

Abstract
The newly emerged coronavirus severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) that caused the COVID-19 pandemic, is the closest relative of SARS-CoV with high genetic similarity. The papain-like protease (PLpro) is an important SARS-CoV/SARS-CoV-2 nonstructural protein that plays a critical role in some infection processes such as the generation of the functional replication complex, maturation of crude polyproteins, and regulation of the host antiviral immune responses. Therefore, the research to discover SARS-CoV-2 PLpro inhibitors could be a sensible strategy to obtain therapeutic agents for the treatment of COVID-19. Aiming to find SARS-CoV/SARS-CoV-2 PLpro inhibitors, various high throughput screenings (HTS) have been performed over the past two decades. Interestingly, the result of these efforts is the identification of hit/lead compounds whose structures have one important feature in common, namely having a chalcone-amide (N-benzylbenzamide) backbone. Structure-activity relationship (SAR) studies have shown that placing an (R)-configurated methyl group on the middle carbon adjacent to the amide group creates a unique backbone called (R)-methyl chalcone-amide, which dramatically increases PLpro inhibitory potency. Although this scaffold has not yet been introduced by medicinal chemists as a specific skeleton for the design of PLpro inhibitors, structural considerations show that the most reported PLpro inhibitors have this skeleton. This review suggests the (R)-methyl chalcone-amide scaffold as a key backbone for the design and development of selective SARS-CoV-2 PLpro inhibitors. Understanding the SAR and binding mode of these inhibitors in the active site of SARS-CoV-2 PLpro can aid the future development of anti-COVID-19 agents.
Keywords: SARS-CoV-2, Papain-like protease (PLpro), Chalcone amide (N-benzylbenzamide), COVID-19, X-ray crystal structure
Graphical abstract
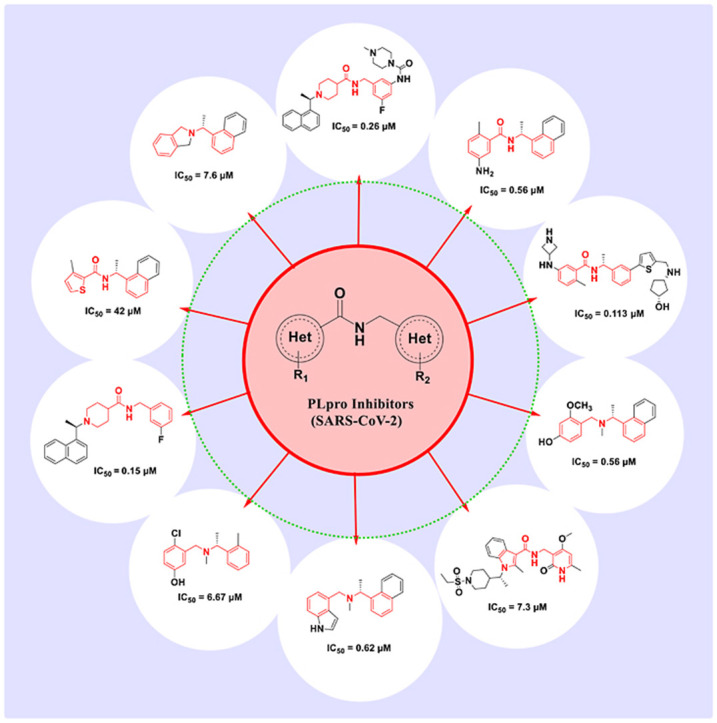
1. Introduction
Coronaviruses are a special family of viruses that cause mild to severe respiratory infections [1]. During the last two decades, some new mutated strains of these viruses such as SARS-CoV in 2002, and Middle East respiratory syndrome coronavirus (MERS-CoV) in 2012, caused disturbing respiratory diseases in different regions world [2,3]. In December 2019, a new type of coronavirus called SARS-CoV-2 causes the COVID-19 pandemic was discovered in Wuhan, China, which was much more contagious than previous species, infecting all countries of the world in a short period of time. Scientific evaluations immediately revealed that this virus has approximately 80% genomic similarity with SARS-CoV and about 50% similarity with MERS-CoV [4]. This finding was used as a basis for targeted actions against the virus and using the results of previous experiences and research on SARS-CoV.
Due to the development of several vaccines and drugs against COVID-19, public concern about the disease has diminished significantly today, but the SARS-CoV-2 still has one of the highest morbidity and mortality rates among viruses worldwide. In addition, variants that are resistant to current medications are likely to emerge in the coming years. Therefore, efforts to find the exact pathogenicity of the SARS-CoV-2, and also to find better drugs with more efficacy and safety are still an urgent need.
The molecular architecture of SARS-CoV-2 has been well described in some recent studies [5,6]. In summary, the structure of this virus is composed of 4 important structural proteins called spike protein (S), envelope protein (E), membrane protein (M), and nucleocapsid (N) [7]. These proteins are mostly responsible for the physical protection and appropriate binding of the virus to the host cells, as well as protecting the virus genome. The SARS-CoV-2 genome encodes important overlapping open-reading frames (ORFs) that are the source of viral proteins [8]. After endocytosis and the entry of the SARS-CoV-2 into the host cell, the virus genome is released and then translated into some polyproteins (pp1a and pp1ab) using the facilities of the host cell especially ribosomes [9,10]. Proteolytic cleavages and processing of these crude polyproteins by 3-chymotrypsin-like protease (3CLpro) and PLpro results in the production of sixteen mature nonstructural proteins (nsp1-16), which are used for the production of new viruses in the next steps [6,11].
Recently, several studies have also well described the biochemistry and function of the SARS-CoV-2 PLpro [[12], [13], [14]]. According to these studies, PLpro is a multifunction protein that plays a critical role in several important processes related to SARS-CoV-2 infection. In summary, PLpro can recognize the tetrapeptide LXGG motif and converts crude polyproteins to some NSPs (nsp1, nsp2, and nsp3), generating the functional replication complex, and also acting as an evasion mechanism against host antiviral immune responses. Biophysical evaluations also show that this enzyme plays a role in the hydrolysis of ubiquitin chains as well as the removal of interferon-stimulated gene 15 modifications [[14], [15], [16], [17]].
Based on findings related to viral genetic material, coronaviruses have one of the largest known viral RNA genomes, which is approximately 30 kb in length (full-length genomic RNA of SARS-CoV-2 is 29,903 nt) [18]. Sequencing of PLpro in SARS-CoV-2 and SARS-CoV shows that these enzymes have about 83% similarity. Although this sequence identity is not as similar to the 3CLpro enzymes in these viruses (sequence identity for 3CLpro is about 96%), it is very significant. On the other hand, the existence of more differences in sequence identity provides a chance that previous compounds with less efficacy in the inhibition of SARS-CoV PLpro be more effective in inhibiting SARS-CoV-2 PLpro. These proteins (3CLpro and PLpro) functionally are cysteine-protease enzymes that have very important roles in virus replication [2,19].
The schematic appearance of SARS-CoV-2 and its structural proteins, genome organization, and the role of PLpro in the maturation of some non-structural proteins and some infectious processes are summarized in Fig. 1 . As shown, PLpro inhibitors can potentially disrupt the infectivity of SARS-CoV-2 by blocking some of the critical functions.
Fig. 1.

The schematic appearance of SARS-CoV-2 along with structural proteins, genome organization, and the role of proteases PLpro and 3CLpro in the maturation of non-structural proteins.
As the starting steps for finding compounds with SARS-CoV/SARS-CoV-2 PLpro inhibitory activity, various HTS have been performed over the past two decades. The first identified SARS-CoV PLpro inhibitors were reported in 2008 by the Ghosh team from an HTS effort on 50,080 compounds, a few years after the advent of the virus (in 2002) [20]. Following this study, optimization of newly identified lead structures has led to the achievement of highly potent PLpro inhibitors with the sub-micromolar IC50 and low toxicity (discussed below). Careful attention to the structure of these compounds and subsequent optimized structures reveals they all have a “chalcone-amide backbone”. Structurally, chalcone amide is the analogue of the chalcone scaffold in which the carbon atom in the alpha position with is replaced by the nitrogen atom (Fig. 2 ). Although this scaffold has been used in various studies, it has not yet been considered or introduced as a special scaffold for the design of SARS-CoV/SARS-CoV-2 PLpro inhibitors.
Fig. 2.

The general structure of SARS-CoV/SARS-CoV-2 PLpro inhibitors, and structural correlation and similarity between scaffolds chalcone, chalcone-amide, and (R)-N-(1-phenylethyl)benzamide.
Today, many viral targets and small molecules have been identified for combating COVID-19, each having its own benefits [6,[21], [22], [23], [24], [25]]. Due to the key role of the PLpro in SARS-CoV-2 infection and replication [26], this enzyme and its inhibitors were considered an important topic for the writing of this review article. More precisely, this study aims to review the structural evaluations and inhibitory effects of small molecules containing chalcone-amide scaffold (as a privileged backbone) on this enzyme. Further structural evaluations and careful study of the binding mode of these compounds at the PLpro active site can inspire researchers to lead optimization better.
2. Antiviral activity of chalcone-amide-based structures
“Chalcone-amide” is an old term in organic/medicinal chemistry that has been commonly used instead of “N-benzylbenzamide” scaffolding. Chalcone amides are particular analogues of chalcones obtained by isosteric replacement of the carbon atom in the α position (in the intermediate chain) with a nitrogen atom. Some old studies have investigated the inhibitory effects of chalcone amide-based compounds against human rhinovirus (HRV) [27,28]. The results of these studies clearly show that compounds with chalcone amide-backbone have significantly stronger anti-HRV activity compared to corresponding compounds with chalcone-backbone (Fig. 3 ).
Fig. 3.

