

Abstract
Dengue is one of the most typical viral infection categorized in the Neglected Tropical Diseases (NTDs). It is transmitted via the female Aedes aegypti mosquito to humans and majorly puts risk to the lives of more than half of the world. Recent advancements in medicinal chemistry have led to the design and development of numerous potential heterocyclic scaffolds as antiviral drug candidates for the inhibition of the dengue virus (DENV). Thus, in this review, we have discussed the significance of inhibitory and antiviral activities of nitrogen, oxygen, and mixed (nitrogen-sulfur and nitrogen-oxygen) heterocyclic scaffolds that are published in the last seven years (2016–2022). Furthermore, we have also discussed the probable mechanisms of action and the diverse structure-activity relationships (SARs) of the heterocyclic scaffolds. In addition, this review has elaborately outlined the mechanism of viral infection and the life cycle of DENV in the host cells. The wide set of heterocycles and their SARs will aid in the development of pharmaceuticals that will allow the researchers to synthesize the promising anti-dengue drug candidate in the future.
Keywords: Neglected tropical diseases, Dengue virus, Heterocyclic scaffolds, Antiviral medication, Surface activity, Structure-activity relationship
Graphical abstract

1. Introduction
Dengue fever has been a significant disease of concern in tropical and subtropical countries since the twentieth century, particularly since the end of World War II. According to a survey report, data collected and interpreted that approximately three hundred and ninety million people are infected, and forty thousand people die annually from this viral infection. Presently, Asia and Latin America are regions of concern due to the high number of deaths among kids, youths, and aged people caused by dengue [1]. The arboviral infection caused by this inanimate viral particle spreads through infected arthropods. It is transmitted by two distinguished female mosquitoes named Aedes aegypti and Aedes albopictus [2,3]. Both the mosquito species perform the role of viral transmitting agents that carry the viral RNA to the human body. DENV is associated with the Flaviviridae family which is composed of a positive (+ve) sense of single-stranded ribonucleic acid (ssRNA). Around seventy diverse virus types with enveloped ssRNA, from the Flaviviridae family, are located inside mosquitoes and ticks. These arthropods are effective and capable vectors that generate severe illness in human beings [4]. Flavivirus is liable for causing high mortality and morbidity rates throughout the planet. Besides DENV, several others in this family also continue to severely infect human beings. These include Japanese encephalitis virus (JEV), Yellow fever virus (YFV), West Nile virus (WNV), Tick-borne encephalitis viruses (TBEV), Zika virus (ZIKV), and St. Louis encephalitis virus (SLEV) [2,5]. Based on the structural arrangement of the surface antigen of the DENV, it is differentiated into four serotype varieties. These are DENV-1, DENV-2, DENV-3, and DENV-4. These serotypes induce specific antibodies when an infection has occurred [4,6]. Furthermore, the latest serotype of the dengue virus is DENV-5, which has been reported in the year 2013 in October [7,8]. Currently, there is a lack of adequate drug candidates to inhibit the DENV from its viral infection. Moreover, dengue vaccines in different nations have encountered extremely adverse responses. Therefore, an urgency exists to discover potential and effective drugs to treat DENV and contain its mobility [9]. Over the past few years, several investigations have revealed that heterocyclic compounds have potent antiviral activity in inhibiting DENV.
Heterocyclic compounds are organic compounds that have a cyclic ring system and one or more heteroatoms. Usually, heteroatoms are nitrogen (N), oxygen (O), and sulfur (S). They have already been employed in various biological domains due to their significance in the inhibition of numerous infections [10,11]. Moreover, these heterocyclic compounds are also discovered as primary scaffolds in numerous biological micro-molecules like hemoglobin (Hb), deoxyribonucleic acid (DNA), ribonucleic acid (RNA), and vitamins, hormones, and many more. It is important to note that these structures are also found in many United States-Food and Drug Administration (US-FDA)-approved drug molecules to treat various diseases. Significant and potential biological efficacies such as anti-fungal, anti-inflammatory, anti-bacterial, anti-viral, anti-tumor, anti-diabetic, anti-allergic, anti-oxidant, anti-convulsant, inhibitory effects in enzymatic reactions have been noted in the N-, O-, and mixed (N, S- and N, O-containing) heterocyclic scaffolds [[12], [13], [14], [15], [16], [17], [18]]. The advancement of therapeutic compounds in the treatment of Dengue over the last seven years is illustrated in Fig. 1 .
Fig. 1.

Timeline showing the advancement of therapeutic compounds in the treatment of Dengue from 2015 to 2021.
This review covers the vast range of significant inhibitory effects and antiviral activity of many heterocyclic scaffolds including N-, O-, and mixed (N, S- and N, O-containing) in dengue treatment. The current review focuses on the potential mechanisms of action and diverse SAR of compounds containing N-, O-, and mixed (N, S- and N, O-containing) heterocyclic scaffolds such as Triazoles, Pyrazoles, Imidazoles, Pyridines, Pyrazines, Pyrimidines, Indoles, Quinolines, Quinazolines, Coumarins, Thiazoles or Benzothiazoles, Thiazolidinones, Benzothiazines, and Oxadiazoles. The review will be beneficial to the researchers working in the field of organic chemistry, medicinal chemistry, and pharmacology.
2. Mechanism of viral infection
The transmission vector for DENV is by the female Aedes mosquitoes, of which A. aegypti is the popular vector while A. albopictus species is another competent vector. A blood meal from a dengue patient during the first week of viremia infects the DENV vectors [19]. Having bit an infected patient, DENV passes into the bloodstream and attaches itself to a cell surface of the new vector via a potential receptor (Fc receptor) (Fig. 2 ). The spherical structure of DENV has an outer shell of proteins surrounding an envelope of capsid proteins. These capsid proteins envelope the viral genome. Having attached to a receptor, the virus enters the cell via the endosomal pathway [20]. This leads to the transport of DENV via endosomes. Endosomal acidification [21] leads to the fusion of the viral envelope with the endosomal membrane [22] within the cell. The viral nucleocapsid is released into the mosquito cell cytosol. The viral nucleocapsid uncoats itself releasing the positive single-stranded RNA genome. The RNA assumes multiple linear and circular conformations which interact with the host cell proteins. The structures are specific for invertebrate and vertebrate host cells. A specific RNA conformation, usually stem-loop in the untranslated regions (UTRs), hijacks the host protein factory. This leads to viral genome replication. The viral polypeptide is a large polyprotein complex. This complex is processed and cleaved by the host cell endoplasmic reticulum giving rise to ten DENV proteins. These viral proteins help in the assembly of replicated viral genomes in the rough endoplasmic reticulum forming immature virions. These immature viruses are transported through the Golgi complex, maturing them into the infectious form, and releasing them to further infect other cells [22].
Fig. 2.

The detailed life cycle of DENV.
3. Detailed life cycle of DENV inside the cell cytoplasm
Like other pathogenic viruses, DENV adopts multiple steps in its life cycle (Fig. 2) [23] to infect its hosts. The life cycle consists of viral entry, genome replication, protein processing, and virion assembly, followed by its release. DENV is a Flavivirus, its life cycle is activated by the normal cellular endocytosis of a host cell. This is mediated by the fusion of the viral envelope with the host cell membrane, leading to the formation of a small DENV-containing endosomal pouch. Research shows that two conditions favor the viral escape from the endosome. A region of low pH surrounds the endosome, while a high negative charge on the endosomal membrane facilitates the process. A low pH activates a viral glycoprotein which mediates the fusion of the viral and endosomal membrane, discharging the nucleocapsid. Other processes, which remain yet to be confirmed, govern the nucleocapsid disassembly and subsequent discharge of viral ssRNA into the cytoplasm [24]. Multiple studies have shown that DENV may utilize a single or multiple cell surface receptor. A specific receptor is yet to be determined. The identified receptors include the dendritic cell-specific intracellular adhesion molecule-3-grabbing non-integrin (DC-SIGN). In humans, DENV exploits the c-type lectin domain containing 5A (CLEC5A) receptor of the macrophage [[25], [26], [27]]. Common glycoprotein receptors such as heparan sulfate and mannose receptors are also exploited [28,29]. After the endocytic process and endosomal acidification, following viral genome release, the viral ssRNA acts as a messenger RNA (mRNA). Utilizing the host protein machinery, the viral ssRNA is translated giving a precursor polyprotein, which is processed and cleaved into ten viral proteins (3 structural and 7 nonstructural proteins). The non-structural proteins mediate the synthesizing process of a complementary negative (-ve) strand, giving a double-stranded RNA (dsRNA). This intermediate strand performs the role of a template for the (+ve) strand synthesizing process for new viral RNA. This process is repeated with multiple rounds of transcription and translation to produce a multitude of new DENV particles [30]. Post protein production and genome replication, the viral structural proteins control new particle assembly. In the endoplasmic reticulum (ER), capsid (C) and precursor-Membrane (prM) proteins ensure the formation and unidirectional transport of pre-mature viral particles across the vesicular system. The capsid proteins utilize the host ER membrane and glycoproteins to form the viral envelope. Pre-mature viral particles drive via a secretory route and the Trans-Golgi network (TGN), which retains acidic conditions. The prM is a pH-dependent (acidic) precursor membrane protein which has furin cleavage sites. It utilizes the host furin protease giving rise to the mature (M) protein during the final stage of viral maturation and release from the host cell surface [31]. Studies conducted on prM have shown its importance in shielding the viral envelope proteins and preventing their premature fusion and viral secretion [[32], [33], [34]]. This leads to a successful viral maturation sheathed with the host vesicular membrane and budding off from the host cell.
4. Common symptoms of dengue infection
DENV causes fever and flu-like symptoms. In infected individuals, the symptoms appear for four to five days and last for approximately ten days after an infected mosquito bite. These individuals suffer from high fever (40 °C), severe headaches, musculoskeletal pain, vomiting, swollen glands, rash, or fatigue. Normal dengue fever may however develop and turn fatal into dengue haemorrhagic fever (DHF) if the patient is not given due care after the appearance of symptoms. DHF appears three to seven days post-normal dengue fever. It has characteristic symptoms of abdominal pain, decrease in basal temperature (<38 °C), bleeding gums, fatigue, and continuous vomiting with or without blood. This occurs as a result of systemic inflammation due to plasma leakage, accumulation of fluid, difficulty in breathing, organ dysfunction (liver augmentation and cardiovascular system (CVS) collapse), and reduced oxygen delivery [35].
5. Challenges in anti-dengue drug discovery
As of yet, a specific treatment or vaccine for dengue does not exist. Thus, options of care and precaution have been laid down as alternatives. Given the inflammatory conditions and lack of treatment options, patients must take plenty of rest, stay hydrated and use pain killers such as acetaminophen, a popular non-steroidal anti-inflammatory drug (NSAID). Other pain killers such as aspirin, naproxen, and ibuprofen may cause bleeding and hence should be avoided. Patients must avoid any future mosquito bites during the first week of infection. This curtails the spread of DENV. The above-mentioned medications may not be effective for DHF. In the event of an early clinical diagnosis of DHF, the existing treatment option of fluid replacement therapy must be started. Despite progressive research, there remains a lack of a definite treatment and vaccine for DENV. Thus, the disease continues to spread and affect people worldwide. Hence, there is an increasing ever need for dengue-specific therapeutic molecules. The US-FDA has previously approved the first dengue vaccine developed at Sanofi, named Dengvaxia in 2019. The vaccine prevents re-infection in administered individuals against all four serotypes of the DENV. Yet, the side effects caused by Dengvaxia has limited its use. To date, there exists no approved scientific treatment option for dengue and antivirals against DENV.
The main challenges in dengue treatment start from the discovery of potential drug candidates and their development, which needs to be safe and effective in curing the infection in the long term. In the process of drug discovery and its development, numerous challenges need to be addressed such as designing the drug, preclinical experiments, and the clearance of clinical trial guidelines. Drug repurposing is an important aspect of developing new drug candidates. The purpose of drugs can be identified by targeting the three mechanistic processes, namely targeting the host proteins, viral proteins, and antibodies. Of the three, selective targeting of the host cellular proteins, which may be intracellular at times, is more challenging than developing drug molecules targeting the viral proteins. Despite the feasibility of targeting viral proteins, the lack of studies on various viral infection processes of the host body and the interaction of viral proteins with the host cellular pathway are key limitations in drug development against them. In antibody-mediated viral targeting, an antibody should be designed such that it effectively opsonizes and neutralizes all the DENV serotypes. However, in current practices, there exist different antibody types which target the four serotypes. Administering an antibody therapy via an intravenous route is laborious and may be painful to the patients. Moreover, designing a novel antibody, along with the facilities required for its production raises the cost of antibody-mediated protection, limiting its affordability and popularity. Acute dengue infection is of limited duration and not chronic in nature. It usually lasts not more than a week. Hence, a drug candidate needs to be responsive and specific in curtailing and clearing the infection rapidly [36,37].
Furthermore, animal models need to be developed to study the infection mechanism and drug efficacy. This is a major challenge in the pre-clinical setup to study anti-DENV molecules since except for humans, most DENV hosts have limited infectivity with fast clearance. To overcome this limitation, a group of scientists investigated the infectivity by developing a murine model (AG129 mouse). This model expressed similar infectivity profiles as human hosts for DENV, however, they had a reduced number and duration of viremia [38]. Given the rapid decline in viremia in acute dengue patients, dosing to the patient in the early stages of infection for clinical trials is another major limitation.
In addition, current diagnostic methods, fail to differentiate between DENV fever and other common febrile diseases. Distinguishing primary or secondary infection is limited due to insufficient data generated from Immunoglobin M/Immunoglobin G (IgM/IgG) measurements. Also, given the acute nature of the disease, the viral load remains high during the initial days of infection and gradually reduces within a week. The limited technological and diagnostic methods limit researchers with the inability to assess how and when the viral number is reduced in response to the drugs. Another aspect of DENV infection is that infection profile varies between demographic, and also between age groups. The majority of the infected population exists in South-East Asian countries with both pediatric and adult patients. Patients having severe infection profiles differ in their physiology and immune strengths in comparison to patients with acute infections. Hence, clinical trials need to address pediatric patients as well as adjust doses suitable to patients with comorbidities [39].
6. Inhibitory sites of dengue virus
The DENV gets entered into the host/targeted cell through the endocytosis process. Once it enters the targeted cell, it becomes fused with the endosomal membrane (EM) with the help of a pH-dependent process. Then, it releases its viral ssRNA into the cytoplasm in the host cell, where the translation process occurs of the polyprotein [40,41]. Polyproteins act as a precursor that helps to cleave both host and viral protease, which further leads to the replication cycle of the RNA inside the cell. Generally, polyproteins consist of three structural proteins and seven non-structural proteins. The structural proteins include C protein, prM protein, and envelope (E) protein. On the other hand, the non-structural proteins contain NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5 (Fig. 3 ). [42]. The molecular activity of structural and non-structural proteins is shown in Table 1, Table 2 It is possible to inhibit dengue infection by deactivating the molecular activities of these structural and non-structural proteins.
Fig. 3.

