

Abstract
Camptothecin is a chemotherapeutic drug widely used to treat various cancers. Ophiorrhiza pumila is an ideal plant model for the study of camptothecin production, with various advantages for studying camptothecin biosynthesis and regulation. The DNA-binding WRKY transcription factors have a key regulatory role in secondary metabolite biosynthesis in plants. However, little is currently known about their involvement in camptothecin biosynthesis in O. pumila. We identified 46 OpWRKY genes unevenly distributed on the 11 chromosomes of O. pumila. Phylogenetic and multiple sequence alignment analyses divided the OpWRKY proteins into three subfamilies. Based on spatial expression and co-expression, we targeted the candidate gene OpWRKY6. Overexpression of OpWRKY6 significantly reduced the accumulation of camptothecin compared with the control. Conversely, camptothecin accumulation increased in OpWRKY6 knockout lines. Further biochemical assays showed that OpWRKY6 negatively regulates camptothecin biosynthesis from both the iridoid and shikimate pathways by directly downregulating the gene expression of OpGES, Op10HGO, Op7DLH, and OpTDC. Our data provide direct evidence for the involvement of WRKYs in the regulation of camptothecin biosynthesis and offer valuable information for enriching the production of camptothecin in plant systems.
Introduction
As the leading cause of death globally, cancer can cause nearly 10 million deaths in a year, posing a severe threat to human health [1]. Treatment options for cancer include surgery, anti-cancer medicines, and radiation therapy, administered alone or in combination [2]. Camptothecin exhibits remarkable antitumor activity by inhibiting topoisomerase I enzyme, and its two derivatives (topotecan and irinotecan) are widely used as anti-cancer chemotherapeutic drugs [2, 3]. As a naturally occurring plant monoterpene indole alkaloid (MIA), camptothecin accumulates in various distantly related plants, including Camptotheca acuminata (Nyssaceae), Nothapodytes nimmoniana (Icacinaceae), and Ophiorrhiza pumila (Rubiaceae) [4–6]. Compared with woody camptothecin-producing plants, O. pumila has the advantages of a short growth cycle, high camptothecin content, and easy genetic transformation, which render it an ideal model plant for the study of camptothecin biosynthesis and regulation [1, 7].
Similar to other MIAs, camptothecin in O. pumila is synthesized from the important precursor strictosidine supplied by tryptamine in the shikimate pathway and secologain in the iridoid pathway (Fig. 1) [6, 8, 9]. In the shikimate pathway, tryptophan decarboxylase (OpTDC) catalyzes tryptamine production from tryptophan [10]. The iridoid pathway includes nine consecutive enzymatic steps initiated by geranyl diphosphate synthase (GPPS) and ending with the reduction of loganin to secologanin catalyzed by secologanin synthase (OpSLS) [11–13]. Strictosidine synthase (OpSTR) then catalyzes the formation of strictosidine from tryptamine and secologanin. Finally, strictosidine is converted to camptothecin through various biochemical reactions such as oxidation, reduction, cyclization, and isomerization [14]. Interestingly, the biosynthesis of strictosidine in O. pumila is distinct from that in C. acuminata [9, 15], in which strictosidinic acid serves as the final precursor. Still, the transcriptional regulation of camptothecin biosynthesis remains largely unknown in the camptothecin-producing plant O. pumila.
Figure 1.
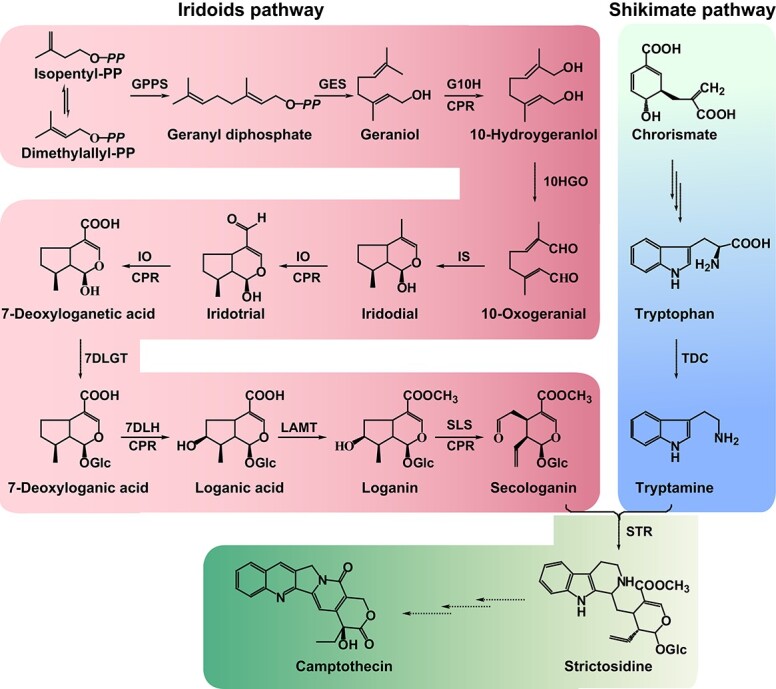
The putative camptothecin biosynthetic pathway in O. pumila. The three dashed arrows indicate that the camptothecin biosynthetic pathway in camptothecin-producing plants is still unknown.
As a large family of transcription factors (TFs), the WRKYs have been reported to participate in various plant developmental and physiological processes and responses [16, 17]. The name WRKY refers to a highly conserved domain with nearly 60 amino acids that contains the signature WRKYGQK sequence and a non-typical zinc-finger-like motif (C2H2 or C2HC) [18, 19]. Several variants of the WRKYGQK motif have been reported, including WRKYGEK and WRKYGKK. Also, atypical WRKY motifs have been suggested, containing the consensus sequence W(R/K)(K/R)Y [20]. WRKY proteins were initially classified into three groups based on the number of WRKY domains and the type of zinc finger [18]. WRKY group I proteins contain two WRKYGQK motifs and a C2H2 zinc finger, whereas group III proteins contain one WRKYGQK motif and a C2HC zinc finger [19, 21]. Group II proteins contain one WRKY domain and a C2H2-type zinc finger, and they can be further classified into five subgroups according to the presence of short conserved structural motifs [22]. Recently, WRKY proteins have been found to regulate the biosynthesis of plant secondary metabolites, including various alkaloids. Their mode of action lies in their ability to bind the W-box in the promoters of their target genes [23–25]. For example, HpWRKY44 was reported to regulate betalain biosynthesis in Hylocereus polyrhizus fruit by transcriptionally activating cytochrome P450-like (HpCytP4501) expression [26]. CjWRKY1 acts as a transcriptional activator and positively regulates the biosynthesis of benzylisoquinoline alkaloids [27]. Both CrWRKY1 in Catharanthus roseus and OpWRKY2 in O. pumila positively regulate monoterpenoid indole alkaloid biosynthesis by directly affecting the expression of the TDC biosynthetic gene [7, 16]. OpWRKY1 was shown to act as a negative regulator of camptothecin production in O. pumila hairy roots. Developmental changes in O. pumila hairy roots caused by overexpression of OpWRKY3 resulted in increased accumulation of camptothecin, also promoting the transcription of OpCPR [28, 29]. In general, the WRKY transcription factors play an essential role in regulating the biosynthesis of specialized metabolites.
