

Abstract
Within the cytoplasm of mammalian cells is a protein called cyclic GMP-AMP synthase (cGAS), which acts to defend against infection and other threats to the host. cGAS operates in this manner through its ability to detect a molecular occurrence that should not exist in healthy cells—the existence of DNA in the cytosol. Upon DNA binding, cGAS synthesizes cyclic GMP-AMP (cGAMP), a cyclic dinucleotide that activates the endoplasmic reticulum-localized protein stimulator of interferon genes (STING). STING-mediated signaling culminates in host defensive responses typified by inflammatory cytokine and interferon expression, and the induction of autophagy. Studies over the last several years have established a consensus in the field of the enzymatic activities of cGAS in vitro, as it relates to DNA-induced production of cGAMP. However, much additional work is needed to understand the regulation of cGAS functions within cells, where multiple sources of DNA can create a problem of self and non-self discrimination. In this review, we provide an overview of how the cGAS-STING pathway mediates innate immune responses during infection and other cellular stresses. We then highlight the recent progress in the understanding of the increasingly diverse ways that this DNA-sensing machinery is regulated inside cells, including how cGAS remains inactive to host-derived DNA under conditions of homeostasis.
Keywords: cGAS, STING, innate immune system, antiviral response
Graphical Abstract
The cGAS-STING pathway plays a central role in health and disease. In this review, we describe the current understanding of the molecular mechanisms that control activity of this pathway.
Introduction
Cells are equipped with protein-based machineries that link the detection of microorganisms to innate immune responses that prevent pathogen infections1. Central to the function of these host-defensive molecular machines are members of the pattern recognition receptor (PRR) superfamily. PRRs recognize conserved microbial molecules that are known as pathogen-associated molecular patterns (PAMPs), with the most common being nucleic acids and microbial cell-wall components1. In the case of DNA sensing, several proteins have been identified as PRRs2.
The endosome-localized Toll-like receptor 9 (TLR9) is responsible for the detection of unmethylated CpG-containing DNA that entered cells from the extracellular space3. Among intracellular DNA sensors are absent in melanoma 2 (AIM2), which induces inflammasome activation in response to cytosolic double-stranded (ds) DNA4, and the enzyme cyclic GMP-AMP synthase (cGAS), which recognizes B-form DNA independent of its sequence through contacts with the sugar-phosphate backbone5, 6. Upon dsDNA binding, cGAS produces 2’3’-cyclic-GMP-AMP (2’3’-cGAMP, hereafter cGAMP), a second messenger molecule that binds to the protein stimulator of interferon genes (STING, also known as MITA, MPYS, and ERIS) to induce innate immune responses7–9. These cGAS-STING-mediated responses include the transcriptional induction of antiviral cytokines such as type I interferon (IFN) and proinflammatory cytokines, as well as the induction of autophagy and cell death. Due to its lack of sequence-specificity10, cGAS has the capacity to recognize dsDNA from any source, including that from viruses, bacteria and endogenous host-derived DNA. cGAS deficiencies in cells and mice are associated with alterations in a variety of infectious and non-infectious diseases, with the latter including cancer and autoinflammatory disorders11. As such, there is an increasing interest in understanding the regulators of cGAS-STING signaling events in natural and therapeutic settings.
In this review, we will first overview the cGAS-STING signaling pathway and the consequent cellular responses, with a focus on the molecular mechanisms by which cGAS is activated by DNA. We will further discuss how cGAS activation is regulated within the cells by subcellular compartmentalization and post-translational modifications, which helps us understand how the endogenous DNA escapes cGAS detection.
The Overview of cGAS-STING Signaling
cGAS is an enzyme that contains latent nucleotidyltransferase (NTase) activity. In vitro studies have demonstrated that binding to dsDNA or RNA-DNA hybrids stimulates cGAS enzymatic activity, resulting in the synthesis of cGAMP from ATP and GTP5 (Figure 1). Within cells, cGAMP serves as a second messenger that binds to the endoplasmic reticulum (ER)-resident protein STING. STING also has the ability to detect bacteria-derived cyclic dinucleotides (e.g. cyclic di-AMP, cyclic di-GMP, and 3’3’-cGAMP)12–15. Binding to cGAMP or bacterial cyclic dinucleotides triggers conformational changes in STING, followed by its trafficking from the ER to the ER-Golgi intermediate compartment (ERGIC) and the Golgi apparatus16–18. As in the case of many other ER proteins, STING trafficking is mediated by vesicles formed by the GTPase SAR1 and the COPII complex18. Upon reaching at the ERGIC and Golgi, STING is palmitoylated and oligomerizes into a signaling platform that recruits downstream kinases and other molecular complexes to initiate the cytokine transcription and other innate immune responses19, 20, which are described in detail below.
Figure 1. Overview of cGAS activation and cGAS-STING signaling.

cGAS binds to both pathogen-derived and host organelles-derived DNA in the cytosol and micronuclei, which then forms cGAS-DNA 2:2 complexes. These complexes undergo phase separation to further assemble into larger oligomers with liquid-like biophysical properties. These cGAS-DNA condensates allow efficient catalytic action of cGAS, which turns ATP and GTP into cGAMP. cGAMP binds to STING on the ER membrane, which in turn traffics to the ERGIC/Golgi and oligomerizes. Activated STING recruits TBK1 that phosphorylates STING at its C-terminal tail (CTT). IRF3 is recruited to the pLxIS motif among the phosphorylated residues at the CTT of STING, then gets phosphorylated by TBK1. Phosphorylated IRF3 dimers and translocates to the nucleus, where it induces the transcription of type I IFN and proinflammatory cytokines in collaboration with NF-κB, which is also activated by TBK1 and IKKε downstream of STING.
Transcriptional and non-transcriptional host defensive responses induced by cGAS-STING
Within the Golgi apparatus, palmitoylated STING oligomers interact with the kinase TANK-binding kinase 1 (TBK1)19. This interaction between STING and TBK1 dimer is mediated by the PLPLRT/SD domain present in the cytoplasmic C-terminal tail (CTT) of STING, leading to TBK1 auto-phosphorylation and activation21, 22. Activated TBK1 phosphorylates STING at Serine 366 (S366, S365 in mice) within a sequence also present in its C-terminus called a pLxIS motif (p, hydrophilic residue; x, any residue; S, phosphorylation site)22, 23. This TBK1-mediated phosphorylation of STING is dependent on the oligomerization, as the TBK1 protein cannot phosphorylate the STING dimer it is bound to due to the large space between the TBK1 active site and S366 of directly bound STING molecule22. The transcription factor interferon regulatory factor 3 (IRF3) is then recruited to the phosphorylated pLxIS motif, where is in turn phosphorylated by TBK122, 23. Activated IRF3 then dimerizes and translocates to the nucleus, where it induces the transcription of type I IFN genes in collaboration with NF-κB24. Type I IFN then elicits an antiviral state of cells in an autocrine or paracrine manner, through binding to its receptor IFN-α/β receptor (IFNAR)25. The IFNAR1-IFNAR2 complex activates the Janus Kinase (JAK)-signal transducer and activator of transcription (STAT) pathway, leading to the transcriptional induction of interferon-stimulated genes (ISGs) that target multiple stages in the viral life cycle25.
As mentioned above, NF-κB is another transcription factor that is activated downstream of STING. Although the activation of NF-κB is dependent on TBK1 and IKKε, it does not require IRF3 activation26. In addition to synergizing with IRF3 to induce type I IFN, NF-κB induces the transcription of inflammatory cytokines and chemokines. Mouse STING mutants that contain a defective pLxIS motif (STING-S365A), which lack the IRF3 binding site and do not activate IRF3, revealed that STING induces IRF3- and IFN-independent antiviral responses27–29. These IRF3-independent factors that are upregulated during these responses include CXCL1, CXCL2, 4-BBL1, and COX2, whose expression is dependent on NF-κB27. Moreover, STING-mediated NF-κB activation in dendritic cells upregulates the surface expression of major histocompatibility complex II (MHC-II) and costimulatory molecules such as CD80 and CD8627. These events are considered hallmark activities needed to stimulate T cell-mediated inflammatory responses30. The IRF3-independent transcriptional responses are important for host defense, as the STING-S365A mice are resistant to herpes simplex virus 1 (HSV-1) infection27–29 or develop polyarthritis in DNA clearance-defective DNase II−/− mice even in the absence of IRF3 activation31.
In addition to the transcription of type I IFN, cGAS-STING signaling has been implicated in other innate immune responses. Autophagy is one such non-transcriptional consequences of STING activation18. Evolutionarily more ancient than the IFN genes, autophagy is a cellular process that was first described to maintain cell homeostasis under conditions of starvation32. Under these conditions, intracellular contents are encapsulated into membrane-bound structures called autophagosomes, which are delivered to lysosomes and degraded. The degraded materials can then be recycled for use by the cell as a means of self-sustenance. Autophagy also operates as a host-defense mechanisms to encapsulate cytosol-localized pathogens, leading to their eventual delivery to and destruction within lysosomes33. During infections with the DNA virus HSV-1 or Mycobacterium tuberculosis (Mtb), DNA from these pathogens can activate cGAS34–37. The resulting cGAMP production leads to STING-dependent autophagy of the pathogens and lysosomal degradation35, 38. Mechanistically, the STING-containing ERGIC serves as a source of the hallmark processes in autophagosome formation—the lipidation of the protein LC318. LC3 lipidation is dependent on autophagy protein 5 (ATG5) and WD repeat domain phosphoinositide-interacting protein 2 (WIPI2) but not on Unc51-like autophagy activating kinase 1 (ULK1), ULK2, or Beclin 1, the components in the conventional autophagy18. A STING mutant lacking its C-terminal activation domain still triggers autophagy, suggesting that STING induces autophagy in a IRF3-independent manner18. STING-induced autophagy also functions as negative feedback of cGAS-STING pathway, as TBK1 activates p62/SQSTM1-dependent autophagy targets STING for lysosomal degradation39. Because STING orthologs in Xenopus tropicalis and the sea anemone Nematostella vectensis induce autophagy without IRF3 activation or I IFN transcription18, autophagy induction is thought to be the primordial function of cGAS-STING signaling.
