR19v1.5). The matched v2 and v1.5 products were manufactured sequentially at full scale, using the same CD4+ and CD8+ selected T cells as starting material. The manufacturing time comparing v2 to v1.5 products was similar for both CD8+ products (11.5 vs. 9.5 days, P=0.25) and CD4+ products (9.5 vs. 9.5 days, P=1; Wilcoxon signed-ranks test).
A comparison of antigen-specific cytokine production between SCRI-CAR19v2 and SCRI-CAR19v1.5 CD8+ products showed that v2 products exhibited a higher percentage of TNFα expressing CD8+ CAR T cells in response to CD19 antigen stimulation compared to v1.5 (Figure 5A), and of those cells expressing TNFα, the MFI was higher with v2 products in 2 out of the 3 products that had the comparison available. This finding for the SCRI-CAR19v2 versus SCRI-CAR19v1.5 CD8+ products was similar to that seen for the SCRI-CAR19v2 versus SCRI-CAR19v1 comparison in terms of MFI, but was distinct in that the percentage of cells expressing TNFα was also higher for the SCRI-CAR19v2 products. The IFNγ findings were more subtle, with some subjects showing a higher percentage of IFNγ+ CD8+ CAR T cells, with the MFI of one subject being substantially higher as well. Although the trends of TIM-3 and LAG-3 were mixed, the percentage of PD-1+ CD8+ CAR T cells was markedly decreased in SCRI-CAR19v2 CD8+ products compared to SCRI-CAR19v1.5 CD8+ products (Figure 5B) similar to the SCRI-CAR19v2 versus SCRI-CAR19v1 comparison. Activation, as assessed by CD69 percentage and MFI, varied among the CD8+ CAR T–cell products and did not appear as a consistent difference between the SCRI-CAR19v2 and SCRI-CAR19v1.5 participant-matched samples, suggesting similar levels of phenotypic activation but lower levels of exhaustion in the SCRI-CAR19v2 CD8+ products. Finally, SCRI-CAR19v2 CD8+ products had a notable increase in the CCR7+ population as well as CD127+ population (Figure 5C), markers associated with engraftment fitness [18–20]. The CD4+ products demonstrated similar TNFα and PD-1 findings to the CD8+ products, and the percentage and MFI of CD69 expression was again mixed (Figure 5D, E). Similar to the CD8+ products, PD-1 expression was decreased in all four of the SCRI-CAR19v2 CD4+ products compared to the SCRI-CAR19v1.5 CD4+ products, and CCR7 and CD127 were increased in three out of the four SCRI-CAR19v2 CD4+ products (Figure 5F).