Genomic arrangement of DENV.
Table 1.
Molecular activity of structural proteins.
| Structural proteins | ||
|---|---|---|
| Structural proteins | Molecular functions | References |
| C protein |
|
[[43], [44], [45], [46], [47]] |
| prM protein |
|
[[48], [49], [50]] |
| E protein |
|
[51,52] |
Table 2.
Molecular activity of non-structural proteins.
| Non-structural proteins | ||
|---|---|---|
| Non-structural protein | Molecular functions | References |
| NS1 |
|
[[53], [54], [55], [56]] |
| NS2A |
|
[[57], [58], [59]] |
| NS2B |
|
[[60], [61], [62], [63]] |
| NS3 |
|
[[64], [65], [66], [67], [68]] |
| NS4A |
|
[[69], [70], [71]] |
| NS4B |
|
[[72], [73], [74], [75]] |
| NS5 |
|
[[76], [77], [78]] |
The C protein helps in the compact binding of the virus to the cellular membrane of the host cell, and the E protein triggers the fusion process between the host cellular membrane and the viral spike proteins. Similarly, non-structural proteins perform a significant function in the replication process of viral ssRNA inside the host cells. Several investigations suggest that there are some possible targets in Table 1, Table 2 in the inhibition of structural and non-structural proteins of DENV.
7. Nitrogen (N)-containing heterocyclic scaffolds for the inhibition of dengue virus
7.1. Triazoles
Triazoles are the 5-membered heterocyclic compounds enclosed by three nitrogen atoms and two carbon atoms in the ring system [79]. These are classified as aromatic heterocyclic compounds as they follow Huckel's rule. There exist two isomeric forms of triazoles: 1,2,3-triazoles and 1,2,4-triazoles. In recent years, a huge amount of studies have been devoted to the biological and pharmacological activities of both the triazole-derivatives [80,81]. Substituted triazole-based heterocyclic compounds display a wide array of biological activity among the numerous N-containing heterocyclic compounds. The noteworthy biological activity can be attributed to the structural and electronic properties of the triazole moiety [82]. The variations of the triazole ring substitution have resulted in upgraded effectiveness and reduced toxicity in various drug candidates [83]. Substituted triazole-based heterocycles have several biological activities such as anti-proliferative, anti-convulsant, anti-microbial, anti-neoplastic, anti-viral, analgesic, anti-inflammatory, anti-cancer, and anti-malarial activities [[84], [85], [86], [87], [88], [89]].
However, there has been a significant account of the anti-dengue action by 1,2,3-triazole-based compounds [90]. For instance, Vernekar et al. (2015) reported a 5′-silylated 3′-azidothymidine derived 1,2,3-triazole nucleoside to inhibit dengue and WNV. The authors revealed that the substituted triazole-based compounds display anti-viral properties by binding to the methyl transferase (MTase) inhibitor. The utilization of molecular modeling and S-Adenosylmethionine (SAM) binding assay further supported their findings. The researchers conducted their investigation by observing the action of Renilla luciferase present in the DENV-subgenomic replicon on the baby hamster kidney (BHK) cell line. The authors have approached a three-step structure-based modification on the substituted-triazole moiety. The first set of modifications was obtained by protecting the 5′-OH position with various silyl protecting groups (Table 3 ). The second-base modification was obtained by substituting different groups on the 3′-position of the triazole ring system (Table 4 ) [91].
Table 3.
Effect of 5′-OH silylated protecting group of the substituted-triazole moiety for the inhibition of DENV.
| Compound No. | Heterocyclic Scaffolds | %Inhibition | %Cell Viability |
|---|---|---|---|
| 1 | 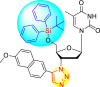 |
100% | 11% |
| 2 | 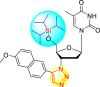 |
98% | 53% |
| 3 | 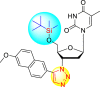 |
85% | 86% |
| 4 | 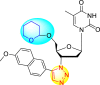 |
21% | 92% |
| 5 | 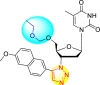 |
13% | 95% |
| 6 | 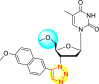 |
0% | 100% |
| 7 | 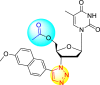 |
0% | 97% |
Table 4.
Effect of 3′-substitution on the triazole molecular backbone for the inhibition of DENV.
| Compound No. | Heterocyclic Scaffolds | %Inhibition | %Cell Viability |
|---|---|---|---|
| 8 | 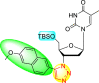 |
85% | 86% |
| 9 | 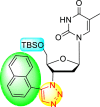 |
99% | 60% |
| 10 | 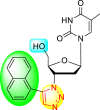 |
0% | 90% |
| 11 | 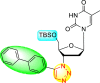 |
99% | 47% |
| 12 | 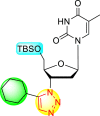 |
75% | 93% |
| 13 | 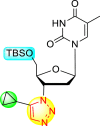 |
64% | 93% |
| 14 | 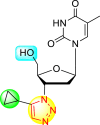 |
47% | 100% |
| 15 | 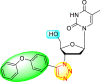 |
0% | 100% |
| 16 | 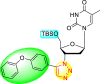 |
95% | 73% |
| 17 | 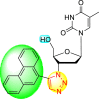 |
21% | 100% |
From the first set of observations, seven diverse 5′-OH silylated protecting groups were examined against the DENV strain at a concentration of 10 μM (Table 3). The SAR revealed that the bulkier silylated protecting groups such as tert-Butyldiphenylsilyl (TBDPS) 1, triisopropylsilyl (TIPS) 2, and tert-butyldimethylsilyl (TBS) 3 inhibited 85–100% against the DENV while the non-silylated protecting groups such as tetrahydropyran THP 4, ethoxymethyl 5, methyl 6, acetyl 7 did not produce significant anti-dengue activities (Table 3). Thus, it has been confirmed that the bulkiness of the protecting groups performs a significant function in DENV inhibition. However, the authors could not acquire the precise mechanism underlying the inhibition. They further extended their work by altering the substituents on the 3′-position of the triazole ring (Table 4). They observed that the 5′-TBS triazole moieties did not display cytotoxicity at 10 μM. The use of bulkier and aromatic side groups produced enhanced anti-dengue activities. However, in the case of an unprotected 5′-OH group, there is no significant inhibition with bulkier substituents at the 5′-position of the triazole group for the scaffolds 8–17 (Table 4). Thus, based on the SAR analysis, it is confirmed that the protection of the –OH group by hindered silyl substituents and the presence of bulkier groups on the triazole scaffold is essential for the anti-dengue action (Fig. 4 ).
Fig. 4.

SAR of the triazole substituents responsible for showing anti-dengue activities.
Vishvakarma et al. (2019) [92], proposed a piece of theoretical evidence that illustrates the biological inhibition of DENV. With the support of in-silico methodology, the authors determined to target the non-structural protein ns2B-nsP3 protease. The authors obtained a set of 138 heterocyclic scaffolds from the ZINC database and screened them for various physiological and bioavailability constraints. The various heterocyclic scaffolds taken in their studies include imidazole, triazole, oxazole, thiadiazole, and thiazolidine. Out of the 138 scaffolds, only 4 were taken for further confirmations, of which 18 and 19 had the 1,2,4-triazole core (Fig. 5 ). However, 18 was the most potent heterocyclic moiety having the highest binding affinity of −141.595 kJ/mol. The use of Density functional theory (DFT) and Molecular dynamics simulation (MDS) supported their observations. The literature search revealed that the active site of the ns2B-nsP3 protease was found to be His51, Asp75, Ser135, Gly153, Gly151, Pro132, Val154, and Leu128. Interestingly, the four scaffolds targeted the identical cavity, the functional area of the ns2B-nsP3 protease. The computational analysis reported H-bonding, electrostatic and hydrophobic interactions taking place between the heterocyclic scaffold and the ns2B-nsP3 protease. The major interactions for compound 18 are ARG-55, ASP-58, LYS-1061, GLU-54, SER-75, ILE-76, and GLY-96. For compound 19 are ARG-55, ASP-58, LEU-74, ALA-56, LYS-1061, GLU-54, SER-75, ILE-76 and GLY-96. Based on the SAR analysis, the presence of halogens on the benzene ring and different amide linkages have contributed to the binding affinity to the ns2B-nsP3 protease. The existence of two halogen atoms enhances the binding affinity slightly (Fig. 5).
Fig. 5.

SAR of the 1,2,4-Triazole scaffolds reported by Vishvakarma et al. for the inhibition of ns2B-nsP3 protease.
7.2. Pyrazoles
Pyrazoles are one of the most vital nitrogen-containing 5-membered heterocyclic scaffolds. These are aromatic and comprise two nitrogen atoms and three carbon atoms [93,94]. Pyrazoles are considered weak bases (pKb = 11.5) and belong to the alkaloids [95]. The distinctive properties and the pharmacological activity of the pyrazole scaffolds make them an important scaffold in pharmaceutical and medicinal chemistry [96,97]. Pyrazoles display extensive biological performances such as anti-viral, anti-cancer, anti-diabetic, anti-bacterial, anti-inflammatory, anti-fungal, and so on [[98], [99], [100]]. The variety of pyrazole-containing scaffolds showing anti-dengue activities is discussed in the upcoming sections.
Saudi et al. (2016) synthesized a few pyrazine-2,3-dicarboxamide and phthalamide substituted to pyrazole scaffold [101]. The authors successfully synthesized and evaluated their anti-viral activity against DENV and YFV. Based on the SAR (Fig. 6 ), it was observed that the scaffolds containing pyrazine-2,3-dicarboxamide 20 displayed lesser potency (half-maximal effective concentration (EC50) = 26.5 μM) than the scaffold containing the phthalamide substituent 24 (EC50 = 0.5 μM). Interestingly, the presence of electron-donating groups at the para-position of the two Ph-rings of the 2,3-dicarboxamide 21 displayed the highest potency (EC50 = 0.5 μM). Contrastingly, in the case of phthalamide group 24 one electron-donating (-Me) and one-electron (-F) withdrawing group demonstrated the highest potency (EC50 = 0.5 μM) (Table 5 ). Based on the SAR, the existence of electron-donating groups decreased the potency of the DENV while the electron-withdrawing group or unsubstituted phenyl tends to increase the potency of the synthesized scaffolds (Fig. 6).
Fig. 6.

SAR of the p-substituents on the Ph-ring of the pyrazole substituents for the inhibition of DENV.
Table 5.
Inhibition activity and cytotoxicity values of pyrazine 2,3-dicarboxamide and phthalamide-based pyrazole scaffolds.
| Compound No. | Heterocyclic Scaffolds | EC50 (μM) | CC50 (μM) | SI |
|---|---|---|---|---|
| 20 | 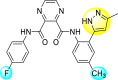 |
26.5 | >116 | >4 |
| 21 | 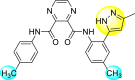 |
0.5 | >117 | >235 |
| 22 | 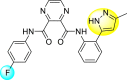 |
2.2 | >120 | >55 |
| 23 | 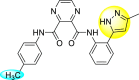 |
3.4 | 24 | 7 |
| 24 | 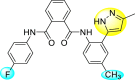 |
0.5 | >117 | >235 |
| 25 | 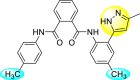 |
29 | >117 | >4 |
| 26 | 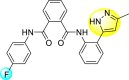 |
>120 | >120 | 1 |
| 27 | 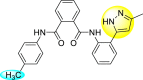 |
14.1 | >122 | >9 |
Lee et al. (2017) introduced a few diarylpyrazolylquinoline heterocyclic skeletons (Table 6 ), which displayed anti-dengue activity against the DENV2 serotype [102]. The anti-DENV action was investigated in Huh-DV-Fluc cells at a concentration of about 1 or 10 μM and compared to the reference compound ribavirin. Furthermore, they investigated both the in-vitro and in-vivo activity of the most potent compounds and found that the heterocyclic scaffold 28 inhibited about 15% of DENV2 at a concentration of 10 μM (Table 6). The inhibition activity is greater than its fluoro-substituent 29 and methoxy-substituent 30. Further, the inhibition activity was more than that of its positional-isomer 30. The positional isomers of pyrazole-derivatives are given in Fig. 8. Additionally, at 10 μM concentration, the inhibition activity of heterocyclic scaffold 31 was found to be approximately 25% on the DENV2 serotype. The values obtained are greater than those obtained for scaffolds 30 and 32. On the contrary, the inhibition increased even further when the phenyl ring was linked to the N3-position of the pyrazole ring with the introduction of –OCH3 and the –SO2NH2 groups at the 4-position of the phenyl ring of the compounds 37–42 (Table 6). From the various heterocyclic scaffolds, compound no. 42 demonstrated the highest inhibition of the DENV2 in the Huh-7 cells, followed by compound no. 39 and 36 (Fig. 7 ). Compound no. 42 (R1 = OMe, R2 = SO2NH2) displayed 85% inhibition while Compound no. 39 (R1 = OMe, R2 = OMe) displayed 66% inhibition and compound no. 36 (R1 = OMe, R2 = F) displayed 55% inhibition. The authors also determined the cytotoxicity of the cells by Cell Proliferation Kit II (XTT) assay at concentrations of 20 μM and 200 μM for 3 days. The results of compounds 36, 39, and 42 (Fig. 7) showed low cytotoxicity with cell viability greater than 50% at 200 μM concentration. On advanced experimentation on the inhibition activity of the compounds 36, 39, and 42; compound 42 displayed the lowest EC50 value of 0.81 and is the most potent among other heterocyclic scaffolds. Surprisingly, it was also more effective against the other serotypes of the DENV. Thus, based on SAR (Fig. 7), we conclude that the R2-position is the crucial position responsible for the inhibition of the DENV2 in the Huh-7 cells. The existence of –SO2NH2 at the R2-position exhibited the highest inhibition towards DENV. So, to obtain efficient inhibition, interchanging the substituents at R2-position may be beneficial to the researchers to report promising scaffolds for the inhibition of the DENV (Table 6).
Table 6.
Table showing the cytotoxicity values and anti-dengue activity of selected diarylpyrazolylquinoline scaffolds.
| Compound No. | Heterocyclic Scaffolds | %Inhibition of DENV2 |
%Cell Viability of Huh-7 cell |
||
|---|---|---|---|---|---|
| 1 μM | 10 μM | 20 μM | 200 μM | ||
| 28 | 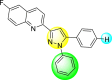 |
5.21 3.1 | 15.31 2.3 | 124.10 7.48 | 115.31 11.43 |
| 29 | 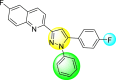 |
3.58 1.7 | 6.14 2.1 | 88.26 9.46 | 89.72 1.71 |
| 30 | 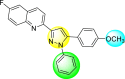 |
4.14 2.3 | 7.15 1.9 | 95.56 5.07 | 93.68 5.75 |
| 31 | 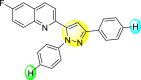 |
3.02 1.8 | 5.13 1.4 | 125.54 6.35 | 98.29 7.59 |
| 32 | 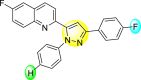 |
11.23 2.8 | 25.39 3.4 | 88.13 1.84 | 87.49 7.61 |
| 33 | 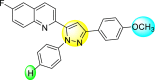 |
5.31 1.6 | 8.09 2.3 | 103.99 3.75 | 98.90 9.26 |
| 34 | 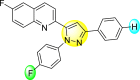 |
4.36 1.7 | 8.31 1.5 | 97.82 3.68 | 91.56 3.85 |
| 35 | 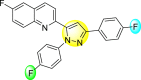 |
8.91 1.3 | 22.35 1.7 | 95.32 2.61 | 86.51 4.51 |
| 36 | 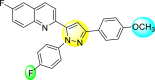 |
41.42 2.4 | 55.32 1.8 | 98.75 2.87 | 95.37 3.64 |
| 37 | 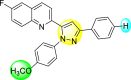 |
7.81 2.5 | 20.68 1.8 | 115.73 3.28 | 97.35 8.21 |
| 38 | 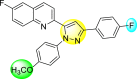 |
6.12 2.1 | 15.83 3.2 | 89.08 8.16 | 52.55 1.89 |
| 39 | 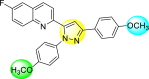 |
46.87 5.3 | 65.71 4.1 | 95.81 4.82 | 98.40 16.44 |
| 40 | 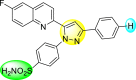 |
4.52 2.7 | 11.81 1.9 | 89.22 4.03 | 83.64 2.68 |
| 41 | 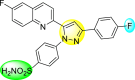 |
5.71 1.7 | 12.75 1.8 | 91.12 2.04 | 42.97 2.47 |
| 42 | 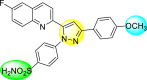 |
58.96 2.8 | 85.34 6.7 | 83.71 2.52 | 89.53 2.41 |
Fig. 8.