With the mining of large-scale genome data, genome-wide identification and characterization of the WRKY transcription factors have been performed in a variety of plants, including Arabidopsis thaliana [21], Oryza sativa [30], Glycine max [31], Populus trichocarpa [32], Vitis vinifera [33], Pyrus bretschneideri [34], Moringa oleifera [35], and Santalum album [36]. However, there is little information on the identification of WRKY transcription factors and the functional characterization of camptothecin biosynthesis regulation in camptothecin-producing medicinal plants. The recent high-quality genome assembly of O. pumila provides an opportunity for genome-wide identification, characterization, and investigation of physiological functions of WRKY genes [1].
Here, forty-six OpWRKY genes were identified in the O. pumila genome, and their chromosomal locations, collinearity, classification, evolution, and expression patterns were analyzed. Based on our biochemical assays, OpWRKY6 was characterized as a negative regulator of OpGES, Op10HGO, Op7DLH, and OpTDC genes. OpWRKY6 binds to their promoters and simultaneously regulates the iridoid and shikimate pathways to reduce camptothecin production. Our identification and characterization of WRKY TFs in O. pumila and the functional characterization of OpWRKY6 as a regulator of camptothecin production provide novel insights and tools for re-engineering medicinal species enriched in valuable natural products.
Results
Identification and characterization of WRKY genes in the O. pumila genome
To identify candidate WRKY genes, an HMM search was performed against the O. pumila genome using the WRKY domain (PF03106), and 46 OpWRKY genes were obtained. OpWRKY1–3 were reported previously, and OpWRKY4–46 were named based on their positions on the O. pumila genome sequence (Table S1). Using the online CDD and SMART programs, the integrity of the WRKY domain was confirmed. Further characterization included analysis of subcellular localizations, amino acid lengths, molecular weights (MWs), and isoelectric points (pIs) (Table S1). Subcellular localization predictions indicated that all 46 proteins were found in the nucleus. Protein sequence lengths ranged from 195 (OpWRKY46) to 785 (OpWRKY41) amino acids, corresponding to MWs between 21.84 kDa (OpWRKY46) and 86.03 kDa (OpWRKY41), with an average of 44.08 kDa. The pIs varied from 4.77 (OpWRKY32) to 9.66 (OpWRKY33). All the data suggested a high variability among WRKY genes in the O. pumila genome. In addition, the number of WRKY family members in this study was much higher than that of WRKY members annotated in a previous transcriptome sequencing (Table S1 and S2). The present study provides a more comprehensive analysis of candidate OpWRKY genes.
As illustrated in Fig. 2a, 46 OpWRKY genes were disproportionately distributed across the 11 chromosomes of O. pumila. For example, OpWRKY genes were most abundant on Chr 9, with 9 genes, and least abundant on Chr 3, with only one gene (Fig. S1). Syntenic blocks within the O. pumila genome were examined to identify relationships among the OpWRKY genes and potential gene duplication events (Fig. 2a). Seven OpWRKY gene pairs were found in the O. pumila genome and were located on different chromosomes, indicating that segmental duplications in these regions probably contributed to expansion of the OpWRKY family.
Figure 2.

Chromosome-level analyses of the OpWRKY gene family in the O. pumila genome assembly. a. Chromosomal locations of OpWRKY genes and their synteny are illustrated by the circos diagram. Colored lines indicate similarity. b–e. Synteny analysis of WRKY genes between O. pumila and A. thaliana, O. sativa, V. vinifera and C. eugenioides, respectively.
Four comparative syntenic maps of O. pumila were constructed at the genome-wide level with four representative species, including one monocot (O. sativa) and three dicots (Arabidopsis, V. vinifera, and Coffea eugenioides) (Fig. 2b–e). A total of 40 OpWRKY genes showed syntenic relationships with those in C. eugenioides, followed by Arabidopsis (34), V. vinifera (18), and O. sativa (15) (Table S3). There were 53, 22, 19, and 63 orthologous pairs between the other four species (Arabidopsis, O. sativa, V. vinifera, and C. eugenioides). Collinear pairs were identified with the four other species (with 6 OpWRKY genes, OpWRKY1, 3, 22, 34, 35, and 44), suggesting that these orthologous pairs may have existed before the ancestral divergence.
Classification and phylogenetic relationships of OpWRKY genes
Multiple sequence alignments were performed to identify the structural features of the OpWRKY proteins. The majority of the 46 OpWRKY proteins (36/46, 78.3%) contained a single conserved WRKY domain, and the remaining 10 proteins (21.7%) contained two conserved WRKY domains (Fig. 3 and Table S4). In the WRKY domains, the WRKYGQK motif was relatively conserved across all OpWRKY family members, but five OpWRKYs differed at one residue, resulting in a WRKYGKK domain, similar to reports from other species such as tomato [37], Sesamum indicum [38], and Cucumis sativus [39]. All OpWRKY proteins had the C-X4–7-C-X22–23-H motif, forming C2HC or C2H2 zinc-finger structures.
Figure 3.

Protein sequence alignment of all OpWRKYs in O. pumila. The conserved WRKY domains are underlined, and the conserved zinc finger domains are highlighted in black boxes with asterisks.
The conserved WRKY domains from OpWRKYs and AtWRKYs were used to construct a phylogenetic tree and were classified into three groups (Fig. S2) [18]. Ten members belonged to Group I and contained two WRKY domains. Group II consisted of 31 OpWRKY proteins and was further divided into 5 subgroups, containing 2, 5, 14, 4, and 6 members in subgroups IIa–IIe, respectively. Five members of group III were found in the O. pumila genome, all of which contained a WRKYGQK domain and a C2HC-type zinc finger motif.
Gene structure and conserved motif analysis of OpWRKY genes
Ten conserved motifs in the WRKY proteins of O. pumila were identified and are shown in Table S5. Motifs 1 and 2 were widely distributed in all OpWRKY proteins, corresponding to the conserved WRKY domain. Motifs 3, 4, and 9 were only found in the type I group. Motifs 6, 7, and 8, were only detected in group II; motifs 6 and 8 were only found in IIa and IIb; and motif 7 was only detected in IIc (Fig. 4b). The majority of proteins that belong to the same class based on similar motif compositions may have similar functions.
Figure 4.
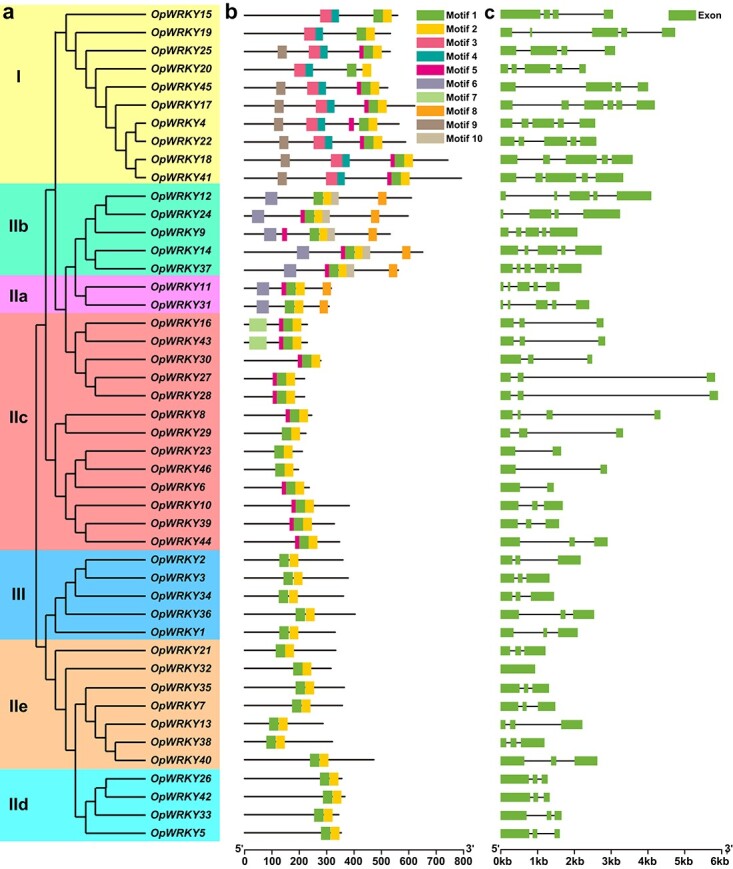
Gene structure and conserved motif analysis of OpWRKY genes. a. Phylogenetic analysis of OpWRKYs. b. Motif compositions of OpWRKY proteins. Ten motifs are shown in different colors, and their detailed information is provided in Table S5. c. Exon-intron structures of OpWRKY genes. Blue boxes and black lines represent exons and introns, respectively.