Overall, cGAS-STING pathway induces IFN and other cytokine responses as well as cytokine-independent autophagy, all of which play significant roles in restricting pathogens.
cGAS-STING activities that impact cell viability and mitosis
cGAS-STING mediated induction of diverse cell death pathways
In addition to the aforementioned activities that mediate host defensive type I IFN and autophagy responses, recent studies have revealed cGAS-STING activities that impact cells in unexpectedly diverse manners. For example, several instances of STING-mediated induction of programed cell death have been reported40–49 (Table 1). T cells that experience intense STING activation trigger type I IFN-independent apoptosis by inducing the transcription of BH3-only proteins and other proapoptotic genes through the activities of IRF3 and p5340. STING also induces transcription-independent T cell apoptosis by disrupting calcium homeostasis and therefore sensitizing cells to ER stress and the unfolded protein responses (UPR), as revealed by a study using an autoinflammatory disease-associated STING mutant41. A gain-of-function STING mutant N154S (N153S in mouse) develops lung inflammation, myeloid cell expansion, and T cell cytopenia in IRF3-lacking mice42, and the T cell death caused by this mutant was abrogated by the ER stress inhibitors41. More recent data have indicated that STING signaling renders tumor cells more sensitive to apoptosis induced by reactive oxygen species (ROS)43. This process may be mediated by the actions of ROS-metabolizing ISGs43, which impact the extent of cellular damage that can be inflicted by oxidation.
Table 1.
cGAS-STING-dependent cell death. Summary of cGAS-STING-dependent cell death described in the main text.
| Cell Death | Cell Types | Stimulation / Model | References |
|---|---|---|---|
| Apoptosis | T cells | Small molecule STING agonist | 38 |
| STING gain-of-function mutant (N154S in human, N153AS in mice) | 39, 40 | ||
| HNSCC cells | DNA-damaging agent, radiation | 41 | |
| Necroptosis | L929 fibroblasts | Sendai virus (SeV), murine gammaherpesvirus-68 (MHV68) | 42 |
| HT29 colon cancer cells | SMAC mimetic (LBW-242) + pan-caspase inhibitor (z-VAD-fmk) | 43 | |
| Mice (in vivo) | Ischemia reperfusion (I/R) injury | 44 | |
| Pyroptosis | BMDMs | Chlamydia trachomatis, Francisella novicida | 45, 46 |
| BLaER human monocytes | Horse testis (HT)-DNA lipofection | 47 |
HNSCC, Head and neck squamous cell carcinomas; BMDM, bone marrow-derived macrophage; BLaER, tamoxifen-inducible derivative of the RCH-ACV B-cell leukemia cell line.
In addition to apoptosis, cGAS-STING signaling promotes other forms of cell death. STING activation induces type I IFN and TNFα secretion. These cytokines can act synergistically to stimulate the necroptosis-inducing kinases receptor interacting kinase 1 (RIPK1)- and RIPK3 in neighboring cells44. STING-dependent necroptosis is not only induced by DNA viruses such as murine gammaherpesvirus 68 (MHV68), but also by mitochondrial DNA (mtDNA) that has been released into the cytosol44, 45. Indeed, a mtDNA-STING-cell death pathway is required for the ischemia reperfusion (I/R) injury-induced intestinal barrier disruption in vivo46.
Pyroptosis, the inflammatory cell death commonly caused by the actions of inflammasomes50, is also positively regulated by cGAS-STING activation. cGAS-STING signaling promotes inflammasome assembly and activation in several experimental contexts, resulting in pyroptosis through type I IFN-dependent and -independent mechanisms. Type I IFN upregulates the expression of ISGs such as guanylate-binding proteins (GBPs) and immunity-related GTPases (IRGs)51. These factors can rupture cytosolic bacterial membranes to expose bacterial DNA to the protein AIM2. DNA-bound AIM2 then seeds the assembly of an inflammasome that stimulates pyroptosis52. Type I IFN also has been reported to induce the expression of the bacterial LPS receptor caspase-11, which can stimulate the NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3) inflammasome by Gram-negative bacteria53. Indeed, cGAS and STING are required for cell death in murine macrophages that occurs during infections with Chlamydia trachomatis and Francisella novicida47, 48. A study using BLaER1 human monocytes revealed that cytosolic DNA detection by cGAS causes the translocation of STING to lysosomes, where STING induces cell death via lysosomal rupture49. This lysosomal cell death (LCD) elicits pyroptosis by potassium (K+) efflux-induced NLRP3 activation.
Altogether, activation of cGAS and STING leads to various forms of cell death in both pathogenic and sterile inflammation, with remaining unclarities as to how the cells determine which forms of cell death to induce in a certain context.
cGAS-STING mediated DNA repair and Cellular Senescence
DNA repair machineries, including homologous recombination (HR) and non-homologous end joining (NHEJ), are crucial for genomic stability and cell viability but also must be tightly regulated, because excessive DNA repair can lead to undesired chromosomal rearrangement54. cGAS has been implicated in the regulation of DNA repair, either as a positive and negative regulator55–57. Negative regulation of HR is mediated by chromatin-bound cGAS in the nucleus56. DNA-bound cGAS oligomerizes and therefore compacts DNA into higher-order state, which is resistant to RAD51 recombinase-mediated strand invasion56. This STING-independent role of cGAS accelerates the generation of micronuclei and cell death under severe genomic injury caused by irradiation. On the contrary, recently reported positive regulation of DNA repair is mediated by cytosolic DNA-bound cGAS57. Upon exposure of cells to ionizing radiation, DNA damage-induced cytosolic DNA leakage stimulates cGAS-STING signaling, leading to TBK1-mediated phosphorylation of phosphoribosyl pyrophosphate synthetases 1 and 2 (PRPS1/2)57. Phosphorylated PRPS1 and 2, in turn, promote DNA synthesis and repair. These opposing roles of cGAS in DNA repair indicate that cGAS functions in a context-dependent fashion, including cell types and the magnitude of DNA damages, which warrants future investigations.
Cellular senescence defines the irreversible cell cycle arrest caused by various cellular and environmental stresses, including inflammation and aging58. In the case of DNA damage, the cGAS-STING pathway induces a senescence-associated secretory phenotype (SASP), the hallmark phenotype of senescence in which cells secrete a variety of proteins including inflammatory cytokines, chemokines, growth factors, and proteases59–61. SASP is not only the hallmark of senescence, but is also an amplifier of this process. The requirement for cGAS-STING in cellular senescence has been revealed by both in vitro and in vivo studies using ionizing radiation and other DNA-damaging agents59–61. In response to these agents, wild type cells arrest their growth rates, whereas cGAS-deficient cells continue to proliferate and do not exhibit SASP. The anti-proliferative roles of cGAS-STING may serve as anti-cancer mechanisms, as cGAS expression levels positively correlate with the survival of human lung adenocarcinoma patients59.
In summary, cGAS-STING signaling upon activation induces not only cytokine responses but also other consequences affecting cellular homeostasis. Below, we will address the types of DNA ligands that activate cGAS and STING.
Sources of substrate DNA that activate cGAS-STING signaling
Exogenous DNA as an IFN-inducing cGAS substrate
As expected from the role of cGAS in the innate immune responses, microbial DNA serves as an IFN-inducing cGAS ligand (Figure 1). Several DNA viruses carrying dsDNA activate cGAS-STING signaling in in vitro and in vivo studies. These DNA viruses include adenovirus62, vaccinia virus (VACV)34, and African swine fever viruses (ASFV)63, and herpesviruses such as cytomegalovirus (CMV)64, 65, MHV6834, and HSV-166. cGAS-STING pathway has also been implicated in the cellular responses to some RNA viruses. RNA retroviruses such as human immunodeficiency virus 1 (HIV-1), murine leukemia virus (MLV), Simian immunodeficiency virus (SIV), and human T lymphotropic virus type 1 (HTLV-1), activate cGAS by providing cytosolic DNA as reverse transcription intermediates (RTIs)67. In addition to those retroviruses, cGAS-STING signaling is involved in the responses to RNA viruses such as West Nile virus (WNV) and dengue virus, although cGAS senses the mtDNA leaked during viral infection, not the viral nucleic acids34, 68.