Positional isomers of the trisubstituted pyrazole scaffolds.
Fig. 7.

SAR of the selected pyrazole-containing heterocyclic scaffolds showing maximum inhibition against the Huh-7 cell infected with DENV2.
Zamei et al. (2019) investigated a fluorinated-pyrazoline scaffold 43 (Fig. 9 ) for the inhibition of DENV2. The authors obtained the fluorinated-pyrazoline scaffold with the aid of a one-pot, three-component reaction in Monowave 50 [103]. With the application of molecular docking (MD) and MDS, the inhibition site of the DENV2 was determined. The authors investigated that the existence of fluorine analogs in the molecular backbone successfully inhibited the NS2B or NS3 site of the serine protease of the DENV (Fig. 9). The computational studies indicated that the scaffold could form several types of bonds, such as hydrogen bonding (His51, Arg74), van der Waals (Asp75), and hydrophobic (His51, Arg74) interactions with the DENV2 protease with a binding affinity (−57.1 kcal/mol). However, they could not support their results with experimental proofs. Based on SAR analysis (Fig. 9), the authors achieved enhanced inhibition in the existence of fluoro-substituent on one of the phenyl rings. If several other fluoro-substituents or other halogens are introduced, the inhibition activity may increase at a considerable rate.
Fig. 9.
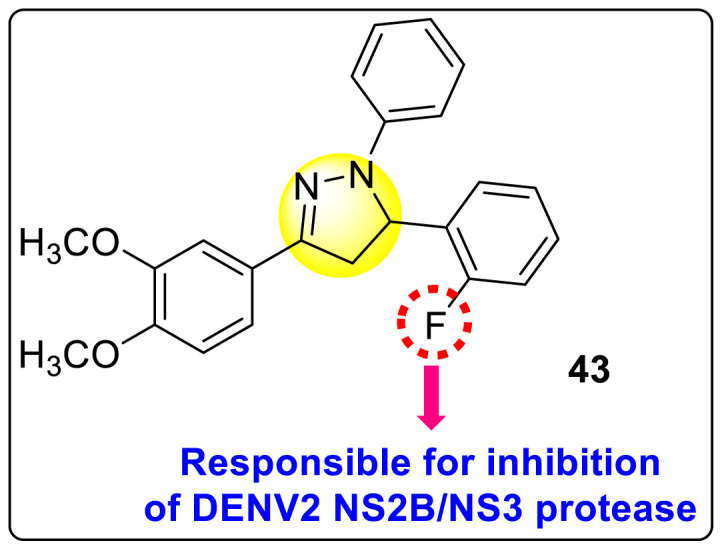
SAR of the fluorinated-Pyrazoline scaffold showing inhibition against DENV2 serotype.
7.3. Imidazoles
Imidazole belongs to the class of five-membered, aromatic heterocyclic scaffolds. The skeletal structure of imidazole comprises three carbon atoms and two nitrogen atoms that are not vicinal with each other. Imidazoles are highly polar and amphoteric in nature [104]. These compounds are vastly explored in various pharmaceutical and agrochemical industries due to their bioactivity [[105], [106], [107]]. The substituted-imidazole scaffolds display a wide array of biological activity, and these have been explored, like anti-cancer, anti-viral, anti-analgesics, anti-inflammatory, anti-HIV, anti-microbial, and many more [105,[108], [109], [110]].
Sucipto et al. (2018) proposed a Cu(II)-imidazole complex for anti-dengue inhibition for the first time. Based on their strategy, the [Cu (2,4,5-triphenylimidazole)2]n derivative was found to be effective in inhibiting the DENV2 serotype infecting Vero cells [111]. The proposed complex showed reduced toxicity 50% cytotoxic concentration (CC50 = 44.174 μg/ml) and high anti-DENV activity half-maximal inhibitory concentration (IC50 = 2.3 μg/ml) against the DENV2 infected Vero cells. On screening the metal-free imidazole scaffold, the inhibition rate was enhanced (IC50 = 0.13 μg/ml), but its cytotoxicity was higher (CC50 = 5.03 μg/ml) for the Vero cells. After a couple of years, the authors further investigated the inhibition of the DENV-2 serotype with a modification in the imidazole complex. The authors introduced [Cu(2,4,5-triphenyl-1H-imidazole)2(H2O)2].Cl2 complex. Experimental results displayed that the complex showed cytotoxicity (CC50) of about 98.62 μg/ml and inhibition rate (IC50) of nearly 300.36 μg/ml against the Vero cells. The selective index (SI) data showed a value of 1.96, confirming that the complex inhibits the DENV replication with minimum cytotoxicity for the Vero cells [112].
Continuing their effort to synthesize various anti-DENV inhibitory complexes, a similar group further advanced their research by chelating different metal ions with the 2,4,5-triphenylimidazoline (TPI) ligand 44 ( Table 7 ). In this case, the authors took three metal ions: cobalt, iron, and zinc. The screening of the metal complexes to the DENV-3 serotype presents that the Zn-TPI and Fe-TPI 46 displayed the lowest cytotoxic effect with increasing concentrations with cell viability greater than 50%. The Co-TPI 45 displayed 43% inhibition of DENV3 replication at a minimum concentration of 6.25 μg/ml. The other complexes, such as zinc and iron, showed 54.9% and 56% inhibition, respectively. Thus, the above values indicate that Fe-TPI 46 and Zn-TPI are among the most potent inhibitors of the DENV-3 viral strains [113]. The results are displayed in Table 7. In contrast to the above results, further studies on the Zinc complex of the above imidazole-containing ligand exhibited high levels of toxicity (CC50 = <100 μg/ml) in the Vero cells. In conclusion, Zn(II)-2,4,5-triphenylimidazole complex cannot be further evaluated to investigate DENV-2 inhibition [114].
Table 7.
Data showing the inhibition activity and the cytotoxicity values of the metal complexes of 2,4,5-triphenylimidazole complexes.
| Compound No. | Heterocyclic Scaffolds | IC50 (μg/ml) | CC50 (μg/ml) | SI |
|---|---|---|---|---|
| 44 | 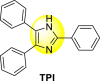 |
1.46 | 36.75 | 25.17 |
| 45 | 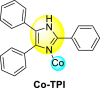 |
−56.29 | 509.14 | −9.04 |
| 46 | 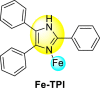 |
98.66 | 1231.71 | 12.48 |
Okano et al. (2019) synthesized various imidazole-based nucleosides and scrutinized their anti-dengue inhibition. Their study revealed that 5-ethynyl-(1-β-d-ribofuranosyl) imidazole-4-carboxamide (EICAR) 47 and 5-ethynyl-(1-β-d-ribofuranosyl) imidazole-4-carbonitrile derivative (EICNR) 48 displayed anti-viral activity (Table 8 ). Unfortunately, the cytotoxicity of the selected compounds was high. So, to overcome this challenge, the authors switched to 4′-thio and 4′-seleno derivatives on the backbone of the selected compounds. Surprisingly, the 4′-thioEICAR 51 and 4′-thioEICNR 49 exhibited greater inhibition against the DENV RNA-dependent RNA polymerase (RdRp) without any cytotoxicity than ribavirin 53 on the viral BHK21 cell line [115]. The results of the viral inhibition and the cytotoxicity values are listed in Table 8. From the SAR analysis, it was found that the existence of cyano substituent on the imidazole ring enhances the inhibitory activity (Fig. 10 ). The amide-substituent tends to decrease the inhibition slightly. Moreover, the rate of inhibition got enhanced when the substituent on the ribofuranosyl is oxygen. When the substituent is a sulfur-atom or a selenium-atom, the rate decreases in order.
Table 8.
Data showing the inhibition activity and the cytotoxicity values of the imidazole-based nucleoside scaffolds.
| Compound No. | Heterocyclic Scaffolds | IC50 (μM) | CC50 (μM) | SI |
|---|---|---|---|---|
| 47 | 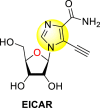 |
0.27 | 1.24 | 4.59 |
| 48 | 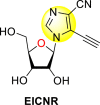 |
0.55 | 3.99 | 7.25 |
| 49 | 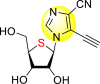 |
6.57 | >50 | >7.67 |
| 50 | 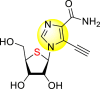 |
14.63 | >50 | >3.42 |
| 51 | 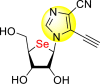 |
22.29 | >50 | >2.24 |
| 52 | 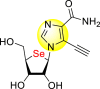 |
>20 | >50 | – |
| 53 | Ribavirin | 6.66 | 48.98 | 7.35 |
Fig. 10.

SAR of the imidazole-based nucleoside scaffolds.
7.4. Pyridines
Pyridine, the most common 6-membered aromatic heterocyclic compound consists of five carbon atoms and one nitrogen atom with a structural formula C5H5N. Pyridine is a basic and planar molecule; the electron cloud is delocalized over the ring system, thus, obeying Huckel's rule of aromaticity [116]. In the last few decades, pyridines have been explored in almost every field [117], especially in medicinal chemistry, where it is biological implication is immense [118,119]. The anti-dengue activity of various pyridine-analogs is discussed in the subsequent sections.
Xu et al. (2016) investigated a pyridoxine-based small molecule that inhibits the RdRp DENV polymerase [120]. The authors constructed the small molecule inhibitor named DMB220 (5-benzenesulfonylmethyl-3-hydroxy-4-hydroxymethyl-pyridine-2-carboxylic acid hydroxyamide) 54 (Fig. 11 ) with the aid of mechanism-based drug synthesis. The mechanism-based drug modeling is based on the chelation of the divalent metal ions to the active site of the viral enzymes. The researcher devised the first-ever protocol aimed at the chelation of the divalent metal ions to the functioning portion of the RdRp of the DENV polymerase. Thus, on screening DMB220, a wide range of inhibitory action was observed on all the four types of DENV RdRp enzymes. The utilization of enzymatic assay proved that DMB220 was proactive against all 4 serotypes of DENV with an inhibition of the RdRp polymerase to be about 50% at a very low micromolar concentration having an IC50 of 5–6.7 μM. Through cellular assay, the anti-dengue activity of DMB220 was determined. The results revealed a 50% effectiveness against the DENV infection with an EC50 of less than 3 μM (Table 9 ). Furthermore, the researchers proved that the DENV RdRp S600T variant displayed a three-fold hyper-susceptible action to DMB220, as previously shown to resist the nucleoside inhibitors. Based on the SAR studies, the existence of the –OH and –NHOH group attached to the pyridine ring of the DMB220 molecule, is involved in the chelation of the divalent metal ions present on the active site of DENV RdRp polymerase (Fig. 11).
Fig. 11.

Structure of the DMB220 molecule.
Table 9.
Data showing the anti-dengue activity of DMB220 against the four serotypes of dengue.
| DENV Serotypes | IC50 (μM) | EC50 (μM) |
|---|---|---|
| DENV1 | 5.7 0.9 | 2.7 0.4 |
| DENV2 | 6.0 0.4 | 2.8 0.6 |
| DENV3 | 6.7 1.0 | 2.7 0.6 |
| DENV4 | 5.0 0.9 | 2.2 0.5 |
7.4.1. Fused pyridines
Pu et al. (2018) proposed several isothiazolo [4,3-b] pyridine scaffolds that are active against the cyclin G-associated kinase [121]. The authors optimized a variety of groups at position-3 of the isothiazolo [4,3-b] pyridines 55–62 (Table 10 ) and obtained that the scaffolds display a broad cyclin G-associated kinase (GAK) inhibition (kd) and a modest anti-viral activity. Further, these scaffolds are active in the human dendritic cells with enhanced anti-DENV activity. The authors carried forward the anti-DENV activity in the Huh-7 cells against the DENV2 serotype. The anti-DENV activity (EC50 and EC90) was conducted with the luciferase assay, while the cytotoxicity (CC50) was taken through the alamarBlue assay. The results are discussed in Table 10. A similar strategy has been adopted by Martinez-Gualda et al. (2021) through the variation in the substituents at the 3-position of the isothiazolo [3,4-b] pyridines. Their research showed that the substitution of Phenyl and N-Piperidine at the 3-position displayed an enhanced affinity towards GAK in a nanomolar concentration [122]. The improved series of substituents demonstrated low IC50 on the nM scale. Molecular docking analysis revealed that carboxamide residue is involved in an H-bond interaction with the Lys69 residue of the ATP binding site of the GAK protein. The SAR revealed that the existence of electron-donating substituents attached to the isothiazole ring exhibited greater inhibition activity. Further, the attachment of a carboxamide side-group at the 3-N-piperidinyl position leads to enhancement in the affinity towards GAK (Fig. 12 ). Hence, –NH2 substituent on the phenyl ring and the various substituents on the fused isothiazole ring contribute significantly to the inhibition of DENV.
Table 10.
Data showing the GAK inhibition and DENV inhibition of a series of isothiazolo [4,3-b] pyridines.
| Compound No. | Heterocyclic Scaffolds | Kd | EC50 | EC90 | CC50 |
|---|---|---|---|---|---|
| 55 | 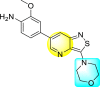 |
0.0089 | 1.844 | 8.05 | 17 |
| 56 | 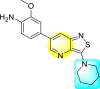 |
0.97 | 5.72 | >10 | >10 |
| 57 | 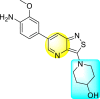 |
0.25 | 0.18 | 0.56 | 2.09 |
| 58 | 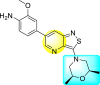 |
0.089 | 0.82 | 1.76 | >25 |
| 59 | 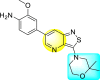 |
0.035 | 0.8416 | 3.92 | 4.14 |
| 60 | 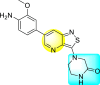 |
0.052 | 4.03 | >10 | >10 |
| 61 | 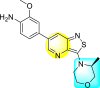 |
0.018 | 0.47 | 1.34 | 6.32 |
| 62 | 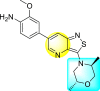 |
0.011 | 0.70 | 2.15 | >10 |
Fig. 12.

