

Abstract
The MYC oncogene is frequently amplified in triple negative breast cancer (TNBC). Here, we show that MYC suppression induces immune-related hallmark gene set expression and tumor-infiltrating T cells in MYC-hyperactivated TNBCs. Mechanistically, MYC repressed stimulator of interferon genes (STING) expression via direct binding to the STING1 enhancer region, resulting in downregulation of the T-cell chemokines CCL5, CXCL10 and CXCL11. In primary and metastatic TNBC cohorts, tumors with high MYC expression or activity exhibited low STING expression. Using a CRISPR-mediated enhancer perturbation approach, we demonstrated that MYC-driven immune evasion is mediated by STING repression. STING repression induced resistance to PD-L1 blockade in mouse models of TNBC. Finally, a small molecule inhibitor of MYC combined with PD-L1 blockade elicited a durable response in immune-cold TNBC with high MYC expression, suggesting a strategy to restore PD-L1 inhibitor sensitivity in MYC-overexpressing TNBC.
Keywords: MYC, STING, Immune evasion, Immunotherapy
Introduction
Triple negative breast cancer (TNBC) accounts for approximately 15% of invasive breast cancers and lacks expression of estrogen receptor (ER), progesterone receptor (PR) and amplification of HER2 (ERBB2). Partly due to limited targeted therapies, TNBC is considered the most virulent subtype of breast cancer (1). A subset of TNBCs exhibits an immunogenic phenotype, in contrast to other breast cancer subtypes. The higher genomic instability and mutational burden of these TNBCs cause a higher propensity to propagate neoantigens, which leads to activation of the adaptive immune response (2). Consequently, the tumor microenvironment (TME) of these TNBCs is typically enriched with tumor-infiltrating lymphocytes (TILs) (3). Moreover, programmed cell death 1-ligand 1 (PD-L1) is overexpressed in TNBC relative to other subtypes and is significantly associated with the presence of TILs (3). Given these immunogenic traits of TNBC, immune checkpoint blockade (ICB) targeting the PD-1/PD-L1 axis has improved clinical outcomes in early and advanced TNBCs (4–6). Unfortunately, only a fraction of TNBCs that express PD-L1 benefit from the ICBs in combination with chemotherapy. Therefore, further elucidation of predictive biomarkers of response/resistance is required to improve patient selection and combination strategies with ICB.
MYC is associated with proliferation, differentiation, apoptosis, self-renewal and cellular energetics in various types of cancer (7). MYC is amplified in ~40% of TNBCs, in which it has been causally associated with cancer progression and resistance to chemotherapy (8,9). Evidence suggests that MYC regulates anti-tumor immunity, for instance, by regulating major histocompatibility complex (MHC) I natural killer (NK) ligands, which promote NK-cell tolerance in lymphomas (10). MYC also globally regulates chemokines that affect the TME. Co-activation of MYC and KRAS orchestrates an immunosuppressive TME via IL23 and CCL9 in lung cancer (11). Similarly, MYC activation in KRAS-driven pancreatic ductal adenocarcinoma results in suppression of the type I interferon (IFN) signaling, which potentiates evasion from NK-cell–mediated immunity (12). Finally, MYC activation induces expression of immune checkpoint proteins such as CD47 and PD-L1 which, in turn, suppress antitumor immune response in multiple cancer types (13,14). However, the role of MYC toward TNBC immune evasion has not been extensively studied.
Stimulator of interferon genes (STING) is an adapter transmembrane protein that plays a critical role in the activation of the innate immune response (15). STING pathway activation is triggered by the recognition of cytosolic double-stranded DNA (dsDNA) by cGAS [cyclic guanosine monophosphate (GMP)-adenosine monophosphate (AMP) synthase], which catalyzes the synthesis of cyclic GMP-AMP (cGAMP) (16). Upon binding of cGAMP, STING works as a scaffold to recruit and phosphorylate tank-binding kinase 1 (TBK1) and IFN regulatory factor 3 (IRF3), which, in turn, activate transcription of type I IFN and other chemokines (17). The cGAS-STING pathway has also been implicated in antitumor immunity (18,19). STING activation promotes tumor infiltration of innate or adaptive immune cells and is associated with favorable response to PD-1 blockade (20). Conversely, MET-mediated STING downregulation attenuates response to immunotherapy in lung cancers (21). Repression of STING is driven epigenetically in various cancer types, including KRAS-mutated lung cancers in which LBK1 loss leads to silencing of the STING1 promoter (18).
In this study, we explored the role of MYC toward TNBC immune evasion in TNBC. We show that hyperactive MYC suppresses immune-activating gene expression signatures and tumor-infiltrating T cells via epigenetic repression of STING. MYC-induced STING repression resulted in resistance to PD-L1 blockade. Finally, MYC inhibition restored sensitivity to PD-L1 blockade in immune-cold TNBC, suggesting a novel therapeutic strategy for this subtype of breast cancer.
Materials and Methods
Cell lines and inhibitors
MDA-MB-436 Tax-R and SUM159PT Tax-R cells were generated as described (9). Briefly, both cell lines were treated with increasing concentrations of taxol up to 50 nM for 6 months. MDA-MB-436 Doxo-R and SUM159PT Doxo-R cells were generated by culture in the presence of increasing concentration of doxorubicin starting at 10 nM up to 250 nM. 4T1 (#ATCC® CRL-2539), Py230 (#ATCC® CRL-3279) and Jurkat (#ATCC® TIB-152) cells were purchased from ATCC in 2019 or 2020. 293FT (#R70007) cells were purchased from Invitrogen in 2016. Every cell line was authenticated by the short-tandem repeat (STR) method and tested for Mycoplasma contamination. MDA-MB-436 and 293FT cells were maintained in DMEM (ThermoFisher Scientific, #41965039)/10% FBS (ThermoFisher Scientific, #10082147)/1% antibiotic-antimycotic (AA; ThermoFisher Scientific, #15240062); SUM159PT cells in DMEM/F-12/5% FBS, 0.5 μg/mL hydrocortisone (Sigma, #H0888) and 1% AA; 4T1 and Jurkat cells in RPMI1640 (ThermoFisher Scientific, A1049101)/10% FBS/1% AA; Py230 cells in F-12K/5% FBS/1% AA. All cell lines were maintained at 37°C in 5% CO2. For experiments with drug-resistant cells, cells were removed from drug for at least 24 h prior to treatment. MYCi975 was purchased from MedChemExpress (#HY-129601). Doxorubicin and taxol were from Selleckchem (#S1208 and #S1150, respectively).
Animal experiments
Animal experiments have been conducted in accordance with the approval of the University of Texas Southwestern Medical Center Institutional Animal Care and Use Committee (IACUC). 2×106 Py230 cells stably expressing doxycycline-inducible Sting1 shRNA were suspended in IMEM and growth factor reduced Matrigel (BD Biosciences, #354230) at a 1:1 ratio and orthotopically injected into the mammary fat pad of 8-week old C57BL/6 female mice (ENVIGO, C57BL/6NHsd). Once tumors reached ≥100 mm3, mice were randomized to treatment with vehicle {0.9% NaCl (Sigma, #S9888)} or doxycycline (10 mg/kg/daily i.p.; Sigma, #D5207). After a week, tumors were harvested for analysis of tumor-infiltrating immune cells. 1×106 4T1 cells were orthotopically injected into 8-week-old BALB/c female mice (Jackson Laboratory, #000651). Once tumors reached ≥100 mm3, mice were randomized to treatment with vehicle {PBS with 10% DMSO (Sigma, #D8418) and 20% TWEEN-80 (Sigma, #P1754)} or MYCi975 (50 mg/kg/twice/day i.p.). After a week, tumors were harvested for analysis of tumor-infiltrating immune cells. For the PBMC-humanized mouse model, 1×106 SUM159PT parental and Tax-R cells were orthotopically injected into 8-week old female NOD/SCID mice (Jackson Laboratory, #005557). After a week, 1×107 hPBMCs (ALLCELLS) were injected i.v. Two weeks later, SUM159PT tumors were harvested for analysis of tumor-infiltrating immune cells.
For studies using PD-L1 blockade, MYCi975 (50 mg/kg, twice daily i.p.) was given for 2 days followed by atezolizumab (Selleckchem, #A2004) 100 mg/day x2. This 4-day cycle was repeated 5 times. C57BL/6 and BALB/c mice bearing Py230 tumors and 4T1 tumors, respectively, were treated with 12.5 mg/kg of a mouse PD-L1 antibody (Bio X Cell, #BE0101) and control IgG (Bio X Cell, #BE0090).
Characterization of tumor-infiltrating immune cells
Tumors were disaggregated in PBS/2% FBS and then incubated with PBS containing 2% FBS, 100 μg/mL collagenase A (Sigma, #10103586001), and 100 μg/mL DNase (STEMCELL, #07900) for 1.5 h at 37°C. Digested cell suspensions were passed through a 70-μm strainer. After centrifugation, cells were resuspended in PBS/2% FBS and, counted. 5×106 cells were labeled in 100 μL PBS containing 2% FBS, 2 μL FC blocker (BD Bioscience, #564220 for human and #553142 for mouse) and 0.4 μL eBioscience™ Fixable Viability Dye eFluor™ 506 (ThermoFisher Scientific, #65-0866-14) for 10 min at 4°C. Next, the following antibodies were next added: Pacific Blue™ anti-human CD45 (#304021), PerCP/Cyanine5.5 anti-human CD3 (#300328), Alexa Fluor® 700 anti-human CD4 (#357418), FITC anti-human CD8a (#300906), FITC anti-mouse CD8a (#100706), PerCP/Cyanine5.5 anti-mouse CD45.2 (#109828), Brilliant Violet 605™ anti-mouse CD4 (#100548), all from BioLegend; And BV786 Hamster Anti-Mouse CD3e, from BD Biosciences (#564379). Thirty min after incubation, cells were resuspended with PBS/2% FBS and analyzed by flow cytometry.
Plasmids
Human STING1 or mouse Sting1 sgRNAs were cloned into pX458 (Addgene, #48138) as described (22). Briefly, the pX458 plasmid digested by BbSI was annealed with primers including the scaffold for guide RNA; Guide sequences were 5’-GTGCACTCTCATTGTACAGG-3’ (Human STING1, top), 5’- GCTTCTGGAGAACGGGCAGC-3’ (Human STING1, bottom), 5’- CAGAATCTCGTAGACGCTGT-3’ (mouse Sting1, top), and 5’-GTGTCAGTTCTAGATTGAGT-3’ (mouse Sting1, bottom). pTY-GFP and pTY-STING1 were kindly provided from Dr. Zhijian Chen at University of Texas Southwestern Medical Center(23). Doxycycline-inducible mouse Sting1 shRNAs were purchased from Horizon Discovery (#V3SM11256-08EG72512). Mouse Sting1 open reading frame (ORF) in the gateway PLUS shuttle was from GeneCopoeia (#GC-Mm22462-CF). Human CXCR3 ORF in pENRT221 was obtained as a part of the Ultimate™ ORF Lite human cDNA collection (Life Technologies). ORFs were cloned into pLenti6.3/V5-DEST (ThermoFisher Scientific, #V53306) using Gateway™ LR Clonase™ II Enzyme Mix (ThermoFisher Scientific, # 11791020).
siRNA transfection
Cells seeded in 60-mm dishes were transfected with 40 pmol of siRNAs using Lipofectamine RNAiMAX transfection reagent (ThermoFisher Scientific, #13778075) according to the manufacturer’s protocol. Control siRNA (#AM4635), human MYC siRNAs (#1299001-VHS40785, #1299001-VHS40789) and mouse Myc siRNAs (#AM16708-156407, #AM16708-156405) were purchased from ThermoFisher Scientific.