Exon-intron organization analysis of OpWRKY genes was also performed. The number of exons in the OpWRKYs ranged from one to six. Among the 46 OpWRKY genes, the majority (56.5%) were spliced with three exons and two introns (Fig. 4c). OpWRKY genes with similar structures clustered together, such as subgroup IId and group III with three exons and two introns or subgroup IIa with five exons and four introns. Group I and subgroup IIb contained 4–6 exons, subgroup IIc contained 2–4 exons, and subgroup IIe contained 3 exons, except for OpWRKY32 (Fig. 4c). These results demonstrated that OpWRKYs exhibited diverse intron/exon structures.
In addition, cis-acting elements were analyzed in a 3000-bp regulatory region upstream of the ATG (promoter). Hormone-responsive (556), light-responsive (499), and stress-responsive (289) boxes were found in the promoter regions of OpWRKY genes (Table S6). Hormone-related elements for methyl jasmonate (MeJA) (186), abscisic acid (ABA) (155), ethylene (81), gibberellin (GA) (57), salicylic acid (SA) (50), and auxin (29) were also detected in the promoters of the OpWRKY genes. A number of stress-related promoter regions were identified in OpWRKY genes, including anaerobic induction (122), wound induction (57), drought (55), low-temperature (30), and defense (25) boxes. Meristem-related (49), circadian-related (19), and endosperm expression (13) cis-acting elements were also identified in the OpWRKY promoters. These results suggest that various cis-acting promoter elements may regulate OpWRKY genes during growth and stress responses.
Co-expression network analysis of OpWRKY genes
Using the expression data, we performed hierarchical clustering and generated heat maps to visualize the expression of OpWRKYs and camptothecin biosynthesis genes in different tissues (Fig. S3). As illustrated in Fig. S3a, some OpWRKY genes had higher transcript accumulation in one or more tissues. For example, OpWRKY3, OpWRKY6, OpWRKY9, OpWRKY10, OpWRKY14, OpWRKY20, OpWRKY23, OpWRKY24, OpWRKY38, OpWRKY40, and OpWRKY45 showed the highest expression levels in the root, consistent with most camptothecin biosynthesis genes (Fig. S3b). In addition, the expression levels of ten OpWRKY genes (OpWRKY2, 7, 12, 13, 16, 26, 32, 39, 42, and 43) were higher in leaves than in stems and roots. These results indicated that OpWRKY genes have diverse tissue-specific expression patterns, suggesting a non-specific tissue-dependent role.
A co-expression network was constructed between OpWRKYs and nine important camptothecin biosynthesis genes. As illustrated in Fig. 5, twenty OpWRKYs were strongly correlated with camptothecin biosynthesis genes. OpWRKY6 and OpWRKY40 were strongly positively correlated with 5 and 6 camptothecin biosynthesis genes, respectively. In addition, OpWRKY15 was strongly negatively correlated with five camptothecin biosynthesis genes. Furthermore, the transcript level of OpWRKY6 was much higher in roots than in leaves compared with other OpWRKY genes. Overall, this result showed that OpWRKY6 may be involved in regulating camptothecin and its precursor biosynthesis.
Figure 5.
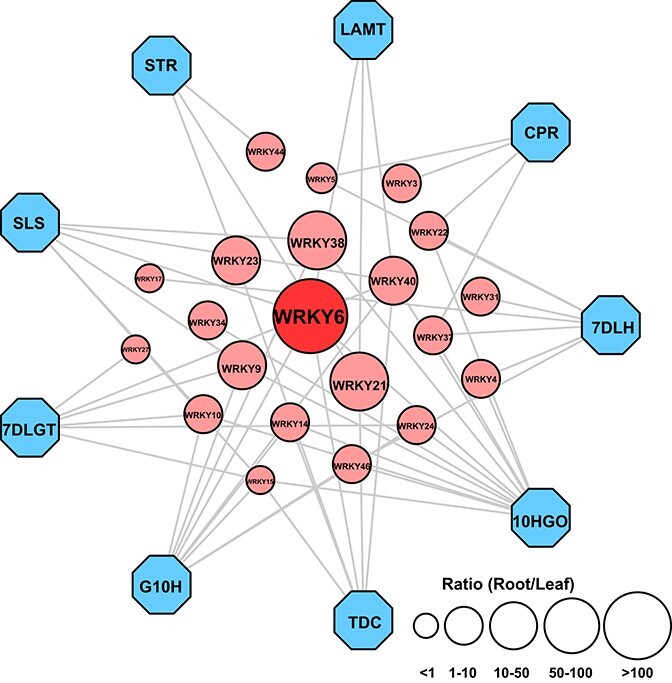
Correlation analysis of OpWRKY genes with camptothecin biosynthetic pathway genes in O. pumila. Blue octagons and red circles represent camptothecin biosynthetic pathway genes and OpWRKY genes, respectively. Larger circles indicate higher expression levels of OpWRKY genes in roots compared with leaves. Edges are drawn when the linear correlation coefficient is >0.8 with p-value <0.05.
Next, OpWRKY6 with a 705-bp open reading frame (ORF) encoding 234 amino acids was isolated and functionally characterized. Protein sequence analyses showed that OpWRKY6 shared 68.84%, 58.10%, and 53.27% identity with PtWRKY43 (AZQ19338.1), PaWRKY19 (TKS06231.1), and TcWRKY56 (EOX95854.1), respectively. A phylogenetic tree was constructed, showing that OpWRKY6 clustered with AtWRKY43 in group IIc (Fig. S4) and contained a single conserved WRKY domain (Fig. S5). A subcellular localization assay of OpWRKY6-YFP fusion protein indicated that it was exclusively localized to nuclei in Nicotiana benthamiana leaf cells. By contrast, the control transformed with pHB-YFP showed fluorescence distributed throughout the cell (Fig. S6). These results demonstrated that OpWRKY6 regulates transcription as a TF in the nucleus.