SAR of the isothiazolo [4,3-b] pyridines for the inhibition of GAK and DENV.
Wouters et al. (2019) proposed a series of isothiazolo [4,3-b] pyridine analogs 63–74 (Table 11 ), showing a great affinity to the GAK. The authors took advantage of the well-known Sonogashira and Suzuki coupling reactions to introduce a substituent connected with the carbon at 3-position [123]. Their research demonstrated that substituting a phenyl-analog can enhance the binding affinity and, thus, is crucial for showing anti-viral activities. GAK is a cellular regulator of the host adapter proteins (AP)-1 and AP-2. The GAK is associated with clathrin and is responsible for the intracellular regulation of the DENV at the primary and later stages of the viral lifespan [124]. Upon substituting the different C-based substituents at the 3-position, the authors found that the 3,4-dimethoxy group showed the highest inhibition at a very low nanomolar concentration. Moreover, inhibition is further affected by the hydrophobic interactions and hydrogen-bond between compound 71 and the ATP binding pocket of GAK. With the help of luciferase assays and AlamarBlue assays, the EC50 values and the CC50 values were determined. The value indicates that compound 71 showed an average anti-dengue activity with a minimum cytotoxic value (Table 11). The SAR (Fig. 13 ) showed the existence of electron-donating substituents on the phenyl ring of the fused isothiazole scaffold enhanced the inhibition activity. But, in the case of electron-withdrawing substituents, the results marked a decrease in the inhibition. Further, different electronic groups on the fused isothiazole ring exhibit inhibition towards DENV. Thus, the substituents on the phenyl ring and the isothiazole ring are crucial for showing anti-dengue activity.
Table 11.
Inhibition activity of isothiazolo [4,3-b]pyridine analogs against GAK.
| Heterocyclic Scaffolds | Kd (μM) | Heterocyclic Scaffolds | Kd (μM) |
|---|---|---|---|
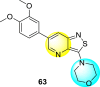 |
0.052 | 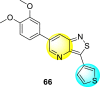 |
2 |
 |
0.77 | 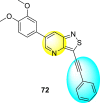 |
>30 |
 |
0.52 | 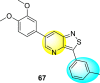 |
0.041 |
 |
0.86 | 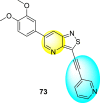 |
>30 |
 |
0.4 | 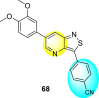 |
7.6 |
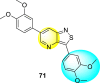 |
>30 | 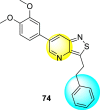 |
3.2 |
Fig. 13.

SAR of isothiazolo [4,3-b] pyridines.
Verdonck et al. (2019) proposed a synthetic strategy and SAR for a diverse 3,5-disubstituted pyrrolo [2,3-b] pyridines [125]. The strategy has been applied for the inhibition of adaptor-associated protein kinase 1 (AAK1) displaying anti-DENV activity. AAK1 is a switch for the host-adapter proteins [126], which are interconnected with the clathrin—the kinase assists in regulating numerous distinct RNA viruses. Upon optimizing the synthetic strategy, the authors established that the pyrrolo [2,3-b] pyridines exhibited an enhanced inhibition against the DENV in-vitro and human primary DCs. Based on the SAR, the proposed compounds demonstrated enhanced anti-viral inhibition against the DENV strains with a low nano-molar binding affinity towards AAK1 kinase. The compounds 75 and 76 (Fig. 14 ) showed the most potent inhibition against the DENV-infected human dendritic cells. Based on the structural orientations, the presence of pyridine and fused pyridine rings are very essential for showing maximum inhibition.
Fig. 14.

Examples of 3,5-disubstituted pyrrolo [2,3-b] pyridines inhibiting AAK1 and DENV.
Martinez-Gualda et al. (2020) proposed a SAR (Fig. 15 ) based on a similar kind of substituted isothiazolo [4,3-b]pyridines against the GAK [127]. However, in this strategy, the 3-position of the isothiazolo [4,3-b]pyridines has been kept fixed by substituting with a cis-2,6-dimethylmorpholine group. The structural variations were performed at the 5-and 6-position. At 6-position, many linkers were introduced between the phenyl ring 77 using Suzuki coupling, and their structural-activity relationship was studied. Further, in some cases, (Ar = phenyl) 78, the substitution of the Ph-ring at the 5-position was also performed (Fig. 15). Upon optimizing the different ligands, the 3,4-dimethoxyphenyl-group at position-5 produced the best inhibition at a lower nanomolar concentration (IC50 = 0.1–0.5 μM). Thus, it was established that the existence of substituted morpholine group at 3-position and aryl substituent at 6-position is responsible for exhibiting GAK inhibition.
Fig. 15.

SAR of the substituted isothiazolo [4,3-b]pyridines for the GAK inhibition.
7.5. Pyrazines
Pyrazines are one of the important N-containing heterocyclic compounds. They belong to the class of 1,4-diazines [128]. The molecular backbone of pyrazine contains two nitrogen atoms and four carbon atoms which constitute a six-membered aromatic heterocyclic ring. Pyrazines display a broad spectrum of biological activities like anti-fungal, anti-microbial, anti-cancer, anti-depressant, and so on [[129], [130], [131]]. They are most frequently used in the perfumery, pharmaceutical industries, and agrochemicals in the form of intermediates [132,133]. In the last few years, a few of them have been utilized to inhibit DENV and are discussed in the upcoming sections (Fig. 11). Saudi et al. (2016) optimized a series of fourteen pyrazine-2,3-dicarboxamide substituting it with the pyrazole backbone 79–86 (Table 12 ) [101]. The optimized compounds were screened against the DENV and YFV activity. The results displayed that the compounds showed a better inhibition against the DENV with an EC50 value in the range of 0.5–3.4 μM. However, the heterocyclic scaffolds 80 and 82 exhibited the maximum inhibition having an EC50 value of 0.5 μM and a SI value greater than 235. The results are discussed in Table 12. Based on SAR (Fig. 16 ), the electron-donating, as well as electron-withdrawing substituents, play a critical role in the rate of inhibition. Moreover, pyrazine moiety contributes significantly to the inhibition activity.
Table 12.
Optimized structures of a few pyrazine-2,3-dicarboxamide exhibiting DENV inhibitions.
| Compound No. | Heterocyclic Scaffolds | EC50 | CC50 | SI |
|---|---|---|---|---|
| 79 | 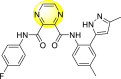 |
26.5 | >116 | >4 |
| 80 | 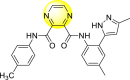 |
0.5 | >117 | >235 |
| 81 | 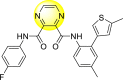 |
4.6 | >116 | >25 |
| 82 | 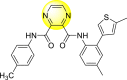 |
0.5 | >117 | >235 |
| 83 |  |
2.2 | >120 | >55 |
| 84 |  |
3.4 | 24 | 7 |
| 85 | 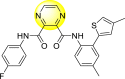 |
2.1 | >116 | >55 |
| 86 | 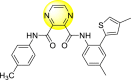 |
<0.9 | 26 | >29 |
Fig. 16.

SAR of the pyrazine-2,3-dicarboxamide substituted pyrazole scaffolds for the inhibition of DENV.
7.6. Pyrimidines
Pyrimidines and their derivatives are largely found in diverse natural products and also constitute the building blocks in living organisms [134]. They exist in the DNA and RNA of living organisms [135]. They possess a broad range of biological products due to their pharmacological and medicinal significance [136]. The pyrimidine scaffold contains four carbon atoms and two nitrogen atoms. Thus, constituting a six-membered aromatic heterocyclic structure. Pyrimidines display a diverse spectrum of biological functions such as cyclooxygenase (COX) inhibitor, anti-inflammatory, anti-cancer, anti-viral, analgesics, anti-allergic, and many more [135,137,138]. The last few years have witnessed several anti-dengue activities of pyrimidine-based analogs, which are discussed in the upcoming works of literature.
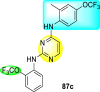
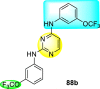
Clark et al. (2016) developed certain disubstituted-pyrimidine analogs that were found to intercept the access of the DENV through the inhibition of the viral kinase. The researchers uncovered that an allosteric ABL kinase inhibitor called GNF-2 suppresses DENV replication via a polypharmacological pathway. The polypharmacological pathway is facilitated via the ABL kinase and the viral E protein. The GNF-2 obstructs the access of DENV via the direct interaction with the E protein of the DENV. Further, the GNF-2 suppresses the effect of Abl kinases that cause the entry of the DENV at the post-entry stage. The researchers further explored that the GNF-2 containing, biotin-conjugated, and fluorophore-conjugated analogs coordinate with the Glycoprotein E of the DENV in the pre-fusion conformation. Thus, this interaction of the GNF-2 and the glycoprotein E on the virion surface helps suppress the entry of the DENV [139]. Upon optimizing different substituents on the pyrimidine core, the authors found that the 2,4-disubstitution with the –OCF3 group at the C-4 position; resulted in enhanced inhibitory activity than the 2,6-disubstitution (Fig. 17 ). A few selected scaffolds are presented in Table 13 , which demonstrates the maximum inhibition against the four DENV serotypes from the numerous reported scaffolds. Compounds 87a, 88a, and 88d displayed powerful inhibition towards DENV1 and DENV2 with a 90% inhibitory concentration (IC90) value between 1 and 5 μM. However, the rest exhibited maximum inhibition towards DENV1. However, 87c and 87d exhibited maximum inhibition towards DENV4 and DENV3, respectively. Based on the SAR analysis (Fig. 17), the existence of electron-donating and electron-withdrawing groups on the substituted pyrimidine scaffold plays a contributing role in the inhibition activity. Further, the existence of the –OCF3 group at a different position on the phenyl ring affects the inhibition activity.
Fig. 17.

SAR of disubstituted pyrimidine analogs against the DENV strains.
Table 13.
Inhibition activity of disubstituted pyrimidine analogs against the DENV strains.
| Heterocyclic Scaffolds | Inhibition Activity (IC90) | Heterocyclic Scaffolds | Inhibition Activity (IC90) |
|---|---|---|---|
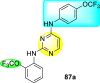 |
DENV1 = 4 μM DENV2 = 5 μM |
 |
DENV1 = 1 μM DENV2 = 5 μM |
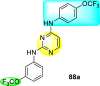 |
DENV1 = 4 μM | 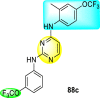 |
DENV1 = 1 μM |
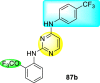 |
DENV4 = 5 μM | 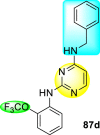 |
DENV1 = 1 μM |
 |
DENV3 = 5 μM | 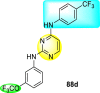 |
DENV1 = 3 μM DENV2 = 5 μM |
McGuigan et al. (2016) synthesized and reported tritylated and 4,4′-dimethoxytritylated-scaffolds of pyrimidine and purine nucleosides. The authors worked out in vitro evaluation of the reported scaffolds against the DENV and YFV [140]. The utilization of the anti-dengue assay demonstrated that the tritylated fludarabine analog expressed the maximum inhibition of the dengue viral strains. The results displayed a 100% inhibition of DENV at an optimum concentration of about 10 μM. However, the mechanism of viral inhibition is still unclear, and studies on the mechanism are yet to be reported. Leal et al. (2019) reported thirty novel heterocyclic compounds enclosing the pyrimidine core to suppress the DENV activity [141]. Out of the thirty compounds, a few of them 90–95 (Table 14 ) displayed the maximum DENV inhibition. However, only two scaffolds, 90 and 91, demonstrated suppression activity against all the DENV serotypes (Table 14). The optimized results revealed that the anti-dengue inhibition is prominent when the length of the side chain is greater. The presence of a heterocyclic compound in the side-chain has little effect on the suppression of viral activity (Fig. 18 ). The compounds 90 and 91 (Table 15 ) also displayed greater stability and improved solubility in a different medium. Experimental and molecular dynamics simulation studies very well support these results. Thus, we can conclude from the SAR analysis (Fig. 18) that the presence of the Piperidine ring and the length of the side-chain on the pyrimidine scaffolds enhance dengue inhibition. However, the existence of heterocyclic scaffolds as side-groups posed a decrease in the inhibition activity.
Table 14.
Inhibition activity and cytotoxicity values of the most potent scaffolds.
| Compound No. | Heterocyclic Scaffolds | EC50 | CC50 | SI |
|---|---|---|---|---|
| 90 | 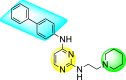 |
0.8 ± 0.2 | 18.1 ± 1.0 | 23 |
| 91 | 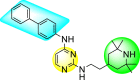 |
0.8 ± 0.2 | 17.3 ± 0.8 | 22 |
| 92 | 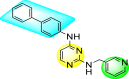 |
7.4 ± 1.0 | 40 ± 1.2 | 5 |
| 93 | 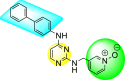 |
9.1 ± 1.0 | 30.2 ± 1.5 | 3 |
| 94 |  |
4.6 ± 0.9 | 25.1 ± 1.2 | 5 |
| 95 |  |
19.9 ± 1.3 | – | – |
Fig. 18.