Chemical structures of chalcone-based compound Ro 09–0410 (1) as a potent anti-human rhinovirus (HRV) and some of its analogues (compounds 2–4) have a chalcone-amide backbone [28].
Histone methyltransferase EZH2/1 inhibitors are pharmacological agents that can effectively inhibit infections of viral pathogens [29]. As shown in Fig. 4 , the structure of many important EZH1/2 inhibitors contains chalcone-amide-like backbones (compounds 5–14). The CPI-169 (compound 9) is an EZH2-inhibitor recently found to have significant inhibitory activity against the SARS-CoV-2 PLpro enzyme with IC50 = 7.3 μM [30].
Fig. 4.

Chemical structures of some well-known EZH1/2 inhibitors (compounds 5–14) contain a chalcone-amide-like backbone.
Some recent studies show that chalcone-amide derivatives have strong inhibitory effects against tubulin polymerization and thus show anti-tumor effects [31]. However, some well-known tubulin polymerization inhibitors such as shikonin also have recently been identified as interesting SARS-CoV-2 inhibitors [[32], [33], [34]]. Although it is not possible to comment at this time due to a lack of sufficient information, the correlation between these biological activities (inhibition of tubulin polymerization and anti-SARS-CoV-2 activity) could be further explored in the future.
At present, there are many potent and safe antiviral drugs and lead compounds with a chalcone-amide backbone (Fig. 5 , compounds 15–25). In particular, the presence of this scaffold in the structure of HIV-1 integrase inhibitors has been repeatedly observed [35,36]. Entrectinib (compound 25) is an FDA-approved anti-cancer drug recently proposed by Garcia and co-workers as a SARS-CoV-2 inhibitor in human lung tissue cells [37].
Fig. 5.

Chemical structures of some approved antiviral drugs and lead structures (compounds 15–25) with the chalcone-amide-like backbone; DAN: DrugBank accession number.
Recently, Fearon and co-workers have released the X-ray crystal structure of a new compound with a chalcone-amide-like backbone bonded in the active site of SARS-CoV-2 3CLpro (PDB code: 5RH8) which created remarkable interactions including two conventional hydrogen bonds with residues Asn142 and Glu166 (compound 22, Fig. 5 and Table 1 ). The achieving and development of anti-SARS-CoV-2 agents with simultaneously inhibitory activity against both PLpro and 3CLpro could further raise hopes of finding effective drugs for the treatment of COVID-19.
Table 1.
The list of PDB codes related to released X-ray crystallographic structures of SARS-CoV/SARS-CoV-2 PLpro bonded with various chalcone-amide-based inhibitors, along with their release dates, resolution, sequence length, and interacting amino acids of the active sites.
| N | PDB code | Virus type | Release date | Resolution | Sequence Length | Co-crystalized ligand | Involved amino acids |
|---|---|---|---|---|---|---|---|
| 1 | 7E35 | SARS-CoV-2 (C112S mutant) | 2021-03-17 | 2.40 Å | 315 | 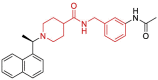 |
Leu163, Gly164, Asp165, Pro248, Pro249, Tyr265, Tyr269 |
| 2 | 7LBS | SARS-CoV-2 (Wild type) | 2021-02-24 | 2.80 Å | 316 | 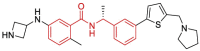 |
Leu162, Asp164, Glu167, Pro248, Tyr264, Gly266, Asn267, Tyr268, Gln269 |
| 3 | 7LBR | SARS-CoV-2 (Wild type) | 2021-02-24 | 2.20 Å | 316 | Leu162, Asp164, Pro248, Tyr264, Gly266, Asn267, Tyr268, Gln269 | |
| 4 | 7LLF | SARS-CoV-2 (Wild type) | 2021-02-24 | 2.30 Å | 316 | Leu162, Asp164, Pro248, Tyr264, Gly266, Tyr268, Gln269, Tyr273 | |
| 5 | 7LLZ | SARS-CoV-2 (Wild type) | 2021-02-24 | 2.90 Å | 316 | Leu162, Asp164, Glu167, Pro248, Tyr264, Tyr268, Gln269, Tyr273 | |
| 6 | 7LOS | SARS-CoV-2 (Wild type) | 2021-02-24 | 2.90 Å | 316 | Leu162, Asp164, Glu167, Pro248, Tyr264, Asn267, Tyr268, Gln269 | |
| 7 | 7CMD | SARS-CoV-2 (Wild type) | 2020-09-02 | 2.59 Å | 319 | 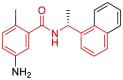 |
Leu162, Asp164, Pro248, Tyr264, Tyr268, Gln269, Tyr273 |
| 8 | 7KRX | SARS-CoV-2 (C111S mutant) | 2020-12-02 | 2.72 Å | 318 | 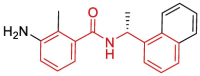 |
Asp164, Glu167, Pro247, Pro248, Tyr268, Gln269 |
| 9 | 7KOJ | SARS-CoV-2 (C111S mutant) | 2020-11-18 | 2.02 Å | 318 | 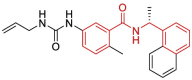 |
Asp164, Pro247, Pro248, Tyr264, Tyr268, Gln269, Tyr273 |
| 10 | 7KOK | SARS-CoV-2 (C111S mutant) | 2020-11-18 | 2.00 Å | 318 | 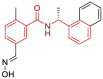 |
Asp164, Pro247, Pro248, Tyr264, Tyr268, Gln269, Tyr273 |
| 11 | 7CJM | SARS-CoV-2 (Wild type) | 2020-09-02 | 3.20 Å | 316 | 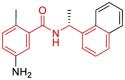 |
Asp164, Pro247, Pro248, Tyr264, Tyr268, Gln269 |
| 12 | 7JRN | SARS-CoV-2 (Wild type) | 2020-08-26 | 2.48 Å | 323 | 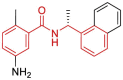 |
Leu162, Asp164, Pro247, Pro248, Tyr264, Tyr268, Gln269, Tyr273 |
| 13 | 7JN2 | SARS-CoV-2 (Wild type) | 2020-08-12 | 1.93 Å | 318 | 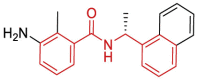 |
Asp164, Pro247, Pro248, Tyr268, Gln269 |
| 14 | 7JIR | SARS-CoV-2 (C111S mutant) | 2020-08-05 | 2.09 Å | 318 | 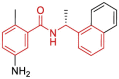 |
Asp164, Pro247, Pro248, Tyr264, Tyr268, Gln269, Tyr273 |
| 15 | 7JIT | SARS-CoV-2 (C111S mutant) | 2020-08-05 | 1.95 Å | 318 | 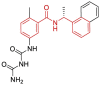 |
Asp164, Glu167, Pro247, Pro248, Tyr264, Tyr268, Gln269, Tyr273 |
| 16 | 7JIV | SARS-CoV-2 (C111S mutant) | 2020-08-05 | 2.05 Å | 318 | 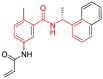 |
Asp164, Glu167, Pro247, Pro248, Tyr264, Tyr268, Gln269 |
| 17 | 7JIW | SARS-CoV-2 (Wild type) | 2020-08-05 | 2.3 Å | 318 | 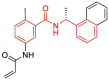 |
Asp164, Glu167, Pro247, Pro248, Tyr264, Tyr268, Gln269 |
| 18 | 5S2J | SARS-CoV-2 (Wild type) | 2021-01-13 | 1.11 Å | 169 (Chains A, B) | 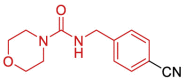 |
His138, Val142, Asp145 |
| 19 | 5RUW | SARS-CoV-2 (Wild type) | 2020-12-16 | 1.0 Å | 169 (Chains A, B) | 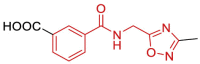 |
Val24, Gly48, Val49, Ala52, Lys55, Ala56, Phe156 |
| 20 | 5RV2 | SARS-CoV-2 (Wild type) | 2020-12-16 | 1.01 Å | 169 (Chains A, B) | 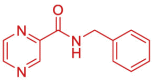 |
Asp22, Gly48, Val49, Ala52, Phe156 |
| 21 | 5S2T | SARS-CoV-2 (Wild type) | 2021-01-13 | 1.11 Å | 169 (Chains A, B) | 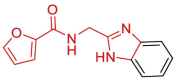 |
Asp22, Val24, Glu25, Val49, Ala52, Phe156 |
| 22 | 4OW0 | SARS-CoV (Wild type) | 2014-04-23 | 2.10 Å | 316 | 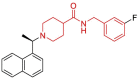 |
Gly164, Asp165, Pro248, Pro249, Tyr265, Tyr269, Gln270, Tyr274 |
| 23 | 4OVZ | SARS-CoV (Wild type) | 2014-04-23 | 2.50 Å | 316 | 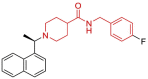 |
Gly164, Asp165, Pro248, Pro249, Tyr265, Tyr269, Gln270 |
| 24 | 3MJ5 | SARS-CoV (Wild type) | 2010-06-30 | 2.63 Å | 316 | 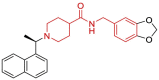 |
Leu163, Gly164, Asp165, Pro248, Pro249, Tyr265, Tyr269, Tyr274 |
| 25 | 3E9S | SARS-CoV (Wild type) | 2008-10-07 | 2.50 Å | 318 | 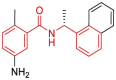 |
Leu163, Asp165, Pro248, Pro249, Tyr265, Tyr269, Gln270, Tyr274 |
3. X-ray crystallographic studies
X-ray structural studies elegantly disclosed PLpro structure and molecular interactions in the active site of this enzyme with inhibitors in detail. Some recent studies have well described the active site of SARS-CoV-2 PLpro along with the most important interacting amino acids [[38], [39], [40]]. PLpro is a large SARS-CoV-2 nonstructural protein with 1945 amino acids, comprised of four subdomains namely N-terminal ubiquitin-like (Ubl2, β1-3), the finger (β4-7), palm subdomain (β8-13), and the thumb (α2-7). Based on the molecular insights obtained from crystallographic studies, the conserved catalytic triad of the SARS-COV-2 PLpro-active site contains Cys111, His272, and Asp286 residues. In addition, the active site contains a portion with conserved cysteines including Cys189, Cys192, Cys224, and Cys226 under the finger subdomain [38].
Today, wild-type and some subsequent mutants of SARS-CoV-2 PLpro are structurally well-characterized, and several X-ray structures of them are available in the protein data bank (https://www.rcsb.org) which are useful for structure-based drug discovery attempts. As can be seen from the structures in Table 1, the most co-crystal inhibitors bonded at the active site of the SARS-CoV/SARS-CoV-2 PLpro have a chalcone-amide backbone.
Studies on the SARS-CoV-2 genome sequence indicate 79–80% identity with the SARS-CoV genome [41]. Sequencing of PLpro in these viruses also indicates about 83% sequence identity [14]. This sequence difference makes the interacting residues in the SARS-CoV PLpro active site slightly different from interacting residues in SARS-CoV-2 PLpro active site.
As shown in Fig. 6 (A-1 and B-1), the PLpro enzyme in both viruses has shallow active sites in the same location. This shallowness prevents the molecules from penetrating well into the protein, so it can be seen in co-crystallized structures where inhibitors commonly are attached to the surface (Fig. 6, A-2 and B-2). The list of the most important residues of SARS-CoV/SARS-CoV-2 PLpro active sites that play an important role in the interaction with non-covalent chalcone-amide-based inhibitors are shown in Table 1. In the case of the SARS-CoV-2 (wild-type), the most important interacting residues are Leu162, Asp164, Glu167, Pro247, Pro248, Tyr264, Tyr268, Tyr273, Asn267, and Gln269. For SARS-CoV, the most important interacting residues are very similar to those in SARS-CoV-2 (with slight variation in sequence and amino acid numbers) and include Leu163, Gly164, Asp165, Pro248, Pro249, Tyr265, Tyr269, Tyr274, and Gln270. This similarity has led to SARS-CoV PLpro inhibitors such as GRL0617 having similar inhibitory effects against SARS-CoV-2 PLpro. This is a great advantage for using the previous SAR findings and lead/hit optimizations related to SARS-CoV PLpro inhibitors for designing targeted and optimal new studies.
Fig. 6.