OpWRKY6 negatively regulates camptothecin production in O. pumila
OpWRKY6 overexpression (OE) transgenic hairy root lines were generated to examine the involvement of OpWRKY6 in camptothecin biosynthesis regulation. Thirteen independent positive transgenic lines were verified by genomic PCR, and the positive rate for the OpWRKY6-OE lines was 52.4% (Fig. 6a). The OpWRKY6 transcript levels were 2.9- to 169.6-fold higher in the OpWRKY6-OE lines than in the empty vector (EV) control lines, and three OpWRKY6-OE lines (OE-2, OE-9, and OE-32) with different higher expression levels were selected for further analysis (Fig. 6b). Compared with the control lines, OpWRKY6-OE transgenic hairy roots showed no significant differences in phenotype or biomass (Fig. S7). HPLC analysis indicated that the concentration of camptothecin in OpWRKY6-OE transgenic hairy roots was clearly decreased (Fig. 6c). Moreover, the camptothecin production of transgenic hairy roots and medium was significantly lower in the overexpression lines (Fig. 6d).
Figure 6.

Overexpression of OpWRKY6 in O. pumila hairy roots. a. Identification of positive OpWRKY6-OE hairy root lines. b. The expression levels of OpWRKY6 in the OpWRKY6-OE lines. OpUBQ was used as the internal reference gene. c, d. The content (c) and production (d) of camptothecin in the OpWRKY6-OE lines. e. The transcript levels of camptothecin biosynthesis genes in the OpWRKY6-OE lines. All data represent the means ± SD of three biological replicates. Student’s t-test: *p < 0.05; **p < 0.01.
The expression levels of 12 camptothecin biosynthesis genes were analyzed in the transgenic lines. The camptothecin biosynthesis-related genes Op10HGO, OpGES, OpG10H, and Op7DLH showed a significant decrease in expression (qPCR analysis) in the OpWRKY6-OE transgenic lines (Fig. 6e). In addition, the transcript levels of OpIO, OpSLS, and OpTDC were lower in OpWRKY6-OE9 and OE32 transgenic lines. Notably, the transcript levels of 12 camptothecin biosynthesis-related genes were significantly lower in the OpWRKY6-OE32 transgenic line compared with the other two lines. These qPCR results demonstrated that OpWRKY6 may control the biosynthesis of camptothecin, probably mainly by affecting the expression of OpGES, Op10HGO, Op7DLH, and OpTDC.
To further elucidate the function of the OpWRKY6 TF in camptothecin biosynthesis, we attempted to knock out OpWRKY6 of O. pumila using the CRISPR/Cas9 system. Sixty-nine independent lines were generated, with 18 positive lines (20.3% positive rate) (Fig. S8). Three independent lines were selected (KO-19, KO-32, and KO-37). The sequencing chromatograms suggested that the transgenic KO-32 line was homozygous, with mutations in both alleles at the same DNA locus (Fig. 7a). The KO-19 line was found to have biallelic mutations (two distinct variations), and the KO-37 line had a heterozygous mutation (wild-type/single mutation). The expression of OpWRKY6 was significantly lower in the transgenic KO-19 line, slightly lower in the KO-32 line, and not significantly different from the control in the KO-37 line (Fig. 7b). The phenotypes and biomass of OpWRKY6-KO transgenic hairy roots were not significantly different from those of the wild-type control (Fig. S9). HPLC analysis indicated that the content and production of camptothecin were significantly higher in the OpWRKY6-KO lines compared with the control (Fig. 7c, d). The increased accumulation of camptothecin in the knockout lines was consistent with the upregulation of OpGES, Op10HGO, Op7DLH, OpLAMT, OpSLS, and OpTDC (Fig. 7e). These results demonstrated that OpWRKY6 was a TF that negatively regulated camptothecin biosynthesis.
Figure 7.

Knockout of OpWRKY6 in O. pumila hairy roots. a. Genomic OpWRKY6 DNA sequences from different OpWRKY6-KO lines were detected by DNA sequencing. b. The expression levels of OpWRKY6 in the OpWRKY6-KO lines. c, d. The content (c) and production (d) of camptothecin in the OpWRKY6-KO lines. e. The expression levels of camptothecin biosynthesis genes in the OpWRKY6-KO lines. All data represent the means ± SD of three biological replicates. Student’s t-test: *p < 0.05; **p < 0.01.
OpWRKY6 acts as a negative regulator of camptothecin biosynthesis genes
Dual-luciferase assays were performed to verify that OpWRKY6 negatively regulates camptothecin biosynthesis genes. proOpGES, proOpG10H, proOp10HGO, proOp7DLH, proOpLAMT, proOpSLS, proOpCPR, proOpTDC, and proOpSTR promoter regions were used to drive the luciferase (LUC) gene as fusion reporters. Overexpression of OpWRKY6 under the control of the 35S promoter was used as an effector (Fig. 8a). OpWRKY6 repressed the promoters of OpGES, Op10HGO, Op7DLH, and OpTDC compared with controls as measured by changes in LUC luminescence (Fig. 8b). These results showed that OpWRKY6 transcriptionally downregulates these four camptothecin biosynthesis genes.
Figure 8.

Dual-luciferase (Dual-LUC) assays showed the suppression effect of OpWRKY6 on the promoters of camptothecin biosynthesis genes. a. Schematic diagrams of the effector and reporter plasmids used in Dual-LUC assays. REN, Renilla luciferase internal reference gene. LUC, firefly luciferase reporter gene. b. Dual-LUC assay in N. benthamiana leaf cells using the constructs shown in (a). The empty vector pHB-YFP was used as the control. The relative LUC activity was normalized to the reference Renilla (REN) luciferase. Error bars indicate the SD (n = 3). Student’s t-test: *p < 0.05.
The promoter sequences of camptothecin biosynthesis-related genes were analyzed to better understand the OpWRKY6 mode of action. The OpGES promoter contained two W-box cis-elements, W1 and W2, that were 1563 and 441 bp upstream of the translation start site (ATG), respectively (Fig. 9a). The Op10HGO promoter contained five W-boxes, W1–W5, which were 741, 673, 614, 605, and 60 bp upstream of the ATG, respectively (Fig. 9b). Three W-boxes (W1, W2, and W3) were found in the promoter region of Op7DLH (Fig. 9c), whereas a single W-box was identified in the OpTDC promoter (Fig. 9d). In all cases, OpWRKY6 was able to bind the W-box in the promoter region, but not the mutant W-box, and the empty vector served as a negative control. Consequently, our results revealed that OpWRKY6 negatively regulates camptothecin biosynthesis by directly binding to W-boxes in the promoter regions of OpGES, Op10HGO, Op7DLH, and OpTDC, thereby repressing their expression (Fig. 10).
Figure 9.

OpWRKY6 protein could directly bind to the W-box in the promoters of four camptothecin biosynthesis genes, as shown by yeast one-hybrid (Y1H) assays. a–d. Schematic diagrams of the OpGES, Op10HGO, Op7DLH, and OpTDC promoters. The positions of potential W-boxes are shown as red triangles. Y1H assays were repeated three times.
Figure 10.