Figure 5. Participant-matched SCRI-CAR19v2 versus SCRI-CAR19v1.5 CAR Product Profiles.
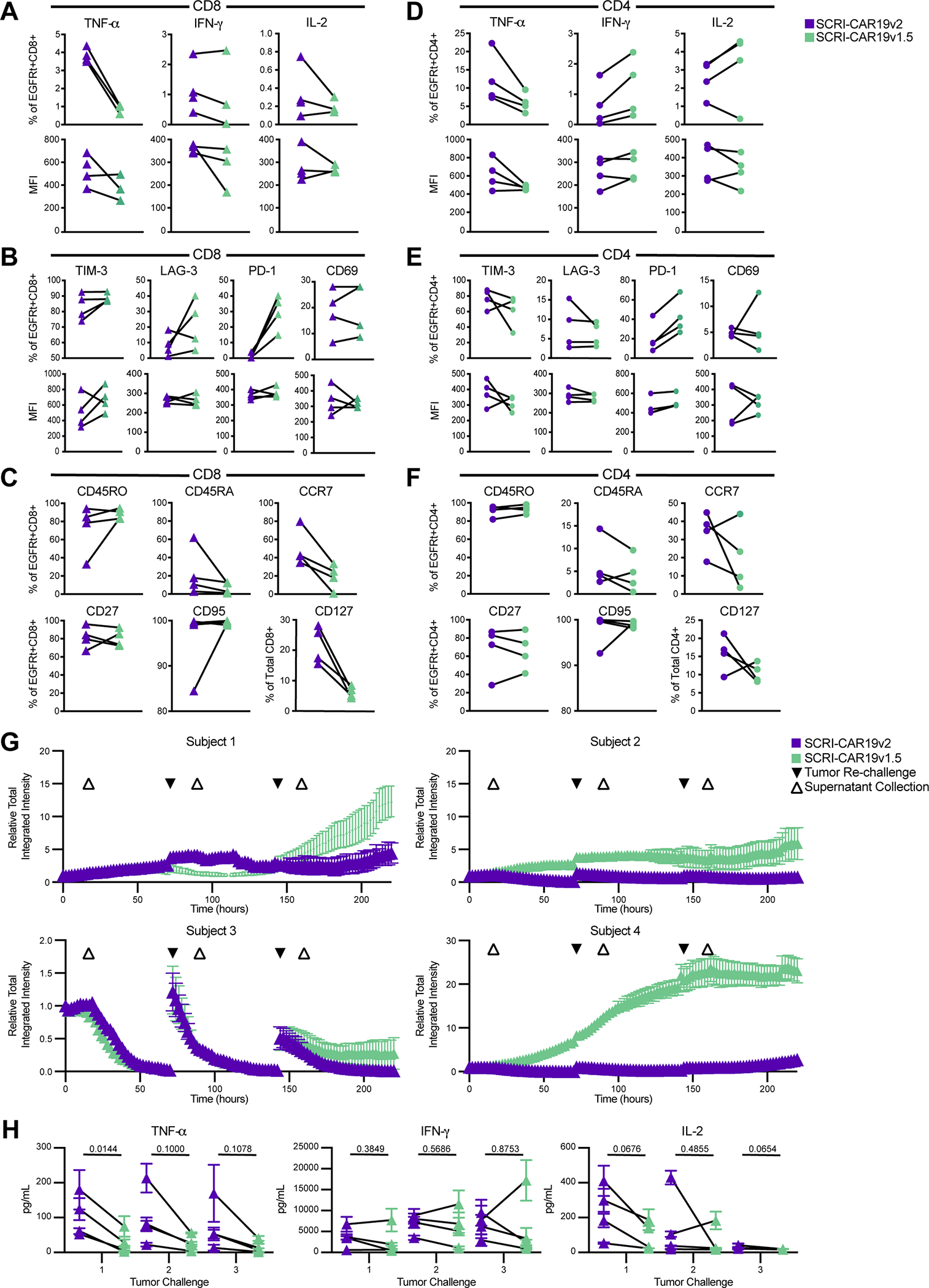
Participant-matched CD8+ and CD4+ final products were generated for four PLAT-02 subjects. Following 18h stimulation with CD19-positive K562 cells, cytokine production was determined by intracellular cytokine staining. The cytokine production of stimulated cells as a percent of the CAR+ (EGFRt+) and the MFI of the cytokine levels is shown for both CD8+ cells (A) and CD4+ cells (D). Phenotypic attributes of CAR+ (EGFRt+) T cells in unstimulated SCRI-CAR19v2 and SCRI-CAR19v1.5 final product–matched pairs were assessed by flow cytometry. (B) Percent of CD8+EGFRt+ and (E) CD4+EGFRt+ cells expressing markers of activation or exhaustion as well as the MFI of the positive cells. (C) Percent of CD8+EGFRt+ and (F) CD4+EGFRt+ cells expressing markers of T-cell differentiation in the CD8+ and CD4+ final products. No significant difference was found in SCRI-CAR19v2 versus SCRI-CAR19v1.5 comparisons from panels A-F as determined by Wilcoxon test. (G) In vitro recursive tumor killing profiles of the four matched product pairs (50/50 mix of CD4+ and CD8+ products), when challenged 3x with K562-CD19-mCherry tumor cells. Increasing intensity indicates an increased growth of the mCherry tumor, inverse to tumor killing. Relative Total integrated intensity normalized per well at timepoint 0h, measured by an Incucyte live-cell imager every 2h. Plots represent the mean red Relative Total Integrated Intensity of triplicate wells for each SCRI-CAR19v1.5 and SCRI-CAR19v2 product. Vertical bars show SEM. Tumor re-challenges added at 72h and 144h (black triangles). Supernatant collections taken 18h after each challenge for cytokine analysis (white triangles). (H) Cytokine concentrations in supernatant of tumor killing assay 18h post-challenge for each product. Triangles represent the mean cytokine concentration in pg/mL of triplicates for a subject’s SCRI-CAR19v2 (purple) or SCRI-CAR19v1.5 (green) product. Vertical bars show SEM. Means associated with the same subject are connected (black lines). P-values reported are from paired t-tests using n=4 subject pairs.