DNA from bacteria also activate cGAS-STING signaling. Microbial DNA released from extracellular bacteria such as Pseudomonas aeruginosa, Klebsiella pneumoniae, and Staphylococcus aureus, and intracellular bacteria such as Listeria monocytogenes, Francisella spp., Neisseria gonorrhoeae, Mtb, and Rickettsia parkeri bind to cGAS to elicit type I IFN responses37, 52, 69–72. While these DNA-induced IFN responses are beneficial to some facultative bacteria such as Listeria and Francisella, as shown in vivo71, 73, obligate bacteria such as Rickettsia are sensitive to IFN-mediated killing, and cytosolic DNA released from lysed Rickettsia subpopulation induces inflammasome-mediated host cell death that masks cGAS-STING-dependent IFN production to protect the remaining population52. Some bacteria utilize secretion systems and toxins to destabilize phagosomal membranes and release their contents, including DNA, into the cytosol. For example, Mtb uses its ESX-1 secretion system, a subtype of Type VII Secretion System (T7SS), to deliver virulence factors into macrophages37. These secretion systems are important for cGAS activation, as their mutations lead to weak type I IFN responses during infection37.
In addition to DNA, some bacteria produce cyclic dinucleotides (CDNs) that directly activate STING without stimulating cGAS. c-di-GMP is synthesized from two GTP monomers by the act of bacterial diguanylate cyclase, and regulates bacterial metabolism, virulence, and biofilm formation74. STING recognizes c-di-GMP to restrict bacterial growth by inducing type I IFN and other cytokine responses15. Similarly, c-di-AMP produced by bacteria such as Chlamydia trachomatis stimulates STING75. However, during an infection, most of these bacteria also release DNA that is sensed by cGAS. The relative contribution of bacterial DNA and CDN to host inflammatory responses is largely undefined.
Lastly, cGAS has been implicated in the defense against parasites and fungi. For example, cGAS responds to the genomic DNA of Plasmodium falciparum, and cGAS-deficient mice permit higher parasite burden after infection than wild type mice76. The cGAS-STING pathway also contributes to the type I IFN responses in Aspergillus fumigatus induced keratitis, although whether cGAS directly binds to fungal DNA is unclear77.
Self-DNA as an IFN-inducing cGAS substrate
In addition to DNA from microbes, self-DNA can access the cytoplasm and activate cGAS (Figure 1). During mitosis within cells with the genomic instability, which is a hallmark of many cancers78, DNA may segregate from the main chromosome to form perinuclear compartments called micronuclei. Nuclear envelope (NE) rupture after mitosis makes the DNA in micronuclei accessible to the cGAS immunosurveillance, therefore leading to the production of type I IFN responses79–81. This DNA damage-induced exposure of nuclear DNA to cGAS in the cytosol also occurs during senescence, anti-mitotic chemotherapy, and dysregulation of epigenetic modification such as DNA hypomethylation that leads to skin inflammation59, 60. Recently, however, Flynn et al. have proposed that chromatin bridges rather than micronuclei are the platforms of cGAS activation during mitotic errors, as IFN-inducing abilities of the antimitotic drugs correlated with the generation of cGAS-coated chromatin bridges, not micronuclei82. These findings provide important insights that not all DNA damages activate cGAS in a same manner, and the source of cGAS ligand depends on the context of DNA damages. Cytosolic DNA, when not optimally metabolized, stimulates the cGAS-STING pathway. Loss-of-function mutations in the genes encoding nucleases that degrade cytosolic DNA, such as three prime repair exonuclease 1 (TREX1), lead to cGAS-STING-dependent production of ISGs, which can lead to the development of an autoimmune disease called Aicardi–Goutières syndrome (AGS)83.
As mentioned in the section above, mtDNA is also a cGAS ligand. The perturbation of mitochondrial function either by infectious or non-infectious stimuli leads to mitochondrial damages that release mtDNA into the cytosol. RNA viruses such as measles virus (MeV), which do not possess cGAS ligands, induce mtDNA-dependent cGAS activation in addition to activating RNA sensing pathways84. During intrinsic apoptosis, Bax/Bak-mediated mitochondrial outer membrane permeabilization (MOMP) triggers the cytosolic release of mtDNA that activates cGAS and STING, which is inhibited by apoptotic caspases (caspase-9 and caspase-3/7) that are downstream of MOMP85, 86. The mechanism by which these caspases inhibit cGAS-STING activation is likely via the cleavage of cGAS, as described later87. Recent studies have found that inflammatory cytokines such as TNFα and IL-1β treatment leads to mtDNA release into cytosol, which triggers cGAS-dependent type I IFN responses88, 89. Also, even in the absence of exogenous stimuli, heterozygous knockout of the mtDNA-binding protein transcription factor A, mitochondrial (TFAM) in murine cells leads to the cytosolic release of mtDNA, resulting in the production of ISGs that made cells protective against HSV-1 infection90.
Extracellular self-DNA can also stimulate cGAS. Macrophages and other myeloid cells internalize self-DNA from the extracellular space through phagocytosis. In principle, phagocytosed cargo should be degraded in lysosomes, with luminal DNA being hydrolyzed down to single nucleotides by DNases present in the intra-lysosomal environment. Early studies, prior to the discovery of cGAS or STING, revealed that mice and cells deficient in the lysosomal enzyme DNase II are prone to express high levels of IFNs and other ISGs91. Subsequent work revealed that STING pathway activation was responsible for these responses92. These results suggested that in the absence of efficient lysosomal nuclease activity, DNA can somehow leak into the cytosol and activate cGAS-STING dependent responses.
Additional instances of extracellular DNA activating cGAS-STING have been reported. For example, non-apoptotic cell death caused by tissue injury and tumor irradiation can release DNA into the extracellular space, as can the production of DNA-containing neutrophil extracellular traps (NETs). NETs are released by neutrophils during a cell death process called NETosis, and consist of chromatin and antimicrobial molecules93. The abundance of extracellular DNA at the sites of inflammation or tissue injury may result in a phenocopy of DNase II deficiency. In this regard, phagocytes that internalize NETs or chromosomal fragments released at sites of irradiation may contain such high amounts of lysosomal DNA that DNase II is overwhelmed, resulting in DNA leakage in the cytosol and the stimulation of cGAS94. Consistent with this idea, myeloid cells are the primary source of cGAS-mediated IFN responses at sites of tissue injury94. Overall, there are several sources of DNA that can activate the cGAS-STING pathway. This promiscuity of DNA detection raises the question of how this inflammatory network is regulated to ensure efficient distinction between self and non-self DNA. In the next sections, we discuss our current knowledge of how DNA-induced cGAMP synthesis by cGAS is regulated.
The biophysical mechanisms of cGAS activation and cGAMP synthesis
cGAS is a 522 amino-acid protein that consists of a basic unstructured N-terminal domain, and a conserved C-terminal Mab21 domain with nucleotidyltransferase (NTase) activity. While cGAS monomers cannot synthesize cGAMP, cGAS dimers formed upon interactions with the sugar phosphate backbone of dsDNA display enzymatic activity. Binding of a parallel-aligned pair of dsDNA to the two DNA-binding sites (A-site and B-site) on each of two cGAS molecules stabilizes 2:2 cGAS-DNA complex, accompanied with the cGAS conformational changes that generate ATP and GTP pockets near the catalytic residues95, 96. The cGAS dimerization and conformational changes occur independently on the sequence of dsDNA, but neither single stranded DNA (ssDNA) nor RNA induces cGAS conformational changes and subsequent cGAMP synthesis although they can bind to cGAS6, 10. The cGAS dimers further align adjacently to form a ladder-like structure97. In addition, a newly discovered DNA binding site in human cGAS (C-site) allows multivalent interaction between cGAS and DNA to form the mesh-like structure98. The formation of these higher order cGAS-DNA complexes requires the certain length of DNA (> 45 bp), and the shorter DNA does not lead to human cGAS oligomerization and efficient cGAMP production99. Mouse cGAS, on the other hand, can oligomerize upon binding to short DNA and produce cGAMP, as the amino acids in the DNA binding sites allow more stable interaction with DNA99. Indeed, when the residues at this position are substituted for those in mouse cGAS, human cGAS gains the ability to synthesize cGAMP in response to short DNA99.
Recently, it has been discovered that cGAS-DNA complexes further assemble into micrometer-sized condensed liquid droplets through the process called phase separation100. These higher-order complexes have liquid-like properties—the droplet formation is reversible, and the droplets can fuse to form larger spherical units. These membrane-less granules consisting of cGAS and DNA provide a platform for cGAMP production, where cGAS encounters its substrates more efficiently. This liquid droplet formation is dependent on the concentration of cGAS and DNA100, suggesting that cGAS is activated only when cytosolic DNA levels reach a certain threshold, such as in the case of infection or cellular stresses with DNA damage. Moreover, under the physiological salt concentration, the formation of cGAS liquid droplets is dependent on intracellular ion concentration, mostly zinc100. Indeed, addition of zinc (Zn2+) to cultured cells has been reported to activate cGAS to induce type I IFN responses in the presence of dsDNA treatment101. Zn2+ is not the only ion that contributes to cGAS activation. Divalent cations such as magnesium (Mg2+) and manganese (Mn2+) are required as cofactors of cGAS that catalyze the conversion of ATP and GTP into cGAMP14. Mn2+ directly activates cGAS even in the absence of dsDNA by inducing the noncanonical conformational changes of cGAS to mimic DNA-activated cGAS, which has a unique η1 helix in the catalytic pocket allowing substrate entry and cGAMP synthesis101.
Regulation of DNA sensing and cGAS activation within cells
Despite the increasing knowledge about the mechanism of cGAS activation in vitro, the intracellular behavior of cGAS is more complicated and remains an active area of investigation. Here, we will focus on the subcellular distribution and posttranslational modification of cGAS as the major factors that regulate DNA sensing and activation of cGAS within cells (Figure 2).