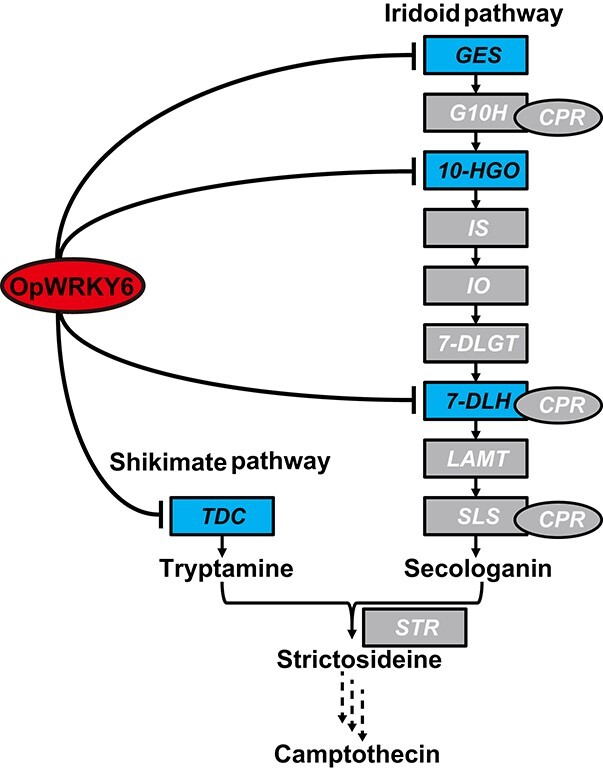
Model of the regulation of camptothecin biosynthesis by OpWRKY6 in O. pumila. The OpWRKY6 transcription factor negatively regulates camptothecin biosynthesis by directly binding and inhibiting biosynthesis genes in the shikimate pathway (OpTDC) and iridoid pathway (OpGES, Op10HGO, and Op7DLH).
Discussion
WRKY proteins are a class of transcriptional regulators found in higher plants [25]. To date, systematic and comprehensive genome-wide analyses of the WRKY family have been performed in a variety of plants [17]. However, systematic identification and functional characterization of WRKY proteins have rarely been performed in camptothecin-producing plants. The genome of O. pumila has been published, providing useful tools for genome-wide analysis of the OpWRKY gene family [1]. In the present study, we identified 46 OpWRKY genes based on the O. pumila genome sequence, similar to the 47 WRKY genes in Ricinus communis [40], 49 in Coffea canephora [41], and 45 in Hordeum vulgare [42]. By contrast, more WRKY members were reported in Glycine max, Malus domestica, O. sativa, Arabidopsis, and V. vinifera (Table S7). Out of the 46 OpWRKY genes, only 14 were found in the genome data and not in the transcriptome data of O. pumila roots and hairy roots (Table S1 and S2). This difference can be explained by low gene expression or by the presence of pseudogenes. Conversely, two OpWRKY genes (OpWRKY12 and 21) were found only in the transcriptome data [7], possibly caused by alternative splicing or incomplete genome assembly. In addition, only 34 WRKY genes were identified in a previous O. pumila genome using the blast_rbh.py script. Our comprehensive analysis of the OpWRKY gene family therefore provides more meaningful data for exploring the characteristics and functions of WRKY family members.
WRKYs can be classified into three groups based on the motif features of the zinc finger and the number of WRKY domains. Our data show that the 46 OpWRKYs are divided into three distinct groups, among which 31 OpWRKYs were assigned to group II, accounting for the largest proportion (67.4%). These results were similar to reports from A. thaliana [18], cucumber [39], and C. canephora [41]. In cucumber, 41 of 61 CsWRKY proteins were assigned to group II, and in C. canephora, 34 (69.39%) CcWRKY proteins belonged to group II. Moreover, variations in the conserved WRKYGKK motif were observed in OpWRKY16, OpWRKY27, OpWRKY28, OpWRKY30, and OpWRKY43 proteins from the WRKY IIc subgroup. Similar cases have been found in other plant species such as C. sativus (CsWRKY10 and CsWRKY47), Ananas comosus (AcWRKY14, AcWRKY23, AcWRKY27, and AcWRKY43), and M. oleifera (MoWRKY24) [35, 39, 43]. Because changes in the WRKYGQK pattern may influence normal interactions of OpWRKYs with target genes, the binding specificities and functions of these five OpWRKY proteins may warrant further study. In addition, OpWRKY genes with similar gene structures and motif compositions always clustered in the same class, such as groups IId and III that contained three exons and two introns (Fig. 4). These points supported the phylogenetic results based on motif compositions, conserved protein architecture, and similarity in gene structures.
WRKY TFs have been found to play important roles in plant development and defense response. In A. thaliana, AtWRKY41 is an important regulator of abscisic acid insensitive 3 (ABI3) expression, conferring reduced primary seed dormancy [44]. Three WRKY transcription factors (AtWRKY46, AtWRKY54, and AtWRKY70) have been reported to participate in BR-regulated plant growth by cooperating with BES1 to regulate BR signaling [45]. In this study, the tissue expression profiles of OpWRKYs showed that most OpWRKY genes had diverse tissue-specific expression patterns; for example, OpWRKY6 was highly expressed in roots. In addition, hormone-responsive (556), light-responsive (499), and stress-responsive (289) boxes were found in the promoter regions of OpWRKY genes (Table S6). These findings revealed that the OpWRKY genes may be regulated by various cis-acting elements in their promoters during growth and stress responses.
Emerging experimental data demonstrate that WRKY transcription factors can regulate a variety of plant secondary metabolites, including terpenoids, phenylpropanoids, and alkaloids [25]. In Artemisia annua, AaWRKY9 was reported to positively regulate the expression of AaDBR2 and AaGSW1 and increase artemisinin accumulation [46]. SlWRKY73 activates the expression of three monoterpene synthase genes in Solanum lycopersicum [47]. In H. polyrhizus, HpWRKY44 transcriptionally activates HpCYP450-like1, which regulates betalain biosynthesis [26]. In C. roseus, CrWRKY1 was shown to regulate the monoterpenoid indole alkaloid pathway by activating the central pathway gene CrTDC [16]. In O. pumila, we reported the involvement of three Group III WRKY TFs in the regulation of camptothecin biosynthesis. Analysis of OpWRKY3 transgenic hairy roots indicated that OpWRKY3 regulated camptothecin production by affecting the development of hairy roots [28]. OpWRKY1 negatively regulates camptothecin biosynthesis by binding to the OpCPR promoter, whereas OpWRKY2 acts as a positive regulator of camptothecin biosynthesis by activating the expression of OpTDC. However, other WRKY subfamilies with important roles in camptothecin biosynthesis have not been reported.
We constructed a co-expression network between all OpWRKYs and nine key camptothecin biosynthesis genes. These results indicated that OpWRKY6 showed strong transcriptional overlap with five camptothecin biosynthesis genes (Fig. 5). Multiple sequence alignment and phylogenetic tree analysis revealed that OpWRKY6 clustered with AtWRKY43 from group IIc. OpWRKY6 expression was tightly negatively correlated with transcript levels of camptothecin biosynthesis genes and camptothecin production in OpWRKY6-OE and OpWRKY6-KO transgenic hairy root lines (Fig. 6 , 7). Moreover, based on in vivo and in vitro biochemical experiments, OpWRKY6 can selectively bind to W-box promoter elements in four camptothecin biosynthesis genes (OpGES, Op10HGO, Op7DLH, and OpTDC). These enzymes may control the flow of iridoid and tryptamine metabolism in camptothecin biosynthesis. Taken together, our results show that the OpWRKY6 TF acts as a negative regulator of camptothecin biosynthesis in O. pumila by directly regulating biosynthesis genes in the iridoid and shikimate pathways (Fig. 10). Therefore, our results reveal the molecular mechanism by which OpWRKY6 regulates camptothecin biosynthesis and provide a strategy for increasing camptothecin content.