The X-ray crystal structure of SARS-CoV PLpro (A-1) and SARS-CoV-2 PLpro (B-1), and their shallow active sites at a similar location on the protein surface (graphic images prepared by Molegro Molecular Viewer 6.5 software); A-2 and B-2: X-ray co-crystal structures of SARS-CoV PLpro (A-2, PDB code: 3E9S) and SARS-CoV-2 PLpro (B-2, PDB code: 7CMD) bonded with well-known inhibitor GRL0617 (graphic images were analyzed by Discovery Studio Visualizer v4.5.).
Table 1 summarizes the properties of some of these chalcone-amide-based inhibitors in complex with SARS-CoV/SARS-CoV-2 PLpro along with the most important interacting residues.
4. Inhibitory effects of chalcone-amides against SARS-CoV and SARS-CoV-2
Since human exposure to SARS and MERS coronavirus epidemics over the past two decades, many chemical structures have been identified that have shown significant inhibitory effects against these viruses. Among these diverse structures, compounds with chalcone-amide scaffolds with inhibitory effects against viral PLpro are among the most promising compounds ever reported. The first identified chalcone-amide-based compound with a remarkable inhibitory effect against SARS-CoV PLpro was identified in an extensive fluorescence-based HTS on a structurally diverse library by 50,080 compounds, performed by Ghosh and co-workers [20]. In this effort, a racemic compound with a chalcone amide backbone was identified as a hit structure that had significant selective effects in inhibiting the SARS-CoV PLpro enzyme with IC50 = 20.1 μM (Fig. 7 , compound 26). Compound 27 was the second hit compound identified from this HTS that represented PLpro inhibitory activity with IC50 = 59.2 μM. These compounds were used as the initial leads for the design and development of the first and second generations of SARS-CoV-PLpro inhibitors in subsequent studies (Fig. 7). Paying attention to the structure of these compounds shows the existence of some similarities. As highlighted in red, a chalcone amide backbone is evident in the structure of the hit compound 26. Compound 27 also has a similar backbone in which the phenyl ring adjacent to the carbonyl group is replaced by a piperidine moiety. On the other hand, in the structure of both compounds, there is a naphthyl moiety that is attached to a nitrogen atom through a short linker. Naphthalene is a versatile platform in medicinal chemistry that is commonly used to increase the lipophilicity of molecules and facilitate penetration into the blood-brain barrier (BBB) [42,43]. These structural features have been investigated extensively in the subsequent optimizations and SAR studies discussed below.
Fig. 7.

Chemical structures of the first identified hit compounds 1 (26) and 2 (27) having chalcone-amide and pseudo-chalcone amide skeletons in HTS as the most active SARS-CoV-2 PLpro inhibitors among 50,080 screened structures.
In a later report by Ghosh et al., the synthesis routes, X-ray crystal structure (PDB code: 3E9S), molecular modeling, antiviral evaluation, and SAR of these noncovalent SARS-CoV-PLpro inhibitors were discussed in detail [44]. After evaluations of optically pure enantiomers, the stereochemical preference was established for the R-isomer, and this enantiomer was used as an optimized lead compound in the subsequent investigations (Fig. 8 , compound 28). Kinetic studies have also shown that this lead compound inhibits the SARS-CoV-PLpro through a reversible and non-covalent mechanism. Structure-based modifications and lead optimization led to the achievement of far more potent inhibitors than lead compounds. Among newly produced compounds, compound 30 (IC50 = 0.56 ± 0.03 μM and EC50 = 14.5 ± 0.8 μM), compound 31 (IC50 = 0.46 ± 0.03 μM and EC50 = 6.0 ± 0.1 μM), and compound 32 (IC50 = 1.3 ± 0.1 μM and EC50 = 5.2 ± 0.3 μM) showed best anti PLpro activities and antiviral effects in SARS-CoV infected Vero E6 cells (Fig. 8).
Fig. 8.

Chemical structures of the hit compound 26 and its enantiomers 28 and 29, and potent non-covalent SARS-CoV-PLpro inhibitors 30–32, were obtained from the lead-optimization [44].
In this study, to detail understand the ligand-binding site interactions responsible for the inhibitory potency of GRL0617, the structure of PLpro complexed with this molecule was described using X-ray crystallography. As shown in Fig. 9 and Table 1, the most important interacting residues with the GRL0617 in the SARS-CoV PLpro active site are Leu163, Asp165, Pro248, Pro249, Tyr265, Tyr269, Gln270, and Tyr274. The most important interactions of GRL0617 in the active site are related to the amide moiety, where the carbonyl group forms a strong hydrogen bond interaction with the –NH– part between residues Tyr269 and Gln270. In addition, the nitrogen of the amide moiety forms another strong hydrogen bond interaction with the –OH of Asp165 residue. The naphthyl nucleus also placed in a hydrophobic pocket (formed by the side chains of the Pro248 and Pro249, and aromatic rings of the Tyr265 and Tyr269) creates five important interactions including three Pi-alkyl interactions with the pyrrolidine moiety of prolines residues, as well as two Pi-Pi T-shaped interactions with the phenyl ring of the Tyr269. It seems that the R configuration of the methyl group is important for the orientation and placement of the naphthyl scaffold in a lipophilic space for creating this number of interactions. Methyl substitution on the A-ring causes three Pi-alkyl interactions with Leu163, Tyr265, and Tyr274 residues (Fig. 9).
Fig. 9.

X-ray crystal structure of GRL0617 (compound 30) in the active site of SARS-CoV PLpro (PDB code: 3E9S), and showing the interactions with residues Leu163, Asp165, Pro248, Pro249, Tyr265, Tyr269, Gln270, and Tyr274 (graphic image was analyzed by Discovery Studio Visualizer v4.5.).
Although SAR findings show that placing an amine group in the meta position of this ring (ring A) significantly affects the activity and increases its potency by more than 4 folds (compare the inhibitory potency of compound 33 vs. GRL0617 in Fig. 2), no effective interaction was observed for this substitution in the PLpro active site based on the crystal structure. Since the active site of the PLpro is shallow, this group is positioned at the opening of the active site cleft and is at a far distance to form interactions with proper residues such as Tyr269. However, this group may be effective by forcing the ligand into a specific orientation or conformation in the active site. These molecular insights gained from X-ray structural evaluations led researchers to perform some modifications on the benzamide fraction (A-ring) for creating effective interactions in this part of the molecule by placing longer branches containing polar substitutions instead of the -NH2 group located in the meta position of the phenyl ring. Accordingly, compounds 31 and 32 containing longer groups were produced, which are the most effective compounds in this study (Fig. 8).
SAR analysis of these structures revealed several important features summarized as follows (Fig. 10 ): First, the change in the direction of the naphthyl linkage from 2-naphthyl to 1-naphthyl leads to a significant increase in potency (compounds 28 and 33). Second, the addition of any substitution to the nitrogen in the intermediate chain (compound 34), as well as the addition of the substitution on the carbon adjacent to the naphthyl ring in the intermediate chain (compound 35 with a gem-dimethyl group), results in a significant reduction in potency. Third, replacing or changing the position of the methyl group at the 1-position of the benzamide ring leads to a significant reduction in potency (compounds 36–39).
Fig. 10.

Occurrence of significant changes in the anti-SARS-CoV PLpro activity induced by structural modifications during SAR studies [44].
In this study, two sets of compounds were synthesized in which the naphthyl moiety is connected from two positions 1 and 2 (Scheme 1 , general structures 40 and 41). Synthetically, the targeted structures are synthesized by coupling naphthyl-ethylamine derivatives and various designed benzoic acids in the final step. Modifying the alternatives at different structural positions, especially on the benzamide ring, results in diverse structures. Because the hit compound 26 is a racemic structure, the researchers synthesized the corresponding chiral derivatives utilizing (R/S)-naphthyl-ethylamine reagents to assess the stereospecific recognition of the most effective compounds.
Scheme 1.