Figure 2. Regulation of cGAS activity in cells.
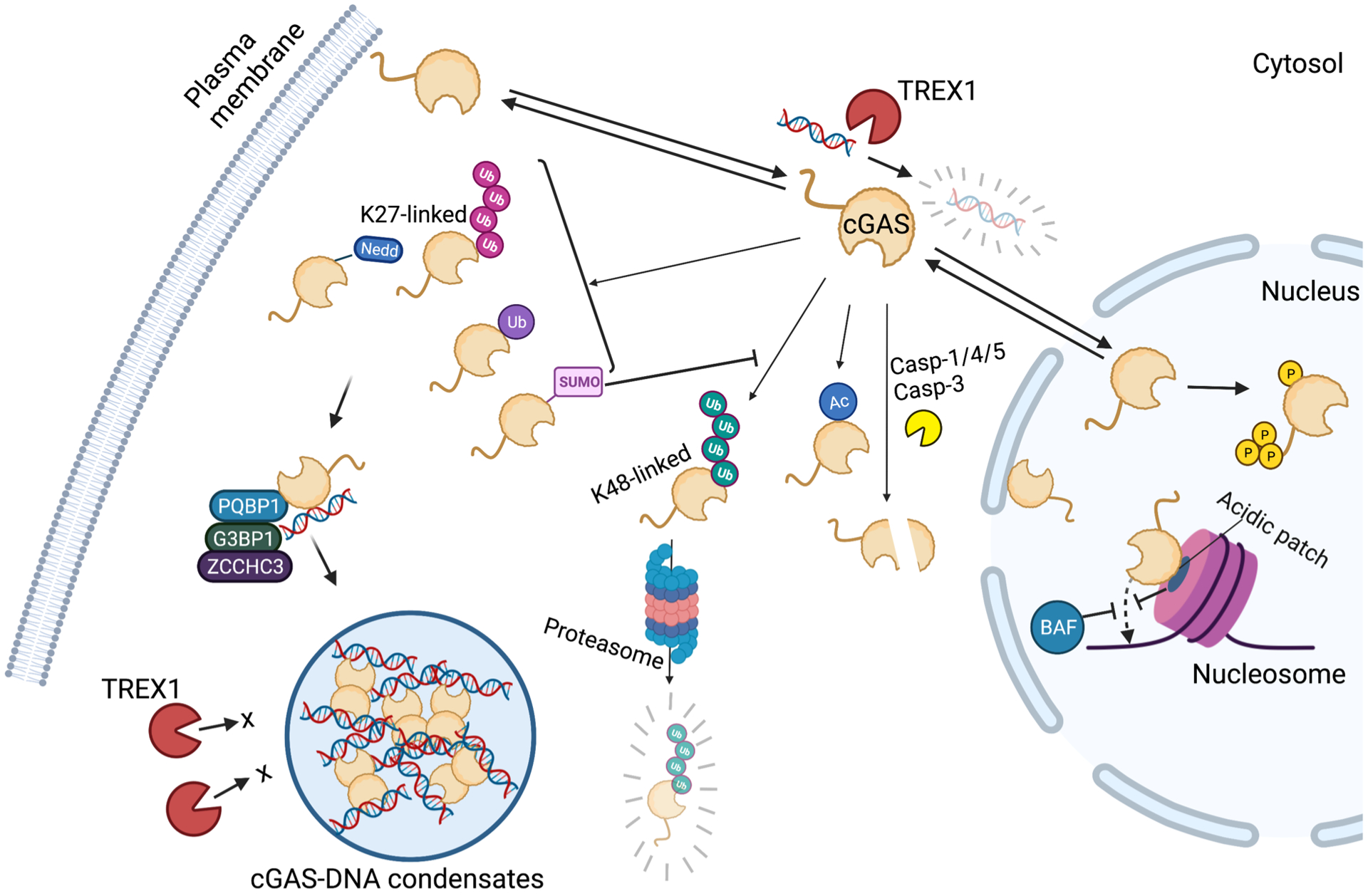
cGAS activation is positively and negatively regulated by various mechanisms in cells. cGAS is localized in not only in cytosol but also in the nucleus and on the plasma membrane, which restricts the access to the cytosolic DNA. cGAS reactivity to the chromosomal DNA is restricted in the nucleus. cGAS tethering to the acidic patch of nucleosomes and BAF binding to DNA prevents cGAS from binding DNA. Besides these mechanisms restricting cGAS accessibility to DNA, cGAS is hyperphosphorylated at its N-terminal region and catalytic activity is attenuated. Post-translational modification such as K27-linked polyubiquitination, monoubiquitination, Neddylation, and SUMOylation positively regulate cGAS activity, whereas K48-linked polyubiquitination, acetylation, and Caspase-mediated cleavage of cGAS negatively regulate the activity. Moreover, cofactor proteins including PQBP1, G3BP1, and ZCCHC3 promote DNA binding, oligomerization, and phase separation of cGAS. Phase separation-induced condensates restricts TREX1-mediated degradation of DNA and therefore accelerate cGAMP synthesis.
Subcellular localization of cGAS
cGAS was originally thought to reside in the cytosol to avoid nuclear self-DNA detection5. Despite this general idea, soon after cGAS was identified, Knipe and colleagues reported that cGAS can be detected in the nucleus of human fibroblasts and keratinocytes102. Other studies have reported cGAS localization in micronuclei, chromatin bridges, and the nucleus. cGAS activation in micronuclei and chromatin bridges are discussed in the section above. Yang et al. revealed that cGAS translocates from the cytosol to the nucleus during mitosis and relocates to the cytosol to react with DNA fragments in the presence of DNA damage59. Liu et al. found that cGAS translocates from cytosol to the nucleus upon DNA damage to suppress DNA repair55. More recently, Sun et al. identified a nuclear export signal (NES) within cGAS 169–174 a.a. (LEKLKL), which is responsible for chromosome maintenance region 1 (CRM1)-dependent cGAS translocation from the nucleus to the cytosol upon DNA stimulation103. Other groups reported that cGAS does not translocate to the nucleus in response, but is rather a nuclear resident protein104. Finally, work from our lab examining a tagged allele of cGAS revealed different localization patterns of this protein in different cells105. For example, cGAS was localized to the plasma membrane in macrophages, but was distributed in the cytosol and/or the nucleus in non-phagocytic cells105. It is unclear why different groups have found cGAS to be localized to distinct regions of the cell, and much more work needs to be done to understand the behavior of cGAS within cells.
The collective data described above could be used to suggest that cGAS is a nuclear protein under several experimental (and perhaps physiological) conditions. However, this conclusion would have significant functional implications for immunosurveillance, as nuclear cGAS is well-recognized to be an inactive protein. Elegant work from Gentili et al. has shown that the nucleus-localized pool of cGAS is ~200-fold less responsive to DNA than extra-nuclear cGAS106. Within the nucleus, the protein barrier-to-autointegration factor 1 (BAF) operates as the negative regulator of cGAS sensing of chromatin DNA upon nuclear rupture107. BAF outcompetes cGAS for DNA binding, thereby inhibiting the formation of cGAS-DNA oligomers in the nucleus. Moreover, while cGAS is able to bind to histone-containing DNA and uncoated DNA, only the latter is able to stimulate cGAMP production efficiently106, 108, 109. Cryo-electron microscopy revealed the mechanism underlying chromatin-mediated inhibition of cGAS activity, as an acidic patch of the histone H2A-H2B contacts cGAS and inhibits cGAS oligomerization upon DNA binding110–114. Thus, an increasing body of evidence supports the idea that the exclusive localization of cGAS to the nucleus would result in the inactivation of its ability to drive type I IFN responses to infection. As such, it is likely that an extra-nuclear pool of cGAS exists and may be responsible for pathogen detection.
cGAS has been reported to be present on the plasma membrane of macrophages, through interactions between the basic N-terminal domain of cGAS and phosphatidylinositol-4,5-bisphosphate (PtdIns(4,5)P2; also known as PIP2)105. The deletion of the cGAS N-terminal domain released cGAS to the cytosol, resulting in the detection of self-DNA and aberrant type I IFN induction. These data suggest that the N-terminal domain of cGAS is required to prevent self-DNA recognition, perhaps by tethering this protein to the cell surface. The finding that N-terminus of cGAS is required to prevent self-DNA reactivity was recently validated by Li et al., who further demonstrated that the self-DNA detected by cGAS is likely to be mtDNA115. Indeed, N-terminal deletion mutants of cGAS were found to be localized to the microchondria115. These data are interesting to consider in the context of the in vitro behaviors of cGAS, which suggested that DNA-induced liquid droplet formation is potentiated by the cGAS N-terminal domain. As droplet formation in vitro is associated with enhanced cGAMP production, one would have expected that deletion of a driver of droplet formation (the N-terminal domain) would impair IFN responses within cells. Yet the opposite results were obtained independently105, 115. More work is needed to understand the relative importance of the cGAS N-terminal domain in liquid droplet formation and localization within cells.