SAR of the substituted pyrimidine scaffolds for the inhibition of DENV.
Table 15.
Anti-dengue activity of the two most potent compounds against the four dengue serotypes.
| Compound No. | EC50 (μM) |
|||
|---|---|---|---|---|
| DENV-1 | DENV-2 | DENV-3 | DENV-4 | |
| 90 | 0.87 ± 0.1 | 0.85 ± 0.2 | 0.56 ± 0.2 | 2.5 ± 1.1 |
| 91 | 0.58 ± 0.1 | 0.81 ± 0.2 | 0.39 ± 0.8 | 0.87 ± 0.3 |
7.6.1. Fused pyrimidines
Wan et al. (2019) reported a new [1,2,4]Triazolo [1,5-a]pyrimidine analog (Fig. 19 ) that is found to suppress the NS5-RdRp DENV protein. The newly developed compound inhibited the DENV2 proliferation and DENV2-induced inflammation [142]. Through a biophysical assay, the authors verified the mechanism between the most potent analog 89 (Fig. 19) and DENV RdRp protein. Through the application of luciferase assay, the inhibition value (IC50) was found to be 1.28 ± 0.2 μM. The cell-based cytopathic assay revealed that compound 89 displayed an EC50 value of 4.5 ± 0.08 μM.
Fig. 19.
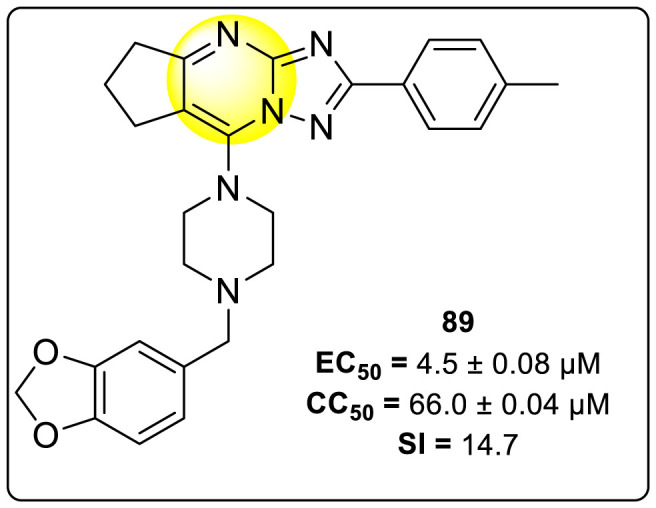
Structure of the most potent inhibitory compound for the inhibition of RdRp DENV-2 serotype.
Further, immunofluorescence assay and quantitative reverse transcription-polymerase chain reaction (qRT-PCR) supported the evidence that compound 89 inhibited DENV RNA. Furthermore, the authors observed that the treatment with compound 89 against the DENV, has significantly reduced the structural E protein activity and the non-structural NS1 proteins with an increase in concentration from 2.5 to 10 μM. Moreover, compound 89 reduced the inflammation in the host cells, which was caused due to the DENV serotype 2. Thus, causing no effect on the defense of the host cells, i.e., the Janus activated kinase (JAK) or STAT-signaling mechanism. The utilization of molecular docking revealed that the best binding affinity for compound 89 is −8.02 kcal/mol. The docking results indicate that the concerned compound is involved in a few hydrogen-bonding and hydrophobic interactions in the RdRp protein pocket. Thus, the presence of a fused pyrimidine scaffold having other heterocyclic rings attached to it exhibits potent inhibition activity (Fig. 19).
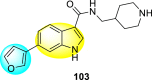
7.7. Indoles
Indoles are one of the vital compounds obtained mostly from natural sources [143]. Indoles are also known as benzopyrroles due to the benzene ring fused with the pyrrole nucleus [144]. Due to their strong binding affinity to many receptors of the various inhibitory sites of pathogenic proteins, indole scaffolds are the most important component in pharmacological compounds [145]. This property is responsible for various clinical and therapeutic applications [144,146]. Indoles display a broad spectrum of biological activity. However, the role of indoles in blocking the DENV is rarely reported. The last few years have witnessed several indole-derivatives discussed in the upcoming sections.
Gobi et al. (2018) reported four indoline-based Schiff base compounds to suppress the larvicidal activity of Aedes aegypti mosquitoes [147]. Aedes aegypti are the main carriers of dengue, zika fever, chikungunya, and yellow fever virus. The incorporation of indole core in Schiff base formation has been an extensive and renovating work. Schiff base compounds are regarded as versatile compounds that display many biological activities [[148], [149], [150]]. The authors proposed that the compounds display a great mortality rate against the larvae of the Aedes aegypti mosquitoes. Sousa et al. (2019) reported six indole-derivatives 92–97 (Table 16 ) that showed larvicidal activity against the larvae of Aedes aegypti mosquitoes [151]. Out of the six indoline derivatives, scaffolds 92, 93, 95, and 96 displayed the most potential larvicidal control. However, compound 93 (Table 16) displayed the maximum inhibition of the larvae of Aedes aegypti. The results further revealed that the proposed heterocyclic scaffolds are non-toxic toward other non-target entities (RAW 264.7 macrophages, Vero cells, C. elegans N2, and G. mellonella) at the larvicidal concentrations. The larvicidal activity persists for almost 30 days of treatment. The SAR of the larvicidal activity in Fig. 20 indicated that the existence of chlorine and bromine as the side groups of the indole-ring hinders the growth of larvae competently. Conversely, the existence of the –COOH group increases the growth of larvae.
Table 16.
Larvicidal activity of the indole-derivatives.
| Compound No. | Heterocyclic Scaffolds | LC50 (μM) | LC90 (μM) |
|---|---|---|---|
| 92 |  |
9.49 | 15.76 |
| 93 |  |
5.88 | 9.50 |
| 94 |  |
>464.57 | >464.57 |
| 95 | 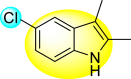 |
19.91 | 30.13 |
| 96 | 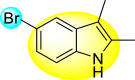 |
10.64 | 14.74 |
| 97 |  |
>853.6 | >528.51 |
Fig. 20.

SAR of the indole-derivatives showing inhibition against the larvae of Aedes aegypti mosquitoes.
Bardiot et al. (2018) investigated several indole-based scaffolds that very efficiently inhibited DENV serotype-2 [152]. The authors carried out broad SARs, in which compound 98 (Fig. 21 ) was found to be the most powerful and selective inhibitor of the DENV2 in the nanomolar to micromolar range. Furthermore, the authors discovered that the physiochemical properties and metabolic stabilities of rats and human microsomes improved significantly. Moreover, separation of the racemic mixtures based on the chirality displayed selectivity for one of the enantiomers. The SAR (Fig. 21) further revealed that the presence of the 3,5-dimethoxy group on the aniline ring displayed a 10-fold enhancement of inhibition of the DENV (EC50 = 0.007 μM). Surprisingly, 2,5-and 2,3-dimethoxyaniline exhibited inhibition in the sub-micromolar range. Extending their investigation by replacing one of the methoxy groups with either alkyl or fluoro-substituents decreased the potency of the viral inhibition. Thus, the presence of the methoxy-group at the meta-position is responsible for the maximum and selective inhibition. Further, the existence of monocyclic or bicyclic aryl groups is the preferred choice for displaying inhibition activity.
Fig. 21.

The most potent inhibitor of DENV2.
Qian et al. (2022) reported several tricyclic analogs containing indoline and imidazolidone scaffolds for the inhibition of DENV infection. Upon optimizing different analogs, only two compounds, 99 and 100 (Fig. 22 ), exhibited potent inhibition against the dengue-virus serotype-2. The authors established that the proposed compounds are active in inhibiting DENV2 serotypes [153]. The results were confirmed by recognizing the E protein via the application of immunofluorescence assay on the antibody of the DENV E protein and with the help of the western blotting technique. The mechanistic study revealed that compounds 99 and 100 displayed moderate activity against the RdRp enzymes. However, SPR imaging further confirms a strong affinity of the compounds 99 and 100 towards the NS5-RdRp. Compound 100 was determined to show the maximum inhibition against DENV2. The outcomes are supported by the in-silico molecular docking, which shows that they bind to the NS5-RdRp through hydrogen-bonding and hydrophobic stabilization interactions. The SAR further confirms that p-nitro hydrazine substituents significantly enhance the anti-dengue activity. Further, the presence of silylating protecting groups confirms to be the most contributing factor to the suppression of dengue replication (Fig. 22).
Fig. 22.

SAR of the most potent tricyclic indoline and imidazolidone fused scaffolds showing inhibition against the DENV.
Nie et al. (2021) investigated several inhibitors containing the indole-core. The compounds efficiently inhibited the Flavivirus NS2B-NS3 protease. Of the various synthesized indole-containing scaffolds, only a few 101–104 showed the maximum inhibition (Table 17 ). However, scaffolds 101 and 103 showed the maximum inhibition against the DENV-2 protease. The SAR describes the presence of the furyl-group as the substituent that performs a significant role in enhancing the anti-dengue activity (Fig. 23 ). Likewise, the existence of cyclohexyl or piperidine substituents at the R′-position displayed improved inhibition. However, other substituents did not show much enhancement compared to the furyl group. Further, studies on the kinetics of the enzymes show that compound 98 exhibits a non-competitive mode of inhibition [154].
Table 17.
Effect of the selected indole-scaffolds against the inhibition of DV2pro.
| Heterocyclic Scaffolds | Inhibition Activity(IC90) | Heterocyclic Scaffolds | Inhibition Activity (IC90) |
|---|---|---|---|
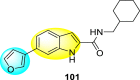 |
DV2pro = 1.6 μM | 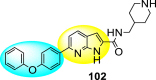 |
DV2pro = 3.1 μM |
 |
DV2pro = 6.5 μM | 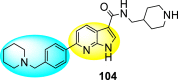 |
DV2pro = 9.2 μM |
Fig. 23.

SAR of the indole-containing scaffolds against DV2pro.
7.8. Quinolines
Quinolines are bicyclic-nitrogen-containing heterocyclic compounds. Quinolines are found in several natural products, mostly Cinchona alkaloids [155]. They display a broad array of biological and pharmacological activities. Several biological activities of quinolines are anti-malarial, anti-convulsant, cardiotonic, anti-fungal, anti-inflammatory, analgesic, and anthelmintic [156]. The oral absorption and inhalation of human beings are non-toxic, with a logP value of 2.04 [157]. Derivatives of quinolines are of great practical importance to synthetic organic chemists and various industries [155]. It is the most crucial heterocyclic analog for the application of drug discovery and also in the context of medicinal chemistry [[158], [159], [160], [161]].
Huang et al. (2021) reported several 4-anilinoquinoline analogs to suppress the DENV [162]. Of the synthesized compounds, only a few 105–109 (Table 18 ) showed potent inhibition against the DENV. The inhibition activity (EC50) of the lead compounds was in the range of 0.63–0.69 μM. However, most compounds displayed limited cytotoxicity slightly greater than 10 μM. The mechanism of inhibition is still under investigation. However, the authors conclude that the binding affinity of the lead compounds to the multiple protein targets could suppress DENV replication. Based on the SAR analysis, the existence of electron-withdrawing groups and more specifically bromine as the substituent as the R-group on the quinoline scaffold showed an increase in the inhibition. Conversely, the existence of electron-donating groups and heterocyclic scaffolds as the R′-position displayed improved inhibition (Fig. 24 ). Thus, switching the electronic substituents on the quinoline scaffold and substituted aniline can significantly affect the inhibition activity.
Table 18.
Inhibition activity and cytotoxicity of the selected 4-anilinoquinoline derivatives.
| Compound No. | Heterocyclic Scaffolds | EC50 | EC90 | CC50 |
|---|---|---|---|---|
| 105 | 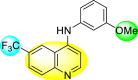 |
0.82 | 1.5 | >10 |
| 106 | 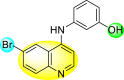 |
0.63 | 3.2 | >10 |
| 107 | 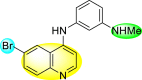 |
0.24 | 0.69 | 5.3 |
| 108 | 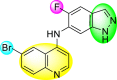 |
0.69 | 4.0 | >10 |
| 109 | 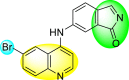 |
0.64 | 1.5 | >10 |
Fig. 24.

SAR of the 4-anilinoquinoline analogs against the inhibition of DENV.
7.9. Quinazolines
Quinazolines are N-containing bicyclic, aromatic heterocyclic scaffolds. The molecular backbone comprises benzene and pyrimidine scaffolds fused collectively to form a whole aromatic ring structure. The molecular formula of quinazoline is C8H6N2. Quinazolines possess a broad range of biological and medicinal significance. These are the widely used scaffolds often explored by medicinal chemists to synthesize various drugs responsible for numerous diseases [163]. The most significant biological activities they have been explored are anti-cancer, anti-tuberculosis, anti-viral, anti-bacterial, anti-hypertensive, anti-diabetes, anti-obesity, and many more [[163], [164], [165], [166], [167], [168]].
Venkatesham et al. (2017) investigated two substituted-aminoquinazoline compounds 100 and 111 (Fig. 25 ) for the anti-DENV activities [169]. The outcomes showed that compound 110 posed the maximum inhibition among the other synthesized aminoquinazoline derivatives (EC50 and SI value of 2.6 μM and 2, respectively). The SAR showed that the scaffolds 110 and 111 interact with the binding pocket through hydrophobic interactions. The employment of molecular docking proved that the proposed compounds inhibited the NS2B-protease successfully. Further, the existence of the electron-releasing group at the para-position of the phenyl ring improved the inhibition as compared to the electron-withdrawing group (Fig. 25). Moreover, the cytotoxicity values got decreased with the above replacement. This implies that the electron-releasing groups can significantly boost the DENV-inhibition. This paves the way for a further outline of anti-DENV compounds in the future. Thus, electron-releasing groups at the p-position possess better inhibition ability as compared to the m-position. Further, the existence of side-groups at 2- and 4-position of the aminoquinazoline derivatives are vital for showing inhibition activity.
Fig. 25.

Structures of aminoquinazoline-derivatives inhibiting DENV.
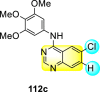
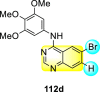
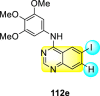
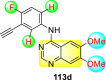
Saul et al. (2020) synthesized several anilinoquinazoline compounds 112a-112j (Table 19 ) for the inhibition of dengue strains [170]. The authors optimized numerous groups and obtained that the substitution with chloro 112c and bromo 112d groups at the 6-position of the trimethoxy anilinoquinazoline scaffold posed the maximum effective inhibition. On the other hand, no inhibition or cytotoxicity is observed upon substituting 6-methyl, 6-fluoro 112b, or 6,7-difluoro groups on the backbone of the anilinoquinazoline derivative. Interestingly, the interchange of the 6-fluoro group to 6-chloro 112c or 6-bromo 112d, or 6-iodo 112e groups enhanced the inhibition activity to almost 10 times. Further, it was observed that when there is an increase in the electronegativity and the size of the substituent group is introduced, a fall in the activity takes place. Interchanging halogen groups to position-7 also marked a downfall in the inhibition. On the contrary, substitution with the cyano-group 112g at position-7 led to a 5-fold increase in the inhibition, while substitution of methylsulfone 112j had no effect. To sum up, 6-iodo-4-((3,4,5-trimethoxyphenyl)amino)quinoline-3-carbonitrile 112e displayed the maximum inhibition (EC50 = 0.079 μM) towards the dengue viral strain with some degree of cytotoxicity (Fig. 26 ). The various optimized structures of anilinoquinazoline are illustrated in Table 19. The authors further explored the anilinoquinazoline scaffolds based on the above results by introducing an acetylene group on the phenyl ring 113a-113g (Table 20 ) [171]. The SAR in Fig. 26 reveals that the existence of electron-withdrawing groups on R1-position enhances the inhibition activity. Moreover, all halogen substituents except the fluoro-group cause an increase in the inhibition rate. Conversely, the existence of electron-withdrawing substituents decreases the inhibition activity present on the R2-position. Thus, R1 and R2 groups on the trimethoxy anilinoquinazoline scaffolds play a vital role in the inhibition of dengue serotype. In the case of acetylene-substituted anilinoquinazoline scaffolds (Fig. 27 ) R4 and R5 positions are significant for greater inhibition. The presence of acetylene further strengthens the rate of inhibition.
Table 19.
Trimethoxy Anilinoquinazoline scaffolds showing inhibition to DENV.
| Heterocyclic Scaffolds | Inhibition Activity (EC50) (μM) | Heterocyclic Scaffolds | Inhibition Activity (EC50) (μM) |
|---|---|---|---|
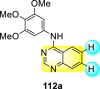 |
2.8 | 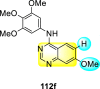 |
>10 |
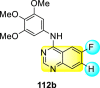 |
1.0 | 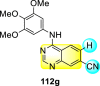 |
0.97 |
 |
1.0 | 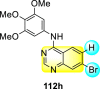 |
2.7 |
 |
1.4 | 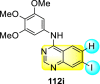 |
2.0 |
 |
2.9 | 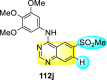 |
>10 |
Fig. 26.