The synthesis routes and general structure of synthesized compounds 40 and 41 have diverse substitutions in various structural situations [44].
As represented in the HTS performed in the previous study (Fig. 7), the second important SARS-CoV PLpro inhibitor has a chalcone-amide-like backbone bearing a piperidine moiety that can exhibit inhibitory effects with IC50 = 59 μM (hit compound 2). Accordingly, another lead optimization performed by Arun K. Ghosh et al., and the second series of SARS-CoV PLpro inhibitors (general structure 42) were designed and synthesized through structural modification of this hit compound [45]. The biological activities of these structures were evaluated using the assessment of PLpro inhibitory activity and anti-SARS-CoV effects against infected Vero E6 cells. Also, the protein-ligand X-ray structure of PLpro enzyme bonding with the most active synthesized compound 44 was released with 3MJ5 PDB code.
The co-crystal structure of PLpro in complex with compound 44 demonstrated that this compound interacts in a pattern almost different from that of GRL0617 at the active site of the enzyme (Fig. 11 ). Although the naphthyl nucleus in this compound produces exactly the same hydrophobic interactions (as the naphthyl nucleus of the GRL0617) with Tyr-265 and Tyr-269 aromatic rings and Pro248 and Pro249 side chains, the amide moiety produces only one hydrogen bond derived from the interaction of –NH– with the carboxylic group of Tyr269 residue. The piperidine moiety forms two Pi-alkyl interactions with the residues Tyr265 and Tyr269, but its nitrogen does not participate in any interaction independently. The chiral methyl group here creates two Pi-alkyl interactions with the phenyl rings of the Tyr265 and Tyr274 residues. Another important point in the protein-ligand X-ray structure of compound 44 was that the 1,3-benzodioxole scaffold is located outside the active site cleft, while SAR studies show that this part affects activity. Molecular modeling studies have shown that oxygen atoms in the dioxole moiety can increase the inhibitory activity due to the formation of hydrogen bonds with the Gln270 carboxamide side chain [45].
Fig. 11.

X-ray crystal structure of compound 44 in the active site of SARS-CoV-2 PLpro (PDB code: 3MJ5), and showing the most important interactions with residues Leu163, Gly164, Asp165, Pro248, Pro249, Tyr265, Tyr269, and Tyr274 (graphic image was analyzed by Discovery Studio Visualizer v4.5.).
Among the structures synthesized in this study, enantiomeric compound 43 (IC50 = 0.32 μM, and EC50 = 9.1 μM) and compound 44 (IC50 = 0.56 μM, and EC50 = 9.1 μM) were identified as the most active compounds for the inhibition of SARS-CoV PLpro and viral replication. As shown in Fig. 12 , the location of the chiral carbon in these structures is outside the chalcone-amide scaffold. SAR evaluations showed that the different spatial arrangement (R/S-configuration) of the substations in this chiral center did not significantly affect the IC50 and EC50 values. However, for the optimized compounds derived from hit compound 26, the difference in the configurations on the chiral center which is located inside the chalcone-amide backbone causes a significant difference in the amounts of enzyme inhibition and antiviral activity (Fig. 12, compounds 28 and 29). From these results, it can be concluded that the configuration of this chiral center (carbon adjacent to nitrogen amide in the chalcone-amide scaffold) has a key effect on the potency of this category of PLpro inhibitors.
Fig. 12.

Chemical structure and anti-SARS-CoV PLpro activity of most active chalcone-amide-like compounds 43 and 44 containing a piperidine scaffold, derived from optimization of hit compound 27. The location of the chiral center containing methyl group inside or outside the chalcone-amide backbone has a significant effect on the PLpro-inhibitory activity [45].
SAR analysis of PLpro inhibitors derived from compound 27 (hit compound 2) reveals some important relationships between their chemical structures and inhibitory activities, the most important of which are as follows: First, the binding of the naphthyl moiety from position 1 causes a significant increase in the inhibitory activity of the compounds (similar to structures derived from hit compound 1). Second, the configuration of the chiral center adjacent to the naphthyl ring (outside of the pseudo-chalcone-amide backbone) has no significant effect on the potency of the compounds. Third, the presence of a methyl group in the chiral center adjacent to the naphthyl ring causes a significant increase in the potency of the tested compounds. The simultaneous presence of two methyl groups in this region causes a greater reduction in the activities. Fourth, replacing the piperazine ring (instead of piperidine) causes a loss of activity (compound 55, Fig. 13 ). This sharp decrease in activity could be related to the structural constraints created by the replacement of carbon with nitrogen in the ring. The new nitrogen binds to the amide group and forms a urea moiety that tends to be flat and imposes a limit on flexibility. Fifth, the presence of methoxy and dioxolane substitutions in positions 3 and 4 of the farther phenyl ring (B-ring) causes a significant increase in potency compared to these substitutions in position 2 (Fig. 13). The molecular modeling studies indicated that methoxy oxygen at the meta or para positions was available to form a hydrogen bond with the Gln270 carboxamide side chain, but this is difficult for methoxy oxygen at the ortho position. However, X-ray structures (PDB code: 3MJ5) displayed that the phenyl ring carrying these substitutions is located outside the active site cleft and does not participate in the active site interactions (of course, Gln-270 is the most available residue for this part of the molecule).
Fig. 13.

The effect of bonding from positions 1 or 2 of the naphthyl moiety and establishing a chiral center by placing a methyl group on the methylene carbon between the naphthyl ring and the piperidine moiety, on the SARS-CoV PLpro inhibitory effects of compounds 49–55 [45].
Another structure-guided study for optimization of SARS-CoV PLpro inhibitors having chalcone-amide backbones was performed by Yahira M. Báez-Santos et al. [46]. In this study, by considering compound 43 as the lead, a number of novel noncovalent PLpro inhibitors (general structure 56, Fig. 14 ) were designed and synthesized, and their antiviral activities were assessed using SARS-CoV infected Vero E6 cells. Among newly synthesized compounds, eight compounds 57–64 were found to have potent SARS-CoV PLpro inhibitory activity with sub-micromolar IC50 ranging from 0.15 to 0.63 μM. Other complementary evaluations showed that these new compounds are not cytotoxic, don't bind to human serum albumin (HSA), and have remarkable metabolic stability. High-resolution crystal structures of the most active compounds 62 and 63 bound to the SARS-CoV PLpro active site show some aspects of ligand binding mode, and provide valuable guidance for further structural optimization of PLpro inhibitors. These X-ray crystal structures were released in 4OVZ and 4OW0 PDB codes for compounds 62 and 63, respectively. Based on the molecular insights obtained from crystallographic studies, it was found that these compounds interact in a pattern similar to that of compound 44 at the SARS-CoV PLpro active site.
Fig. 14.

Chemical structure and effects of some potent SARS-CoV PLpro inhibitors on enzyme inhibition (IC50), viral replication (EC50), and cytotoxicity (CC50) [46].
Continuing efforts to find SARS-CoV 3CLpro/PLpro inhibitors, a highly extensive HTS was conducted in 2009 against the NIH molecular libraries sample collection containing more than 293,000 diverse compounds. This HTS led to the identification of a novel structural class of noncovalent SARS 3CLpro inhibitors. Although the newly identified 3CLpro inhibitors were named dipeptide inhibitors, the presence of the chalcone amide-like backbone is evident in their structures. As shown in Fig. 15 , compound 65 is an initial lead compound identified in this HTS that in addition to inhibiting 3CLpro (IC50 = 2.2 μM), has the remarkable ability to inhibit the PLpro with IC50 = 34 μM (Further details of this HTS can be found at http://mli.nih.gov/mli/mlpcn/).
Fig. 15.

Chemical structure of hit compound 65 and novel structural SARS-CoV inhibitors compounds 66–68 derived from HTS and subsequent lead optimization.
A study later published in 2013 by Jon Jacobs et al. described the results of this screening campaign and subsequent efforts to optimize the identified inhibitors [47]. Due to the stronger initial effect of identified lead compound 65 on 3CLpro inhibition compared to PLpro, the researchers performed lead optimization focusing on finding 3CLpro-inhibitors. These efforts resulted in the identification of a selective 3CLpro inhibitor compound 68 (R-enantiomer) with IC50 = 1.5 μM. In this study, the effect of different enantiomers on the PLpro enzyme was not characterized. As shown in Fig. 15, a racemic mixture of the lead compound 65 has a significant inhibitory effect against PLpro (IC50 = 34 μM). Due to the presence of a chalcone-amide-like backbone in this structure, evaluation of the PLpro inhibitory effects of the pure enantiomers (compounds 67 and 68) seems to help to better understand the SAR of these compounds. Considering the effectiveness of lead compound 65 on the inhibition of both enzymes, paying more attention to the PLpro inhibitory effects of these inhibitors can lead to the achievement of valuable agents with inhibitory ability on both SARS-CoV-2 3CLpro and PLpro enzymes.
Given the differences in the amino acid sequence of SARS-CoV PLpro with the amino acid sequence of PLpro in other viruses such as MERS-CoV, there is some evidence that these chalcone amides act selectively against SARS-CoV PLpro. In a study, Hyun Lee et al. examined the inhibitory effects of the most active compounds reviewed above (30, 32, 43, and 44) on MERS-CoV PLpro and found that neither was effective against MERS-CoV PLpro (Fig. 16 ). By comparing the structures and amino acid sequence alignment of both enzymes, the researchers reported that the two residues Tyr269 and Gln270, which are responsible for binding inhibitors to SARS-CoV PLpro, have been replaced with Thr274 and Ala275 in MERS-CoV PLpro [48]. This difference makes it difficult for inhibitors to create similar binding interactions at the active site of both enzymes. Due to the great similarity of the amino acid sequence of SARS-CoV PLpro with SARS-CoV-2 PLpro, the above chalcone-amide inhibitors show excellent effects on the inhibition of SARS-CoV-2 PLpro, which will be discussed below.
Fig. 16.