A recent study by Zhou et al. revealed that cGAS-DNA phase separation is not required for the intrinsic ability of cGAS to produce cGAMP in vitro116. Rather, phase separation may be important to restrict DNA degradation by exonuclease TREX1. Cell-free studies demonstrated that TREX1 is restricted to the outer shell of a cGAS-DNA containing liquid droplet. It is likely that the phase separation resists other negative regulators than TREX1, as the droplet-deficient cGAS mutant signals weaker than wild type cGAS even in the absence of TREX1116. In addition to cytosolic DNA, TREX1 has been reported to degrade micronuclear DNA to prevent cGAS activation117. The access to the ruptured micronuclei is achieved by ER localization of TREX1 and does not depend on DNA-binding function of TREX1. It is unclear whether cGAS-DNA phase separation occurs in micronuclei to resist TREX1-mediated DNA degradation.
Altogether, DNA sensing by cGAS is regulated not only by the physical separation of receptor and ligand within cells, but also by several safeguard mechanisms offered by DNA- or cGAS-binding proteins in the nucleus and the cytosol. These may be the main factors that underline the difference of cGAMP synthetic activity of cGAS in vitro and in cells. However, there is still a lack of consensus about cGAS localization, possibly due to the difference in the experimental settings such as cell types and imaging protocols used in each study. Future investigation that compares these factors will shed more light on the cell biology of cGAS.
Post-translational modifications of cGAS
In addition to regulation by subcellular localization, interaction with other molecules regulate cGAS through post-translational modifications (PTMs). Phosphorylation of cGAS has been reported to inhibit cGAS activity. Akt phosphorylates cGAS at S305 in human cGAS (S291 in mouse cGAS) within the C-terminal catalytic domain, thereby negatively regulating cGAMP synthesis in response to exogenous dsDNA118. Cyclin-dependent kinase 1 (CDK1)-cyclin B complex phosphorylates the same serine residues of cGAS upon mitotic entry to suppress cGAMP synthesis upon self-DNA in the nucleus119. These residues are dephosphorylated by type 1 phosphatase (PP1) upon mitotic exit to allow cGAS reactivity to exogenous DNA in the cytosol. Furthermore, multiple serine residues within the N terminus of cGAS are phosphorylated by Aurora kinase B (AurB) and other kinases during mitosis115. This hyperphosphorylation, along with chromatin tethering, blocks cGAS activation by self-DNA in the nucleus during cell cycle transition115. In addition to serine phosphorylation, cGAS is phosphorylated at tyrosine residues. B-lymphocyte kinase (BLK) phosphorylates cGAS at Y215, thereby inhibits the cGAS nuclear translocation during DNA damage mentioned in the previous section55.
Ubiquitination and other ubiquitin-like protein conjugation events affect cGAS-STING signaling. K27-linked polyubiquitination of cGAS by ER-resident E3 ligase RNF185 positively regulates cGAS-mediated innate immune responses to HSV-1 infection120. Additionally, E3 ligase TRIM56-induced monoubiquitination of cGAS at K335 is necessary for cGAS oligomerization upon DNA binding121. Moreover, cGAS is subject to K48-linked polyubiquitination at K271 and K414, which targets cGAS for proteasomal degradation. As a positive feedback loop of cGAS-STING signaling, this K48-linked ubiquitination is inhibited by tripartite motif (TRIM) E3 ligases. TRIM38 conjugates small ubiquitin-like modifier (SUMO) to cGAS that inhibits DNA binding and K48-linked polyubiquitination of cGAS122, while TRIM14 recruits deubiquitinating enzyme (DUB) USP14 to cleave K48-linked ubiquitin chains of cGAS123.
Neddylation is the covalent conjugation of neural precursor cell expressed, developmentally downregulated 8 (NEDD8), another ubiquitin-like protein. cGAS is neddylated on K231 and K421 by the roles of E2 enzyme Ube2m and E3 ligase Rnf111, inhibition of which abolishes type I IFN induction during HSV-1 infection124.
Acetylation of cGAS has been reported to modulate cGAS-STING innate immune signaling. cGAS K384, K394, and K414 are acetylated in the steady state, and DNA binding of cGAS stabilizes the interaction with histone deacetylase 3 (HDAC3), which deacetylates cGAS to activate the signaling125. Aspirin, a non-steroidal anti-inflammatory drug (NSAID), inhibits cGAS activation by directly binding and acetylation on these residues. Treatment of aspirin suppresses inflammation in AGS patient cells and in an AGS mouse model, suggesting cGAS acetylation prevents self-DNA-induced cGAS-STING signal activation. On the other hand, the cGAS acetylation in the N-terminal domain at K47, K56, K62, and K83 by the lysine acetyltransferase 5 (KAT5) potentiates cGAS-STING signaling during DNA virus infections126.
The cleavage of cGAS also modulates is inflammatory activities in cells. The canonical and non-canonical inflammasome activation leads to caspase-1- and caspase-11-mediated cGAS cleavage, respectively, to dampen cGAS-STING signaling127. Similarly, activated caspase-3 cleaves cGAS during apoptosis to suppress DNA-induced cytokine responses, keeping apoptotic cells immunologically silent87.
Besides post-translational modification, several proteins enhance cGAS-DNA complex formation by physically interacting either the enzyme or the substrate. GTPase-activating protein SH3-binding protein 1 (G3BP1) was identified as a protein that interacts with cGAS in the cytosol to promote DNA binding and subsequent activation128. Recently, the interaction with G3BP1 has been found to promote primary condensation of cGAS even in the resting cells, which primes cGAS for its rapid response to free DNA129. Although G3BP1 is required for stress granule (SG) assembly and therefore plays an important role in RNA-sensing innate immune signaling, cGAS activation by G3BP1 is independent on the SG assembly128.
In addition, polyglutamine binding protein 1 (PQBP1) and CCHC-type zinc-finger (ZF) protein ZCCHC3 are co-sensors of cGAS, which bind to both DNA and cGAS to promote cGAS recognition of DNA130, 131. While PQBP1 is specific receptor for the reverse-transcribed retroviral DNA such as HIV-1, ZCCHC3 binds to viral DNA such as HSV-1 and synthetic dsDNA in the cytosol. Because the affinity of cGAS alone for DNA is low and the recognition is promiscuous95, 99, these co-receptors are essential for optimal cGAS-DNA complex formation. Absence of these co-receptors in the nucleus may explain the low reactivity of cGAS to the self-DNA in the nucleus in addition to the presence of BAF as mentioned above, which needs further examination. Moreover, PQBP1 interacts with extrinsic tau, transmissible neurodegenerative disease protein, and then triggers cGAS-STING-dependent NF-κB activation132. As this finding bridges cGAS-STING signaling to tau-related neurodegenerative disorders, identification of cGAS-STING signaling regulators that are known to play important roles in a disease pathogenesis will indicate the unexpected involvement of cGAS and STING in that disease.
Altogether, cGAS-STING signaling is regulated by various mechanisms in cells including subcellular localization and post-translational modifications. In the next section, we will discuss how cGAS-STING pathway is targeted by pathogens.
Viral targeting of cGAS as an immune evasion strategy
Viruses and bacteria utilize several strategies to inhibit cGAS-STING signaling including the regulation of transcriptional levels and degrading CDNs, but here we focus on the modifications of cGAS and its activation reported for some viral proteins. Some DNA and RNA viruses target cGAS for degradation. The NS2B protease complex of dengue virus (DENV) cleaves cGAS to block its activation by mtDNA released from stressed mitochondria during infection133. Other viruses take indirect strategies to degrade cGAS. NS1 protein of Zika virus (ZIKV) stabilizes caspase-1, which cleaves cGAS as described above134. F17 protein in poxviruses binds to Raptor and Rictor, regulators of mammalian target of rapamycin complexes mTORC1 and mTORC2, to hyperactivate mTOR leading to the proteasomal degradation of cGAS135.
Other viral proteins physically perturb DNA sensing and activation of cGAS. VP22 and UL37 from HSV-1136, 137, UL31, UL42, and UL83 from human cytomegalovirus (HCMV)138–140, and ORF52 and LANA from Kaposi’s sarcoma-associated herpesvirus (KSHV)141, 142, ORF9 from Varicella-Zoster virus (VZV) are known examples of cGAS-inhibiting viral proteins143. Among these viral proteins, VP22, ORF52, and other structurally similar homolog tegument proteins have been recently found to compete DNA binding and phase separation with cGAS, thereby inhibiting cGAS-DNA phase separation144. In contrast, the HSV-1 UL37 tegument protein inhibits cGAS through PTM. UL37 deamidates N210 within the activation loop of human cGAS, which blocks cGAMP synthesis by impinging on the catalytic site137. This deamidation residue of cGAS is conserved in mice but not in many other non-human primates, which accounts for the species-specific permissiveness of HSV-1.
Conclusion
cGAS function has been implicated as a driver of inflammation in a variety of cellular processes. Studies from diverse laboratories have unified our understanding of the intrinsic enzymatics that govern DNA-induced cGAS activities. However, much less unanimity exists regarding the regulation of these intrinsic activities within cells. The cell biology of the cGAS-STING pathway will therefore likely remain at the forefront of research in this area. The open questions in this regard include how diverse the regulation of cGAS activation is in different cell types and different species. These questions are even more demanded by our recent discovery of three classes of cGAS with different self-DNA reactivity in mammals145. Also, the mechanisms of cGAS activation by non-self- and self-DNA in cells are regarded to be the same, but there is still a possibility that these are different, which warrants further investigation. Since the dysregulation of cGAS binding of self-DNA and subsequent aberrant cytokine production underlies a wide variety of diseases, future studies will likely be focused on filling the gap between our knowledge of cGAS activities in vitro and those within cells.