Viral transduction
1 μg of each plasmid was co-transfected with 0.75 μg of psPAX2 (2nd generation lentiviral packaging plasmid; Addgene, #12260) and 0.5 μg of pMD2.G (VSV-G envelope expressing plasmid; Addgene, #12259) into 293FT cells using Lipofectamine2000 (ThermoFisher Scientific). Cell medium was changed to fresh medium 24 h post-transfection, and cells were collected 48 h later. For viral transduction, virus-containing medium was applied to host cells with 8 μg/mL polybrene (Sigma, #H9268). Cells stably transduced with pTY-GFP, pTY-STING1, and mouse Sting1 sgRNAs were selected with 1 μg/mL puromycin (Sigma, #P9620). 2 μg/mL blasticidin (ThermoFisher Scientific, #A1113903) was used for selection of cells transduced with pLenti6.3/V5-DEST-CXCR3, -mouse Sting1 and -LacZ.
ELISA
Cells (1×105) were seeded in 6 well-plates and grown for 24 h in serum-free media. 50 μL of conditioned media (CM) were collected and then subjected to enzyme-linked immunosorbent assay (ELISA). ELISA was performed with an ELISA kit detecting CCL5 (Invitrogen, #EHRNTS), CXCL10 (Invitrogen, #KAC2361) and CXCL11 (Invitrogen, #EHCXCL11) as described in the manufacturer’s protocol.
Transwell migration assays
Serum-free conditioned media (24 h) was placed on the bottom chamber of a transwell plate (Sigma-Aldrich, #CLS3421). Jurkat-CXCR3 cells (5×105) were plated in the top chamber and cells migrating to the bottom chamber after 4 h were counted using a Z2 coulter counter analyzer (Beckman Coulter). An antibody neutralizing CCL5 (R&D systems, #MAB678-SP), CXCL10 (R&D systems, #MAB266-SP) CXCL11 (R&D systems, #MAB672-SP), IFN alpha (R&D systems, #211001), or IFN beta (R&D systems, #MAB814-SP) was added to the CM in the bottom chamber.
RNA-sequencing analysis
MDA-MB-436 Tax-R, SUM159PT Tax-R, and 4T1 cells were transfected with MYC siRNA or control siRNA for 48 h. Py230 cells stably expressing doxycycline-inducible Sting1 shRNA were grown with or without 200 ng/mL doxycycline for 48 h. Cells were then harvested and RNA was extracted using Maxwell® RSC simplyRNA Cells Kit (Promega). The absence of RNA degradation and contamination was confirmed by agarose gel electrophoresis and RNA was quantitated using the 2100 bioanalyzer instrument (Agilent). mRNA was enriched from total RNA using the NEBNext® Poly(A) mRNA Magnetic Isolation Module (NEW ENGLAND BioLabs) and library preparation was conducted using the NEBNext® Ultra™ II RNA Library Prep Kit for Illumina® (NEW ENGLAND BioLabs). Libraries were sequenced on Novaseq6000 machines using paired-end 150 base-pair sequencing. In the raw fastq files, the ends of sequences with remaining adapter or quality scores <25 were trimmed. Any sequence less than 35bp was removed. The trimmed fastq files were aligned to the selected reference genome (hg38) using HiSAT2 (24). After marking duplicates using SAMBAMBA, genes, transcripts and exons were counted using featureCounts (25) and StringTie (26). DESeq2 package (27) was used for analysis of differentially expressed genes. Each experiment was conducted in two or three biological replicates.
Gene set enrichment analysis (GSEA)
GSEA was conducted with the javaGSEA interface from the Broad Institute (http://software.broadinstitute.org/gsea/index.jsp). Normalized counts generated using DESeq2 package were used as input data. The h.all.v7.2.symbols.gmt [Hallmarks] was used as gene sets database (28). For GSEA of mouse cells, the chip platform was replaced with the Mouse_Gene_Symbol_Remapping_Human_Orthologs_MSigDB.v7.2.chip. A gene set enrichment with FDR q-value <0.05 was considered statistically significant.
Analysis of STING1 mRNA and MYC hallmark gene signature in the cohort of Hutchinson et al.
Targeted exome sequencing and whole-transcriptome sequencing were performed for paired primary and metastatic specimens from patients with TNBC (Hutchinson, et al. TNBC cohort, n=41) as described previously (29). The same analysis methods were applied herein to this dataset (n=41) to determine STING1 mRNA expression and the relative expression score of two MYC hallmark gene signature sets (28) in paired primary and metastatic TNBCs, and p-values were calculated from paired t-tests. To determine the relationship between STING1 mRNA and MYC hallmark gene signature scores, Spearman correlation coefficients were calculated.
Immunohistochemistry (IHC)
Tissue microarrays (TMAs) of formalin-fixed, paraffin-embedded (FFPE) tumor blocks from 76 patients with primary TNBCs that were collected at Instituto Nacional de Enfermedades Neoplásicas in Lima, Peru under institutionally approved protocol INEN 10–018 after treatment with neoadjuvant chemotherapy were used for IHC. FFPE blocks were stored at room temperature. TMA sections were subjected to antigen retrieval with citrate buffer (pH 6; Agilent #S2369) in a decloaking chamber (Biocare). Endogen peroxidase was blocked with 3% hydrogen peroxide (ThermoFisher, #H325) and protein block sol (Agilent, #X0909). TMA sections were then incubated overnight at 4°C with a MYC antibody (Abcam, #Ab32072, dilution 1:600) or STING antibody (Protein Tech, #1985-1-AP, dilution 1:1600). The visualization system was Envision (Agilent, #K4011) with 3,3′-diaminobenzidine (DAB) as the chromogen (Agilent, #K3468). Hematoxylin was applied as counterstain. Tonsil and a MYC-overexpressing breast cancer biopsy were used as positive controls for STING and MYC, respectively. FFPE tissues were obtained according to Vanderbilt University Medical Center protocol IRB030747 and stored at room temperature. Whole sections were digitally acquired using an AxioScan Z1 slide scanner (Carl Zeiss) at 20x. Automated semiquantitative scoring was performed by a blinded expert pathologist using QuPath software (30). Color deconvolution stains were set from a representative area. Cell segmentation was determined by hematoxylin staining. An object classification was trained to differentiate tumor and stroma STING- and MYC-positive cells.
RT-qPCR
RNA was extracted from cells using the Maxwell RSC simplyRNA Cells Kit (Promega) according to the manufacturer’s protocol. cDNA was synthesized using the iSCRIPT cDNA synthesis Kit (Bio-Rad) and then subjected to PCR with PowerUpTM SYBRTM Green Master Mix (ThermoFisher Scientific) using the QuantStudio3 Real-Time PCR System (ThermoFisher). All primers were purchased from QIAGEN: GAPDH (PPH00150F), CCL5 (PPH00703B), CXCL10 (PPH00765E), CXCL11 (PPH00506A), STING1 (PPH18521A), mouse Gapdh (PPM02946E), mouse Sting1 (PPM29247A), mouse Ccl5 (PPM02960F), mouse Cxcl10 (PPM02978E) and mouse Cxcl11 (PPM03192C).
Chromatin immunoprecipitation (ChIP)-qPCR
Cells grown in 150-mm dishes were washed with PBS and crosslinked with pre-warmed (37°C) media containing 1% formaldehyde (Sigma, #F8775) 10 min at room temperature. The crosslinking reaction was stopped by adding 1.25 M glycine (Sigma, #410225) and cells were sonicated using the Bioruptor Plus sonication device (Diagenode) at 4°C. Sonicated chromatin was adjusted to a final concentration of 200 mM NaCl and incubated at 65°C overnight. The following day, the chromatin was incubated with RNAse A (QIAGEN, #19101) for 30 min at 37°C, followed by Proteinase K (ThermoFisher Scientific, #25530049) for 1 h at 42°C. DNA was purified using QIAquick PCR Purification Kit (QIAGEN, #28106). DNA shearing was checked on an agarose gel, with an average fragment size of 200–750 bp. For ChIP, chromatin was eluted with ChIP dilution buffer {0.01% SDS buffer, 1.1% Triton X-100, 1.2 mM EDTA, 167 mM NaCl, 16.7 mM Tris-HCl, pH 8.0–8.1, and protease inhibitors (Protease Inhibitor Cocktail, Roche)} after preclearing with Gammabind G Sepharose beads (GE Healthcare, #17-0885-01) that had been washed three times with ChIP-dilution buffer and pre-blocked in 0.5% BSA (Sigma, #A9418) at 4°C for 1 h. The precleared chromatin was next incubated overnight at 4°C with primary antibody. The following day, Gammabind G Sepharose beads were added to the antibody pulldowns for 2 h at 4°C. Beads were then washed once in Buffer I (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, 150 mM NaCl and protease inhibitors, pH 8.0–8.1), once in Buffer II (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, 500 mM NaCl, and protease inhibitors), once in Buffer III {0.25 M LiCl (Sigma, #L9650), 1% NP40 (Sigma, #NP40), 1% Na deoxycholate (Sigma, #D6750), 1 mM EDTA, 10 mM Tris-HCl, pH 8.0–8.1} and twice in TE buffer, pH 8.0–8.1. Immune complexes were eluted with Elution Buffer {0.1 M NaHCO3 (Sigma, #S5761), 1%SDS} at 65°C for 10 min. Antibody pulldowns and sonicated chromatin (as input controls) were adjusted to 200 mM NaCl and incubated at 65°C overnight. The following day, sequential incubations with RNase A for 30 min at 37°C, and Proteinase K for 1 h at 42°C were performed. DNA was purified using QIAquick PCR Purification Kit. Precipitated and input DNA was analyzed by qRT-PCR or used to construct sequencing libraries. For ChIP-qPCR, the enrichment was shown as percent of input. ChIP-qPCR results were reproduced in two or more independent experiments. For ChIP, we used the following primary antibodies: mouse anti-Myc (Santa Cruz Biotechnology, #sc-40x), normal mouse IgG (Santa Cruz Biotechnology, #sc-2025), rabbit anti-histone H3 acetyl K27 (Abcam, #ab4729), and normal rabbit IgG (Cell Signaling Technology, #2729). Sequences of primer pair were designed based on the nucleotide sequences identified by MYC ChIP-sequencing (R1: Forward 5’-ATCCAGCTTGTAGTAAGTGCTCG-3’ and reverse 5’-GCTGTAGTGTCCCTAGCTGGT-3’; R2: Forward 5’-GCCCAGATTGTGCCACTCTA-3’ and reverse 5’-CAGGCTGGTCTTGAATTCTTGA-3’; R3: Forward 5’-TGACACACCCAGAATAGCATCC-3’ and reverse 5’-GCCCTTCTCTGAGCTGTAGTG-3’).
ChIP-sequencing analysis
Libraries from 5–10 ng ChIP DNA were prepared using KAPA Hyper Library Preparation Kit (KAPABIOSYSTEMS, #KK8504). Precipitated DNA was quantitated on the Qubit® 4 Fluorometer (Invitrogen). Samples were end-repaired, 3’ ends-adenylated and barcoded with multiplex adapters, followed by size selection with Ampure XP beads (BECKMAN COULTER, #A63881) and PCR amplification. Samples were validated on the Agilent Tapestation 4200, normalized, pooled, and run on the Illumina NextSeq 500 using 75 cycle SBS v2.5 reagents. Analysis for ChIP-sequencing was conducted using the UTSW BICF ChIP-seq Analysis workflow (31). Briefly, raw fastq files were trimmed by TrimGalore and then aligned to human reference genome (hg38) using BWA (32). Low-quality reads and duplicate reads were removed from aligned files using Sambamba (33) and Samtools (34). Model-based Analysis of ChIPSeq (MACS) (35) software tool (v.2.1.2) was used to call peaks from the ChIP-sequencing data. All peaks were annotated using ChipSeeker (36) and differential binding activity was calculated using DiffBind (37). ChIP peaks were visualized using IGV (https://igv.org/).
Immunoblot analysis
Cell lysates were prepared in RIPA buffer (ThermoFisher Scientific) containing 1X protease inhibitor (Protease Inhibitor Cocktail, Roche) and phosphatase inhibitor (Roche, #4906837001) for 30 min on ice and then subjected to centrifugation at 14,000 rpm for 10 min. Protein concentration in the supernatants was measured with the Pierce BCA Protein Assay kit (ThermoFisher Scientific, #23227). 30 μg of protein was subjected to SDS-PAGE followed by transfer to nitrocellulose membranes (BIO-RAD, #1704270). Membranes were incubated with primary antibodies followed by rabbit or mouse antibodies conjugated with HRP or fluorescence dyes. Immunoblot images were taken using the Chemidoc MP imaging system (BIO-RAD). All antibodies were purchased from Cell Signaling Technology: cGAS (#83623), STING (#13647), p-TBK1 S172(#5483), TBK1 (#3504), p-IRF3 S386 (#37829), IRF3 (#11094), MYC (#9402), EZH2 (#4905), DNMT1 (#5032), KDM5B (#15327), KDM5C (#5361), and actin (#4970).