SAR of the trimethoxy anilinoquinazoline scaffolds against the inhibition of DENV.
Table 20.
Acetylene-substituted Anilinoquinazoline scaffolds showing inhibition to DENV.
| Heterocyclic Scaffolds | Inhibition Activity (EC50) (μM) | Heterocyclic Scaffolds | Inhibition Activity (EC50) (μM) |
|---|---|---|---|
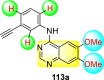 |
6.5 | 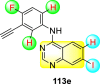 |
9.0 |
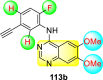 |
>10 | 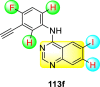 |
7.6 |
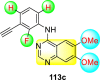 |
>10 | 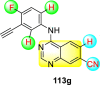 |
5.0 |
 |
>10 | -- |
Fig. 27.

SAR of the acetylene-substituted anilinoquinazoline scaffolds against the inhibition of DENV.
8. O-containing heterocyclic scaffolds for the inhibition of dengue virus
8.1. Coumarins
Coumarins belong to the class of oxygen-containing heterocyclic compounds. The Coumarins are obtained from various natural product sources, bacteria, and fungi and also can be synthesized chemically. These compounds provide various therapeutic applications due to their high stability, solubility, and low toxicity [172]. Coumarins can be called privileged heterocyclic compounds. They display a broad range of biological activities such as anti-viral, anti-microbial, anti-HIV, anti-malarial, and so on [173,174].
Calderón et al. (2017) reported an innovative strategy in which the authors extracted two Coumarin scaffolds 114 and 115 (Fig. 28 ) from the seeds of Mammea americana to study the inhibition of anti-viral infections. The pre-experimental procedure indicated suppression of about 40% (p < 0.01) of the DENV2 infection while the post-experimental procedure exhibited a 50% inhibition (p < 0.01). The scaffolds displayed low toxicity at a concentration [175]. The Coumarin 114 scaffold inhibited DENV2 at concentrations greater than 3.1 μg/ml while Coumarin 115 inhibited DENV2 at a concentration range of 0.8–200 μg/ml. Based on the SAR (Fig. 28), the existence of a cyclic 6-membered structure on coumarin 114 is responsible for greater inhibition as compared to the acyclic structure on coumarin 115.
Fig. 28.

Structure and SAR of two Coumarins derived from the seeds of Mammea americana.
Yusufzai et al. (2018) reported certain 4-thiazolidone [17] and hydrazinyl thiazolyl [16] based Coumarin scaffolds and evaluated their activity with the help of in-silico molecular docking. The results disclose that the synthesized Coumarin derivatives show potential inhibition towards the NS2B/NS3 DENV serine protease. The binding affinity indicates the scaffolds involved in various hydrogen bonding and also some polar interactions in the binding pocket of the serine protease. However, the authors could not bring together the experimental evaluation which needs to be supplied in order to address the Coumarin derivatives against the DENV infection and replication.
9. Mixed heterocyclic scaffolds
9.1. N, S-containing heterocyclic scaffolds for the inhibition of dengue virus
9.1.1. Thiazoles or benzothiazoles
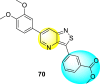
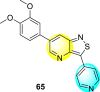
Thiazoles and benzothiazoles are the most important heterocyclic compounds containing nitrogen atoms and sulfur atoms. Thiazoles are rich in natural products, whereas benzothiazoles are rarely obtained from natural products [176]. Benzothiazoles are bicyclic heterocyclic compounds obtained from the fusion of benzene and thiazole cores [177]. These compounds display a broad range of biological activities and are utilized for pharmaceutical and medicinal chemistry purposes [178]. The various biological and pharmacological activities of these scaffolds are anti-bacterial, anti-protozoal, anti-viral, anti-cancer, anti-allergic, gene-modulators, anti-schizophrenia, anti-hypertensive, anti-inflammatory, and so on [176,[179], [180], [181]].
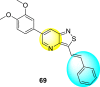
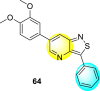
Jadav et al. (2015) synthesized several substituted-thiazole-based scaffolds 116–119 (Table 21 ) to inhibit DENV serotype-2 [182]. The anti-viral inhibitory activity was determined through the help of a virus reduction yield assay. Upon screening the scaffolds, compound 119 was found to be the most powerful inhibitor, with an EC50 value of 1.32 in micro-molar concentration. The application of in-silico molecular docking revealed that the compounds show greater affinity to the binding cavity of the β-OG pocket of the viral protein. Several van der Waals, H-bonding, and hydrophobic stabilization interactions contribute to the inhibition of these compounds against the DENV-2 protein.
Table 21.
Inhibition activity and cytotoxicity values of the few selected thiazole-based compounds in the virus reduction yield assay against DENV2.
| Compound No. | Heterocyclic Scaffolds | EC50 (μM) | CC50 (μM) | SI |
|---|---|---|---|---|
| 116 | 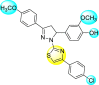 |
7.00 ± 4.16 | 54.6 ± 38.8 | 8 |
| 117 | 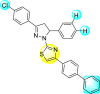 |
8.44 ± 2.02 | 86.8 ± 30.1 | 10 |
| 118 | 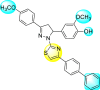 |
3.79 ± 1.22 | 25.6 ± 11.9 | 7 |
| 119 | 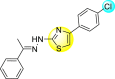 |
1.32 ± 0.41 | 125.0 ± 40.8 | 95 |
Batool et al. (2021) investigated a few N-substituted 1,2-benzoisothiazol-3(2H)-ones 117–123 (Table 22 ) via a one-pot and two-step protocol [183]. Upon screening the synthesized compounds, it was observed that the compounds inhibited the DENV2 NS2B/NS3 protease significantly. The dose-dependent investigation demonstrated that the compounds inhibited the DENV2 protease in micromolar concentration. The utilization of in-silico molecular docking revealed that the compounds bind to the protease in the cavity of the catalytic triad of the NS2B/NS3 protease. The SAR (Fig. 29 ) revealed that the existence of ortho-substituent on the phenyl ring of the benzoisothiazole derivatives 123, 124, 125 shows a better inhibition rate than the para-substituents. However, a potent electron-withdrawing group (-NO2) on the phenyl ring of the benzoisothiazole derivative 120, 121 displayed considerably less DENV inhibition. But, when an electron-donating group is inserted at the ortho-position in the presence of –NO2 at the para-position of the benzoisothiazole derivative, 122 displayed powerful inhibition (Fig. 29). Thus, scaffolds 125 and 126 displayed maximum inhibition (Table 22). The docking outcomes show that the highest potent compounds 125 and 126 exhibited a binding score of −7.6 and −6.2 kcal/mol, respectively. The results further display that the scaffolds bind with DENV through some H-bonding, π-π interactions, and hydrophobic interactions at the binding pocket of the NS3 catalytic pocket. Thus, ortho-substitution and para-substitution effects DENV inhibition considerably. Ortho-substitution displays greater inhibition as compared to the para-substitution.
Table 22.
Several N-substituted 1,2-benzoisothiazol-3(2H)-ones derivatives and their inhibition activity.
| Compound No. | Heterocyclic Scaffolds | %DENV Inhibition | IC50 (μM) |
|---|---|---|---|
| 120 | 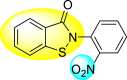 |
72% | 41.80 ± 1.17 |
| 121 | 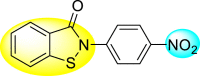 |
67% | 52.70 ± 3.23 |
| 122 | 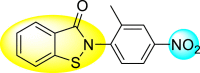 |
92% | 2.56 ± 1.03 |
| 123 | 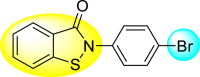 |
80% | 8.24 ± 1.16 |
| 124 | 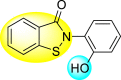 |
89% | 6.66 ± 2.17 |
| 125 | 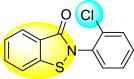 |
94% | 2.01 ± 0.98 |
| 126 | 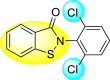 |
94% | 5.28 ± 1.89 |
Fig. 29.

SAR of the N-substituted 1,2-benzoisothiazol-3(2H)-ones derivatives against the inhibition of DENV.
Maus et al. (2021) investigated several new benzothiazole scaffolds to target the allosteric pocket of the DENV NS2B/NS3 protease [184]. The authors synthesized several sequences of Y-shaped benzothiazole scaffolds, but only a few of them, 127–129 (Table 23 ), displayed promising inhibition activity against the DENV. Furthermore, they extended their investigation by truncating numerous benzothiazole compounds 130–134 (Table 24 ). The results exhibited the DENV serotype-2 protease suppression in the short micromolar concentration. The benzothiazole scaffold containing the iodine as the substituent 131 displayed the maximum inhibition having an IC50 value of about 4.38 μM. However, when the iodine is replaced by chlorine 132 and dichloro 134, the inhibition rate got decreases marginally. Surprisingly, when there is no substituent attached to 130, the inhibition rate decreased to about 6-fold (Table 24). The SAR of the truncated benzothiazole scaffolds is presented in Fig. 30 which shows that various substituents present on the phenyl ring of the truncated benzothiazole scaffolds affect greater inhibition. However, the authors also observed that the absence of any substituent decreases the inhibition rate. Upon extending their search for better suppression activity, replacing various heterocyclic compounds with different amino acid linkers posed an excellent inhibition rate.
Table 23.
Inhibition activity of Y-shaped benzothiazole-based heterocyclic scaffolds against DENV2 NS2B/NS3 protease.
| Compound No. | Heterocyclic Scaffolds | % IC50 |
|---|---|---|
| 127 | 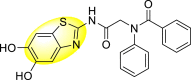 |
56% |
| 128 |  |
45% |
| 129 |  |
58% |
Table 24.
Inhibition activity of truncated benzothiazole-based heterocyclic scaffolds against DENV2 NS2B/NS3 protease.
| Compound No. | Heterocyclic Scaffolds | IC50 (μM) |
|---|---|---|
| 130 | 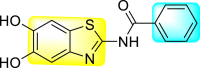 |
26.95 ± 1.61 |
| 131 | 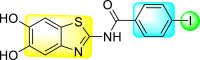 |
4.38 ± 0.38 |
| 132 | 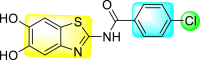 |
5.97 ± 0.50 |
| 133 | 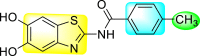 |
6.39 ± 0.20 |
| 134 | 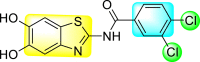 |
4.86 ± 0.33 |
Fig. 30.

SAR of the benzothiazole-based heterocyclic scaffolds against DENV2 NS2B/NS3 protease.
9.1.2. Thiazolidinones
Thiazolidinones are considered a crucial variety of heterocyclic compounds due to their broad array of biological practicalities. These compounds are mostly explored in innumerable pharmaceutical and medicinal chemistry [185,186]. The thiazolidinone scaffold shows numerous biological applications, including anti-bacterial, anti-viral, anti-fungal, anti-diabetic, anti-inflammatory, anti-cancer, and so on [[187], [188], [189]]. Manvar et al. (2015) reported several thiazolidinone and thiadiazole conjugated scaffolds 135–139 ( Table 25 ). The authors utilized these scaffolds to inhibit DENV serotype-2 RdRp in micromolar concentration [190]. The optimization of reaction conditions demonstrated that compounds 135 and 136 show the maximum suppression of the DENV2 RdRp. The application of SAR infers the presence of halogens is significant for the inhibition of DENV2 RdRp (Fig. 31 ). More specifically, the substitution of 3-fluorobenzilidine at the C-5 position of the thiazolidinone scaffold displays the ideal switch. However, the presence of 2-chlorophenyl and 3-fluorophenyl exhibited the maximum suppression. The inhibition activity of various conjugated thiazolidinone-thiazole scaffolds is shown in Table 25. The in-silico molecular docking reveals that the compounds bind to the Thumb pocket-II of DENV serotype-2 RdRp NS5 polymerase. Based on the SAR (Fig. 31), modulation of various substituents on R1 and R2 affects the inhibition activity.
Table 25.
Inhibition activity of some selected conjugated thiazolidinone-thiazole scaffolds against DENV-2 NS5 RdRp polymerase.
| Compound No. | Heterocyclic Scaffolds | IC50 (μM) |
|---|---|---|
| 135 |  |
2.3 ± 0.5 |
| 136 |  |
2.1 ± 0.4 |
| 137 |  |
5.5 ± 0.4 |
| 138 |  |
5.8 ± 0.8 |
| 139 | 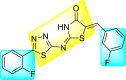 |
7.2 ± 0.3 |
Fig. 31.

SAR of the conjugated thiazolidinone-thiazole scaffolds against DENV-2 NS5 RdRp polymerase.
9.1.3. Benzothiazines
Benzothiazine derivatives are considered one of the most vital compounds in pharmaceutical and medicinal chemistry. These compounds have been presented as various drug intermediates, inhibitors of corrosion, stabilizers in the vulcanization of rubber, and also used as fading items to obstruct the fading process [191]. Apart from various industrial applications, these analogs are important in biological applications. They have been reported as anti-viral, anti-microbial, anti-HIV, anti-hypertensive, immunomodulators, neuroprotective, anti-oxidant, anti-rheumatic, ATP-sensitive potassium channel opener, aldose reductase inhibitors, and many more [[192], [193], [194]].
Cannalire et al. (2018) reported several 2,1-benzothiazine 2,2-dioxides 140–146 (Table 26 ) to suppress DENV3 NS5 RdRp [195]. Based on the SAR, compounds 140 and 141 exhibited the maximum inhibition rate (IC50) of 0.6 and 0.9, respectively, in the micromolar concentration. This implies that the existence of the benzoyl group at the C-4 position and phenoxy substituent at the C-6 position plays a key role in inhibiting the DENV3 NS5 RdRp. The substitution of different halogen groups also contributed to enhancing the inhibition activity of the benzothiazine-derivatives. The compounds with no halogen-substituted to the benzoyl-group posed a decrease in the inhibition activity (Fig. 32 ). Based on the kinetics analysis and microscale thermophoresis analysis, the binding mechanism of other compounds acts by a mixed mechanism of inhibition. This implies that the binding to the RdRp is not dependent on the RNA and GTP substrates. Furthermore, the application of computational studies reveals that compound 140 performs as a non-competitive inhibitor. The most potent compound 140 (Table 26) binds to the N-pocket of NS5 RdRp via an allosteric mechanism. Thus, the substituents at R1 and R2 contribute significantly to the inhibition of DENV3 NS5 RdRp. Strongly electron-donating and electron-withdrawing groups tend to decrease the inhibition. Also, o- or m-substitution at R3 plays a significant role in the inhibition (Fig. 32).
Table 26.
Inhibition activity of some selected 2,1-benzothiazine 2,2-dioxide against DENV3 NS5 RdRp.
| Compound No. | Heterocyclic Scaffolds | IC50 (μM) |
|---|---|---|
| 140 | 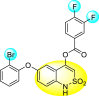 |
0.6 ± 0.1 |
| 141 | 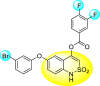 |
0.9 ± 0.1 |
| 142 | 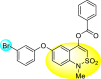 |
2.6 ± 0.4 |
| 143 | 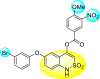 |
4.7 ± 0.7 |
| 144 | 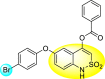 |
4.7 ± 1.0 |
| 145 | 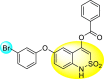 |
7.0 ± 0.7 |
| 146 | 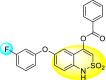 |
7.0 ± 1.0 |
Fig. 32.