We next determined whether the manufacturing changes from SCRI-CAR19v2 to SCRI-CAR19v1.5 impacted the functional performance of the CAR product’s abilities for recursive tumor killing in vitro. Briefly, SCRI-CAR19v2 and SCRI-CAR19v1.5 CD8+ and CD4+ products were mixed at a 1:1 ratio for each subject and co-cultured with CD19-expressing K562 target cells. Consistent with a more favorable phenotype, SCRI-CAR19v2 demonstrated superior recursive tumor cell killing over three tumor challenges compared to SCRI-CAR19v1.5 for three out of four subjects (Figure 5G). Subject 3 appeared to have similar killing over the first two tumor challenges, with some evidence for superior killing with the SCRI-CAR19v2 product on the third challenge. Cytokine secretion measured after each tumor challenge revealed within individual subjects, SCRI-CAR19v2 products had higher TNFα and IL2 release compared to SCRI-CAR19v1.5 products following the first challenge, with similar IFNγ release (Figure 5H). Over successive tumor challenges, these trends continued, but with less amplitude. The noted difference of specific cytokine secretion was similar to the cytokine production measured in the intracellular cytokine secretion assay with the SCRI-CAR19v2 products appearing to have an increase in the percentage of cells capable of secreting TNFα across both CD8+ and CD4+ products.
DISCUSSION
Sustained remission rates following CD19-specific CAR T–cell therapy for pediatric patients with B-ALL are suboptimal, with approximately half of patients relapsing within a year of attaining an initial remission, with additional events occurring beyond one year [3, 5, 6]. We noted that the patient cohort receiving the SCRI-CAR19v2 product had improved LFS with superior duration of functional persistence as well. Persistence of CAR products is a major factor for maintaining remission in patients with B-ALL. Although there may be inherent subject differences that contribute to enhanced persistence, we did not identify any known subject characteristics to account for the differences observed between our SCRI-CAR19v1 and SCRI-CAR19v2 products.
Although phenotypic and functional CAR T–cell product attributes have been associated with durability of engraftment, the T-cell intrinsic features that mechanistically contribute to engraftment fitness are not fully elucidated. In our clinical studies utilizing a 4–1BB:zeta anti-CD19 CAR, CD8+ CAR T cells predominate both short- and long-term engraftment in treated subjects will B-ALL. Although currently ill defined, it is possible that the phenotype of the CD8+ CAR T cells may be more impactful than that of the CD4+ CAR T cells in predicting persistence, and this is in line with our initial findings of CD8+ CAR T–cell products with increased TNF α production and decreased TIM-3 expression being associated with enhanced persistence [7].