Enhancer perturbation
The perturbation system was adopted from a previous report (38). pgRNA-humanized (plasmid #44248) and pHR-SFFV-dCas9-BFP-KRAB (plamid #46911), both required for the perturbation, were purchased from Addgene. Target-specific single guide RNAs (sgRNAs) for the STING enhancer were designed using the UCSC genome browser. The sgRNA sequences used were CCTCCCCCTGTGTCATACCTTGA (hg38/Human, chr5:139,481,141–139,481,163) and CCCACTCCCAGTACTCAGCTCAG (hg38/Human, chr5:139481074-139481096). pgRNA-humanuized was linearized by BstxI (NEW ENGLAND BioLabs, #R0113S). Oligonucleotides containing the sequence of sgRNAs were subcloned into the linearized pgRNA-humanized using NEBuilder® HiFi DNA Assembly (NEW ENGLAND BioLabs, #E2621). The oligonucleotides were synthesized by Integrated DNA technologies (IDT):
5’-CTATAAGTATCCCTTGGAGAACCACCTTGCCTCCCCCTGTGTCATACCTTGAGTTTTAGAGCTAGAAATAGCAAGTTAAA-3’
5’- CTATAAGTATCCCTTGGAGAACCACCTTGCCCACTCCCAGTACTCAGCTCAGGTTTTAGAGCTAGAAATAGCAAGTTAAA-3’. pHR-SFFV-dCas9-BFP-KRAB and pgRNA-humanized-sgRNA were stably transduced into MDA-MB-436 and SUM159PT Tax-R cells. Transduced cells were selected by 1 μg/mL puromycin over 2 weeks, and then sorted by BFP expression.
TCGA Data
Normalized transcriptome data of breast tumors (n=82), which were generated as a part of The Cancer Genome Atlas (TCGA) project and categorized as TNBC (39), were downloaded from cBioPortal (https://www.cbioportal.org/study/summary?id=brca_tcga_pub2015). Hallmark gene set signature scores calculated with normalized transcriptome data by gene set variation analysis (40) were used for the correlation analysis.
Statistical analysis
Analysis for T-tests, Pearson correlation coefficient, and log-rank hazard ratio were performed with GraphPad Prism version 8. Data were represented as mean ± standard deviation. All experiments were conducted at least three times. A p value <0.05 was considered statistically significant. For RNA-sequencing analysis and GSEA, FDR <0.05 was considered statistically significant. R version 3.5.2 and R studio version 1.1.463 were used.
Data availability
RNA-sequencing and ChIP-sequencing data are available on the Gene Expression Omnibus (GEO) under accession number GSE196325. Other data is available upon reasonable request to the authors.
Results
MYC overexpression is associated with immunosuppressive gene expression in TNBC
We previously reported that MYC can be overexpressed in TNBC cells with acquired resistance to taxol (9). In these cells, MYC siRNA reversed taxol resistance. Gene set enrichment analysis (GSEA) of RNA-sequencing data showed that hallmark gene signatures associated with MYC pathway activation are significantly enriched in both MDA-MB-436 and SUM159PT taxol-resistant (Tax-R) cells compared to taxol-sensitive parental cells (Fig. 1A). In addition, hallmark gene signatures indicative of immune activation were suppressed in both MDA-MB-436 and SUM159PT Tax-R cells (Fig. 1A). In line with these findings, we subsequently showed that immune-related gene set signatures and stromal TILs are significantly suppressed in a cohort of metastatic TNBCs treated with chemotherapy compared to matched treatment-naïve primary tumors (29). In this same cohort (hereafter referred as the cohort of Hutchinson et al.), we found that MYC hallmark gene signatures are elevated in metastases compared to matched primary tumors (n=41, Fig. 1B). High MYC hallmark gene set signatures were also associated with low immune activation hallmark gene set signatures in TNBCs in The Cancer Genome Atlas (TCGA; Fig. 1C). These data imply that overexpression of MYC is associated with suppression of immune activation pathways in TNBC.
Figure 1. MYC pathway activation is associated with suppression of immune-related gene expression in TNBC.
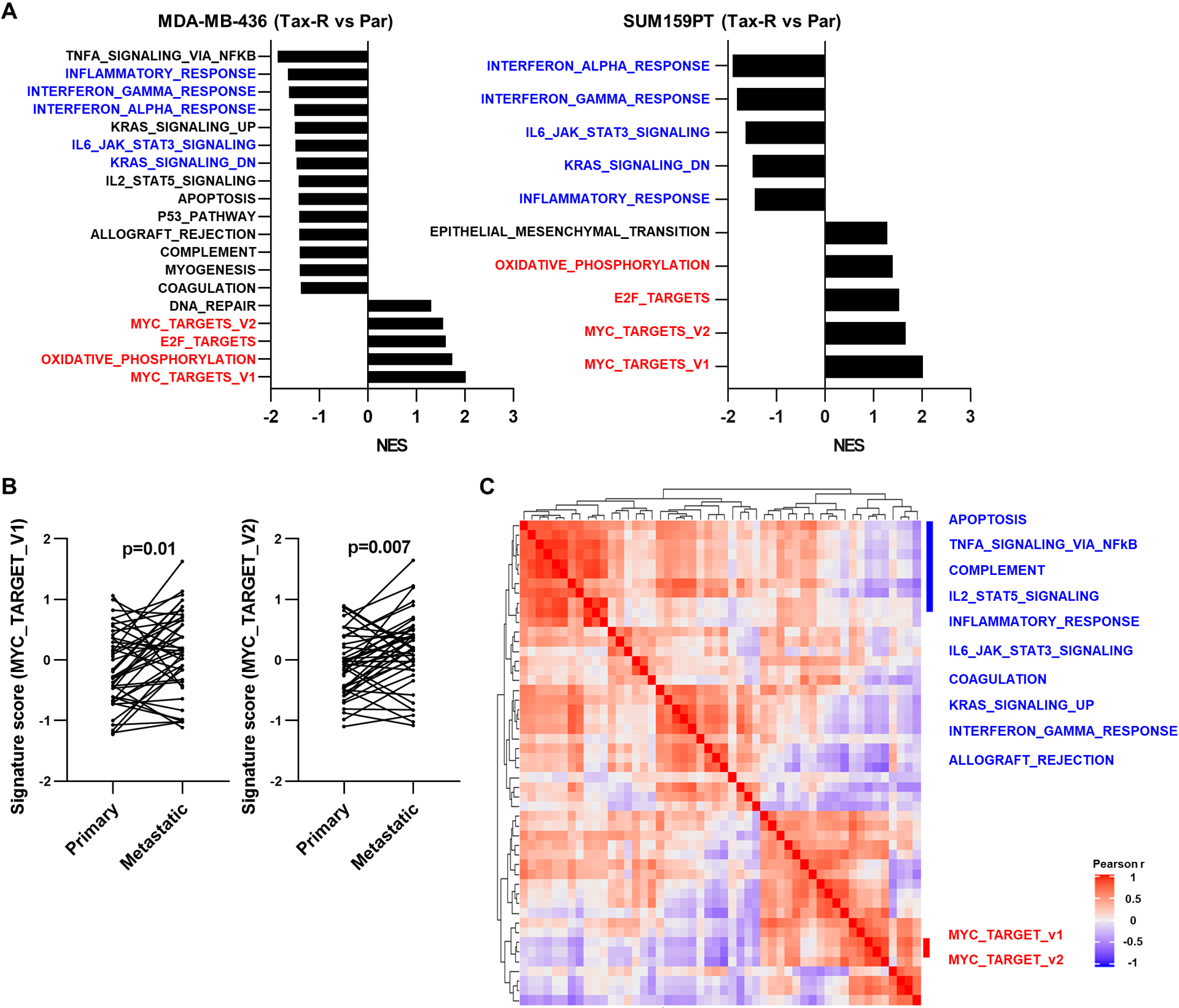
A, Hallmark gene signatures significantly enriched or downregulated in MDA-MB-436 Tax-R and SUM159PT Tax-R cell transcriptomes relative to respective parental cells (FDR<0.05). The x-axis represents the normalized enrichment score (NES). Hallmark gene sets identified in both cell lines are denoted in blue (downregulated) or red (upregulated). B, Hallmark gene signatures of MYC activation assessed from transcriptome data of primary TNBCs and matched metastatic tumors (n=41 pairs; two-tailed paired t-tests). C, Gene set variation analysis (GSVA) correlations with fifty hallmark gene signatures from TNBCs in TCGA. The color key represents Pearson correlation coefficient.
MYC inhibition promotes a T cell–inflamed TME
To test the role of MYC in immunosuppression in TNBC, we used two MYC siRNAs in Tax-R and control MDA-MB-436 and SUM159PT cells. GSEA of RNA-sequencing data confirmed that hallmarks related to MYC activation are significantly downregulated by MYC silencing (Fig. 2A). Ablation of MYC resulted in a significant enrichment of the immune-related hallmark gene signatures (INTERFERON_GAMMA_RESPONSE and INFLAMMATORY_RESPONSE). MYC knockdown also resulted in induction of T-cell chemokines (CCL5, CXCL10 and CXCL11), all of which were significantly downregulated in MDA-MB-436 and SUM159PT Tax-R compared to parental cells (Supplementary Fig. 1A and B).
Figure 2. MYC inhibition promotes a T cell–inflamed tumor microenvironment.