Acknowledgments
We thank members of Kagan Laboratory for the feedback and insightful discussion. This work was supported by NIH grants AI133524, AI093589, AI116550 and P30DK34854 to J.C.K. Figures were created with BioRender.com.
Footnotes
Conflict of Interests
JCK consults for IFM Therapeutics and consults and holds equity in Corner Therapeutics, Larkspur Biosciences and Neumora Therapeutics. None of these relationships influenced the work performed in this study. The other authors declare that they have no competing interests.
References
- 1.Janeway CA Jr, Medzhitov R Innate immune recognition. Annu Rev Immunol 2002; 20: 197–216. [DOI] [PubMed] [Google Scholar]
- 2.Briard B, Place DE, Kanneganti TD. DNA Sensing in the Innate Immune Response. Physiology (Bethesda) 2020; 35: 112–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol 2010; 11: 373–384. [DOI] [PubMed] [Google Scholar]
- 4.Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 2009; 458: 509–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013; 339: 786–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kranzusch PJ, Lee AS, Berger JM, Doudna JA. Structure of human cGAS reveals a conserved family of second-messenger enzymes in innate immunity. Cell Rep 2013; 3: 1362–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu J, Sun L, Chen X, et al. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 2013; 339: 826–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008; 455: 674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang X, Shi H, Wu J, et al. Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Mol Cell 2013; 51: 226–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Civril F, Deimling T, de Oliveira Mann CC, et al. Structural mechanism of cytosolic DNA sensing by cGAS. Nature 2013; 498: 332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Motwani M, Pesiridis S, Fitzgerald KA. DNA sensing by the cGAS-STING pathway in health and disease. Nat Rev Genet 2019; 20: 657–674.. [DOI] [PubMed] [Google Scholar]
- 12.Ablasser A, Goldeck M, Cavlar T, et al. cGAS produces a 2’−5’-linked cyclic dinucleotide second messenger that activates STING. Nature 2013; 498: 380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Diner EJ, Burdette DL, Wilson SC, et al. The innate immune DNA sensor cGAS produces a noncanonical cyclic dinucleotide that activates human STING. Cell Rep 2013; 3: 1355–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gao P, Ascano M, Wu Y, et al. Cyclic [G(2’,5’)pA(3’,5’)p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell 2013; 153: 1094–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burdette DL, Monroe KM, Sotelo-Troha K, et al. STING is a direct innate immune sensor of cyclic di-GMP. Nature 2011; 478: 515–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shang G, Zhang C, Chen ZJ, Bai XC, Zhang X. Cryo-EM structures of STING reveal its mechanism of activation by cyclic GMP-AMP. Nature 2019; 567: 389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 2009; 461: 788–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gui X, Yang H, Li T, et al. Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature 2019; 567: 262–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mukai K, Konno H, Akiba T, et al. Activation of STING requires palmitoylation at the Golgi. Nat Commun 2016; 7: 11932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dobbs N, Burnaevskiy N, Chen D, Gonugunta VK, Alto NM, Yan N. STING Activation by Translocation from the ER Is Associated with Infection and Autoinflammatory Disease. Cell Host Microbe 2015; 18: 157–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao B, Du F, Xu P, et al. A conserved PLPLRT/SD motif of STING mediates the recruitment and activation of TBK1. Nature 2019; 569: 718–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang C, Shang G, Gui X, Zhang X, Bai XC, Chen ZJ. Structural basis of STING binding with and phosphorylation by TBK1. Nature 2019; 567: 394–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu S, Cai X, Wu J, et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 2015; 347: aaa2630. [DOI] [PubMed] [Google Scholar]
- 24.Abe T, Barber GN. Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-κB activation through TBK1. J Virol 2014; 88: 5328–5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol 2014; 32: 513–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Balka KR, Louis C, Saunders TL, et al. TBK1 and IKKε Act Redundantly to Mediate STING-Induced NF-κB Responses in Myeloid Cells. Cell Rep 2020; 31: 107492. [DOI] [PubMed] [Google Scholar]
- 27.Yum S, Li M, Fang Y, Chen ZJ. TBK1 recruitment to STING activates both IRF3 and NF-κB that mediate immune defense against tumors and viral infections. Proc Natl Acad Sci USA 2021; 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yamashiro LH, Wilson SC, Morrison HM, et al. Interferon-independent STING signaling promotes resistance to HSV-1 in vivo. Nat Commun 2020; 11: 3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu J, Dobbs N, Yang K, Yan N. Interferon-Independent Activities of Mammalian STING Mediate Antiviral Response and Tumor Immune Evasion. Immunity 2020; 53: 115–126 e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sharpe AH, Freeman GJ. The B7-CD28 superfamily. Nat Rev Immunol 2002; 2: 116–126. [DOI] [PubMed] [Google Scholar]
- 31.Li T, Yum S, Li M, Chen X, Zuo X, Chen ZJ. TBK1 recruitment to STING mediates autoinflammatory arthritis caused by defective DNA clearance. J Exp Med 2022; 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science 2000; 290: 1717–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shahnazari S, Brumell JH. Mechanisms and consequences of bacterial targeting by the autophagy pathway. Curr Opin Microbiol 2011; 14: 68–75. [DOI] [PubMed] [Google Scholar]
- 34.Schoggins JW, MacDuff DA, Imanaka N, et al. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature 2014; 505: 691–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Watson RO, Bell SL, MacDuff DA, et al. The Cytosolic Sensor cGAS Detects Mycobacterium tuberculosis DNA to Induce Type I Interferons and Activate Autophagy. Cell Host Microbe 2015; 17: 811–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Collins AC, Cai H, Li T, et al. Cyclic GMP-AMP Synthase Is an Innate Immune DNA Sensor for Mycobacterium tuberculosis. Cell Host Microbe 2015; 17: 820–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wassermann R, Gulen MF, Sala C, et al. Mycobacterium tuberculosis Differentially Activates cGAS- and Inflammasome-Dependent Intracellular Immune Responses through ESX-1. Cell Host Microbe 2015; 17: 799–810. [DOI] [PubMed] [Google Scholar]
- 38.Liang Q, Seo GJ, Choi YJ, et al. Crosstalk between the cGAS DNA sensor and Beclin-1 autophagy protein shapes innate antimicrobial immune responses. Cell Host Microbe 2014; 15: 228–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Prabakaran T, Bodda C, Krapp C, et al. Attenuation of cGAS-STING signaling is mediated by a p62/SQSTM1-dependent autophagy pathway activated by TBK1. EMBO J 2018; 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gulen MF, Koch U, Haag SM, et al. Signalling strength determines proapoptotic functions of STING. Nat Commun 2017; 8: 427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu J, Chen YJ, Dobbs N, et al. STING-mediated disruption of calcium homeostasis chronically activates ER stress and primes T cell death. J Exp Med 2019; 216: 867–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Warner JD, Irizarry-Caro RA, Bennion BG, et al. STING-associated vasculopathy develops independently of IRF3 in mice. J Exp Med 2017; 214: 3279–3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hayman TJ, Baro M, MacNeil T, et al. STING enhances cell death through regulation of reactive oxygen species and DNA damage. Nat Commun 2021; 12: 2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schock SN, Chandra NV, Sun Y, et al. Induction of necroptotic cell death by viral activation of the RIG-I or STING pathway. Cell Death Differ 2017; 24: 615–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen D, Tong J, Yang L, et al. PUMA amplifies necroptosis signaling by activating cytosolic DNA sensors. Proc Natl Acad Sci USA 2018; 115: 3930–3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang X, Wu J, Liu Q, et al. mtDNA-STING pathway promotes necroptosis-dependent enterocyte injury in intestinal ischemia reperfusion. Cell Death Dis 2020; 11: 1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Webster SJ, Brode S, Ellis L, et al. Detection of a microbial metabolite by STING regulates inflammasome activation in response to Chlamydia trachomatis infection. PLoS Pathog 2017; 13: e1006383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Meunier E, Wallet P, Dreier RF, et al. Guanylate-binding proteins promote activation of the AIM2 inflammasome during infection with Francisella novicida. Nat Immunol 2015; 16: 476–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gaidt MM, Ebert TS, Chauhan D, et al. The DNA Inflammasome in Human Myeloid Cells Is Initiated by a STING-Cell Death Program Upstream of NLRP3. Cell 2017; 171: 1110–1124 e1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vanaja SK, Rathinam VA, Fitzgerald KA. Mechanisms of inflammasome activation: recent advances and novel insights. Trends Cell Biol 2015; 25: 308–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.MacMicking JD. IFN-inducible GTPases and immunity to intracellular pathogens. Trends Immunol 2004; 25: 601–609. [DOI] [PubMed] [Google Scholar]