Conclusion
In this work, we first provided a systemic analysis of OpWRKY genes and then identified 46 OpWRKYs based on the O. pumila genome sequence. We identified OpWRKY6 using co-expression network analysis between all OpWRKYs and camptothecin biosynthesis genes. OpWRKY6 serves as a negative regulator of camptothecin biosynthesis by directly binding to and inhibiting the promoters of OpGES, Op10HGO, Op7DLH, and OpTDC. This genome-wide diversity analysis of WRKY genes in O. pumila provides new insights for future functional characterization of the regulation of plant specialized metabolites such as monoterpenoid indole alkaloids.
Materials and methods
Identification of candidate OpWRKY genes in the O. pumila genome
The O. pumila genome was downloaded from the O. pumila Genome DataBase, and putative OpWRKY TF genes in O. pumila were identified using HMMER3.2 with the WRKY domain (Pfam, PF03106). Subsequently, two online programs, CDD and SMART, were used to manually confirm whether all candidate OpWRKY proteins contained conserved WRKY domains. The amino acid lengths, MWs, and pIs of all OpWRKY proteins were determined using the ExPASy website, and their subcellular localizations were predicted with PSORT software [48].
Chromosomal locations of OpWRKYs and gene duplications in the O. pumila genome
The chromosomal distributions of OpWRKY genes were displayed using TBtools software. MCScanX and BLASTP were used to analyze gene duplication events of OpWRKY genes in the O. pumila genome [49]. The synonymous relationships between OpWRKY genes and A. thaliana, O. sativa, C. eugenioides, and V. vinifera WRKY genes were displayed with the TBtools program.
Protein sequence alignment and phylogenetic analyses
Protein sequence alignments of all conserved WRKY domains were performed with Clustal software. WRKY domain regions of O. pumila and A. thaliana were aligned after retrieval from the TAIR database. A neighbor-joining phylogenetic tree was constructed using MEGA 7.0 with 1000 bootstrap replicates [50].
Gene structure and motif composition analysis
The coding sequences (CDSs) of OpWRKYs and their corresponding genomic sequences were used to predict exon-intron structures. Exon-intron structures of OpWRKY genes were visualized using GSDS v2.0 software [51]. The conserved motifs of OpWRKY proteins were investigated using the MEME v5.1.1 online program [52]. A schematic diagram of gene structures and conserved motifs was displayed and re-edited with TBtools software.
Promoter cis-acting element analysis
The 3000-bp sequences upstream of the translation start codons of the OpWRKY genes were submitted to the PlantCARE database [53] to analyze the promoter regions. The binding elements for OpWRKY genes were searched by analyzing the 3000-bp promoter sequences of 12 genes encoding key enzymes of the camptothecin biosynthetic pathway using the PlantPAN 3.0 database [54].
Co-expression network of OpWRKYs and camptothecin biosynthesis genes
To obtain candidate OpWRKYs for the regulation of camptothecin biosynthesis, the tissue expression patterns of all OpWRKYs and camptothecin biosynthesis genes were used to construct a co-expression network using the PCC (Partial Correlation Coefficients) method in the R platform between each set of variables to calculate Pearson correlation coefficients [55]. The correlation network was displayed using Cytoscape software (version 3.7.2) [56].
Subcellular localization assay
First, Agrobacterium tumefaciens GV3101 strains harboring the pHB-YFP or pHB-OpWRKY6-YFP plasmids were separately infiltrated into leaves of N. benthamiana. Then, the YFP signals were observed using a confocal laser microscope (Carl Zeiss, Germany) after two days of infiltration. Nuclei were stained with 20 μg/mL 4, 6-diamidino 2-phenylindole (DAPI, Sigma) before observation.
Generation of OpWRKY6 transgenic O. pumila hairy roots
OpWRKY6 overexpression lines of O. pumila hairy roots were generated using the recombinant pHB-OpWRKY6-YFP vector. An OpWRKY6 knockout vector was constructed using the CRISPR/Cas9 system as reported previously [13]. The empty pHB-YFP and pCAMBIA1300 vectors were used as controls for overexpression and knockout of OpWRKY6, respectively. All plasmids were isolated and transferred into A. tumefaciens strain C58C1, which was then used to transform O. pumila stems to generate hairy roots as previously described (Fig. S10) [15, 28]. Positive transgenic lines (OpWRKY6-OE and OpWRKY6-KO) were verified by PCR using the relevant primers (Table S8). PCR products of OpWRKY6-KO lines were cloned into the pMD19T vector (Takara), and the plasmids of individual colonies were sequenced by the Sunya company (Hangzhou, China). Positive transgenic hairy roots of OpWRKY6-OE and OpWRKY6-KO were cultured in B5 liquid medium under dark conditions at 25°C and 120 rpm for 40 days.
RNA extraction and qRT-PCR analysis
Samples of roots, stems, and leaves were collected from 2-month-old sterile O. pumila plants to determine the tissue expression patterns of all OpWRKY genes. Transgenic hairy roots were collected to analyze the expression of OpWRKY6, OpTDC, OpGES, OpG10H, Op10HGO, Op7DLH, OpCPR, Op7DLGT, OpIO, OpIS, OpLAMT, OpSLS and OpSTR in OpWRKY6-OE and OpWRKY6-KO lines. Total RNA was isolated from the three different tissues and OpWRKY6-OE and OpWRKY6-KO transgenic lines, and qRT-PCR was performed with three biological replicates as described previously [7]. All gene-specific primers used in this study are listed in Table S8. The relative gene expression levels in different samples were calculated relative to that of OpUBQ (Ubiquitin) using the 2−ΔΔCt method.
Determination of camptothecin in O. pumila hairy roots
Samples of different hairy root lines (OpWRKY6-OE, OpWRKY6-KO, and empty vector controls) were freeze-dried and ground into powder. Extraction of camptothecin from transgenic hairy root lines and their corresponding media were performed as described previously [7]. High-performance liquid chromatography (HPLC) was used to determine the concentration of camptothecin.
Dual-luciferase assays
The recombinant pHB-OpWRKY6-YFP vector was used as an effector to determine the regulatory role of the OpWRKY6 TF in the camptothecin biosynthetic pathway. For the reporter constructs, the promoters of the OpTDC, OpGES, OpG10H, OpCPR, Op10HGO, Op7DLH, Op7DLGT, OpIO, OpIS, OpLAMT, OpSLS, and OpSTR biosynthesis genes were inserted into the pGREEN0800-LUC vector and separately transformed into A. tumefaciens GV3101 strains. The empty pHB-YFP vector was used as the control. Infiltration into leaves of N. benthamiana and detection of luciferase activities were carried out as described previously [7]. Three biological replicates were performed for each combination.
Yeast one-hybrid assays
To explore the regulatory mechanism of OpWRKY6 in camptothecin biosynthesis, yeast one-hybrid assays (Y1H) were carried out as described previously [57]. First, the ORF sequence of OpWRKY6 was fused to the pB42AD effector vector. Three tandem copies of the W-box or a mutant-W-box from the promoter regions of camptothecin biosynthesis genes were separately inserted into the pLacZ reporter vector. Then, EGY48 yeast strains were prepared with different combinations of effector and reporter constructs and cultured on selective medium (SD/−Ura/−Trp/+X-gal) for 2 days.
Supplementary Material
Contributor Information
Can Wang, Laboratory for Core Technology of TCM Quality Improvement and Transformation, The Third Affiliated Hospital, School of Pharmaceutical Sciences, Zhejiang Chinese Medical University, Hangzhou, Zhejiang, 310053, China; Key Laboratory of Exploration and Utilization of Aquatic Genetic Resources Conferred by Ministry of Education, Shanghai Ocean University, Shanghai, 201306, China.