Several published reports have sought to correlate clinical performance of CAR T–cell products with measurable features of the product at the end of manufacture. Fraietta et al. have analyzed CAR T–cell products and outcomes in the setting of chronic lymphocytic leukemia (CLL). They observed that products from patients who achieved a CR following CD19-specific CAR T–cell infusion exhibited markers of early memory differentiation (CD27+PD-1−CD8+ CAR T cells expressing high levels of the IL6 receptor); in contrast, products from PR and NR (no response) patients had increased frequencies of T cells with terminal effector differentiation. Additionally, products from patients with CR and PR had a lower percentage of CD8+ T cells that were PD-1+, PD-1+TIM-3+ and PD-1+LAG-3+ [16]. Our findings are consistent with these findings in that the SCRI-CAR19v2 CD8+ products had significantly fewer CD8+ CAR T cells expressing PD-1, TIM-3 and LAG-3. Rossi et al. investigated CAR T–cell products for outcomes in patients with non-Hodgkin lymphoma (NHL) and found that CAR products that contained polyfunctional T-cell subsets were associated with improved clinical response [15]. It is not clear how product attributes differentially affect clinical performance across the spectrum of B-cell malignancies. For patients with both CLL and NHL, only a subset of treated subjects get into remission. Additionally, for NHL patients, persistence may not be as important as it is in ALL patients. Therefore, the ideal attributes of a CAR T–cell product designed for treatment of ALL may be unique from those for treatment of CLL and NHL, where the focus is more on the longevity of persistence rather than initial remission. Our group will continue to focus on product profiling and validate which phenotypes/signatures are predictive of clinical performance in the setting of pediatric B-ALL.
Additional differences were detected in the comparison of SCRI-CAR19v2 and SCRI-CAR19v1 products aside from those attributes that we had previously identified as being associated with persistence durability. SCRI-CAR19v2 CD4+ products were characterized by increased production of multiple effector cytokines, including IL4, IL5, IL13, IL17A, IL17F, IL22, TNFα and TNFβ; SCRI-CAR19v2 CD8+ products had elevated IL2, IL5 and IL13 compared to SCRI-CAR19v1 CD8+ products. As mentioned earlier, others have shown that polyfunctional products in NHL have been associated with both improved clinical responses and higher toxicity, and this finding was more impressive within CD4+ CAR T cells than CD8+ CAR T cells for the NHL patient population [15]. It is unknown if this CD4+ polyfunctional profile also helps to promote long-term functional persistence in our ALL population. Given that many of these cytokines have been implicated in T-cell proliferation and expansion, we speculate that increased secretion of these cytokines may function in an autocrine fashion to boost CAR T–cell performance and/or function in a paracrine fashion to establish an environment that better supports CAR T–cell functional persistence in vivo. The number of infused subjects in this study is small; therefore, it will be important to replicate these analyses in the larger SCRI- CAR19v1.5 phase 2 cohort to validate whether specific product characteristics are associated with enhanced clinical attributes.
Similar to changes in therapeutic performance, it is likely that manufacturing changes can lead to changes in toxicity. Shah et al. have recently shown that CD4/CD8 selection of apheresis products prior to initiation of manufacturing leads to a change in potency and thus affects the toxicity profile of a product [21]. In addition to showing enhanced clinical responses, Rossi et al. showed that grade 3 and higher CRS and neurotoxicity were associated with polyfunctional IL17A-producing T cells [15]. Although the overall rates of CRS and neurotoxicity were similar between SCRI-CAR19v1 and SCRI-CAR19v2 treated patients, an observed earlier onset of symptoms may be related to a hastened onset of CAR T–cell expansion with higher cytokine production at earlier timepoints. Unfortunately, the pre-determined schedule for collection of correlative analyses post-infusion did not allow us to fully investigate the tempo of CAR T–cell expansion or in vivo cytokine profiles at earlier timepoints to confirm this.
The single event of fatal cerebral edema following infusion of SCRI-CAR19v2 invoked concern for an association with the SCRI-CAR19v2 manufacturing platform, as cerebral edema had not been encountered with SCRI-CAR19v1. Around the time of the fatal event, the licensing agreements were modified to allow for the use of the CTS Dynabead CD3/CD28 stimulation reagent. Using our best judgement at the time and prior to completion of the analyses reported in this manuscript, further treatment with SCRI-CAR19v2 was abandoned and the phase 2 portion of our trial was re-initiated with the SCRI-CAR19v1.5 product. Based on our current knowledge, we have no evidence that the use of the ExpAct reagent in manufacturing was associated with the cerebral edema event.