Chemical structures of the most active SARS-CoV PLpro inhibitors 30, 32, 43, and 44 did not show inhibitory effects on the MERS-CoV PLpro enzyme.
In a recent structure-based drug design study, Jerzy Osipiuk et al. determined the high-resolution structure of SARS-CoV-2 PLpro and some new PLpro inhibitors that are structurally related to the lead compound GRL0617 (Fig. 17 ) [39]. Biological evaluations showed that these compounds could effectively prevent the activity of PLpro in vitro with IC50 ranging from 5.1 to 16.8 μM, and some of them can suppress SARS-CoV-2 replication in cell culture assays (Vero E6 cells). This study released several high-resolution crystal structures including the structure of wild-type (WT) PLpro with PDB code of 6WZU, the active site of cysteine mutant C111S in which a single sulfur atom was replaced by oxygen with PDB codes 6WRH and 6XG3, and the complexes of PLpro with some of the mentioned noncovalent chalcone-amide-based inhibitors including PDB codes 7JIW (complex of compound 71 and SARS-CoV-2 PLpro, wild type), 7JIV (complex of compound 71 and SARS-CoV-2 PLpro, mutant C111S), 7JIT (complex of compound 72 and SARS-CoV-2 PLpro, mutant C111S), and 7JIR (complex of GRL0617 and SARS-CoV-2 PLpro, mutant C111S). Evaluation of the interactions and bonds at the active site indicates that these compounds bind in the same site (located 8–10 Å away from the catalytic cysteine) and in a very similar manner.
Fig. 17.

A: Chemical structure and production path of SARS-CoV-2 PLpro inhibitors compounds 70–74; B: Removal of carbon-containing (R)-methyl group severely reduces the PLpro inhibitory activity in compound 75.
The chemical structure and production path of these inhibitors (compounds 70–75) are shown in Fig. 17. Of the few compounds synthesized in this study, compound 75 is structurally different from the others. In this compound, the methylene carbon adjacent to nitrogen amide (bearing the chiral-methyl group) is removed. This structural change has significantly reduced its SARS-CoV-2 PLpro inhibitory activity, but it has not been completely eliminated. However, a chalcone-amide scaffold is still present in the structure of this compound (Fig. 17, B).
The X-ray crystal structures of SARS-CoV-2 PLpro bounded to these compounds showed that all three structures bind exactly to the same site as observed previously for GRL0617. The design of these compounds is based on the modification of the amine group located in the meta position of the A-ring of GRL0617, as stated in previously reviewed studies that extending this substitution using polar groups has a high potential for achieving new active compounds with stronger interactions in the PLpro active site. The X-ray structure of the mentioned compounds (GRL0617, 71, and 72) co-crystallized in complex with SARS-CoV-2 PLpro along with the most important interactions is shown in Fig. 18 . As shown by the dotted line circle in Fig. 18-A, the X-ray structure here also shows that the amine substitution of the phenyl ring is located in the outer space of the active site and does not interact effectively. Placing a more elongated polar group such as acrylamide instead of the amine group causes the creation of two strong hydrogen bond interactions with the Glu167 and Gln269 residues in this part of the molecule (compound 97, Fig. 18-B). Interestingly, these interactions also occur exactly in the C111S-mutant PLpro active site, which indicates the therapeutic ability of this compound to inhibit different variants of the virus (Fig. 18-C). In the site mentioned on the A-phenyl ring of compound 72, a highly polar chain containing three nitrogen atoms and two carbonyl groups (resembling two interconnected urea moieties) is placed. Although this group may have adverse effects on the pharmacokinetics and drug-likeness characters, based on the crystallographic findings this highly hydrophilic short-chain creates several strong interactions (such as the two conventional hydrogen bonds caused by the interaction of the Tyr268 and Gln269 side chains with the internal carbonyl group) in the outer opening of the SARS-CoV-2 PLpro active site in vitro (Fig. 18-D).
Fig. 18.

X-ray crystal structures of compounds GRL0617 (A. PDB code: 7JIR; mutant C111S), compound 71 (B. PDB code: 7JIW; wild type), compound 71 (C. PDB code: 7JIV; mutant C111S), and compound 72 (D. PDB code: 7JIT; mutant C111S), in the SARS-CoV-2 PLpro active site along with the most important interactions, creates by residues Asp164, Glu167, Pro247, Pro248, Tyr264, Tyr268, Gln269, and Tyr273. In Figures B, C, and D, for better understanding, only interactions of the polar substitutions on the phenyl ring are shown, and other interactions generated by other parts of the molecule are omitted (graphic images were analyzed by Discovery Studio Visualizer v4.5.).
In another new study performed by Armin Welker et al., two structural classes benzamides and isoindolines and some other derivatives (Fig. 19 , general structures 76–79) designed and synthesized as noncovalent SARS-CoV/SARS-CoV-2 PLpro inhibitors by the use of GRL0617 as the lead compound [49]. Inhibitory activity assessments of the new compounds against the PLpro enzyme in both viruses showed that some of them (compounds 80–86) have promising activities with a single-digit micromolar range, more effective against SARS-CoV (Fig. 20 ). In this study, antiviral activity and potential cytotoxicity of some of the most active derivatives such as compounds 87–90 were also evaluated using SARS-CoV-2-infected- and mock-infected Vero E6 cells and MTT assay. In vitro toxicity assessments showed very low cytotoxicity with CC50 values far above the respective EC50 values for the new compounds (Fig. 21 ).
Fig. 19.

General structures of synthesized benzamide derivatives 76 (A), isoindoline derivatives 77 (B), and some other related structures 78 (C) and 79 (D).
Fig. 20.

A: Structural similarity between benzamide and isoindoline derivatives as novel SARS-CoV-2 PLpro inhibitors; A-1: Most active benzamide-derivatives 80–83 derived from optimization of lead compound GRL0617; A-2: Most active isoindoline-derivatives 84–86.
Fig. 21.

Antiviral activity and potential cytotoxicity of the most active benzamide-derivatives 87–89 and isoindoline-derivative 90 were evaluated using SARS-CoV-2-infected Vero E6 cells.
Although isoindoline derivatives were introduced in this study as a new structural class of PLpro inhibitors, their structural properties reveal many similarities between these compounds and chalcone-amide-based PLpro inhibitors (Fig. 20, A). SAR evaluations also showed again that (R)-configuration of the methyl group on internal carbon adjacent to nitrogen amide is highly privileged which could be shown in both benzamide- and isoindoline-derivatives. Another important SAR finding is that inversion of the amide orientation resulted in the loss of anti-PLpro activity in the anilide-derivatives (Fig. 19, C). Also, non-aromatic substitutions as alternatives for the phenyl ring of benzamide moiety were not tolerated (Fig. 19, D).
Zhengnan Shen and collaborators recently performed a comprehensive structural study to aid a robust HTS which yielded interesting results [30]. Among all the 15,370 screened compounds consisting of two chemical libraries, both compounds 9 (CPI-169) and 30 with the highest SARS-CoV-2 PLpro inhibitory activity had a chalcone-amide skeleton (Fig. 22 ). The compound 9 was previously developed as a histone methyltransferase enhancer of zeste homolog 2 (EZH2) inhibitor [50]. Although this compound has been introduced as a new scaffold for the inhibition of SARS-Cov-2 PLpro by the researchers of this study, its structure also contains a chalcone-amide-like scaffold. These HTS-results once again demonstrated the importance of the chalcone-amide backbone as a key scaffold for having SARS-CoV-2 PLpro inhibitory activity. Considering the structures of lead compounds 9 and 30, the researchers developed a structure-based study for finding derivatives with better activities (general structure 91, Fig. 23 ). Lead optimization yielded highly potent non-covalent PLpro inhibitors bearing 2-phenylthiophene moiety with high metabolic stability and low IC50 ranging in sub-micromolar concentrations (Fig. 24 -A, compounds 94–108). Assessing the SAR of these compounds reveals some interesting points. For example, it was once again confirmed that the (R)-configuration for the chiral center (the carbon located in the chalcone-amide scaffold containing methyl group) significantly increased the PLpro inhibitory activity compared to the (S)-configuration (Fig. 24-B). Also, placing piperidine and azetidine substitutions on the -NH2 group located in the meta position of the phenyl ring significantly increases the inhibitory activity (Fig. 24-A and C). Another important finding of SAR studies was that replacing the naphthyl moiety with a 2-phenyl-thiophene scaffold resulted in derivatives with better activities. This finding could change the name of this structural category of compounds from naphthalene-derived PLpro inhibitors to chalcone-amide-based PLpro inhibitors, and also could be the beginning of the production of the third generation of SARS-CoV/SARS-CoV-2 PLpro inhibitors in which the main backbone is (R)-N-(1-(3-(thiophen-2-yl)phenyl)ethyl)benzamide.
Fig. 22.

Chemical structures of the compounds 9 (CPI-169) and 30 (GRL0617) having chalcone-amide/chalcone-amide-like backbones identified in HTS as the most active SARS-CoV-2 PLpro inhibitors.
Fig. 23.

The general structure of the new SARS-CoV-2 PLpro inhibitors 92, designed by the use of GRL0617 as a lead compound.
Fig. 24.