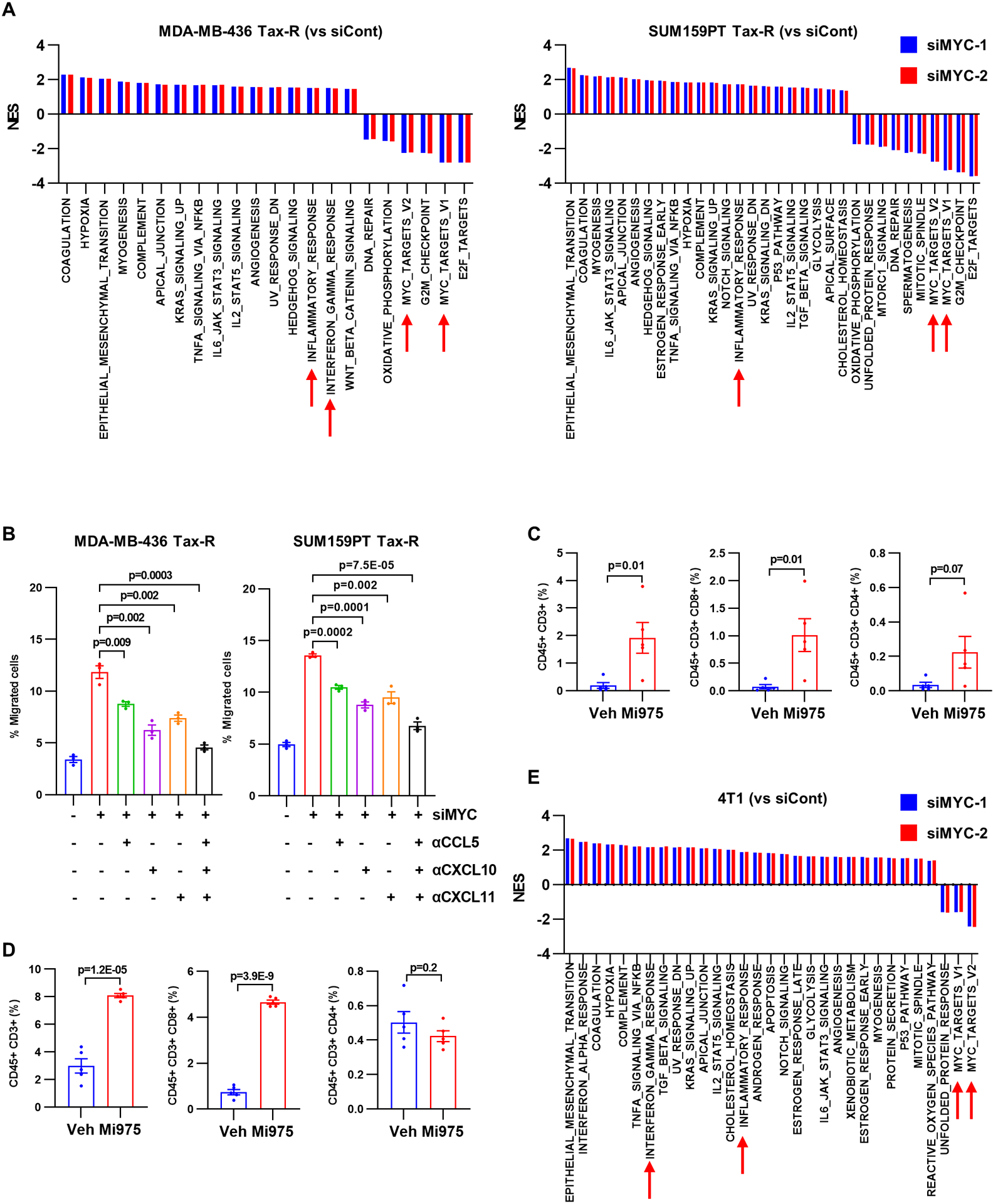
A, Hallmark gene sets significantly enriched or downregulated in MDA-MB-436 and SUM159PT Tax-R cells transfected (24 h) with each of two MYC siRNAs compared to control siRNA (FDR<0.05). B, Transwell migration of Jurkat CXCR3 cells with CM from MDA-MB-436 and SUM159PT Tax-R cells ± neutralizing CCL5, CXCL10 or CXCL11 antibodies. Each bar represents the mean ± SEM (n=3; two-tailed unpaired t-tests). C-D, Flow cytometry quantification of tumor-infiltrating lymphocytes isolated from PBMC-humanized mice bearing SUM159PT Tax-R tumors (C) or immunocompetent mice bearing syngeneic 4T1 tumors (D) treated with or without MYCi975. Each bar represents the mean ± SEM (n=5, n=5; two-tailed unpaired t-tests). E, Hallmark gene sets significantly enriched or downregulated in 4T1 cells transfected with each of two MYC siRNAs compared to control siRNA (FDR<0.05).
To determine the effect of MYC-mediated T-cell chemoattractant downregulation, we performed transwell migration assays with Jurkat cells stably overexpressing CXCR3 (hereafter, Jurkat CXCR3; Supplementary Fig. 1C). Migration of Jurkat CXCR3 cells was reduced by conditioned media (CM) from MDA-MB-436 Tax-R and SUM159PT Tax-R cells compared to CM from parental cells (Supplementary Fig. 1D). Transduction of MYC siRNA into MDA-MB-436 Tax-R and SUM159PT Tax-R cells abrogated the effect of their CM on Jurkat CXCR3 cell migration (Fig. 2B). This increase in cell migration induced by CM from MYC-silenced Tax-R cells was also abrogated by the combination of antibodies neutralizing CCL5, CXCL10 and CXCL11 (Fig. 2B).
We next examined T-cell infiltration in the TME of Tax-R xenografts established in NOD-SCID mice that had been inoculated with human peripheral blood mononuclear cells (hPBMC; hereafter PBMC-humanized model). Consistent with the previous report (41), the majority of tumor-infiltrating CD45+ cells derived from hPBMCs were CD3+ lymphocytes representing T cells in these mice (Supplementary Fig. 1E). Flow cytometry of cells isolated from the xenografts revealed that human CD45+/CD3+ cells, CD45+/CD3+/CD8+ cells, and CD45+/CD3+/CD4+ cells – representing a population of T cells, CD8+ T cells, and CD4+ T cells, respectively – were significantly decreased in Tax-R compared to parental SUM159PT tumors (Supplementary Fig. 1F). In the absence of hPBMCs, the growth rate of Tax-R and parental SUM159PT tumors was similar. However, in presence of hPBMCs, Tax-R tumors grew faster compared to parental tumors (Supplementary Fig. 1G). These data suggest that T cell–mediated immune surveillance is impaired in chemotherapy-resistant tumors compared to drug-sensitive parental tumors.
We next examined the effect of pharmacological inhibition of MYC on T-cell infiltration into the TME using a small molecule MYC inhibitor, MYCi795 (42). Treatment with MYCi975 induced infiltration of human CD45+/CD3+ cells and CD8+ T cells into SUM159PT Tax-R xenograft tumors (Fig. 2C). We also tested the effect of MYCi975 in immunocompetent syngeneic mice bearing 4T1 mouse tumors, which are known to overexpress MYC (43). Similar to SUM159PT tumors, 4T1 tumors were enriched with tumor-infiltrating CD45+/CD3+ and CD8+ T cells upon treatment with MYCi975 (Fig. 2D). MYC ablation by siRNA also resulted in an enrichment of immune-related hallmark gene signatures in 4T1 cells (Fig. 2E). These data suggest that hyperactivation of MYC suppresses T cells in the TME of TNBC.
STING loss is associated with the lack of tumor-infiltrating T cells
We next investigated the combined transcriptome data to identify putative mechanisms by which MYC inhibition elicits a T cell–inflamed TME. We first listed 1) downregulated genes in MDA-MB-436 Tax-R and SUM159PT Tax-R cells compared to respective parental cells and 2) upregulated genes by MYC knockdown in Tax-R cells. Seventy genes overlapped in both lists (Fig. 3A), one of which was STING1, a gene associated with a cell-autonomous immune response (Supplementary Fig. 2A and B). However, genes previously shown to be effectors of MYC-driven immunomodulation, such as CD47, CD274 (13), IL23, CCL9 (11) and HLA (44), were not identified in the overlapping set. STING1 mRNA abundance correlated negatively with MYC hallmark gene signatures and correlated positively with immune-related gene signatures in the TCGA TNBC cohort (Supplementary Fig. 2C). STING mRNA and protein were also lower in MYC-overexpressing MDA-MB-436 Tax-R and SUM159PT Tax-R cells compared to parental drug-sensitive cells (Fig. 3B and C). In line with these results, phosphorylation of TBK1 and IRF3 was suppressed in MDA-MB-436 Tax-R and SUM159PT Tax-R cells (Fig. 3C). MDA-MB-436 and SUM159PT cells with acquired resistance to doxorubicin after long-term drug exposure also showed high expression of MYC and lower expression of STING by immunoblot (hereafter, Doxo-R; Supplementary Fig. 2D–F). cGAS has also been shown to be repressed in various types of cancer (45). However, cGAS expression was not downregulated in Tax-R cells (Fig. 3C).
Figure 3. STING suppression reduces tumor-infiltrating T cells.
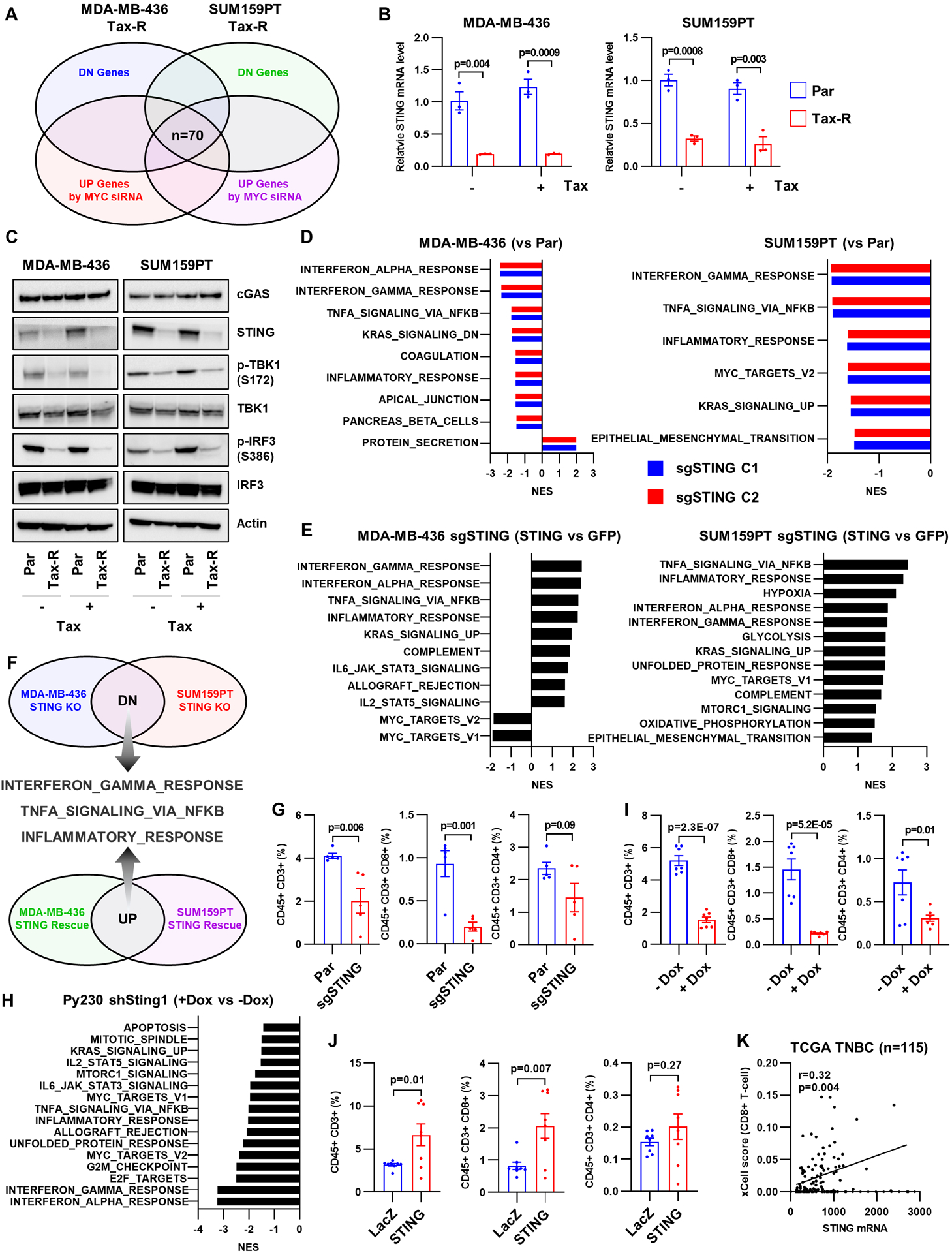
A, A Venn diagram identifying seventy overlapping genes across groups of downregulated genes in Tax-R compared to parental cells (top circles) and upregulated genes in Tax-R cells transfected with MYC siRNA compared to control siRNA-transfected cells (bottom circles). B, RNA extracted from the indicated cells after treatment with 10 nM taxol for 48 h was used for quantification of STING1 mRNA RT-qPCR. Each bar represents the mean ± SEM (n=3; two-tailed unpaired t-tests). C, Lysates from the same cells described in B were subjected to immunoblot analysis with cGAS, STING, phospho-TBK1(serine 172), TBK1, phospho-IRF3 (serine 386), IRF3 and actin antibodies. D, Hallmark gene sets significantly enriched or downregulated in MDA-MB-436 STING1 knockout (KO) and SUM159PT STING1 KO cells compared to parental cells (FDR<0.05). E, Hallmark gene sets significantly enriched or downregulated by the re-expression of STING1 in MDA-MB-436 STING1 KO and SUM159PT STING1 KO cells (FDR<0.05). F, Hallmark gene sets found in E and F were integrated to identify hallmark gene sets regulated by STING. G, Flow cytometry quantification of tumor-infiltrating lymphocytes isolated from PBMC-humanized mice bearing SUM159PT parental and STING1 KO tumors. Each bar represents the mean ± SEM (n=5; two-tailed unpaired t-tests). H, Hallmark gene sets significantly enriched or downregulated in Py230 doxycycline-inducible shSting1 cells treated with 200 ng/mL doxycycline (48h) compared to untreated cells (FDR<0.05). I and J, Flow cytometry quantification of tumor-infiltrating lymphocytes isolated from immunocompetent mice bearing Py230 doxycycline-inducible shSting1 tumors ± 10 mg/kg doxycycline (i.p) for 2 weeks (I) or 4T1 tumors stably expressing LacZ or Sting1 (J). Each bar represents the mean ± SEM (n=7; two-tailed unpaired t-tests). K, CD8+ T cell xCell scores correlations with STING1 mRNA expression in the TCGA TNBC cohort (r, Pearson correlation coefficient).