To elucidate whether the observed product differences were a direct effect of the activation reagent change in manufacturing and independent of discontinuation of the EGFRt enrichment step, we did a limited comparison of products manufactured with ExpAct or CTS beads on an otherwise identical manufacturing platform. At the same time, we sought to use the same starting material for the comparisons to ensure that the differences were not accounted for by starting T-cell populations. Comparing these full scale, participant-matched SCRI-CAR19v1.5 and SCRI-CAR19v2 products, SCRI-CAR19v2 products expressed a higher percentage of phenotypic markers associated with engraftment fitness [18–20] and exhibited lower percentages of cells expressing markers associated with exhaustion and/or attenuated effector function (PD-1, TIM-3, LAG-3). The SCRI-CAR19v2 products also demonstrated superior abilities in comparison to the SCRI-CAR19v1.5 participant-matched products in an in vitro recursive killing assay. The combination of lower expression of exhaustion markers and superior repeated killing abilities suggests that the SCRI-CAR19v2 CAR T cells are less likely to get exhausted in vivo with repeated exposure to antigen. This overall observation was similar to our clinical results comparing SCRI-CAR19v2 with SCRI-CAR19v1, where the in vivo persistence was enhanced with the SCRI-CAR19v2 product in treated subjects. Acknowledging the small numbers of subjects in this matched product comparison, the phenotypic differences seen in the SCRI-CAR19v1 and SCRI-CAR19v2 final product comparison are unlikely to be explained by variations in the starting T-cell populations, as these differences were maintained in the participant-matched analysis. The SCRI-CAR19v2 versus SCRI-CAR19v1.5 product comparison is highly suggestive that a single change of activation reagent at the initiation of product manufacturing can lead to alterations in product phenotype and in vivo function.
In consideration of the impact of the activation reagent on modulating the final product performance, it is important to consider some of the differences between the reagents. CTS Dynabeads are 4.5 um super-paramagnetic beads coupled to anti-CD3 and anti-CD28. ExpAct beads from Miltenyi are antibody coated nanoparticles of 3.5 um in size, loaded with anti-CD28, anti-biotin and CD3–biotin. Due to proprietary information, a comparison of the densities of anti-CD3 and anti-CD28 antibodies on the beads is not available. Previously published analyses demonstrated that PBMCs directly activated with CTS Dynabeads, TransAct and ExpAct beads had similar transduction efficiency and expansion [22]. In contrast, our process development work demonstrated superior CD4+ in vitro expansion with the ExpAct beads over the initial iteration of the TransAct beads. This divergent experience may be related to the separation of CD4 and CD8 T cells into independent cultures.
Variation in bead ratios can lead to divergent culture phenotypes, with higher bead ratios leading to increased cell activation and ultimately activation-induced cell death of effector cells, whereas while lower bead ratios can preserve memory T-cell populations [23]. In the process development work, we attempted to align the bead ratio with the ExpAct beads to allow for similar growth kinetics as the CTS bead manufacturing and to retain similar basic phenotyping. However, due to the different bead to T cell ratios used, it is possible that the decreased activation observed in the CD8+ cultures is a result of the ratio used rather than the bead reagent. Because ExpAct beads are engineered to expand regulatory T cells rather than effector T cells, they may have additional properties that lead to the differences being seen. Further investigation of the T-cell interaction with these beads may uncover the properties that drive divergent T-cell differentiation during the ex vivo manufacturing process.