A: Optimization of the lead compound GRL0617 by adding azetidine substitution on -NH2 group located in the meta position of the phenyl ring, and replacing the naphthyl moiety with the 2-phenyl-thiophene scaffold. The chemical structure of produced highly potent SARS-CoV-2 PLpro inhibitors with sub-micromolar IC50s (structures 94–108), derived from optimization of compound 93; B: Preference of (R)-configuration over (S)-configuration for the chiral center containing methyl group located in the adjacent of NH-amide of chalcone-amide backbone; C: Significant effect of the placing of piperidine substitution on the -NH2 group on increasing the inhibitory activity of chalcone-amide-based PLpro inhibitors 112–114.
Among synthesized compounds, 107 displayed highly potent PLpro inhibitory activity with IC50 = 0.113 μM. The X-ray structure of this inhibitor in complex with PLpro released with the PDB code of 7LBR. Investigation of the interactions of compound 107 in the active site of SARS-CoV-2 PLpro indicates the construction of stronger conventional hydrogen bonds compared to the lead compound GRL0617 (Table 2 ). The most important interactions of the compounds GRL0617 and compound 107 in the active site of SARS-CoV-2 PLpro are shown in detail in Fig. 25 and Table 2. Based on these results, compound 107 creates fourteen strong interactions with eight amino acids Leu162, Asp164, Pro248, Tyr264, Gly266, Asn267, Tyr268, and Gln269. The lead compound GRL0617 also forms almost similar interactions with seven amino acids Leu162, Asp164, Pro248, Tyr264, Tyr268, Gln269, and Tyr273.
Table 2.
The most important interactions of the GRL0617 (PDB code: 7CMD) and compound 107 (PDB code: 7LBR) in the active site of SARS-CoV-2 PLpro were analyzed by Discovery Studio Visualizer v4.5.
| Interaction | Compound 107 |
GRL0617 |
||||
|---|---|---|---|---|---|---|
| Residues | Interacting groups | Distance (Å) | Residues | Interacting groups | Distance (Å) | |
| Conventional H-bond | Gln269 | Carbonyl group | 1.87 | Gly269 | Carbonyl group | 2.37 |
| Asp164 | NH (Amide) | 1.71 | Asp164 | NH (Amide) | 2.79 | |
| – | – | – | Tyr264 | Carbonyl group | 2.66 | |
| Pi-Alkyl | Tyr264 | CH3 (A-Phenyl ring) | 4.84 | Tyr264 | CH3 (A-Phenyl ring) | 4.60 |
| Pro248 | B-Phenyl ring | 4.45 | Pro248 | Naphthyl moiety | 4.51 | |
| Pro248 | Th-ring | 4.97 | Pro248 | Naphthyl moiety | 4.13 | |
| – | – | – | Tyr273 | CH3 (A-Phenyl ring) | 5.45 | |
| Amide-Pi Stacked | Asn267 | Th-ring | 5.12 | – | – | – |
| Pi-Sigma | Asp164 | A-Phenyl ring | 2.59 | Asp164 | A-Phenyl ring | 2.72 |
| – | – | – | Gln269 | A-Phenyl ring | 2.86 | |
| – | – | – | Tyr268 | Naphthyl moiety | 3.01 | |
| Pi-Pi T-Shaped | Tyr268 | A-Phenyl ring | 5.25 | Tyr268 | A-Phenyl ring | 5.08 |
| Tyr268 | B-Phenyl ring | 4.75 | Tyr268 | Naphthyl moiety | 5.02 | |
| – | – | – | Tyr268 | Naphthyl moiety | 5.24 | |
| Carbon | Tyr268 | Carbonyl group | 2.56 | Tyr268 | Carbonyl group | 2.64 |
| Gln269 | Azetidine | 3.00 | – | – | – | |
| Gly266 | CH2 (Adj to Th-ring) | 2.74 | – | – | – | |
| Alkyl | Leu162 | CH3 (A-Phenyl ring) | 4.10 | Leu162 | CH3 (A-Phenyl ring) | 4.36 |
| Pi-Sulfur | Tyr264 | S (Th-ring) | 5.52 | – | – | – |
Fig. 25.

2D and 3D interactions of the compounds GRL0617 (PDB code: 7CMD) and 107 (PDB code: 7LBR) in the active site of SARS-CoV-2 PLpro (graphic images were analyzed by Discovery Studio Visualizer v4.5.).
In order to find and develop the novel SARS-CoV-2 PLpro inhibitors, recently Zilei Xia et al. performed an extent FRET-based high-throughput screening using a library containing 50,240 diverse enamine compounds [51]. As a result of this HTS, two structurally similar new compounds 115 (IC50 = 7.29 ± 1.03 μM) and 116 (IC50 = 6.67 ± 0.05 μM) were identified as the strongest inhibitors. Interestingly, these compounds also have a similar skeleton to the previously identified chalcone-amide-based SARS-CoV-2 PLpro inhibitors, but in the structures, the middle carbonyl group has been removed (Fig. 26 , A). Subsequent lead optimization was investigated through the design and development of two sets of new compounds 117 and 118 using newly identified compounds 115, 116, and GRL0617 as leads (Fig. 26B and C), which results in produce several potent inhibitors 119–126 with IC50 values ranging from 0.56 to 0.90 μM (Fig. 27 , A).
Fig. 26.

Chemical structures of the compounds 115 and 116 as the most active SARS-CoV-2 PLpro inhibitors identified by HTS effort; A: General structure of designed compounds set A (117) having naphthyl moiety; B: General structure of designed compounds set B (118) [51].
Fig. 27.

A: Chemical structures of highly potent SARS-CoV-2 PLpro inhibitors 119–126 with sub-micromolar IC50s; B: Preference of (R)-configuration over (S)-configuration for the chiral center containing methyl group located in the adjacent NH-amide of chalcone-amide backbone, compared for compounds 127 and 128; C: Significant reduction in the potency of chalcone-based structures by placing a methyl group on the nitrogen of amide moiety [51].
Although previous efforts to obtain potent inhibitors have been promising, the discovery of compounds with improved cellular antiviral activity and enzymatic inhibition has not been very satisfactory. For example, although GRL0617 is one of the most potent identified PLpro inhibitors, its antiviral activity is weak (EC50 > 20 μM). To assess the intracellular PLpro inhibitory potency of the new compounds, the researchers of this study performed a cell-based FlipGFP assay that prioritized lead compounds to evaluate their anti-SARS-CoV-2 activity. It was observed that two prioritized compounds 126 (EC50 = 8.89 μM) and 127 (EC50 = 8.32 μM) can effectively inhibit SARS-CoV-2 replication (in Caco-2 hACE2 cells), which was much more significant compared to GRL0617 with EC50 = 25.1 μM.
SAR analysis of the synthesized compounds reveals some new structural relations. For example, considering the structures of compound 126 and the lead compound GRL0617 indicates that the presence of the carbonyl group is not necessary to have an inhibitory effect or even on potency. The absence of the carbonyl group could potentially cause the mentioned nitrogen (as an active secondary amine) to interact easily with the electrophiles in the biological environment and cause adverse interactions. Therefore, in the absence of the carbonyl group (such as the compounds in this study), the presence of a methyl group on central nitrogen can act as an activity balancer. Preparation of the crystal structure of the mentioned compounds in the active site of the PLpro enzyme can increase the molecular insights into the activity of these compounds.
Another important SAR finding was that the crucial role of the methyl group configuration (adjacent to the nitrogen amide) on the potency of the PLpro inhibitors was reaffirmed (Fig. 27, B). Because the researchers may not have considered the structural relationship between the identified lead compounds 115 and 116 with GRL0617, they did not use the SAR findings from previous studies (reviewed in this article). Considering the structure of these compounds (115 and 116) shows that on ring 1 (equivalent to ring A) in the meta position, there is a hydroxyl group that can be considered equivalent to the -NH2 group located in the meta position of the GRL0617 phenyl ring. Based on the structural insights gained from previous SAR findings, placing longer substitutions containing polar ends instead of this hydroxyl group could potentially lead to achieving stronger inhibitors. However, structural modifications performed in this study have led to the acquisition of interesting new PLpro inhibitors with remarkable activity.
Previous SAR evaluations have also shown that placing the alkyl substitutes such as methyl group on the nitrogen of amide significantly reduces PLpro inhibitory activity (Fig. 27, C) [44]. The reason for this decrease in activity can be explained by the findings related to the crystal structures of previous compounds. As shown in Fig. 9, Fig. 11, amide nitrogen can act as a hydrogen bond donor. Therefore, when an alkyl group is placed on it, the possibility of performing this important interaction at the active site of the PLpro is eliminated. However, in the structure of the final derivatives of this study, a methyl group is located on the middle nitrogen (corresponding to nitrogen amide) which effect of its absence/presence on the potency of the synthesized compounds has not been investigated. This issue can be assessed and researched in future evaluations [51].
Ubiquitin-rhodamine 110 (Ub-Rho) is a highly sensitive and expensive fluorogenic activity probe in HTS assays for both human deubiquitinating enzymes (DUBs) and SARS-CoV-2 PLpro (due to the functional similarity of the SARS-CoV-2 PLpro and DUBs) [52]. Recently, Hengyue Shan and co-workers performed an HTS using two probes (Ub-Rho and a self-designed fluorescence resonance energy transfer (FRET)-based fluorogenic probe 7 by these researchers) screened DUB-inhibitors and other proteases. As a result of this screening, some pseudo chalcone amide-based structures including compounds 129–131 and GRL0617 were identified as the best leads for the development of SARS-CoV-2 PLpro inhibitors (Fig. 28 ).
Fig. 28.