SAR of the 2,1-benzothiazine 2,2-dioxide against DENV3 NS5 RdRp.
9.2. N, O-containing heterocyclic scaffolds for the inhibition of dengue virus
9.2.1. Oxadiazoles
Oxadiazoles are 5-membered heterocyclic scaffolds. The structure of oxadiazole contains two nitrogen atoms, one oxygen atom, and two carbon atoms. There exist four possible isomers of Oxadiazoles: 1,2,3-oxadiazoles, 1,2,4-oxadiazoles, 1,3,4-oxadiazoles, and 1,2,5-oxadiazoles. The aromaticity of the oxadiazole scaffolds is significantly lowered due to the replacement of two nitrogen atoms in the place of two CH bonds [196]. Oxadiazoles possess a broad range of therapeutic applications [197]. Besides this, these privileged scaffolds display a broad array of biological applications such as anti-viral, anti-bacterial, anti-tuberculosis, anti-cancer, anti-HIV, anti-convulsant, analgesics, pesticide, and so on [[198], [199], [200], [201]].
Benmansour et al. (2016) reported the synthesis and the anti-dengue activity of numerous 2-phenyl-5-[(E)-2-(thiophen-2-yl)ethenyl]-1,3,4-oxadiazole and 3-phenyl-5-[(E)-2-(thiophen-2-yl)ethenyl]-1,2,4-oxadiazole analogs [15]. A few of the various analogs, 147–153 (Table 27 ), displayed maximum inhibition against the DENV2 RdRp. Upon screening the analogs against the infected cells model, the inhibition activity was acquired in the submicromolar concentration range. The authors screened the compounds against all the DENV cell lines obtained from the clinical isolates. The results displayed that compounds 147, 149, 150, and 151 were the most efficient against all the DENV serotypes (Table 28 ). The SAR (Fig. 33 .) of the oxadiazole-derivatives reveals that the existence of chlorine on the thiophene ring plays a contributing role in the inhibition activity. Further, the existence of chlorine substituents at the p-position enhanced the inhibition activity while chlorine at the m-position decreased the inhibition activity. Thus, p-substituents on the phenyl ring and bromo-substituent on the thiophene are the vital position where the inhibition takes place.
Table 27.
Inhibition activity of various oxadiazole derivatives against the DENV2 RdRp.
| Compound No. | Heterocyclic Scaffolds | IC50 (μM) |
|---|---|---|
| 147 | 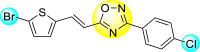 |
2.2 ± 0.1 |
| 148 | 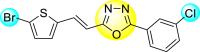 |
6.0 ± 1.0 |
| 149 | 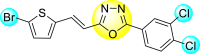 |
3.5 ± 0.5 |
| 150 |  |
4.8 ± 0.4 |
| 151 |  |
4.0 ± 0.5 |
| 152 | 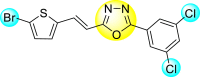 |
11.0 ± 1.8 |
| 153 | 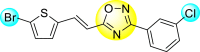 |
9.1 ± 1.3 |
Table 28.
Inhibition activity of the most potent oxadiazole derivatives against the DENV1-4.
| Compound No. | Vero E6 CC50 (μM) | DENV-1 EC50 (μM) | DENV-2 EC50 (μM) | DENV-3 EC50 (μM) | DENV-4 EC50 (μM) |
|---|---|---|---|---|---|
| 147 | >100 | 2.0 | 2.7 | 3.7 | 3.0 |
| 149 | 30 | 5.9 | 4.7 | – | – |
| 150 | >100 | 6.9 | 12 | 9.8 | 10.0 |
| 151 | >100 | 2.5 | 2.6 | 3.0 | 3.0 |
Fig. 33.

SAR of the oxadiazole derivatives for the inhibition of DENV.
Tok et al. (2017) reported various 1,3,4-oxadiazole analogs 152–157 (Table 29 ) to suppress the larvicidal and adulticidal activity of several Aedes aegypti mosquitoes [202]. The outcomes indicated that the synthesized oxadiazole scaffolds did not display larvicidal inhibition; rather, a few of the oxadiazole scaffolds exhibited inhibition towards the adulticidal activity of Aedes aegypti mosquitoes. The mortality rates of a few compounds 152–155 were ≥80% at about 5 μg per mosquito concentration. The application of dose-response bioassay indicated that a few of the compounds 152–155 displayed LD50 values in the range of about 5–10 μg per mosquito. The SAR (Fig. 34 ) directed that substituent on the phenyl ring significantly contributed to the adulticidal activity. For instance, the addition of nitro (–NO2), bromo (-Br), and chloro (-Cl) substituents possesses a greater mortality rate. But, the addition of fluoro-substituent decreased the mortality rate.
Table 29.
Mortality rate and LD50 values of a few selected oxadiazole derivatives against the female Aedes aegypti mosquitoes.
| Compound No. | Heterocyclic Scaffolds | % Mortality rate (5μg/mosquito) | LD50 (μg/mosquito) |
|---|---|---|---|
| 152 | 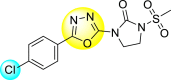 |
83.3 ± 15.3 | 3.08 ± 0.27 |
| 153 | 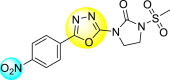 |
86.7 ± 15.3 | 4.96 ± 1.21 |
| 154 | 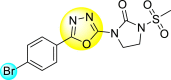 |
86.7 ± 11.5 | 3.55 ± 0.24 |
| 155 | 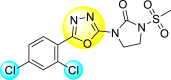 |
86.7 ± 5.8 | 2.39 ± 0.15 |
| 156 | 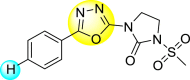 |
73.3 ± 15.3 | – |
| 157 | 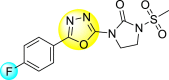 |
70 ± 34.6 | – |
Fig. 34.

SAR of the oxadiazole derivatives against the female Aedes aegypti mosquitoes.
Hamdani et al. (2020) synthesized several S-alkylated and S-alkylphthalimidated scaffolds by fusing 1,3,4-oxadiazole and benzenesulfonamide 158–162 (Table 30 ) [203]. The authors reported the scaffolds to examine their inhibition against the DENV serotype-2. The application of the bioactivity assay demonstrated that compounds 158 and 159 exhibited the highest inhibition, having IC50 values of 13.9 μM and 15.1 μM, respectively. The results of a few of the most potential scaffolds are summarized in Table 30 . The docking analysis revealed that compound 158 possesses the maximum binding affinity (−9.0 kcal/mol). The computational results further illustrate that the compounds bind to the allosteric pocket of the DENV2 NS2B/NS3pro receptor. Further, the presence of the phthalamide moiety displayed several H-bonding interactions with the Trp-83 and Asn-165. Moreover, hydrophobic stabilization has also contributed to the overall affinity of the scaffolds. However, further work is needed to quantify the structure for the fine-tuning of anti-viral activities. The SAR (Fig. 35 ) analysis revealed that the existence of isoindoline-1,3-dione as the substituent of oxadiazole scaffold enhances the inhibition activity. Also, increasing the alkyl chain causes greater inhibition. Moreover, the presence of –CH3 on the phenyl ring causes a decrease in the inhibition while –CF3 enhances the inhibition rate. Thus, the length of the alkyl chain and the substituents on the phenyl ring are the most contributing factors to the inhibition of the DENV.
Table 30.
Inhibition activity and binding scores of the S-alkylated and S-alkylphthalimidated analogs against the DENV-2.
| Compound No. | Heterocyclic Scaffolds | % Inhibition of DENV2 | IC50 (μM) | Docking Score (kcal/mol) |
|---|---|---|---|---|
| 158 |  |
99.9 ± 1.3 | 13.9 ± 1.4 | −9.0 |
| 159 | 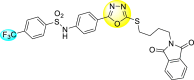 |
77.6 ± 2.1 | 15.1 ± 1.3 | −8.8 |
| 160 | 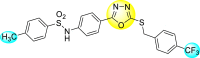 |
83.9 ± 1.3 | 25.2 ± 1.2 | −8.6 |
| 161 | 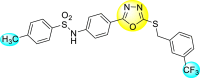 |
80.4 ± 1.7 | 23.9 ± 1.2 | −8.6 |
| 162 | 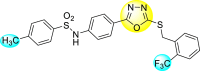 |
80.8 ± 1.0 | 24.0 ± 1.6 | −8.6 |
Fig. 35.