Although these data indicate that the activation reagent used during the manufacturing process affects product phenotype, it is also possible that discontinuing the EGFRt immunomagnetic enrichment step with SCRI-CAR19v2 products may have contributed to the increased clinical potency with SCRI-CAR19v2. This could explain some of the discrepancy seen in the SCRI-CAR19v2 versus SCRI-CAR19v1 comparison, in which only the SCRI-CAR19v1 products underwent enrichment based on EGFRt, that was not recapitulated in the SCRI-CAR19v2 vs SCRI-CAR19v1.5 comparison where neither product underwent enrichment. There are two potential effects of the selection that may impact product phenotype and performance. The EGFRt selection step causes some cell loss, which ultimately extends the time in culture. Additionally, it enriches for cells with the highest level of transgene expression in the final product, as measured by EGFRt MFI at the end of culture [5]. The signaling biology of CAR T cells with the highest expression levels may predispose them to activation-induced cell death or functional exhaustion that limits their proliferative activity upon repeated encounter with CD19+ targets and attenuates their persistence. Eyquem et al. showed, in a preclinical mouse model, that integration of the CAR into the TRAC locus using CRISPR/Cas9 genome editing yields a CAR T–cell product with enhanced performance. This gene modification process generated CAR T cells with decreased MFI of CAR expression when compared to random viral vector integration, and these CAR T cells had lower expression of PD-1, LAG-3 and TIM-3 [24]. Further investigation will help to determine whether including T cells expressing lower levels of the CAR in the final product can enhance clinical in vivo persistence, and it is anticipated that the phase 2 results from infusion of SCRI-CAR19v1.5 will help to clarify this issue.
In aggregate, the SCRI-CAR19v2 platform appears to generate products with less activation-induced differentiation and retained markers associated with engraftment fitness and prolonged in vivo persistence. Our data further indicates that changing ex vivo CAR T–cell manufacturing is sufficient to impart significant alterations in the product’s therapeutic potency without concomitant enhancement of the product’s toxicity profile. Whether this enhanced performance of the SCRI-CAR19v2 product was due to the reagent change, altered bead ratios, duration of ex vivo expansion or mid culture enrichment of transduced cells remains unknown. Moreover, these studies further support defining product attributes that are associated with superior in vivo persistence and eventually engineering these attributes into all products in a systematic manner through interrogation and iteration of manufacturing platforms.
Supplementary Material
SYNOPSIS.
Through analysis of CAR T–cell product features and clinical outcomes, the authors reveal significant performance differences attributable to changes in the manufacturing process, even when products are delivered in defined numbers and CD4:CD8 ratios.
Acknowledgements
We thank the clinical trial participants and their families, referring physicians and care teams, clinical research teams, the Therapeutic Cell Production Core, and the Correlative Studies Lab. We would also like to acknowledge Meredith Jackson for her guidance of the Incucyte assay and analysis.
Funding
We are grateful for the generous funding from a St. Baldrick’s Foundation - Stand Up To Cancer Pediatric Dream Team Translational Research Grant (SU2C-AACR-DT-27-17), the William Lawrence and Blanche Hughes Foundation, Alex’s LemonadeStand, National Institutes of Health award 5U01TR002487-03, the ISREC Foundation (the Swiss Institute for Experimental Cancer Research), National Cancer Institute award P30CA014089 and Juno Therapeutics (A Bristol-Myers Squibb company). Stand Up To Cancer is a division of the Entertainment Industry Foundation. The indicated Stand Up To Cancer grant is administered by the American Association for Cancer Research, the Scientific Partner of SU2C.
Conflict of Interest
A.S.W. has received research support from Kite Pharma, a Gilead Company, and Institut de Recherches Internationales Servier. M.A.P. has been an invited speaker for Novartis, Adaptive Biotechnologies, Miltenyi Biotec and Bellicum Pharmaceuticals, serves on advisory boards at Novartis, Jasper Therapeutics and Mesoblast, receives study support for investigator-initiated studies from Adaptive Biotechnologies and Miltenyi Biotec, and serves on a study steering committee at Novartis. M.C.J. is an inventor and Seattle Children’s Research Institute is an applicant on patents and pending patents related to CAR T cell therapies. He has received consulting fees and grants from and is an inventor on patents licensed to Juno Therapeutics, a Bristol Myers Squibb company. R.A.G. serves on a study steering committee for and is an inventor on a patent licensed to Juno Therapeutics, a Bristol Myers Squibb company, and has served on advisory boards for Novartis.