To examine the potential impact of STING loss in TNBC cells, we generated MDA-MB-436 and SUM159PT cells in which STING1 was silenced by CRISPR genome editing (Supplementary Fig. S3A). Since dsDNA and viral infection can activate STING, we used a transient transfection approach (22). GSEA of the transcriptome of these cells showed that STING1 knock out (KO) reproduced the downregulation of immune-related hallmark gene signatures that was observed in Tax-R cells (Fig. 3D). STING1 silencing also resulted in a reduction of expression and secretion of CCL5, CXCL10 and CXCL11 (Supplementary Fig. S3C and D). CM from these cells inhibited Jurkat CXCR3 cell migration through transwells (Supplementary Fig. S3E). To support a causal role of STING in these changes, STING1 was re-expressed in MDA-MB-436 STING1 KO and SUM159PT STING1 KO cells (Supplementary Fig. S3B). Re-expression of STING significantly restored immune-related hallmark gene signatures (Fig. 3E and F) and rescued migration of CM-treated Jurkat cells (Supplementary Fig. S3F and G). The role of STING in the innate immune response involves induction of type I IFNs (16). However, antibodies neutralizing type I IFNs failed to block the chemotaxis of T cells, which was enhanced by STING1 overexpression (Supplementary Fig. S3G), suggesting that STING induces T-cell chemotaxis independent of the type I IFN signaling in TNBCs.
We next assessed the impact of STING loss in tumor cells on the TME in vivo. STING1 KO resulted in a decrease of infiltrating CD45+/CD3+ and CD8+ T cells in SUM159PT tumors established in PBMC-humanized mice (Fig. 3G). We next determined the role of STING in a syngeneic mouse model to overcome potential limitations of the humanized mouse models (46). We stably transduced Py230 mouse TNBC cells (47), derived from polyomavirus middle T transgenic mammary tumors in C57BL/6 mice, with a doxycycline-inducible Sting1 shRNA (Supplementary Fig. S4A and B). Sting1 knockdown in Py230 cells downregulated immune-related hallmark gene signatures, T-cell chemokines, and tumor infiltrating CD45+/CD3+ and CD8+ T cells (Fig. 3H and I; Supplementary Fig. S4B). Conversely, stable transduction of Sting1 into mouse 4T1 cells led to an increase of tumor infiltrating CD45+/CD3+ and CD8+ T cells (Fig. 3J; Supplementary Fig. S4C).
Finally, we conducted a cell type enrichment analysis of TNBCs in TCGA using xCell (48), a deconvolution analysis that estimates the differential proportions of immune cell populations based on gene expression data. The xCell analysis revealed that STING1 mRNA abundance correlates with CD8+ T cell infiltration (Fig. 3K; Supplementary Fig. S5A). Lack of tumor-infiltrating T cells has been associated with poor outcomes in breast cancers (49). Consistent with this, low STING1 expression was associated with shorter relapse free survival (RFS) of patients with TNBC in the Kaplan-Meier plotter database (Supplementary Fig. S5B).
MYC directly represses STING expression through epigenetic regulation
Given the opposing effects of tumor cell expression of MYC and STING on tumor-infiltrating T cells, we hypothesized that MYC activation suppressed immune activation via direct suppression of STING. MYC knockdown by siRNA restored STING expression in MDA-MB-436 Tax-R, MDA-MB-436 Doxo-R, SUM159PT Tax-R and SUM159PT Doxo-R cells (Fig. 4A and B; Supplementary Fig. S6A and B). Further, pharmacological inhibition of MYC with MYCi975 induced STING expression in these and in 4T1 cells (Fig. 4C; Supplementary Fig. S6C and D). In the cohort of Hutchinson et al., low STING1 mRNA was associated with a high score of MYC hallmark gene signatures (Fig. 4D). Further, in a previously reported cohort of residual TNBCs after neoadjuvant chemotherapy (8), tumoral STING and MYC protein measured by IHC were inversely correlated (Fig. 4E).
Figure 4. MYC binds to a STING1 enhancer and directly represses STING1 expression.
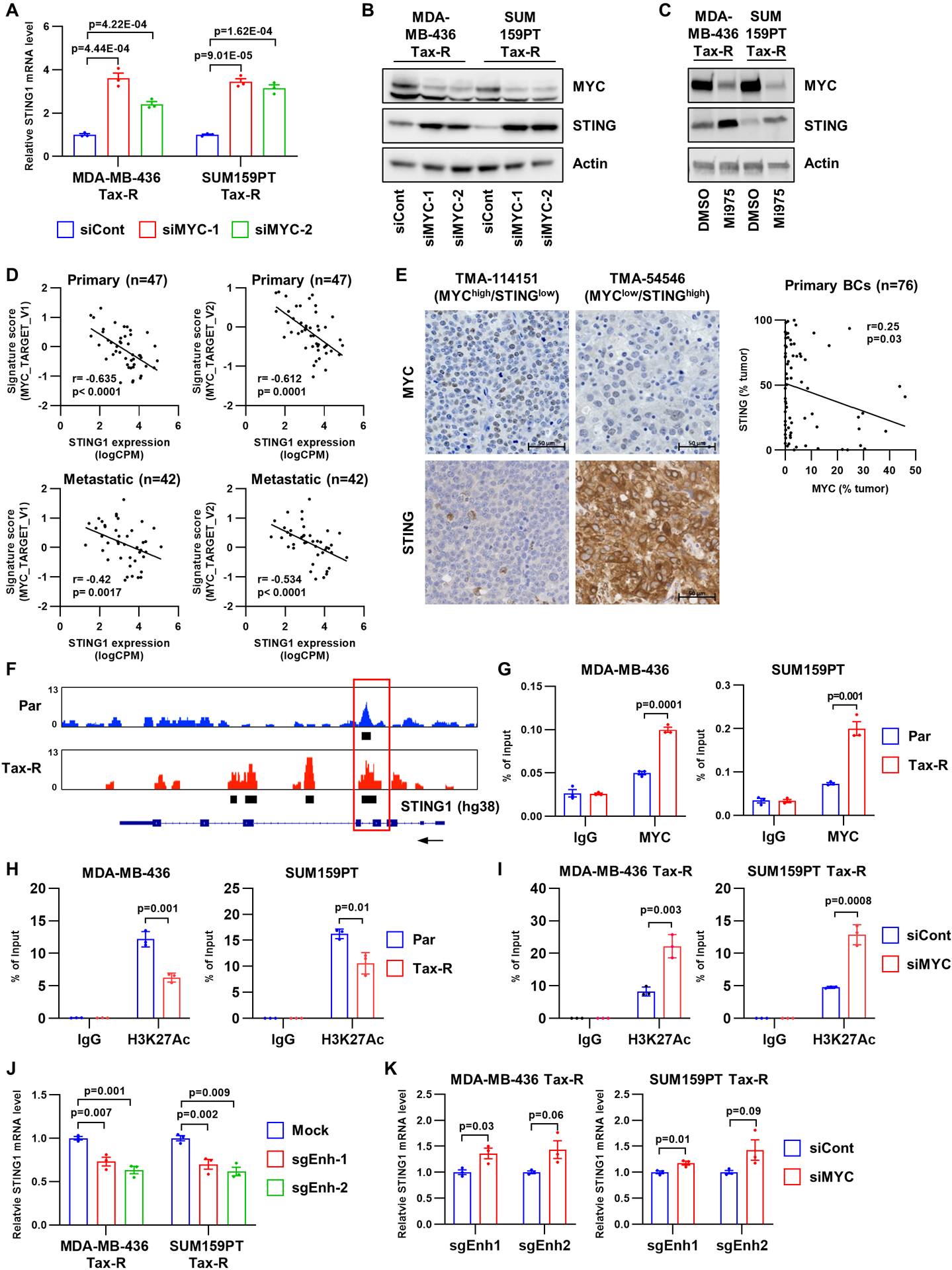
A, MDA-MB-436 Tax-R and SUM159PT Tax-R cells were transfected with control or MYC siRNA. After 48 h, RNA was extracted and subjected to RT-qPCR. Bars represent the mean ± SEM (n=3; two-tailed unpaired t-tests). B, Lysates from the same cells shown in A were subjected to immunoblot analysis with MYC, STING and actin antibodies. C, Lysates from MDA-MB-436 Tax-R and SUM159PT Tax-R cells, which had been treated with 6 μM MYCi975 for 48 h, were subjected to immunoblot analysis with MYC, STING and actin antibodies. D, STING1 mRNA abundance was compared to hallmark MYC activation gene signatures scores in primary and metastatic TNBC tumors (r, Spearman correlation coefficient). E, MYC and STING protein were assessed by IHC in residual TNBCs after neoadjuvant chemotherapy (n=76). Representative IHC images are shown on the left. The scatter plot shows Pearson correlation coefficient of the MYC and STING IHC scores (right). F, Binding of MYC to chromatin regions of STING1 in SUM159PT parental and Tax-R cells by ChIP-sequencing. Black rectangles indicate regions with significant association of MYC. G, ChIP-qPCR validating binding of MYC to STING1 chromatin in the indicated cells. Bars represent the mean ± SEM of enrichment values that were expressed as percent of input (n=3; two-tailed unpaired t-tests). H, ChIP-qPCR of acetylated H3K27 binding to the putative enhancer regions of STING1 in the indicated cells. Bars represent the mean ± SEM (n=3; two-tailed unpaired t-tests). I, ChIP-qPCR of acetylated H3K27 binding to the putative enhancer regions of STING1 in the indicated cells that had been transfected with MYC siRNA or control siRNA. Bars represent the mean ± SEM (n=3; two-tailed unpaired t-tests). J, STING1 RT-qPCR from RNA extracted from MDA-MB-436 Tax-R and SUM159PT Tax-R cells, which were transduced with dCas9-KRAB and sgRNA that recognizes the putative enhancer region of STING1. Each bar represents the mean ± SEM relative to non-transduced cells (n=3; two-tailed unpaired t-tests). K, STING1 RT-qPCR on RNA extracted from cells described in J transfected with MYC siRNA or control siRNA. Bars represent the mean ± SEM (n=3; two-tailed unpaired t-tests).
STING1 has been shown to be epigenetically suppressed in various types of cancer (45). Therefore, we next determined whether MYC suppresses STING in TNBC through an epigenetic mechanism. Using chromatin immunoprecipitation (ChIP)-sequencing with SUM159PT parental and Tax-R cells, we found that MYC binds to several intragenic regions of the STING1 gene (Fig. 4F). Per the encyclopedias of DNA elements candidate cis-regulatory elements [ENCODE cCREs (50)], a STING1 intragenic region between the 4th and 5th exons was predicted to be a cis-regulatory element with significant proximal enhancer-like signature (Supplementary Fig. S6E). Moreover, an E-box was found in the intragenic region (chr5:139,480,883–139,480,888; hg38). ChIP-qPCR assays with a MYC antibody confirmed the direct association of MYC with this intragenic region (Fig. 4G). ChIP-qPCR also showed that this binding was significantly increased in Tax-R cells compared to parental cells. MYC binding to this region was also enhanced in MDA-MB-436 Doxo-R and SUM159PT Doxo-R cells compared to the respective parental cells (Supplementary Fig. S6F). Finally, we ruled out somatic alterations in STING1 in MDA-MB-436 Tax-R and SUM159PT Tax-R cells by qPCR (Supplementary Fig. 6G and H) and exome sequencing.