- 52.Burke TP, Engstrom P, Chavez RA, Fonbuena JA, Vance RE, Welch MD. Inflammasome-mediated antagonism of type I interferon enhances Rickettsia pathogenesis. Nat Microbiol 2020; 5: 688–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Meunier E, Dick MS, Dreier RF, et al. Caspase-11 activation requires lysis of pathogen-containing vacuoles by IFN-induced GTPases. Nature 2014; 509: 366–370. [DOI] [PubMed] [Google Scholar]
- 54.Guirouilh-Barbat J, Lambert S, Bertrand P, Lopez BS. Is homologous recombination really an error-free process? Front Genet 2014; 5: 175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu H, Zhang H, Wu X, et al. Nuclear cGAS suppresses DNA repair and promotes tumorigenesis. Nature 2018; 563: 131–136. [DOI] [PubMed] [Google Scholar]
- 56.Jiang H, Xue X, Panda S, et al. Chromatin-bound cGAS is an inhibitor of DNA repair and hence accelerates genome destabilization and cell death. EMBO J 2019; 38: e102718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu R, Li J, Shao J, et al. Innate immune response orchestrates phosphoribosyl pyrophosphate synthetases to support DNA repair. Cell Metabolism 2021. [DOI] [PubMed] [Google Scholar]
- 58.Aging Campisi J., cellular senescence, and cancer. Annu Rev Physiol 2013; 75: 685–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yang H, Wang H, Ren J, Chen Q, Chen ZJ. cGAS is essential for cellular senescence. Proc Natl Acad Sci USA 2017; 114: E4612–E4620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gluck S, Guey B, Gulen MF, et al. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat Cell Biol 2017; 19: 1061–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dou Z, Ghosh K, Vizioli MG, et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 2017; 550: 402–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lam E, Stein S, Falck-Pedersen E. Adenovirus detection by the cGAS/STING/TBK1 DNA sensing cascade. J Virol 2014; 88: 974–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Garcia-Belmonte R, Perez-Nunez D, Pittau M, Richt JA, Revilla Y. African Swine Fever Virus Armenia/07 Virulent Strain Controls Interferon Beta Production through the cGAS-STING Pathway. J Virol 2019; 93: e02298–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lio CW, McDonald B, Takahashi M, et al. cGAS-STING Signaling Regulates Initial Innate Control of Cytomegalovirus Infection. J Virol 2016; 90: 7789–7797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Paijo J, Doring M, Spanier J, et al. cGAS Senses Human Cytomegalovirus and Induces Type I Interferon Responses in Human Monocyte-Derived Cells. PLoS Pathog 2016; 12: e1005546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Horan KA, Hansen K, Jakobsen MR, et al. Proteasomal degradation of herpes simplex virus capsids in macrophages releases DNA to the cytosol for recognition by DNA sensors. J Immunol 2013; 190: 2311–2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gao D, Wu J, Wu YT, et al. Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science 2013; 341: 903–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sun B, Sundstrom KB, Chew JJ, et al. Dengue virus activates cGAS through the release of mitochondrial DNA. Sci Rep 2017; 7: 3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhou CM, Wang B, Wu Q, et al. Identification of cGAS as an innate immune sensor of extracellular bacterium Pseudomonas aeruginosa. iScience 2021; 24: 101928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hansen K, Prabakaran T, Laustsen A, et al. Listeria monocytogenes induces IFNβ expression through an IFI16-, cGAS- and STING-dependent pathway. EMBO J 2014; 33: 1654–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Storek KM, Gertsvolf NA, Ohlson MB, Monack DM. cGAS and Ifi204 cooperate to produce type I IFNs in response to Francisella infection. J Immunol 2015; 194: 3236–3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Andrade WA, Agarwal S, Mo S, et al. Type I Interferon Induction by Neisseria gonorrhoeae: Dual Requirement of Cyclic GMP-AMP Synthase and Toll-like Receptor 4. Cell Rep 2016; 15: 2438–2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Auerbuch V, Brockstedt DG, Meyer-Morse N, O’Riordan M, Portnoy DA. Mice lacking the type I interferon receptor are resistant to Listeria monocytogenes. J Exp Med 2004; 200: 527–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jenal U, Reinders A, Lori C. Cyclic di-GMP: second messenger extraordinaire. Nat Rev Microbiol 2017; 15: 271–284. [DOI] [PubMed] [Google Scholar]
- 75.Barker JR, Koestler BJ, Carpenter VK, et al. STING-dependent recognition of cyclic di-AMP mediates type I interferon responses during Chlamydia trachomatis infection. mBio 2013; 4: e00018–00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gallego-Marin C, Schrum JE, Andrade WA, et al. Cyclic GMP-AMP Synthase Is the Cytosolic Sensor of Plasmodium falciparum Genomic DNA and Activates Type I IFN in Malaria. J Immunol 2018; 200: 768–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Han F, Guo H, Wang L, et al. The cGAS-STING signaling pathway contributes to the inflammatory response and autophagy in Aspergillus fumigatus keratitis. Exp Eye Res 2021; 202: 108366. [DOI] [PubMed] [Google Scholar]
- 78.Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability--an evolving hallmark of cancer. Nat Rev Mol Cell Biol 2010; 11: 220–228. [DOI] [PubMed] [Google Scholar]
- 79.Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, Greenberg RA. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 2017; 548: 466–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mackenzie KJ, Carroll P, Martin CA, et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 2017; 548: 461–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.de Oliveira Mann CC, Kranzusch PJ. cGAS Conducts Micronuclei DNA Surveillance. Trends Cell Biol 2017; 27: 697–698. [DOI] [PubMed] [Google Scholar]
- 82.Flynn PJ, Koch PD, Mitchison TJ. Chromatin bridges, not micronuclei, activate cGAS after drug-induced mitotic errors in human cells. Proc Natl Acad Sci USA 2021; 118: e2103585118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ablasser A, Hemmerling I, Schmid-Burgk JL, Behrendt R, Roers A, Hornung V. TREX1 deficiency triggers cell-autonomous immunity in a cGAS-dependent manner. J Immunol 2014; 192: 5993–5997. [DOI] [PubMed] [Google Scholar]
- 84.Sato H, Hoshi M, Ikeda F, Fujiyuki T, Yoneda M, Kai C. Downregulation of mitochondrial biogenesis by virus infection triggers antiviral responses by cyclic GMP-AMP synthase. PLoS Pathog 2021; 17: e1009841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.White MJ, McArthur K, Metcalf D, et al. Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell 2014; 159: 1549–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rongvaux A, Jackson R, Harman CC, et al. Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell 2014; 159: 1563–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ning X, Wang Y, Jing M, et al. Apoptotic Caspases Suppress Type I Interferon Production via the Cleavage of cGAS, MAVS, and IRF3. Mol Cell 2019; 74: 19–31 e17. [DOI] [PubMed] [Google Scholar]
- 88.Willemsen J, Neuhoff MT, Hoyler T, et al. TNF leads to mtDNA release and cGAS/STING-dependent interferon responses that support inflammatory arthritis. Cell Rep 2021; 37: 109977. [DOI] [PubMed] [Google Scholar]
- 89.Aarreberg LD, Esser-Nobis K, Driscoll C, Shuvarikov A, Roby JA, Gale M Jr. Interleukin-1β Induces mtDNA Release to Activate Innate Immune Signaling via cGAS-STING. Mol Cell 2019; 74: 801–815 e806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.West AP, Khoury-Hanold W, Staron M, et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature 2015; 520: 553–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kawane K, Ohtani M, Miwa K, et al. Chronic polyarthritis caused by mammalian DNA that escapes from degradation in macrophages. Nature 2006; 443: 998–1002. [DOI] [PubMed] [Google Scholar]
- 92.Ahn J, Gutman D, Saijo S, Barber GN. STING manifests self DNA-dependent inflammatory disease. Proc Natl Acad Sci USA 2012; 109: 19386–19391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Brinkmann V, Reichard U, Goosmann C, et al. Neutrophil extracellular traps kill bacteria. Science 2004; 303: 1532–1535. [DOI] [PubMed] [Google Scholar]
- 94.Apel F, Andreeva L, Knackstedt LS, et al. The cytosolic DNA sensor cGAS recognizes neutrophil extracellular traps. Sci Signal 2021; 14: eaax7942. [DOI] [PubMed] [Google Scholar]
- 95.Li X, Shu C, Yi G, et al. Cyclic GMP-AMP synthase is activated by double-stranded DNA-induced oligomerization. Immunity 2013; 39: 1019–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhang X, Wu J, Du F, et al. The cytosolic DNA sensor cGAS forms an oligomeric complex with DNA and undergoes switch-like conformational changes in the activation loop. Cell Rep 2014; 6: 421–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Andreeva L, Hiller B, Kostrewa D, et al. cGAS senses long and HMGB/TFAM-bound U-turn DNA by forming protein-DNA ladders. Nature 2017; 549: 394–398. [DOI] [PubMed] [Google Scholar]