Chemical structure and anti-SARS-CoV-2 activity of chalcone amide-based lead compound GRL0617 and compounds 129–131 identified by HTS as the most active SARS-CoV-2 PLpro inhibitors, and subsequent optimized potent analogues 132–137 [53].
Further evaluations on identified compounds utilizing various in vitro and in vivo cellular assays resulted in the introduction of some potent and selective SARS-CoV-2 PLpro inhibitors such as compound 137 that not only blocks the suppression of the immune system (exerted by PLpro) but also significantly reduces the PLpro activity. In addition to having this effect, biological evaluations revealed that 137 and its analogues (compounds 135 and 136) containing amide moieties on meta position of B-ring have a potent antiviral activity that can effectively prevent the SARS-CoV-2 replication in human cells with viral replication IC50 values ranging from 0.18 to 0.60 μM. The chemical structures of the optimized compounds 132–137 together with their inhibitory potency against the SARS-CoV-2 PLpro and viral replication are shown in Fig. 28 [53].
5. Conclusion
As stated in the text, the PLpro is a multifunction SARS-CoV-2 cysteine protease that plays a critical role in various viral infection-related processes such as the maturation of crude polyproteins and regulating host antiviral immune responses. Therefore, trying to discover effective PLpro inhibitors by better understanding the structure-activity relationships and then optimizing the identified lead structures seems a reasonable policy to achieve effective drugs for the treatment of COVID-19. As a result of efforts made in different parts of the world, diverse structures have been designed and produced that have significant inhibitory effects against SARS-CoV/SARS-CoV-2 PLpro. In the structure of these inhibitors, some interesting correlations exist that have not been considered so far. Especially, structural comparisons show that most of them have a chalcone-amide backbone. On the other hand, SAR studies show that the placement of the (R)-configured methyl group on the middle carbon adjacent to the amide creates a unique backbone called “(R)-methyl chalcone-amide” that dramatically increases the inhibitory power of PLpro. Since there are many lead structures and accepted antiviral drugs that have chalcone amide-like backbone (such as raltegravir, dolutegravir, cabotegravir, bictegravir, dutacatib, etc.), it seems that placing an (R)-configured methyl group in the mentioned position could make these compounds potential candidates for the treatment of COVID-19.
The choice of “chalcone-amide” word for the backbone structure of SARS-CoV/SARS-CoV-2 PLpro inhibitors summarized in this study is doubtful. The most important reason was that in the second generation of PLpro inhibitors, there is a non-aromatic piperazine scaffold instead of the A-ring. This issue was explained in the text and an attempt was made to use the word “pseudo-chalcone-amide” in these cases. However, most studies in this field are related to the optimization of first-generation compounds with a full chalcone-amide backbone, which seems to create more effective interactions in the active site and have stronger/more promising effects. Importantly, SAR evaluation shows that when this backbone is basically changed and comes out of the chalcone-amide forms (either by expanding the backbone, compressing it, or removing the B-ring), the PLpro inhibitory activity is strongly reduced or completely eliminated. In addition, activity is lost when the chalcone-amide (N-benzylbenzamide) backbone is converted to the “N, 2-diphenylacetamide” backbone by inverting the amide orientation (see general structure 78). These SAR findings suggest that the use of this word (chalcone-amide) is the best possible option for the backbone structure of compounds reviewed in this study.
Although optimization of these chalcone-amide-based compounds has led to achieving the highly potent PLpro inhibitors with the sub-micromolar IC50 values, further efforts appear to be needed to achieve more effective inhibitors with clinically applicable use. In this study, the chalcone-amide backbone was introduced as a unique skeleton for the design and development of SARS-CoV-2 inhibitors. A deep understanding of the SAR and the binding mode of these structures in the SARS-CoV-2 PLpro active site can aid the future development of anti-COVID-19 agents.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Data availability
Information used in the current article was collected from the creditable internet databases such as Elsevier, PubMed, Web of Science, Wiley Online Library, Europe PMC, etc.
References
- 1.Saif L.J., Wang Q., Vlasova A.N., Jung K., Xiao S. 2019. Coronaviruses, Diseases of Swine; pp. 488–523. [Google Scholar]
- 2.Amin S.A., Banerjee S., Gayen S., Jha T. Protease targeted COVID-19 drug discovery: what we have learned from the past SARS-CoV inhibitors? Eur. J. Med. Chem. 2021;215 doi: 10.1016/j.ejmech.2021.113294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kankanamalage A.C.G., Kim Y., Damalanka V.C., Rathnayake A.D., Fehr A.R., Mehzabeen N., Battaile K.P., Lovell S., Lushington G.H., Perlman S. Structure-guided design of potent and permeable inhibitors of MERS coronavirus 3CL protease that utilize a piperidine moiety as a novel design element. Eur. J. Med. Chem. 2018;150:334–346. doi: 10.1016/j.ejmech.2018.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lu R., Zhao X., Li J., Niu P., Yang B., Wu H., Wang W., Song H., Huang B., Zhu N. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395:565–574. doi: 10.1016/S0140-6736(20)30251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yao H., Song Y., Chen Y., Wu N., Xu J., Sun C., Zhang J., Weng T., Zhang Z., Wu Z. Molecular architecture of the SARS-CoV-2 virus. Cell. 2020;183:730–738. doi: 10.1016/j.cell.2020.09.018. e713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yadav R., Chaudhary J.K., Jain N., Chaudhary P.K., Khanra S., Dhamija P., Sharma A., Kumar A., Handu S. Role of structural and non-structural proteins and therapeutic targets of SARS-CoV-2 for COVID-19. Cells. 2021;10:821. doi: 10.3390/cells10040821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Amin S.A., Jha T. Fight against novel coronavirus: a perspective of medicinal chemists. Eur. J. Med. Chem. 2020;201 doi: 10.1016/j.ejmech.2020.112559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsai P.-H., Wang M.-L., Yang D.-M., Liang K.-H., Chou S.-J., Chiou S.-H., Lin T.-H., Wang C.-T., Chang T.-J. Genomic variance of Open Reading Frames (ORFs) and Spike protein in severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) J. Chin. Med. Assoc. 2020;83:725. doi: 10.1097/JCMA.0000000000000387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baranov P.V., Henderson C.M., Anderson C.B., Gesteland R.F., Atkins J.F., Howard M.T. Programmed ribosomal frameshifting in decoding the SARS-CoV genome. Virology. 2005;332:498–510. doi: 10.1016/j.virol.2004.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wong J.P., Damania B. SARS-CoV-2 dependence on host pathways. Science. 2021;371:884–885. doi: 10.1126/science.abg6837. [DOI] [PubMed] [Google Scholar]
- 11.Ziebuhr J., Snijder E.J., Gorbalenya A.E. Virus-encoded proteinases and proteolytic processing in the Nidovirales. Microbiology. 2000;81:853–879. doi: 10.1099/0022-1317-81-4-853. [DOI] [PubMed] [Google Scholar]
- 12.Klemm T., Ebert G., Calleja D.J., Allison C.C., Richardson L.W., Bernardini J.P., Lu B.G., Kuchel N.W., Grohmann C., Shibata Y. Mechanism and inhibition of the papain‐like protease, PLpro, of SARS‐CoV‐2. EMBO J. 2020;39 doi: 10.15252/embj.2020106275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kandeel M., Kitade Y., Fayez M., Venugopala K.N., Ibrahim A. The emerging SARS‐CoV‐2 papain‐like protease: its relationship with recent coronavirus epidemics. J. Med. Virol. 2021;93:1581–1588. doi: 10.1002/jmv.26497. [DOI] [PubMed] [Google Scholar]
- 14.Shin D., Mukherjee R., Grewe D., Bojkova D., Baek K., Bhattacharya A., Schulz L., Widera M., Mehdipour A.R., Tascher G. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature. 2020;587:657–662. doi: 10.1038/s41586-020-2601-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heaton S.M., Borg N.A., Dixit V.M. Ubiquitin in the activation and attenuation of innate antiviral immunity. J. Exp. Med. 2016;213:1–13. doi: 10.1084/jem.20151531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Daczkowski C.M., Dzimianski J.V., Clasman J.R., Goodwin O., Mesecar A.D., Pegan S.D. Structural insights into the interaction of coronavirus papain-like proteases and interferon-stimulated gene product 15 from different species. J. Mol. Biol. 2017;429:1661–1683. doi: 10.1016/j.jmb.2017.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cho C.C., Li S.G., Lalonde T.J., Yang K.S., Yu G., Qiao Y., Xu S., Ray Liu W. Drug repurposing for the SARS‐CoV‐2 papain‐like protease. ChemMedChem. 2022;17 doi: 10.1002/cmdc.202100455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim D., Lee J.-Y., Yang J.-S., Kim J.W., Kim V.N., Chang H. The architecture of SARS-CoV-2 transcriptome. Cell. 2020;181:914–921. doi: 10.1016/j.cell.2020.04.011. e910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., Zhang B., Li X., Zhang L., Peng C. Structure of M pro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582:289–293. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 20.Ratia K., Pegan S., Takayama J., Sleeman K., Coughlin M., Baliji S., Chaudhuri R., Fu W., Prabhakar B.S., Johnson M.E. A noncovalent class of papain-like protease/deubiquitinase inhibitors blocks SARS virus replication. Proc. Natl. Acad. Sci. USA. 2008;105:16119–16124. doi: 10.1073/pnas.0805240105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Valipour M., Zarghi A., Ebrahimzadeh M.A., Irannejad H. Therapeutic potential of chelerythrine as a multi-purpose adjuvant for the treatment of COVID-19. Cell Cycle. 2021;20:2321–2336. doi: 10.1080/15384101.2021.1982509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Valipour M. ACS Pharmacology & Translational Science; 2022. Different Aspects of Emetine's Capabilities as a Highly Potent SARS-CoV-2 Inhibitor against COVID-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xiang R., Yu Z., Wang Y., Wang L., Huo S., Li Y., Liang R., Hao Q., Ying T., Gao Y. Acta Pharmaceutica Sinica B; 2021. Recent Advances in Developing Small-Molecule Inhibitors against SARS-CoV-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ahmad I. The race to treat COVID-19: potential therapeutic agents for the prevention and treatment of SARS-CoV-2. Eur. J. Med. Chem. 2021;213 doi: 10.1016/j.ejmech.2021.113157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.M. Valipour, H. Irannejad, S. Emami, Papaverine, a Promising Therapeutic Agent for the Treatment of COVID‐19 Patients with Underlying Cardiovascular Diseases (CVDs), Drug Development Research. [DOI] [PMC free article] [PubMed]