We next quantified acetylated histone H3 lysine 27 (H3K27Ac), a histone mark representing enhancers with high accessibility. ChIP-qPCR revealed an enrichment of H3K27Ac at the intragenic region of STING1 predicted to be an enhancer (Fig. 4H). H3K27Ac bound to this region was reduced in MDA-MB-436 Tax-R and SUM159PT Tax-R cells compared to parental cells (Fig. 4H). MYC has been shown to repress gene expression through chromatin deacetylation (51,52). Thus, we examined if enhancer-bound MYC reduces H3K27Ac in Tax-R cells. MYC knockdown by siRNA resulted in an induction of H3K27Ac in the enhancer region of Tax-R cells (Fig. 4I). The effect of MYC knockdown on the enrichment of H3K27Ac was superior in the putative enhancer compared to other regions, although MYC knockdown exhibited a broad effect (Supplementary Fig. S6I and J). To further dissect this result, we utilized an enhancer perturbation approach mediated by Cas9 endonuclease dead (dCas9) conjugated with Krüppel associated box (KRAB) (53). Perturbation of the enhancer also resulted in a reduction of STING1 expression in MDA-MB-436 Tax-R and SUM159PT Tax-R cells (Fig. 4J), implying that this region is functionally associated with STING1 transcription. Finally, perturbation of the enhancer abrogated the effect of MYC knockdown on restoring STING1 expression in MDA-MB-436 Tax-R and SUM159PT Tax-R cells (Fig. 4K). These results suggest that MYC directly binds to the enhancer region of STING1, repressing STING1 transcription.
Other epigenetic regulators such as EZH2, DNMT1, KDM5B and KDM5C have been shown to repress STING expression in various cancer types (18,19). However, neither mRNA nor protein of these epigenetic regulators were elevated in Tax-R cells compared to parental cells (Supplementary Fig. S6K and L).
T cell–inflamed TME driven by MYC inhibition is mediated by STING
We next investigated whether STING1 loss is required for MYC-driven immune evasion. To test this, we silenced STING in MDA-MB-436 Tax-R and SUM159PT Tax-R cells using CRISPR. In the STING1 KO cells, MYC ablation by siRNA did not induce STING expression (Fig. 5A). MYC knockdown also failed to induce T-cell chemoattractant expression as well as increased Jurkat CXCR3 cell chemotaxis mediated by CM (Fig. 5B and C). Treatment of PBMC-humanized mice bearing SUM159PT Tax-R STING1 KO tumors with MYCi975 did not increase tumor infiltrating CD45+/CD3+ and CD8+ T cells, whereas this effect was still observed in mice bearing isogenic STING1-intact xenografts (Fig. 5D). We also generated 4T1 STING1 KO cells using CRISPR. Similarly to MDA-MB-436 and SUM159PT Tax-R STING1 KO cells, MYC knockdown did not induce STING1 expression (Fig. 5E), T-cell chemoattractant expression (Fig. 5F), and tumor-infiltrating CD45+/CD3+ and CD8+ T cells (Fig. 5G) in 4T1 STING1 KO cells and tumors. These data strongly suggest that the T cell–inflamed TME elicited by MYC inhibition requires the induction of STING.
Figure 5. T cell–inflamed TME driven by MYC inhibition is mediated by STING.
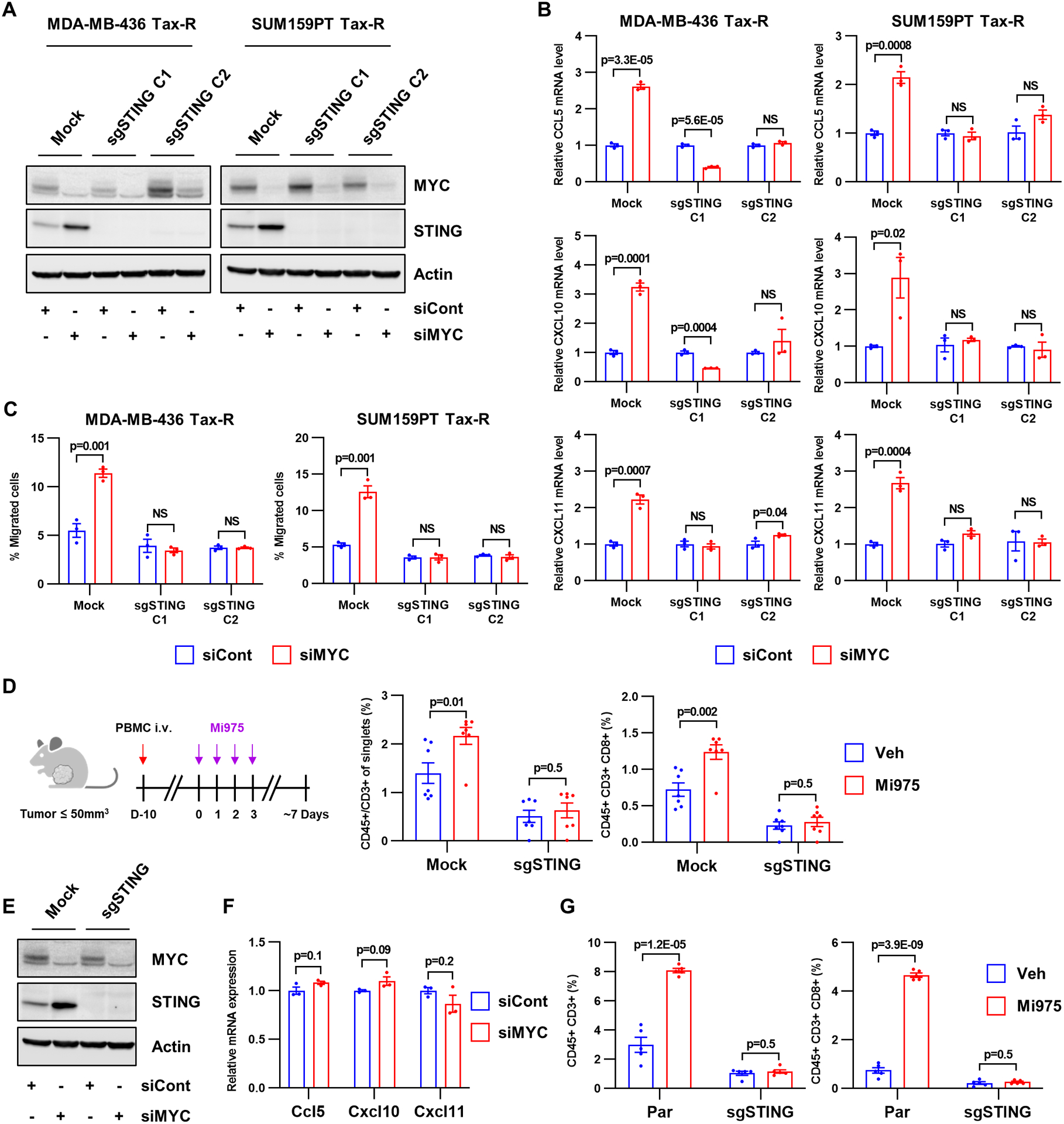
A, Immunoblot analysis for MYC, STING and actin in MDA-MB-436 and SUM159PT Tax-R STING1 KO cells transfected with MYC siRNA or control siRNA. B, RNA was extracted from cells shown in A and subjected to RT-qPCR. Bars represent the mean ± SEM (n=3; two-tailed unpaired t-tests). C, Transwell migration of Jurkat CXCR3 cells. Conditioned media from cells shown in A was added to the bottom wells. Each bar represents the mean ± SEM (n=3; two-tailed unpaired t-tests). D, Flow cytometry quantification of tumor-infiltrating lymphocytes isolated from PBMC-humanized mice bearing established SUM159PT Tax-R or Tax-R STING1 KO tumors treated with vehicle or MYCi975. Bars represent the mean ± SEM (n=5; two-tailed unpaired t-tests). E, 4T1 Sting1 KO cells were transfected with MYC siRNA or control siRNA for 48 h. Cell lysates were prepared and subjected to immunoblot analysis with MYC, STING and actin antibodies. F, RNA was extracted from cells shown in E and subjected to RT-qPCR. Bars represent the mean ± SEM of mRNA relative to cells transfected with control siRNA (n=3; two-tailed unpaired t-tests). G, Flow cytometry quantification of tumor-infiltrating lymphocytes isolated from BALB/c female mice bearing 4T1 parental and Sting1 KO tumors treated with vehicle or MYCi975. Each bar represents the mean ± SEM (n=5; two-tailed unpaired t-tests).
MYC-driven STING repression promotes resistance to immune checkpoint inhibitors
The presence of tumor-infiltrating T cells is one of the key determinants of tumor response to ICB (54). Therefore, we hypothesized that STING1 loss and the associated decrease of tumor-infiltrating T cells promotes resistance to ICB. We first examined the effect of atezolizumab, a PD-L1 inhibitor approved for use in combination with nab-paclitaxel in patients with metastatic TNBC (55), against SUM159PT tumors established in PBMC-humanized mice. Treatment with atezolizumab attenuated growth of SUM159PT parental but not SUM159PT STING1 KO tumors (Fig. 6A–C). Growth of Py230 tumors in syngeneic mice was also delayed upon treatment with an antibody targeting mouse PD-L1. However, Sting1 ablation by the doxycycline-inducible shRNA abrogated the effect of the antibody against Py230 tumors (Fig. 6D–F).
Figure 6. MYC-driven STING repression promotes resistance to immune checkpoint inhibitors.
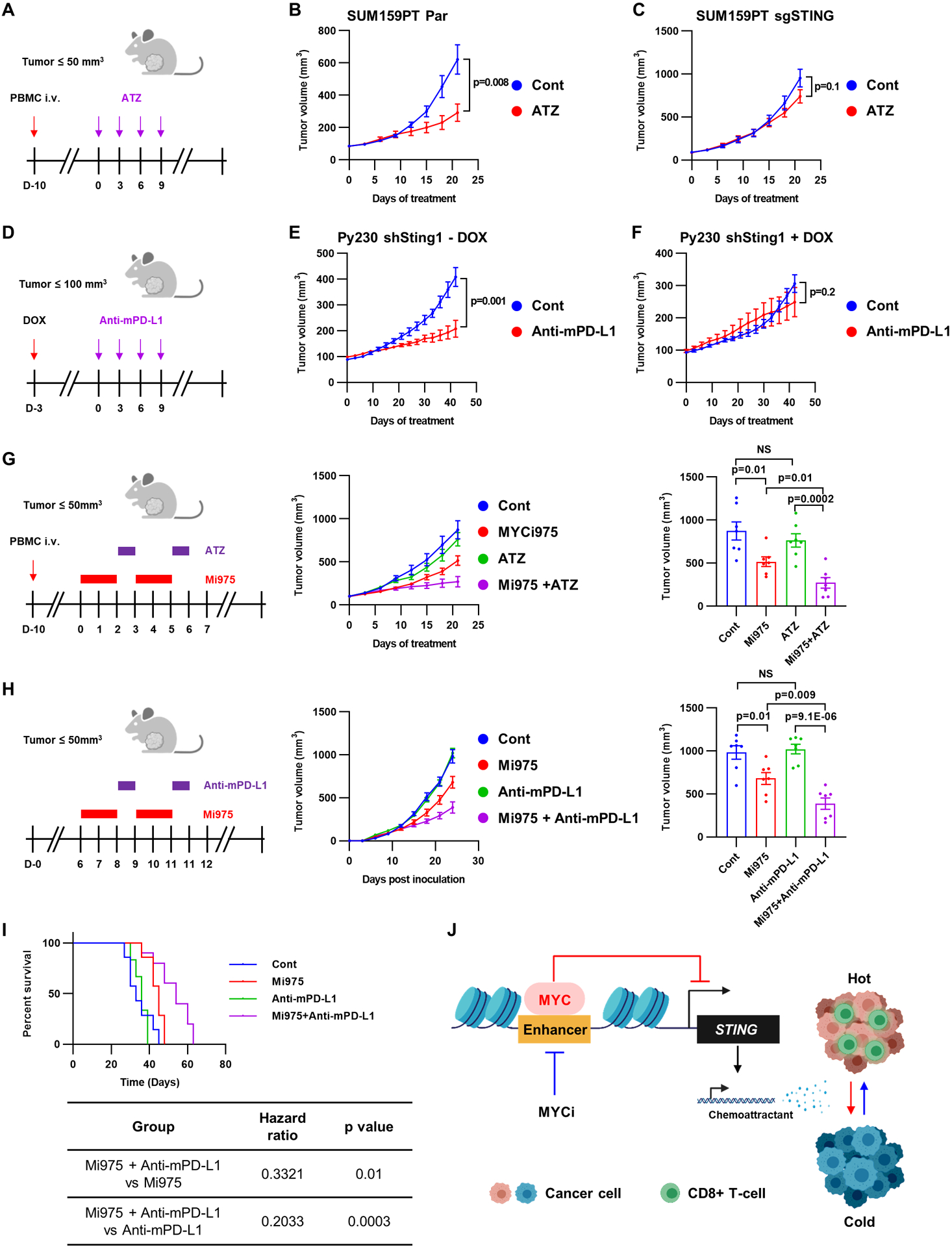
A, Experimental schema for B and C. ATZ, atezolizumab B and C, hPBMCs were inoculated into NOD/SCID female mice bearing established SUM159PT parental (B) or SUM159PT STING1 KO (C) tumors treated with vehicle or atezolizumab. Tumor volumes were serially measured with calipers and monitored for 3 weeks. Bars represent the mean ± SEM of tumor volume (n=7; two-tailed unpaired t-tests). D, Experimental schema for D and E. E and F, Py230 cells transduced with doxycycline-inducible Sting1 shRNA were orthotopically injected into C57BL/6 female mice treated with vehicle (E) or doxycycline (F). Three days after starting doxycycline, mice were randomized for treatment with a mPD-L1 antibody or IgG1 isotype control. Bars represent the mean ± SEM (n=7; two-tailed unpaired t-tests). G, hPBMCs were inoculated into NOD/SCID female mice bearing established SUM159PT Tax-R tumors and randomized for treatment with 1) vehicle, 2) atezolizumab, 3) MYCi975, or 4) atezolizumab plus MYCi975. Tumor volumes were serially measured with calipers and monitored for 3 weeks. Data in middle panel represent the mean ± SEM of tumor volume over time. Bars (in right panel) represent the mean ± SEM of tumor volume at the end of the experiment (n=7; two-tailed unpaired t-tests). H, 4T1 WT cells were orthotopically injected into BALB/c female mice randomized for treatment with 1) vehicle, 2) mPD-L1 antibody, 3) MYCi975, or 4) mPD-L1 antibody plus MYCi975. Data in middle panel represent the mean ± SEM. tumor volume in mm3 over time. Bars (in right panel) represent the mean ± SEM of tumor volume in mm3 at the end of the experiment (n=7; two-tailed unpaired t-tests). I, Overall survival of BALB/c mice bearing 4T1 tumors (n=7/ group; logrank). J, Mechanism by which MYC drives immune-cold TNBCs via STING repression.