- 98.Xie W, Lama L, Adura C, et al. Human cGAS catalytic domain has an additional DNA-binding interface that enhances enzymatic activity and liquid-phase condensation. Proc Natl Acad Sci USA 2019; 116: 11946–11955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhou W, Whiteley AT, de Oliveira Mann CC, et al. Structure of the Human cGAS-DNA Complex Reveals Enhanced Control of Immune Surveillance. Cell 2018; 174: 300–311 e311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Du M, Chen ZJ. DNA-induced liquid phase condensation of cGAS activates innate immune signaling. Science 2018; 361: 704–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhao Z, Ma Z, Wang B, Guan Y, Su XD, Jiang Z. Mn2+ Directly Activates cGAS and Structural Analysis Suggests Mn2+ Induces a Noncanonical Catalytic Synthesis of 2’3’-cGAMP. Cell Rep 2020; 32: 108053. [DOI] [PubMed] [Google Scholar]
- 102.Orzalli MH, Broekema NM, Diner BA, et al. cGAS-mediated stabilization of IFI16 promotes innate signaling during herpes simplex virus infection. Proc Natl Acad Sci USA 2015; 112: E1773–1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sun H, Huang Y, Mei S, et al. A Nuclear Export Signal Is Required for cGAS to Sense Cytosolic DNA. Cell Rep 2021; 34: 108586. [DOI] [PubMed] [Google Scholar]
- 104.Volkman HE, Cambier S, Gray EE, Stetson DB. Tight nuclear tethering of cGAS is essential for preventing autoreactivity. Elife 2019; 8: e47491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Barnett KC, Coronas-Serna JM, Zhou W, et al. Phosphoinositide Interactions Position cGAS at the Plasma Membrane to Ensure Efficient Distinction between Self- and Viral DNA. Cell 2019; 176: 1432–1446 e1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gentili M, Lahaye X, Nadalin F, et al. The N-Terminal Domain of cGAS Determines Preferential Association with Centromeric DNA and Innate Immune Activation in the Nucleus. Cell Rep 2019; 26: 2377–2393 e2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Guey B, Wischnewski M, Decout A, et al. BAF restricts cGAS on nuclear DNA to prevent innate immune activation. Science 2020; 369: 823–828. [DOI] [PubMed] [Google Scholar]
- 108.Lahaye X, Gentili M, Silvin A, et al. NONO Detects the Nuclear HIV Capsid to Promote cGAS-Mediated Innate Immune Activation. Cell 2018; 175: 488–501 e422. [DOI] [PubMed] [Google Scholar]
- 109.Zierhut C, Yamaguchi N, Paredes M, Luo JD, Carroll T, Funabiki H. The Cytoplasmic DNA Sensor cGAS Promotes Mitotic Cell Death. Cell 2019; 178: 302–315 e323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Pathare GR, Decout A, Gluck S, et al. Structural mechanism of cGAS inhibition by the nucleosome. Nature 2020. [DOI] [PubMed] [Google Scholar]
- 111.Michalski S, de Oliveira Mann CC, Stafford C, et al. Structural basis for sequestration and autoinhibition of cGAS by chromatin. Nature 2020; 587(7835): 668–672. [DOI] [PubMed] [Google Scholar]
- 112.Zhao B, Xu P, Rowlett CM, et al. The Molecular Basis of Tight Nuclear Tethering and Inactivation of cGAS. Nature 2020; 587(7835): 673–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Boyer JA, Spangler CJ, Strauss JD, et al. Structural basis of nucleosome-dependent cGAS inhibition. Science 2020; 370(6515): 450–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kujirai T, Zierhut C, Takizawa Y, et al. Structural basis for the inhibition of cGAS by nucleosomes. Science 2020; 370(6515): 455–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Li T, Huang T, Du M, et al. Phosphorylation and chromatin tethering prevent cGAS activation during mitosis. Science 2021; 371(6535): eabc5386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zhou W, Mohr L, Maciejowski J, Kranzusch PJ. cGAS phase separation inhibits TREX1-mediated DNA degradation and enhances cytosolic DNA sensing. Mol Cell 2021; 81: 739–755 e737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mohr L, Toufektchan E, von Morgen P, Chu K, Kapoor A, Maciejowski J. ER-directed TREX1 limits cGAS activation at micronuclei. Mol Cell 2021; 81: 724–738 e729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Seo GJ, Yang A, Tan B, et al. Akt Kinase-Mediated Checkpoint of cGAS DNA Sensing Pathway. Cell Rep 2015; 13: 440–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zhong L, Hu M-M, Bian L-J, Liu Y, Chen Q, Shu H-B. Phosphorylation of cGAS by CDK1 impairs self-DNA sensing in mitosis. Cell Discovery 2020; 6: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wang Q, Huang L, Hong Z, et al. The E3 ubiquitin ligase RNF185 facilitates the cGAS-mediated innate immune response. PLoS Pathog 2017; 13: e1006264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Seo GJ, Kim C, Shin WJ, Sklan EH, Eoh H, Jung JU. TRIM56-mediated monoubiquitination of cGAS for cytosolic DNA sensing. Nat Commun 2018; 9: 613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hu MM, Yang Q, Xie XQ, et al. Sumoylation Promotes the Stability of the DNA Sensor cGAS and the Adaptor STING to Regulate the Kinetics of Response to DNA Virus. Immunity 2016; 45: 555–569. [DOI] [PubMed] [Google Scholar]
- 123.Chen M, Meng Q, Qin Y, et al. TRIM14 Inhibits cGAS Degradation Mediated by Selective Autophagy Receptor p62 to Promote Innate Immune Responses. Mol Cell 2016; 64: 105–119. [DOI] [PubMed] [Google Scholar]
- 124.Li C, Zhang L, Qian D, et al. RNF111-facilitated neddylation potentiates cGAS-mediated antiviral innate immune response. PLoS Pathog 2021; 17: e1009401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Dai J, Huang YJ, He X, et al. Acetylation Blocks cGAS Activity and Inhibits Self-DNA-Induced Autoimmunity. Cell 2019; 176: 1447–1460 e1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Song ZM, Lin H, Yi XM, Guo W, Hu MM, Shu HB. KAT5 acetylates cGAS to promote innate immune response to DNA virus. Proc Natl Acad Sci USA 2020; 117: 21568–21575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wang Y, Ning X, Gao P, et al. Inflammasome Activation Triggers Caspase-1-Mediated Cleavage of cGAS to Regulate Responses to DNA Virus Infection. Immunity 2017; 46: 393–404. [DOI] [PubMed] [Google Scholar]
- 128.Liu ZS, Cai H, Xue W, et al. G3BP1 promotes DNA binding and activation of cGAS. Nat Immunol 2019; 20: 18–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zhao M, Xia T, Xing JQ, et al. The stress granule protein G3BP1 promotes pre-condensation of cGAS to allow rapid responses to DNA. EMBO Rep 2022; 23: e53166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Yoh SM, Schneider M, Seifried J, et al. PQBP1 Is a Proximal Sensor of the cGAS-Dependent Innate Response to HIV-1. Cell 2015; 161: 1293–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lian H, Wei J, Zang R, et al. ZCCHC3 is a co-sensor of cGAS for dsDNA recognition in innate immune response. Nat Commun 2018; 9: 3349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Jin M, Shiwaku H, Tanaka H, et al. Tau activates microglia via the PQBP1-cGAS-STING pathway to promote brain inflammation. Nat Commun 2021; 12: 6565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Aguirre S, Luthra P, Sanchez-Aparicio MT, et al. Dengue virus NS2B protein targets cGAS for degradation and prevents mitochondrial DNA sensing during infection. Nat Microbiol 2017; 2: 17037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zheng Y, Liu Q, Wu Y, et al. Zika virus elicits inflammation to evade antiviral response by cleaving cGAS via NS1-caspase-1 axis. EMBO J 2018; 37: e99347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Meade N, Furey C, Li H, et al. Poxviruses Evade Cytosolic Sensing through Disruption of an mTORC1-mTORC2 Regulatory Circuit. Cell 2018; 174: 1143–1157 e1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Huang J, You H, Su C, Li Y, Chen S, Zheng C. Herpes Simplex Virus 1 Tegument Protein VP22 Abrogates cGAS/STING-Mediated Antiviral Innate Immunity. J Virol 2018; 92; e00841–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Zhang J, Zhao J, Xu S, et al. Species-Specific Deamidation of cGAS by Herpes Simplex Virus UL37 Protein Facilitates Viral Replication. Cell Host Microbe 2018; 24: 234–248 e235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Huang ZF, Zou HM, Liao BW, et al. Human Cytomegalovirus Protein UL31 Inhibits DNA Sensing of cGAS to Mediate Immune Evasion. Cell Host Microbe 2018; 24: 69–80 e64. [DOI] [PubMed] [Google Scholar]
- 139.Fu YZ, Guo Y, Zou HM, et al. Human cytomegalovirus protein UL42 antagonizes cGAS/MITA-mediated innate antiviral response. PLoS Pathog 2019; 15: e1007691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Biolatti M, Dell’Oste V, Pautasso S, et al. Human Cytomegalovirus Tegument Protein pp65 (pUL83) Dampens Type I Interferon Production by Inactivating the DNA Sensor cGAS without Affecting STING. J Virol 2018; 92: e01774–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Wu JJ, Li W, Shao Y, et al. Inhibition of cGAS DNA Sensing by a Herpesvirus Virion Protein. Cell Host Microbe 2015; 18: 333–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Zhang G, Chan B, Samarina N, et al. Cytoplasmic isoforms of Kaposi sarcoma herpesvirus LANA recruit and antagonize the innate immune DNA sensor cGAS. Proc Natl Acad Sci USA 2016; 113: E1034–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Hertzog J, Zhou W, Rigby RE, et al. Varicella-Zoster Virus ORF9 Is an Antagonist of the DNA Sensor cGAS. bioRxiv 2021. doi.org/ 10.1101/2020.02.11.943415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Xu G, Liu C, Zhou S, et al. Viral tegument proteins restrict cGAS-DNA phase separation to mediate immune evasion. Mol Cell 2021; 81: 2823–2837.e9. [DOI] [PubMed] [Google Scholar]
- 145.Mosallanejad K, Zhou W, Govande A, Hancks DC, Kranzusch PJ, Kagan JC. Three functionally distinct classes of cGAS proteins in nature revealed by self-DNA-induced interferon responses. bioRxiv 2022. doi.org/ 10.1101/2022.03.09.483681. [DOI] [Google Scholar]