Xiaolong Hao, Laboratory for Core Technology of TCM Quality Improvement and Transformation, The Third Affiliated Hospital, School of Pharmaceutical Sciences, Zhejiang Chinese Medical University, Hangzhou, Zhejiang, 310053, China.
Yao Wang, Laboratory for Core Technology of TCM Quality Improvement and Transformation, The Third Affiliated Hospital, School of Pharmaceutical Sciences, Zhejiang Chinese Medical University, Hangzhou, Zhejiang, 310053, China.
Itay Maoz, Department of Postharvest Science, ARO, The Volcani Center, HaMaccabim Rd 68, POB 15159, Rishon LeZion, 7528809, Israel.
Wei Zhou, Laboratory for Core Technology of TCM Quality Improvement and Transformation, The Third Affiliated Hospital, School of Pharmaceutical Sciences, Zhejiang Chinese Medical University, Hangzhou, Zhejiang, 310053, China.
Zhigang Zhou, Key Laboratory of Exploration and Utilization of Aquatic Genetic Resources Conferred by Ministry of Education, Shanghai Ocean University, Shanghai, 201306, China.
Guoyin Kai, Laboratory for Core Technology of TCM Quality Improvement and Transformation, The Third Affiliated Hospital, School of Pharmaceutical Sciences, Zhejiang Chinese Medical University, Hangzhou, Zhejiang, 310053, China.
Acknowledgments
This work was supported by the Major Science and Technology Projects of Breeding New Varieties of Agriculture in Zhejiang Province (2021C02074), the National Natural Science Foundation of China (82003889, 31571735, 82073963, 81522049), the Zhejiang Provincial Natural Science Foundation of China (LQ21H280004), the Zhejiang Provincial Ten Thousands Program for Leading Talents of Science and Technology Innovation (2018R52050), the Zhejiang Provincial Program for the Cultivation of High-level Innovative Health Talents, Research Project of Zhejiang Chinese Medical University Research Foundation (2021JKZDZC06, 2020ZR15, 2020ZR13, 2020YKJ06), and the Opening Project of Zhejiang Provincial Preponderant and Characteristic Subject of Key University (Traditional Chinese Pharmacology) of Zhejiang Chinese Medical University (ZYAOX2018009, ZYAOXYB2019009). We appreciate the experimental support from the Public Platform of Medical Research Center, Academy of Chinese Medical Science, Zhejiang Chinese Medical University.
Author Contributions
GK and ZZ conceived and designed the project. CW performed the experiments and analyzed the data. CW and HX wrote the original draft of this paper. YW, GK, WZ, IM, and ZZ revised the paper. All authors have read and approved the final version.
Data Availability
The data generated or analyzed in the present research are included in this published article and its supplemental data files and available from the corresponding author upon reasonable request.
Conflict of interest
The authors declare no conflicts of interest.
Supplementary data
Supplementary data is available at Horticulture Research online.
References
- 1. Rai A, Hirakawa H, Nakabayashi R et al. Chromosome-level genome assembly of Ophiorrhiza pumila reveals the evolution of camptothecin biosynthesis. Nat Commun. 2021;12:405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Martino E, Volpe SD, Terribile E et al. The long story of camptothecin: from traditional medicine to drugs. Bioorg Med Chem Lett. 2017;27:701–7. [DOI] [PubMed] [Google Scholar]
- 3. Sirikantaramas S, Yamazaki M, Saito K. Mutations in topoisomerase I as a self-resistance mechanism coevolved with the production of the anticancer alkaloid camptothecin in plants. Proc Natl Acad Sci U S A. 2008;105:6782–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sirikantaramas S, Meeprasert A, Rungrotmongkol T et al. Structural insight of DNA topoisomerases I from camptothecin-producing plants revealed by molecular dynamics simulations. Phytochemistry. 2015;113:50–6. [DOI] [PubMed] [Google Scholar]
- 5. Ruan Q, Patel G, Wang J et al. Current advances of endophytes as a platform for production of anti-cancer drug camptothecin. Food Chem Toxicol. 2021;151:112113. [DOI] [PubMed] [Google Scholar]
- 6. Yang MQ, Wang Q, Liu Y et al. Divergent camptothecin biosynthetic pathway in Ophiorrhiza pumila. BMC Biol. 2021;19:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hao X, Xie C, Ruan Q et al. The transcription factor OpWRKY2 positively regulates the biosynthesis of the anticancer drug camptothecin in Ophiorrhiza pumila. Hortic Res. 2021;8:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kai GY, Wu C, Gen L et al. Biosynthesis and biotechnological production of anti-cancer drug camptothecin. Phytochem Rev. 2015;14:525–39. [Google Scholar]
- 9. Sadre R, Magallanes-Lundback M, Pradhan S et al. Metabolite diversity in alkaloid biosynthesis: a multilane (Diastereomer) highway for camptothecin synthesis in Camptotheca acuminata. Plant Cell. 2016;28:1926–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Asano T, Kobayashi K, Kashihara E et al. Suppression of camptothecin biosynthetic genes results in metabolic modification of secondary products in hairy roots of Ophiorrhiza pumila. Phytochemistry. 2013;91:128–39. [DOI] [PubMed] [Google Scholar]
- 11. Yamazaki Y, Kitajima M, Arita M et al. Biosynthesis of camptothecin. In silico and in vivo tracer study from [1-13C] glucose. Plant Physiol. 2004;134:161–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Miettinen K, Dong L, Navrot N et al. The seco-iridoid pathway from Catharanthus roseus. Nat Commun. 2014;5:3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shi M, Gong H, Cui L et al. Targeted metabolic engineering of committed steps improves anti-cancer drug camptothecin production in Ophiorrhiza pumila hairy roots. Ind Crop Prod. 2020;148:112277. [Google Scholar]
- 14. Yamazaki M, Mochida K, Asano T et al. Coupling deep transcriptome analysis with untargeted metabolic profiling in Ophiorrhiza pumila to further the understanding of the biosynthesis of the anti-cancer alkaloid camptothecin and anthraquinones. Plant Cell Physiol. 2013;54:686–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cui L, Ni X, Teng X et al. Co-overexpression of geraniol-10-hydroxylase and strictosidine synthase improves anti-cancer drug camptothecin accumulation in Ophiorrhiza pumila. Sci Rep. 2015;5:8227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Suttipanta N, Pattanaik S, Kulshrestha M et al. The transcription factor CrWRKY1 positively regulates the terpenoid indole alkaloid biosynthesis in Catharanthus roseus. Plant Physiol. 2011;157:2081–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen F, Hu Y, Vannozzi A et al. The WRKY transcription factor family in model plants and crops. Crit Rev Plant Sci. 2017;36:311–35. [Google Scholar]
- 18. Eulgem T, Rushton PJ, Robatzek S et al. The WRKY superfamily of plant transcription factors. Trends Plant Sci. 2000;5:199–206. [DOI] [PubMed] [Google Scholar]
- 19. Rushton PJ, Somssich IE, Ringler P et al. WRKY transcription factors. Trends Plant Sci. 2010;15:247–58. [DOI] [PubMed] [Google Scholar]
- 20. Xie Z, Zhang ZL, Zou X et al. Annotations and functional analyses of the Rice WRKY gene superfamily reveal positive and negative regulators of abscisic acid signaling in aleurone cells. Plant Physiol. 2005;137:176–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wu KL, Guo ZJ, Wang HH et al. The WRKY family of transcription factors in Rice and Arabidopsis and their origins. DNA Res. 2005;12:9–26. [DOI] [PubMed] [Google Scholar]