Finally, we tested if therapeutic inhibition of MYC would sensitize MYC-driven immune-cold TNBCs to ICB via induction of STING. PBMC-humanized mice with established SUM159PT Tax-R tumors were treated with atezolizumab in combination with MYCi975 (Fig. 6G). Treatment with the combination suppressed tumor growth more potently than each drug alone (Fig. 6G). Similar to SUM159PT Tax-R tumors, MYC-overexpressing 4T1 tumors did not respond to the mouse PD-L1 antibody (Fig. 6H). However, MYCi975 combined with mouse PD-L1 blockade significantly suppressed the tumor growth and prolonged the survival of mice compared to single-agent treatment (Fig. 6H and I). Altogether, these data support the combination of a MYC inhibitor with PD-L1 blockade as an effective therapeutic strategy in patients with immune-cold, MYC-overexpressing TNBCs.
Discussion
Gene expression profiles of TNBCs have shown that the enrichment of T cell–related signatures associates with a low risk of recurrence and benefit from chemotherapy (56). Abundance of TILs in the tumor microenvironment also predict favorable outcome in TNBC (57,58). Here, we report that immune-related hallmark gene set signatures are negatively correlated with MYC hallmark gene set signatures by interrogating the transcriptome profile of TNBCs that acquired the hyperactivation of MYC, as well as primary and metastatic TNBCs.
Tumor regression upon MYC inactivation has been shown to require the host immune system in various genetically engineered mouse models (59–61), suggesting a role of MYC in the host TME. MYCi975 has been shown to increase tumor infiltration by immune cells and sensitize mouse prostate and lung cancers to PD-1 blockade (42). Consistent with this report, we showed herein that pharmacological and genetic inhibition of MYC induces immune-related gene set signatures and T-cell chemokines which, in turn, foster a T cell–inflamed TME. Finally, we showed that the MYC inhibitor combined with anti-PD-L1 immunotherapy induced a significant anti-tumor effect in immune-cold TNBC models.
Both pharmacologic and genetic inhibition of MYC resulted in an induction of STING1 expression, but not other molecules that have been shown to mediate MYC-driven immune evasion, such as CD274, CD47 (13), IL23, CCL9 (11) and HLA (44). The impairment of the STING pathway by either epigenetic suppression or post-translational modification has been reported in melanoma, colon, lung and breast cancers (18,62,63), which dampens immune surveillance of cancer progression. Here, we show that MYC directly binds to an enhancer region of STING1. MYC inhibition induced the association of the histone marker H3K27Ac with the STING1 enhancer region. Finally, enhancer perturbation clearly demonstrated that this enhancer region plays a role in the repression of STING1 by MYC.
Activation of STING in host immune cells is a critical innate immune sensing machinery for tumor surveillance (64). Evidence suggests that STING1 loss also dampens immune surveillance in a tumor cell autonomous manner, which, in turn, associates with resistance to PD-1 blockade (18,20). In this study, we show that STING1 loss is causally associated with downregulation of immune-related hallmark gene set signatures and T-cell chemokines. These lead to a decrease of tumor-infiltrating T cells and a poor response to PD-L1 blockade. Innate and adaptive responses driven by STING pathway activation have also been shown to depend on type I IFN signaling (16). However, other evidence suggests that the immune response mediated by STING may occur independent of type I IFNs (65–67). In this study, we showed that neutralizing antibodies against type I IFNs do not affect T-cell migration promoted by ectopic expression of STING1, whereas neutralizing antibodies for CCL5, CXCL10 and CXCL11 significantly diminish it. Moreover, transcriptome analyses revealed that type I IFNs were not detectable in TNBC cells, further suggesting that STING induces immunogenic gene expression independent of type I IFNs in TNBC.
Immunotherapy is an effective anti-cancer strategy with curative potential in several cancer types. The initial efficacy of PD-L1/PD-1 blockade in TNBC would benefit from further identification of predictive biomarkers for response or resistance. To date, tumor mutational load (68), PD-L1 expression (69) and a T cell–inflamed TME (70) have been suggested as predictive biomarkers of response to PD-1/PD-L1 blockade. Here, we show that STING downregulation driven by MYC hyperactivation is causally linked to resistance to PD-L1 blockade, suggesting STING loss as a biomarker that predicts resistance to immune checkpoint inhibitors in TNBC (Fig. 6J). Taken together, these data support the testing of MYC inhibitors in combination with PD-L1 blockade as a treatment strategy for immune-cold TNBC with hyperactivation of MYC.
Supplementary Material
Synopsis.
MYC-driven immune evasion is a barrier to treating triple-negative breast cancer (TNBC). MYC hyperactivation epigenetically represses STING to drive immune suppression in TNBC. Combining MYC inhibition with checkpoint blockade increased immune infiltration and antitumor immunity in TNBC.
Acknowledgements
We received the following financial support to conduct this work: UTSW Simmons Cancer Center P30 CA142543, CPRIT RR170061 (C.L.A.), NCI Breast SPORE P50 CA098131, Vanderbilt-Ingram Cancer Center P30 CA68485, Susan G. Komen Breast Cancer Foundation SAB1800010 (C.L.A.), Breast Cancer Research Foundation (C.L.A.), NCI R01CA224899 (C.L.A. and A.B.H.), CPRIT RP150596 (J.L., S.D.B., and V.S.M.), the research fund of Hanyang University (HY-202100000001135; K.-M.L.) and Basic Science Research Program through the National Research Foundation of Korea (NRF) grant funded by Korea government (MSIT) (2021R1F1A1062411 and 2022R1C1C1011387; K.-M.L.).
Authors’ Disclosures
C.L.A. receives or has received research grant support from Pfizer, Lilly, Radius, Bayer, and Takeda, holds stock options in Provista, and serves or has served in a scientific advisory role to Puma Biotechnology, Novartis, Lilly, TAIHO Oncology, Daiichi Sankyo, Merck, AstraZeneca, OrigiMed, Immunomedics, Athenex, Arvinas, and the Susan G. Komen Foundation. All other authors declare no competing interests. A.B.H. receives or has received research grant support from Takeda and Lilly and travel support from Puma Biotechnology.