REFERENCES
- 1.Swann JB and Smyth MJ, Immune surveillance of tumors. J Clin Invest, 2007. 117(5): p. 1137–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blattman JN and Greenberg PD, Cancer immunotherapy: a treatment for the masses. Science, 2004. 305(5681): p. 200–5. [DOI] [PubMed] [Google Scholar]
- 3.Lee DW, et al. , T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet, 2015. 385(9967): p. 517–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maude SL, et al. , Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med, 2014. 371(16): p. 1507–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gardner RA, et al. , Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood, 2017. 129(25): p. 3322–3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maude SL, et al. , Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N Engl J Med, 2018. 378(5): p. 439–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Finney OC, et al. , CD19 CAR T cell product and disease attributes predict leukemia remission durability. J Clin Invest, 2019. 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shah NN and Fry TJ, Mechanisms of resistance to CAR T cell therapy. Nat Rev Clin Oncol, 2019. 16(6): p. 372–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Turtle CJ, et al. , CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest, 2016. 126(6): p. 2123–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Salter AI, et al. , Phosphoproteomic analysis of chimeric antigen receptor signaling reveals kinetic and quantitative differences that affect cell function. Sci Signal, 2018. 11(544). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van der Stegen SJ, Hamieh M, and Sadelain M, The pharmacology of second-generation chimeric antigen receptors. Nat Rev Drug Discov, 2015. 14(7): p. 499–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buchmann S, et al. , Remission, treatment failure, and relapse in pediatric ALL: an international consensus of the Ponte-di-Legno Consortium. Blood, 2022. 139(12): p. 1785–1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roederer M, Nozzi JL, and Nason MC, SPICE: exploration and analysis of post-cytometric complex multivariate datasets. Cytometry A, 2011. 79(2): p. 167–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Terakura S, et al. , Generation of CD19-chimeric antigen receptor modified CD8+ T cells derived from virus-specific central memory T cells. Blood, 2012. 119(1): p. 72–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rossi J, et al. , Preinfusion polyfunctional anti-CD19 chimeric antigen receptor T cells are associated with clinical outcomes in NHL. Blood, 2018. 132(8): p. 804–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fraietta JA, et al. , Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med, 2018. 24(5): p. 563–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Finney HM, et al. , Chimeric receptors providing both primary and costimulatory signaling in T cells from a single gene product. J Immunol, 1998. 161(6): p. 2791–7. [PubMed] [Google Scholar]
- 18.Xu Y, et al. , Closely related T-memory stem cells correlate with in vivo expansion of CAR.CD19-T cells and are preserved by IL-7 and IL-15. Blood, 2014. 123(24): p. 3750–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Klaver Y, et al. , T Cell Maturation Stage Prior to and During GMP Processing Informs on CAR T Cell Expansion in Patients. Front Immunol, 2016. 7: p. 648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oliveira G, et al. , Tracking genetically engineered lymphocytes long-term reveals the dynamics of T cell immunological memory. Sci Transl Med, 2015. 7(317): p. 317ra198. [DOI] [PubMed] [Google Scholar]
- 21.Shah NN, et al. , CD4/CD8 T-Cell Selection Affects Chimeric Antigen Receptor (CAR) T-Cell Potency and Toxicity: Updated Results From a Phase I Anti-CD22 CAR T-Cell Trial. J Clin Oncol, 2020. 38(17): p. 1938–1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang X and Riviere I, Clinical manufacturing of CAR T cells: foundation of a promising therapy. Mol Ther Oncolytics, 2016. 3: p. 16015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kalamasz D, et al. , Optimization of human T-cell expansion ex vivo using magnetic beads conjugated with anti-CD3 and Anti-CD28 antibodies. J Immunother, 2004. 27(5): p. 405–18. [DOI] [PubMed] [Google Scholar]
- 24.Eyquem J, et al. , Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature, 2017. 543(7643): p. 113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated in this study are available within the article and its supplementary data files or available upon request from the corresponding author.