- 22. Eulgem T, Somssich IE. Networks of WRKY transcription factors in defense signaling. Curr Opin Plant Biol. 2007;10:366–71. [DOI] [PubMed] [Google Scholar]
- 23. Yamasaki K, Kigawa T, Inoue M et al. Solution structure of an Arabidopsis WRKY DNA binding domain. Plant Cell. 2005;17:944–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chi YJ, Yang Y, Zhou Y et al. Protein–protein interactions in the regulation of WRKY transcription factors. Mol Plant. 2013;6:287–300. [DOI] [PubMed] [Google Scholar]
- 25. Schluttenhofer C, Yuan L. Regulation of specialized metabolism by WRKY transcription factors. Plant Physiol. 2015;167:295–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cheng MN, Huang ZJ, Hua QZ et al. The WRKY transcription factor HpWRKY44 regulates CytP450-like1 expression in red pitaya fruit (Hylocereus polyrhizus). Hortic Res. 2017;4:17039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kato N, Dubouzet E, Kokabu Y et al. Identification of a WRKY protein as a transcriptional regulator of benzylisoquinoline alkaloid biosynthesis in Coptis japonica. Plant Cell Physiol. 2007;48:8–18. [DOI] [PubMed] [Google Scholar]
- 28. Wang C, Wu C, Wang Y et al. Transcription factor OpWRKY3 is involved in the development and biosynthesis of camptothecin and its precursors in Ophiorrhiza pumila hairy roots. Int J Mol Sci. 2019;20:3996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xu M, Wu C, Zhao L et al. WRKY transcription factor OpWRKY1 acts as a negative regulator of camptothecin biosynthesis in Ophiorrhiza pumila hairy roots. Plant Cell Tissue Organ Cult. 2020;142:69–78. [Google Scholar]
- 30. Ross CA, Liu Y, Shen QJ. The WRKY gene family in rice (Oryza sativa). J Integr Plant Biol. 2007;49:827–42. [Google Scholar]
- 31. Schmutz J, Cannon SB, Schlueter J et al. Genome sequence of the palaeopolyploid soybean. Nature. 2010;463:178–83. [DOI] [PubMed] [Google Scholar]
- 32. He HS, Dong Q, Shao Y et al. Genome-wide survey and characterization of the WRKY gene family in Populus trichocarpa. Plant Cell Rep. 2012;31:1199–217. [DOI] [PubMed] [Google Scholar]
- 33. Wang M, Vannozzi A, Wang G et al. Genome and transcriptome analysis of the grapevine (Vitis vinifera L.) WRKY gene family. Hortic Res. 2014;1:14016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Huang X, Li K, Xu X et al. Genome-wide analysis of WRKY transcription factors in white pear (Pyrus bretschneideri) reveals evolution and patterns under drought stress. BMC Genomics. 2015;16:1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang J, Yang E, He Q et al. Genome-wide analysis of the WRKY gene family in drumstick (Moringa oleifera lam.). Peer J. 2019;7:e7063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yan H, Li M, Xiong Y et al. Genome-wide characterization, expression profile analysis of WRKY family genes in Santalum album and functional identification of their role in abiotic stress. Int J Mol Sci. 2019;20:5676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Huang SX, Gao Y, Liu J et al. Genome-wide analysis of WRKY transcription factors in Solanum lycopersicum. Mol Gen Genomics. 2012;287:495–513. [DOI] [PubMed] [Google Scholar]
- 38. Li D, Liu P, Yu J et al. Genome-wide analysis of WRKY gene family in the sesame genome and identification of the WRKY genes involved in responses to abiotic stresses. BMC Plant Biol. 2017;17:152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chen CH, Chen X, Han J et al. Genome-wide analysis of the WRKY gene family in the cucumber genome and transcriptome-wide identification of WRKY transcription factors that respond to biotic and abiotic stresses. BMC Plant Biol. 2020;20:443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zou Z, Yang L, Wang D et al. Gene structures, evolution and transcriptional profiling of the wrky gene family in castor bean (Ricinus communis L.). PLoS One. 2016;11:e0148243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dong XS, Yang Y, Zhang Z et al. Genome-wide identification of WRKY genes and their response to cold stress in Coffea canephora. Forests. 2019;10:355. [Google Scholar]
- 42. Mangelsen E, Kilian J, Berendzen KW et al. Phylogenetic and comparative gene expression analysis of barley (Hordeum vulgare) WRKY transcription factor family reveals putatively retained functions between monocots and dicots. BMC Genomics. 2008;9:194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Xie T, Chen C, Li C et al. Genome-wide investigation of WRKY gene family in pineapple: evolution and expression profiles during development and stress. BMC Genomics. 2018;19:490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ding ZJ, Yan JY, Li GX et al. WRKY41 controls Arabidopsis seed dormancy via direct regulation of ABI3 transcript levels not downstream of ABA. Plant J. 2014;79:810–23. [DOI] [PubMed] [Google Scholar]
- 45. Chen J, Nolan TM, Ye H et al. Arabidopsis WRKY46, WRKY54, and WRKY70 transcription factors are involved in brassinosteroid-regulated plant growth and drought responses. Plant Cell. 2017;29:1425–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fu X, Peng B, Hassani D et al. AaWRKY9 contributes to light‐ and jasmonate‐mediated to regulate the biosynthesis of artemisinin inArtemisia annua. New Phytol. 2021;231:1858–74. [DOI] [PubMed] [Google Scholar]
- 47. Spyropoulou EA, Haring MA et al. RNA sequencing on Solanum lycopersicum trichomes identifies transcription factors that activate terpene synthase promoters. BMC Genomics. 2014;15:402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wilkins MR, Gasteiger E, Bairoch A et al. Protein identification and analysis tools in the ExPASy server. Methods Mol Biol. 1999;112:531–52. [DOI] [PubMed] [Google Scholar]
- 49. Cheng Y, Yao ZP, Ruan MY et al. In silico identification and characterization of the WRKY gene superfamily in pepper (Capsicum annuum L.). Genet Mol Res. 2016;15:gmr.15038675. [DOI] [PubMed] [Google Scholar]
- 50. Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hu B, Jin J, Guo AY et al. GSDS 2.0: an upgraded gene feature visualization server. Bioinformatics. 2015;31:1296–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bailey TL, Boden M, Buske FA et al. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;37:W202–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lescot M, Dehais P, Thijs G et al. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002;30:325–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chow CN, Lee TY, Huang YC et al. PlantPAN3.0: a new and updated resource for reconstructing transcriptional regulatory networks from ChIP-seq experiments in plants. Nucleic Acids Res. 2019;47:D1155–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wang C, Hao X, Wang Y et al. Genome-wide identification and comparative analysis of the teosinte branched 1/cycloidea/proliferating cell factors 1/2 transcription factors related to anti-cancer drug camptothecin biosynthesis in Ophiorrhiza pumila. Front Plant Sci. 2021;12:746648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Shannon P, Markiel A, Ozier O et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Deng CP, Hao X, Shi M et al. Tanshinone production could be increased by the expression of SmWRKY2 in salvia miltiorrhiza hairy roots. Plant Sci. 2019;284:1–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated or analyzed in the present research are included in this published article and its supplemental data files and available from the corresponding author upon reasonable request.