References
- 1.Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med 2010;363(20):1938–48 doi 10.1056/NEJMra1001389. [DOI] [PubMed] [Google Scholar]
- 2.Brown SD, Warren RL, Gibb EA, Martin SD, Spinelli JJ, Nelson BH, et al. Neo-antigens predicted by tumor genome meta-analysis correlate with increased patient survival. Genome Res 2014;24(5):743–50 doi 10.1101/gr.165985.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wimberly H, Brown JR, Schalper K, Haack H, Silver MR, Nixon C, et al. PD-L1 Expression Correlates with Tumor-Infiltrating Lymphocytes and Response to Neoadjuvant Chemotherapy in Breast Cancer. Cancer Immunol Res 2015;3(4):326–32 doi 10.1158/2326-6066.CIR-14-0133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schmid P, Rugo HS, Adams S, Schneeweiss A, Barrios CH, Iwata H, et al. Atezolizumab plus nab-paclitaxel as first-line treatment for unresectable, locally advanced or metastatic triple-negative breast cancer (IMpassion130): updated efficacy results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 2020;21(1):44–59 doi 10.1016/S1470-2045(19)30689-8. [DOI] [PubMed] [Google Scholar]
- 5.Mittendorf EA, Zhang H, Barrios CH, Saji S, Jung KH, Hegg R, et al. Neoadjuvant atezolizumab in combination with sequential nab-paclitaxel and anthracycline-based chemotherapy versus placebo and chemotherapy in patients with early-stage triple-negative breast cancer (IMpassion031): a randomised, double-blind, phase 3 trial. Lancet 2020;396(10257):1090–100 doi 10.1016/S0140-6736(20)31953-X. [DOI] [PubMed] [Google Scholar]
- 6.Schmid P, Cortes J, Pusztai L, McArthur H, Kummel S, Bergh J, et al. Pembrolizumab for Early Triple-Negative Breast Cancer. N Engl J Med 2020;382(9):810–21 doi 10.1056/NEJMoa1910549. [DOI] [PubMed] [Google Scholar]
- 7.Dang CV. MYC on the path to cancer. Cell 2012;149(1):22–35 doi 10.1016/j.cell.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Balko JM, Giltnane JM, Wang K, Schwarz LJ, Young CD, Cook RS, et al. Molecular profiling of the residual disease of triple-negative breast cancers after neoadjuvant chemotherapy identifies actionable therapeutic targets. Cancer Discov 2014;4(2):232–45 doi 10.1158/2159-8290.CD-13-0286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee KM, Giltnane JM, Balko JM, Schwarz LJ, Guerrero-Zotano AL, Hutchinson KE, et al. MYC and MCL1 Cooperatively Promote Chemotherapy-Resistant Breast Cancer Stem Cells via Regulation of Mitochondrial Oxidative Phosphorylation. Cell Metab 2017;26(4):633–47 e7 doi 10.1016/j.cmet.2017.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Braun J, Felsher DW, Goodglick LA. c-myc, MHCI, and NK resistance in immunodeficiency lymphomas. Ann N Y Acad Sci 1992;651:467–9 doi 10.1111/j.1749-6632.1992.tb24647.x. [DOI] [PubMed] [Google Scholar]
- 11.Kortlever RM, Sodir NM, Wilson CH, Burkhart DL, Pellegrinet L, Brown Swigart L, et al. Myc Cooperates with Ras by Programming Inflammation and Immune Suppression. Cell 2017;171(6):1301–15 e14 doi 10.1016/j.cell.2017.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Muthalagu N, Monteverde T, Raffo-Iraolagoitia X, Wiesheu R, Whyte D, Hedley A, et al. Repression of the Type I Interferon Pathway Underlies MYC- and KRAS-Dependent Evasion of NK and B Cells in Pancreatic Ductal Adenocarcinoma. Cancer Discov 2020;10(6):872–87 doi 10.1158/2159-8290.CD-19-0620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Casey SC, Tong L, Li Y, Do R, Walz S, Fitzgerald KN, et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science 2016;352(6282):227–31 doi 10.1126/science.aac9935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim EY, Kim A, Kim SK, Chang YS. MYC expression correlates with PD-L1 expression in non-small cell lung cancer. Lung Cancer 2017;110:63–7 doi 10.1016/j.lungcan.2017.06.006. [DOI] [PubMed] [Google Scholar]
- 15.Zhang X, Bai XC, Chen ZJ. Structures and Mechanisms in the cGAS-STING Innate Immunity Pathway. Immunity 2020;53(1):43–53 doi 10.1016/j.immuni.2020.05.013. [DOI] [PubMed] [Google Scholar]
- 16.Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013;339(6121):786–91 doi 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 2015;347(6227):aaa2630 doi 10.1126/science.aaa2630. [DOI] [PubMed] [Google Scholar]
- 18.Kitajima S, Ivanova E, Guo S, Yoshida R, Campisi M, Sundararaman SK, et al. Suppression of STING Associated with LKB1 Loss in KRAS-Driven Lung Cancer. Cancer Discov 2019;9(1):34–45 doi 10.1158/2159-8290.CD-18-0689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu L, Cao J, Cai WL, Lang SM, Horton JR, Jansen DJ, et al. KDM5 histone demethylases repress immune response via suppression of STING. PLoS Biol 2018;16(8):e2006134 doi 10.1371/journal.pbio.2006134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu C, Guan J, Lu S, Jin Q, Rousseau B, Lu T, et al. DNA Sensing in Mismatch Repair-Deficient Tumor Cells Is Essential for Anti-tumor Immunity. Cancer Cell 2021;39(1):96–108 e6 doi 10.1016/j.ccell.2020.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Y, Yang Q, Zeng X, Wang M, Dong S, Yang B, et al. MET amplification attenuates lung tumor response to immunotherapy by inhibiting STING. Cancer Discov 2021. doi 10.1158/2159-8290.CD-20-1500. [DOI] [PubMed] [Google Scholar]
- 22.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 2013;8(11):2281–308 doi 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gui X, Yang H, Li T, Tan X, Shi P, Li M, et al. Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature 2019;567(7747):262–6 doi 10.1038/s41586-019-1006-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods 2015;12(4):357–60 doi 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014;30(7):923–30 doi 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 26.Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol 2015;33(3):290–5 doi 10.1038/nbt.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15(12):550 doi 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102(43):15545–50 doi 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hutchinson KE, Yost SE, Chang CW, Johnson RM, Carr AR, McAdam PR, et al. Comprehensive Profiling of Poor-Risk Paired Primary and Recurrent Triple-Negative Breast Cancers Reveals Immune Phenotype Shifts. Clin Cancer Res 2020;26(3):657–68 doi 10.1158/1078-0432.CCR-19-1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bankhead P, Loughrey MB, Fernandez JA, Dombrowski Y, McArt DG, Dunne PD, et al. QuPath: Open source software for digital pathology image analysis. Sci Rep 2017;7(1):16878 doi 10.1038/s41598-017-17204-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barnes SD, Ruess H, Mathews JA, Chen B, Malladi VS. BICF ChIP-seq Analysis Workflow. Zenodo 2020. doi 10.5281/zenodo.3759481. [DOI] [Google Scholar]
- 32.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25(14):1754–60 doi 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tarasov A, Vilella AJ, Cuppen E, Nijman IJ, Prins P. Sambamba: fast processing of NGS alignment formats. Bioinformatics 2015;31(12):2032–4 doi 10.1093/bioinformatics/btv098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009;25(16):2078–9 doi 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol 2008;9(9):R137 doi 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yu G, Wang LG, He QY. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 2015;31(14):2382–3 doi 10.1093/bioinformatics/btv145. [DOI] [PubMed] [Google Scholar]
- 37.Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 2012;481(7381):389–93 doi 10.1038/nature10730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 2013;152(5):1173–83 doi 10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ciriello G, Gatza ML, Beck AH, Wilkerson MD, Rhie SK, Pastore A, et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell 2015;163(2):506–19 doi 10.1016/j.cell.2015.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hanzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 2013;14:7 doi 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pearson T, Greiner DL, Shultz LD. Creation of “humanized” mice to study human immunity. Curr Protoc Immunol 2008;Chapter 15:Unit 15 21 doi 10.1002/0471142735.im1521s81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Han H, Jain AD, Truica MI, Izquierdo-Ferrer J, Anker JF, Lysy B, et al. Small-Molecule MYC Inhibitors Suppress Tumor Growth and Enhance Immunotherapy. Cancer Cell 2019;36(5):483–97 e15 doi 10.1016/j.ccell.2019.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schrors B, Boegel S, Albrecht C, Bukur T, Bukur V, Holtstrater C, et al. Multi-Omics Characterization of the 4T1 Murine Mammary Gland Tumor Model. Front Oncol 2020;10:1195 doi 10.3389/fonc.2020.01195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Versteeg R, Noordermeer IA, Kruse-Wolters M, Ruiter DJ, Schrier PI. c-myc down-regulates class I HLA expression in human melanomas. EMBO J 1988;7(4):1023–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sokolowska O, Nowis D. STING Signaling in Cancer Cells: Important or Not? Arch Immunol Ther Exp (Warsz) 2018;66(2):125–32 doi 10.1007/s00005-017-0481-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Morton JJ, Bird G, Refaeli Y, Jimeno A. Humanized Mouse Xenograft Models: Narrowing the Tumor-Microenvironment Gap. Cancer Res 2016;76(21):6153–8 doi 10.1158/0008-5472.CAN-16-1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Biswas T, Gu X, Yang J, Ellies LG, Sun LZ. Attenuation of TGF-beta signaling supports tumor progression of a mesenchymal-like mammary tumor cell line in a syngeneic murine model. Cancer Lett 2014;346(1):129–38 doi 10.1016/j.canlet.2013.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Aran D, Hu Z, Butte AJ. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol 2017;18(1):220 doi 10.1186/s13059-017-1349-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Aaltomaa S, Lipponen P, Eskelinen M, Kosma VM, Marin S, Alhava E, et al. Lymphocyte infiltrates as a prognostic variable in female breast cancer. Eur J Cancer 1992;28A(4–5):859–64 doi 10.1016/0959-8049(92)90134-n. [DOI] [PubMed] [Google Scholar]
- 50.Consortium EP, Moore JE, Purcaro MJ, Pratt HE, Epstein CB, Shoresh N, et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 2020;583(7818):699–710 doi 10.1038/s41586-020-2493-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim TW, Hong S, Lin Y, Murat E, Joo H, Kim T, et al. Transcriptional Repression of IFN Regulatory Factor 7 by MYC Is Critical for Type I IFN Production in Human Plasmacytoid Dendritic Cells. J Immunol 2016;197(8):3348–59 doi 10.4049/jimmunol.1502385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kurland JF, Tansey WP. Myc-mediated transcriptional repression by recruitment of histone deacetylase. Cancer Res 2008;68(10):3624–9 doi 10.1158/0008-5472.CAN-07-6552. [DOI] [PubMed] [Google Scholar]
- 53.Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE, et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 2013;154(2):442–51 doi 10.1016/j.cell.2013.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017;168(4):707–23 doi 10.1016/j.cell.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N Engl J Med 2018;379(22):2108–21 doi 10.1056/NEJMoa1809615. [DOI] [PubMed] [Google Scholar]
- 56.Callari M, Cappelletti V, D’Aiuto F, Musella V, Lembo A, Petel F, et al. Subtype-Specific Metagene-Based Prediction of Outcome after Neoadjuvant and Adjuvant Treatment in Breast Cancer. Clin Cancer Res 2016;22(2):337–45 doi 10.1158/1078-0432.CCR-15-0757. [DOI] [PubMed] [Google Scholar]
- 57.West NR, Milne K, Truong PT, Macpherson N, Nelson BH, Watson PH. Tumor-infiltrating lymphocytes predict response to anthracycline-based chemotherapy in estrogen receptor-negative breast cancer. Breast Cancer Res 2011;13(6):R126 doi 10.1186/bcr3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Denkert C, von Minckwitz G, Brase JC, Sinn BV, Gade S, Kronenwett R, et al. Tumor-infiltrating lymphocytes and response to neoadjuvant chemotherapy with or without carboplatin in human epidermal growth factor receptor 2-positive and triple-negative primary breast cancers. J Clin Oncol 2015;33(9):983–91 doi 10.1200/JCO.2014.58.1967. [DOI] [PubMed] [Google Scholar]
- 59.Jain M, Arvanitis C, Chu K, Dewey W, Leonhardt E, Trinh M, et al. Sustained loss of a neoplastic phenotype by brief inactivation of MYC. Science 2002;297(5578):102–4 doi 10.1126/science.1071489. [DOI] [PubMed] [Google Scholar]
- 60.Shachaf CM, Kopelman AM, Arvanitis C, Karlsson A, Beer S, Mandl S, et al. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature 2004;431(7012):1112–7 doi 10.1038/nature03043. [DOI] [PubMed] [Google Scholar]
- 61.Felsher DW. MYC Inactivation Elicits Oncogene Addiction through Both Tumor Cell-Intrinsic and Host-Dependent Mechanisms. Genes Cancer 2010;1(6):597–604 doi 10.1177/1947601910377798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Konno H, Yamauchi S, Berglund A, Putney RM, Mule JJ, Barber GN. Suppression of STING signaling through epigenetic silencing and missense mutation impedes DNA damage mediated cytokine production. Oncogene 2018;37(15):2037–51 doi 10.1038/s41388-017-0120-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xia T, Konno H, Barber GN. Recurrent Loss of STING Signaling in Melanoma Correlates with Susceptibility to Viral Oncolysis. Cancer Res 2016;76(22):6747–59 doi 10.1158/0008-5472.CAN-16-1404. [DOI] [PubMed] [Google Scholar]
- 64.Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MY, et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 2014;41(5):830–42 doi 10.1016/j.immuni.2014.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yamashiro LH, Wilson SC, Morrison HM, Karalis V, Chung JJ, Chen KJ, et al. Interferon-independent STING signaling promotes resistance to HSV-1 in vivo. Nat Commun 2020;11(1):3382 doi 10.1038/s41467-020-17156-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wu J, Dobbs N, Yang K, Yan N. Interferon-Independent Activities of Mammalian STING Mediate Antiviral Response and Tumor Immune Evasion. Immunity 2020;53(1):115–26 e5 doi 10.1016/j.immuni.2020.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Parekh NJ, Krouse TE, Reider IE, Hobbs RP, Ward BM, Norbury CC. Type I interferon-dependent CCL4 is induced by a cGAS/STING pathway that bypasses viral inhibition and protects infected tissue, independent of viral burden. PLoS Pathog 2019;15(10):e1007778 doi 10.1371/journal.ppat.1007778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Roszik J, Haydu LE, Hess KR, Oba J, Joon AY, Siroy AE, et al. Novel algorithmic approach predicts tumor mutation load and correlates with immunotherapy clinical outcomes using a defined gene mutation set. BMC Med 2016;14(1):168 doi 10.1186/s12916-016-0705-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Taube JM, Klein A, Brahmer JR, Xu H, Pan X, Kim JH, et al. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin Cancer Res 2014;20(19):5064–74 doi 10.1158/1078-0432.CCR-13-3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014;515(7528):568–71 doi 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-sequencing and ChIP-sequencing data are available on the Gene Expression Omnibus (GEO) under accession number GSE196325. Other data is available upon reasonable request to the authors.