- 26.McClain C.B., Vabret N. SARS-CoV-2: the many pros of targeting PLpro. Signal Transduct. Targeted Ther. 2020;5:1–2. doi: 10.1038/s41392-020-00335-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Al-Nakib W., Tyrrell D.A. A ‘new’generation of more potent synthetic antirhinovirus compounds: comparison of their MICs and their synergistic interactions. Antivir. Res. 1987;8:179–187. doi: 10.1016/0166-3542(87)90072-6. [DOI] [PubMed] [Google Scholar]
- 28.Ninomiya Y., Shimma N., Ishitsuka H. Comparative studies on the antirhinovirus activity and the mode of action of the rhinovirus capsid binding agents, chalcone amides. Antivir. Res. 1990;13:61–74. doi: 10.1016/0166-3542(90)90022-y. [DOI] [PubMed] [Google Scholar]
- 29.Arbuckle J.H., Gardina P.J., Gordon D.N., Hickman H.D., Yewdell J.W., Pierson T.C., Myers T.G., Kristie T.M. Inhibitors of the histone methyltransferases EZH2/1 induce a potent antiviral state and suppress infection by diverse viral pathogens. mBio. 2017;8 doi: 10.1128/mBio.01141-17. e01141-01117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shen Z., Ratia K., Cooper L., Kong D., Lee H., Kwon Y., Li Y., Alqarni S., Huang F., Dubrovskyi O. bioRxiv; 2021. Potent, Novel SARS-CoV-2 PLpro Inhibitors Block Viral Replication in Monkey and Human Cell Cultures. [Google Scholar]
- 31.Zhu H., Li W., Shuai W., Liu Y., Yang L., Tan Y., Zheng T., Yao H., Xu J., Zhu Z. Discovery of novel N-benzylbenzamide derivatives as tubulin polymerization inhibitors with potent antitumor activities. Eur. J. Med. Chem. 2021;216 doi: 10.1016/j.ejmech.2021.113316. [DOI] [PubMed] [Google Scholar]
- 32.Valipour M. Recent advances of antitumor shikonin/alkannin derivatives: a comprehensive overview focusing on structural classification, synthetic approaches, and mechanisms of action. Eur. J. Med. Chem. 2022 doi: 10.1016/j.ejmech.2022.114314. [DOI] [PubMed] [Google Scholar]
- 33.Shao Y.-Y., Yin Y., Lian B.-P., Leng J.-F., Xia Y.-Z., Kong L.-Y. Synthesis and biological evaluation of novel shikonin-benzo [b] furan derivatives as tubulin polymerization inhibitors targeting the colchicine binding site. Eur. J. Med. Chem. 2020;190 doi: 10.1016/j.ejmech.2020.112105. [DOI] [PubMed] [Google Scholar]
- 34.Li J., Zhou X., Zhang Y., Zhong F., Lin C., McCormick P.J., Jiang F., Luo J., Zhou H., Wang Q. Crystal structure of SARS-CoV-2 main protease in complex with the natural product inhibitor shikonin illuminates a unique binding mode. Sci. Bull. 2021;66:661. doi: 10.1016/j.scib.2020.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Serrao E., Odde S., Ramkumar K., Neamati N. Raltegravir, elvitegravir, and metoogravir: the birth of" me-too" HIV-1 integrase inhibitors. Retrovirology. 2009;6:1–14. doi: 10.1186/1742-4690-6-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hajimahdi Z., Zarghi A. Progress in HIV-1 integrase inhibitors: a review of their chemical structure diversity. Iran. J. Pharm. Res. (IJPR): Iran. J. Pharm. Res. (IJPR) 2016;15:595. [PMC free article] [PubMed] [Google Scholar]
- 37.Peralta-Garcia A., Torrens-Fontanals M., Stepniewski T.M., Grau-Expósito J., Perea D., Ayinampudi V., Waldhoer M., Zimmermann M., Buzón M.J., Genescà M. Entrectinib—a SARS-CoV-2 inhibitor in human lung tissue (HLT) cells. Int. J. Mol. Sci. 2021;22 doi: 10.3390/ijms222413592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gao X., Qin B., Chen P., Zhu K., Hou P., Wojdyla J.A., Wang M., Cui S. Crystal structure of SARS-CoV-2 papain-like protease. Acta Pharm. Sin. B. 2021;11:237–245. doi: 10.1016/j.apsb.2020.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Osipiuk J., Azizi S.-A., Dvorkin S., Endres M., Jedrzejczak R., Jones K.A., Kang S., Kathayat R.S., Kim Y., Lisnyak V.G. Structure of papain-like protease from SARS-CoV-2 and its complexes with non-covalent inhibitors. Nat. Commun. 2021;12:1–9. doi: 10.1038/s41467-021-21060-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fu Z., Huang B., Tang J., Liu S., Liu M., Ye Y., Liu Z., Xiong Y., Zhu W., Cao D. The complex structure of GRL0617 and SARS-CoV-2 PLpro reveals a hot spot for antiviral drug discovery. Nat. Commun. 2021;12:1–12. doi: 10.1038/s41467-020-20718-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhou P., Yang X.-L., Wang X.-G., Hu B., Zhang L., Zhang W., Si H.-R., Zhu Y., Li B., Huang C.-L. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Makar S., Saha T., Singh S.K. Naphthalene, a versatile platform in medicinal chemistry: sky-high perspective. Eur. J. Med. Chem. 2019;161:252–276. doi: 10.1016/j.ejmech.2018.10.018. [DOI] [PubMed] [Google Scholar]
- 43.Valipour M., Naderi N., Heidarli E., Shaki F., Motafeghi F., Amiri F.T., Emami S., Irannejad H. Design, synthesis and biological evaluation of naphthalene-derived (arylalkyl) azoles containing heterocyclic linkers as new anticonvulsants: a comprehensive in silico, in vitro, and in vivo study. Eur. J. Pharmaceut. Sci. 2021;166 doi: 10.1016/j.ejps.2021.105974. [DOI] [PubMed] [Google Scholar]
- 44.Ghosh A.K., Takayama J., Aubin Y., Ratia K., Chaudhuri R., Baez Y., Sleeman K., Coughlin M., Nichols D.B., Mulhearn D.C. Structure-based design, synthesis, and biological evaluation of a series of novel and reversible inhibitors for the severe acute respiratory syndrome− coronavirus papain-like protease. J. Med. Chem. 2009;52:5228–5240. doi: 10.1021/jm900611t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ghosh A.K., Takayama J., Rao K.V., Ratia K., Chaudhuri R., Mulhearn D.C., Lee H., Nichols D.B., Baliji S., Baker S.C. Severe acute respiratory syndrome coronavirus papain-like novel protease inhibitors: design, synthesis, protein− ligand X-ray structure and biological evaluation. J. Med. Chem. 2010;53:4968–4979. doi: 10.1021/jm1004489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Báez-Santos Y.M., Barraza S.J., Wilson M.W., Agius M.P., Mielech A.M., Davis N.M., Baker S.C., Larsen S.D., Mesecar A.D. X-ray structural and biological evaluation of a series of potent and highly selective inhibitors of human coronavirus papain-like proteases. J. Med. Chem. 2014;57:2393–2412. doi: 10.1021/jm401712t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jacobs J., Grum-Tokars V., Zhou Y., Turlington M., Saldanha S.A., Chase P., Eggler A., Dawson E.S., Baez-Santos Y.M., Tomar S. Discovery, synthesis, and structure-based optimization of a series of N-(tert-butyl)-2-(N-arylamido)-2-(pyridin-3-yl) acetamides (ML188) as potent noncovalent small molecule inhibitors of the severe acute respiratory syndrome coronavirus (SARS-CoV) 3CL protease. J. Med. Chem. 2013;56:534–546. doi: 10.1021/jm301580n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee H., Lei H., Santarsiero B.D., Gatuz J.L., Cao S., Rice A.J., Patel K., Szypulinski M.Z., Ojeda I., Ghosh A.K. Inhibitor recognition specificity of MERS-CoV papain-like protease may differ from that of SARS-CoV. ACS Chem. Biol. 2015;10:1456–1465. doi: 10.1021/cb500917m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Welker A., Kersten C., Müller C., Madhugiri R., Zimmer C., Müller P., Zimmermann R., Hammerschmidt S., Maus H., Ziebuhr J. Structure‐activity relationships of benzamides and isoindolines designed as SARS‐CoV protease inhibitors effective against SARS‐CoV‐2. ChemMedChem. 2021;16:340–354. doi: 10.1002/cmdc.202000548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gehling V.S., Vaswani R.G., Nasveschuk C.G., Duplessis M., Iyer P., Balasubramanian S., Zhao F., Good A.C., Campbell R., Lee C. Discovery, design, and synthesis of indole-based EZH2 inhibitors. Bioorg. Med. Chem. Lett. 2015;25:3644–3649. doi: 10.1016/j.bmcl.2015.06.056. [DOI] [PubMed] [Google Scholar]
- 51.Ma C., Sacco M.D., Xia Z., Lambrinidis G., Townsend J.A., Hu Y., Meng X., Szeto T., Ba M., Zhang X. ACS Central Science; 2021. Discovery of SARS-CoV-2 Papain-like Protease Inhibitors through a Combination of High-Throughput Screening and a FlipGFP-Based Reporter Assay. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rut W., Lv Z., Zmudzinski M., Patchett S., Nayak D., Snipas S.J., El Oualid F., Huang T.T., Bekes M., Drag M. Activity profiling and crystal structures of inhibitor-bound SARS-CoV-2 papain-like protease: a framework for anti–COVID-19 drug design. Sci. Adv. 2020;6 doi: 10.1126/sciadv.abd4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shan H., Liu J., Shen J., Dai J., Xu G., Lu K., Han C., Wang Y., Xu X., Tong Y. Cell chemical biology; 2021. Development of Potent and Selective Inhibitors Targeting the Papain-like Protease of SARS-CoV-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Information used in the current article was collected from the creditable internet databases such as Elsevier, PubMed, Web of Science, Wiley Online Library, Europe PMC, etc.