SAR of the S-alkylated and S-alkylphthalimidated oxadiazole derivatives against DENV-2.
Adawara et al. (2021) carried out a computational analysis of 25 oxadiazole scaffolds to study the inhibition of DENV [204]. The authors support their findings with the aid of quantitative structure-activity relationship (QSAR) and molecular docking. The computational analysis supported their results against the DENV serotype-2 NS5. The MD simulation outcomes exhibited several hydrogen bonding and hydrophobic stabilizing interactions with the binding pocket of the DENV2 NS5 receptor. The molecular docking results indicated that compounds 164 and 165 (Table 31 ) exhibited the maximum binding affinity value of −19.1 kcal/mol. This indicates that the presence of halogen substituents performs a significant function in the binding to the pocket of the NS5 receptor (Fig. 36 ). Compound 163 exhibited the minimum binding affinity. However, the hydrogen bond energy value of compound 163 was found to be the highest (−3.9 kcal/mol) among the other potent inhibitors. The perceived value of 163 can be ascribed to the existence of the methoxy group, which binds to the pocket efficiently. Thus, this observation can significantly play a key role in designing several oxadiazole-based inhibitors for overpowering the DENV replication (Table 31). The presence of halogens might significantly contribute to the formation of hydrogen bonds, which eventually cause inhibition activity in the DENV serotype-2 (Fig. 36).
Table 31.
Binding affinity values and the H-bond energy of the selected oxadiazole scaffolds.
| Compound No. | Heterocyclic Scaffolds | Binding affinity (kcal/mol) | H-bond energy (kcal/mol) |
|---|---|---|---|
| 163 |  |
−14.7 | −3.4 |
| 164 | 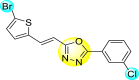 |
−19.1 | −3.1 |
| 165 | 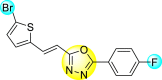 |
−19.1 | −3.3 |
Fig. 36.
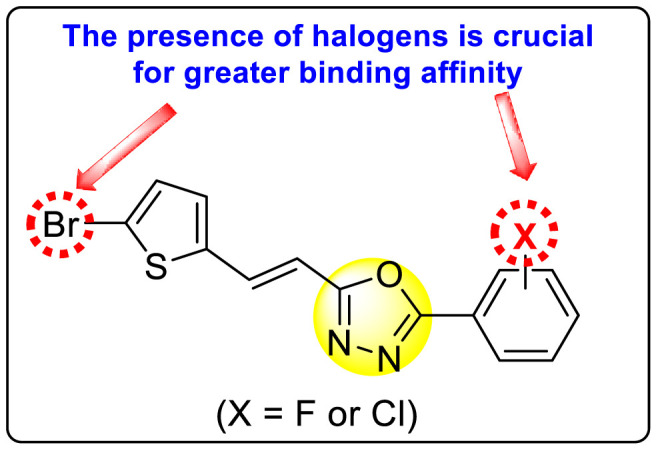
SAR of the oxadiazole scaffolds.
10. Summarizing the SAR analysis of reported heterocyclic scaffolds for inhibiting the DENV
So far, we have elaborately outlined the various heterocyclic scaffolds that significantly inhibit the DENV serotypes. The various scaffolds and the respective SARs assisted in quickly capturing the group or the substituent responsible for inhibiting the DENV. In this section, we have summarized the SARs of each category of the heterocyclic scaffolds. The information will enable the reader to grasp the concepts and the structural patterns of the reported heterocyclic inhibitors. The quick access to the structural patterns will assist the researchers working in medicinal chemistry, organic synthesis, and pharmacology to design and report promising scaffolds that will contribute to the development of DENV vaccines. Altogether, we aim to recollect the multiple pieces of literature and conclusively present them so that the information present in it will enable a boost in the DENV research. The summarized SARs of N-heterocyclic (Table 32 ), O-heterocyclic (Table 33 ), N, S-heterocyclic (Table 34 ), and N, O-heterocyclic (Table 35 ) scaffolds are discussed.
Table 32.
Comprehensive SAR of the reported N-heterocyclic scaffolds.
| N-Heterocyclic Scaffold | SARs | Key Highlights | References |
|---|---|---|---|
| Triazole | 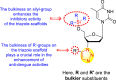 |
|
[91] |
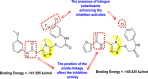 |
|
[92] | |
| Pyrazole | 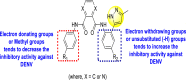 |
|
[101] |
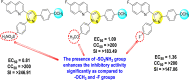 |
|
[102] | |
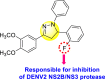 |
|
[103] | |
| Imidazole | 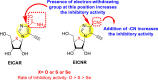 |
|
[115] |
| Pyridine & Fused Pyridine |  |
|
[120] |
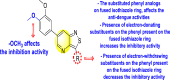 |
|
[124] | |
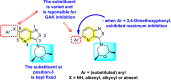 |
|
[127] | |
| Pyrazine | 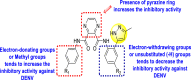 |
|
[101] |
| Pyrimidine | 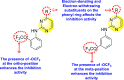 |
|
[139] |
 |
|
[141] | |
| Indole | 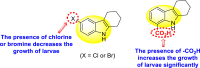 |
|
[151] |
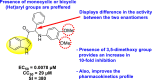 |
|
[152] | |
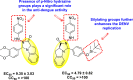 |
|
[153] | |
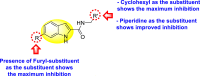 |
|
[154] | |
| Quinoline | 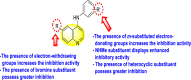 |
|
[162] |
| Quinazoline | 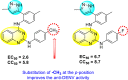 |
|
[169] |
 |
|
[170] | |
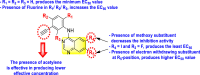 |
|
[171] |
Table 33.
Comprehensive SAR of the reported O-heterocyclic scaffolds.
| O-Heterocyclic Scaffold | SARs | Key Highlights | References |
|---|---|---|---|
| Coumarin | 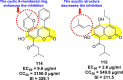 |
|
[175] |
Table 34.
Comprehensive SAR of the reported N, S-heterocyclic scaffolds.
| N, S-Heterocyclic Scaffold | SARs | Key Highlights | References |
|---|---|---|---|
| Thiazoles or Benzothiazoles |  |
|
[183] |
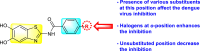 |
|
[184] | |
| Thiazolidinones | 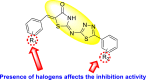 |
|
[190] |
| Benzothiazines | 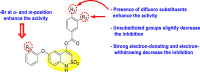 |
|
[195] |
Table 35.
Comprehensive SAR of the reported N, O-heterocyclic scaffolds.
| N, O-Heterocyclic Scaffold | SARs | Key Highlights | References |
|---|---|---|---|
| Oxadiazole |
|
[15] | |
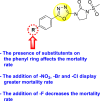 |
|
[202] | |
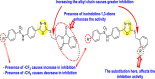 |
|
[203] | |
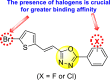 |
|
[204] |
11. Conclusion
In this review, we have rationalized the role of numerous heterocyclic compounds in the inhibition of DENV. Heterocyclic compounds are present in almost all drug categories. The occurrence of nitrogen, oxygen, and sulfur atoms in the molecular structure of the heterocyclic scaffolds have contributed significantly to its numerous therapeutic applications. However, for the discovery of every new drug, the viral organisms adapt to the new drug and deem themselves resilient to the drug. This poses a huge challenge for medicinal chemists and also to pharmacists. The development of heterocyclic compounds provides them a free hand in designing various drug candidates in terms of lipophilicity, solubility, and polarity. Further, the use of computational tools has considerably contributed to the rapid design of drug moieties.
Apart from the numerous benefits of heterocyclic compounds, there are still many concerns that need to be looked upon. As of now, there are no marketed drugs accessible for DENV treatment. The treatment of DENV has many challenges. The foremost challenge of anti- DENV drugs is to be effective against all the serotypes and genotypes. In the majority of the heterocyclic compounds we have discussed, the compounds have exclusively inhibited only some particular serotypes. These challenges need to be looked into by fellow researchers and scientists who are working towards the design and therapy of drugs containing heterocyclic scaffolds. Another obstacle that still needs to be addressed is the evidence regarding the host factors that cause the pathogenesis of DENV infection. This challenge has not been reported to date. Further, we have witnessed the lack of evidence regarding the mechanism of action of numerous drugs. Information on the mechanism pathway can be a major contribution to identifying and designing heterocyclic compounds for better inhibition capability. To address all the above concerns, researchers should aim for the viral proteins rather than the host proteins. By following this strategy, the cytotoxicity or the side-effect of the heterocyclic scaffolds or drug molecules can be overcome.
Furthermore, several studies are being conducted to collect additional data on the inhibitory activity of heterocyclic scaffolds in dengue infection. It will contribute to a better understanding of heterocyclic scaffolds' antiviral activity against the DENV by limiting their cytotoxic effects on the host cell. Achieving these conditions will provide a satisfactory interpretation of the potential therapeutic activities of these heterocyclic scaffolds against dengue infection and lead to the discovery of novel drug candidates leading to the suppression of DENV infectivity and its replication.
Funding
There is no financial funding component associated with the investigation of this work.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
The authors are thankful and do acknowledge their respective organizations for extending adequate resources and facility support.
Abbreviation
- (+ve)
Positive
- (-ve)
Negative
- AAK1
Adaptor-Associated Protein Kinase 1
- AP
Adapter Proteins
- BHK
Baby Hamster Kidney
- C protein
Capsid protein
- CC50
50% Cytotoxic Concentration
- CLEC5AC
type lectin domain containing 5A
- COX
Cyclooxygenase
- CVS
Cardiovascular system
- DCs
Dendritic cells
- DC-SIGN
Dendritic Cell-Specific Intracellular Adhesion molecule-3-Grabbing Non-integrin
- DENV
Dengue Virus
- DFT
Density Functional Theory
- DHF
Dengue Haemorrhagic Fever
- DNA
Deoxyribonucleic acid
- dsRNA
Double-stranded RNA
- E protein
Envelope Protein
- EC50
Half maximal Effective Concentration
- EM
Endosomal membrane
- ER
Endoplasmic Reticulum
- GAK
Cyclin G-Associated Kinase
- GAPDH
Glyceraldehyde- 3-phosphate dehydrogenase
- Hb
Hemoglobin
- IC50
Half maximal inhibitory concentration
- IC90
90% inhibitory concentration
- IFN-α/β
Interferon alpha/beta
- JAK
Janus Activated Kinase
- JEV
Japanese Encephalitis Virus
MAVS Mitochondrial Antiviral Signaling Protein
- MD
Molecular Docking
- MDS
Molecular Dynamics Simulation
- mRNA
messenger RNA
- MTase
Methyl transferase
- NSAID
Non-Steroidal Anti-Inflammatory Drug
- IgM
Immunoglobin M
- IgG
Immunoglobin G
- prM
precursor-Membrane
- QSAR
Quantitative Structure-Activity Relationship
- RdRp
RNA-dependent RNA polymerase
- RNA
Ribonucleic acid
- SAM
S-Adenosylmethionine
- SAR
Structure-Activity Relationship
- SI
Selective Index
- SLEV
St. Louis Encephalitis Virus
- ssRNA
single-stranded Ribonucleic Acid
- STAT
Signal Transducer and Activator of Transcription
- TBDPS
tert-Butyldiphenylsilyl
- TBEV
Tick-borne Encephalitis Viruses
- TBS
tert-butyldimethylsilyl
- TGN
Trans-Golgi Network
- THP
Tetrahydropyran
- TIPS
Triisopropylsilyl
- TLR4
Toll-like receptor-4
- US-FDA
United States-Food and Drug Administration
- UTRs
Untranslated Regions
- WNV
West Nile Virus
- YFV
Yellow Fever Virus.
- ZIKV
Zika Virus.
Data availability
No data was used for the research described in the article.
References
- 1.World Health Organization Dengue and severe dengue. https://www.who.int/news-room/fact-sheets/detail/dengue-and-severe-dengue n.d.
- 2.Guzman M.G., Halstead S.B., Artsob H., Buchy P., Farrar J., Gubler D.J., Hunsperger E., Kroeger A., Margolis H.S., Martí-nez E., Nathan M.B., Pelegrino J.L., Simmons C., Yoksan S., Peeling R.W. Dengue: a continuing global threat. Nat. Rev. Microbiol. 2010;8 doi: 10.1038/nrmicro2460. S7–S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.S A.H., Pujar G.V., Sethu A.K., Bhagyalalitha M., Singh M. Dengue structural proteins as antiviral drug targets: current status in the drug discovery & development. Eur. J. Med. Chem. 2021;221 doi: 10.1016/j.ejmech.2021.113527. [DOI] [PubMed] [Google Scholar]
- 4.Rodenhuis-Zybert I.A., Wilschut J., Smit J.M. Dengue virus life cycle: viral and host factors modulating infectivity. Cell. Mol. Life Sci. 2010;67:2773–2786. doi: 10.1007/s00018-010-0357-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barrows N.J., Campos R.K., Liao K.-C., Prasanth K.R., Soto-Acosta R., Yeh S.-C., Schott-Lerner G., Pompon J., Sessions O.M., Bradrick S.S., Garcia-Blanco M.A. Biochemistry and molecular biology of flaviviruses. Chem. Rev. 2018;118:4448–4482. doi: 10.1021/acs.chemrev.7b00719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wilder-Smith A., Ooi E.E., Horstick O., Wills B. Dengue, Lancet. 2019;393:350–363. doi: 10.1016/S0140-6736(18)32560-1. [DOI] [PubMed] [Google Scholar]
- 7.Mustafa M.S., Rasotgi V., Jain S., Gupta V. Discovery of fifth serotype of dengue virus (denv-5): a new public health dilemma in dengue control. Med. J. Armed Forces India. 2015;71:67–70. doi: 10.1016/j.mjafi.2014.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dennis N. Surprising new dengue virus throws a spanner in disease control efforts. Science. 2013;342:415. doi: 10.1126/science.342.6157.415. 80- [DOI] [PubMed] [Google Scholar]
- 9.Wan Y., Wu W., Zhang J., Li L., Wan Y., Tang X., Chen X., Liu S., Yao X. Tenovin-1 inhibited dengue virus replication through SIRT2. Eur. J. Pharmacol. 2021;907 doi: 10.1016/j.ejphar.2021.174264. [DOI] [PubMed] [Google Scholar]
- 10.Jampilek J. Heterocycles in medicinal chemistry. Molecules. 2019;24:3839. doi: 10.3390/molecules24213839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De A., Sarkar S., Majee A. Recent advances on heterocyclic compounds with antiviral properties. Chem. Heterocycl. Compd. 2021;57:410–416. doi: 10.1007/s10593-021-02917-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mermer A., Keles T., Sirin Y. Recent studies of nitrogen containing heterocyclic compounds as novel antiviral agents: a review. Bioorg. Chem. 2021;114 doi: 10.1016/j.bioorg.2021.105076. [DOI] [PubMed] [Google Scholar]
- 13.Pathania S., Narang R.K., Rawal R.K. Role of sulphur-heterocycles in medicinal chemistry: an update. Eur. J. Med. Chem. 2019;180:486–508. doi: 10.1016/j.ejmech.2019.07.043. [DOI] [PubMed] [Google Scholar]
- 14.Sharma P.K., Amin A., Kumar M. A review: medicinally important nitrogen sulphur containing heterocycles. Open Med. Chem. J. 2020;14:49–64. doi: 10.2174/1874104502014010049. [DOI] [Google Scholar]
- 15.Benmansour F., Eydoux C., Querat G., De Lamballerie X., Canard B., Alvarez K., Guillemot J.C., Barral K. Novel 2-phenyl-5-[(E)-2-(thiophen-2-yl)ethenyl]-1,3,4-oxadiazole and 3-phenyl-5-[(E)-2-(thiophen-2-yl)ethenyl]-1,2,4-oxadiazole derivatives as dengue virus inhibitors targeting NS5 polymerase. Eur. J. Med. Chem. 2016;109:146–156. doi: 10.1016/j.ejmech.2015.12.046. [DOI] [PubMed] [Google Scholar]
- 16.Yusufzai S.K., Osman H., Shaheen M., Basma K., Razik M.A. Synthesis , X-ray crystallographic study , pharmacology and docking of hydrazinyl thiazolyl coumarins as dengue virus NS2B/NS3 serine protease inhibitors. Med. Chem. Res. 2018;27:1647–1665. doi: 10.1007/s00044-018-2179-8. [DOI] [Google Scholar]
- 17.Yusufzai S.K., Osman H., Khan M.S., Razik B.M.A. 4 - thiazolidinone coumarin derivatives as two - component NS2B/NS3 DENV flavivirus serine protease inhibitors : synthesis , molecular docking , biological evaluation and structure – activity relationship studies. Chem. Cent. J. 2018;12:1–16. doi: 10.1186/s13065-018-0435-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shrivastava N., Nawaz F., Ahmed S., Alam O., Hamdard J., Delhi N. Benzimidazole scaffold as anticancer agent: synthetic approaches and structure–activity relationship. Arch. Pharm. (Weinheim). 2017;350 doi: 10.1002/ardp.201700040. 1–80. [DOI] [PubMed] [Google Scholar]
- 19.Gubler D.J., Suharyono W., Tan R., Abidin M., Sie A. Viraemia in patients with naturally acquired dengue infection. Bull. World Health Organ. 1981;59:623–630. [PMC free article] [PubMed] [Google Scholar]
- 20.Mukhopadhyay S., Kuhn R.J., Rossmann M.G. A structural perspective of the Flavivirus life cycle. Nat. Rev. Microbiol. 2005;3:13–22. doi: 10.1038/nrmicro1067. [DOI] [PubMed] [Google Scholar]
- 21.Yu I.M., Zhang W., Holdaway H.A., Li L., Kostyuchenko V.A., Chipman P.R., Kuhn R.J., Rossmann M.G., Chen J. Structure of the immature dengue virus at low pH primes proteolytic maturation. Science. 2008;319:1834–1837. doi: 10.1126/science.1153264. 80- [DOI] [PubMed] [Google Scholar]
- 22.Li L., Lok S.M., Yu I.M., Zhang Y., Kuhn R.J., Chen J., Rossmann M.G. The flavivirus precursor membrane-envelope protein complex: structure and maturation. Science. 2008;319:1830–1834. doi: 10.1126/science.1153263. 80- [DOI] [PubMed] [Google Scholar]
- 23.Obi J.O., Gutiérrez-Barbosa H., Chua J.V., Deredge D.J. Current trends and limitations in dengue antiviral research. Trav. Med. Infect. Dis. 2021;6:180. doi: 10.3390/tropicalmed6040180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heinz F.X., Allison S.L. Flavivirus structure and membrane fusion. Adv. Virus Res. 2003;59:63–97. doi: 10.1016/S0065-3527(03)59003-0. [DOI] [PubMed] [Google Scholar]
- 25.Chen Y., Maguire T., Hileman R.E., Fromm J.R., Esko J.D., Linhardt R.J., Marks R.M. Dengue virus infectivity depends on envelope protein binding to target cell heparan sulfate. Nat. Med. 1997;3:866–871. doi: 10.1038/nm0897-866. [DOI] [PubMed] [Google Scholar]
- 26.Tassaneetrithep B., Burgess T.H., Granelli-Piperno A., Trumpfheller C., Finke J., Sun W., Eller M.A., Pattanapanyasat K., Sarasombath S., Birx D.L., Steinman R.M., Schlesinger S., Marovich M.A. DC-SIGN (CD209) mediates dengue virus infection of human dendritic cells. J. Exp. Med. 2003;197:823–829. doi: 10.1084/jem.20021840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miller J.L., DeWet B.J.M., Martinez-Pomares L., Radcliffe C.M., Dwek R.A., Rudd P.M., Gordon S. The mannose receptor mediates dengue virus infection of macrophages. PLoS Pathog. 2008;4 doi: 10.1371/journal.ppat.0040017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Watson A.A., Lebedev A.A., Hall B.A., Fenton-May A.E., Vagin A.A., Dejnirattisai W., Felce J., Mongkolsapaya J., Palma A.S., Liu Y., Feizi T., Screaton G.R., Murshudov G.N., O'Callaghan C.A. Structural flexibility of the macrophage dengue virus receptor CLEC5A. J. Biol. Chem. 2011;286:24208–24218. doi: 10.1074/jbc.m111.226142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen S.T., Lin Y.L., Huang M.T., Wu M.F., Cheng S.C., Lei H.Y., Lee C.K., Chiou T.W., Wong C.H., Hsieh S.L. CLEC5A is critical for dengue-virus-induced lethal disease. Nature. 2008;453:672–676. doi: 10.1038/nature07013. [DOI] [PubMed] [Google Scholar]
- 30.Clyde K., Kyle J.L., Harris E. Recent advances in deciphering viral and host determinants of dengue virus replication and pathogenesis. J. Virol. 2006;80:11418–11431. doi: 10.1128/jvi.01257-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stadler K., Allison S.L., Schalich J., Heinz F.X. Proteolytic activation of tick-borne encephalitis virus by furin. J. Virol. 1997;71:8475–8481. doi: 10.1128/jvi.71.11.8475-8481.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guirakhoo F., Heinz F.X., Mandl C.W., Holzmann H., Kunz C. Fusion activity of flaviviruses: comparison of mature and immature (prM-containing) tick-borne encephalitis virions. J. Gen. Virol. 1991;72:1323–1329. doi: 10.1099/0022-1317-72-6-1323. [DOI] [PubMed] [Google Scholar]
- 33.Guirakhoo F., Bolin R.A., Roehrig J.T. The Murray Valley encephalitis virus prM protein confers acid resistance to virus particles and alters the expression of epitopes within the R2 domain of E glycoprotein. Virology. 1992;191:921–931. doi: 10.1016/0042-6822(92)90267-S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Y. Structures of immature flavivirus particles. EMBO J. 2003;22:2604–2613. doi: 10.1093/emboj/cdg270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Centers for Disease Control and Prevention (Cdc) Epidemiology of dengue. 2022. https://www.cdc.gov/dengue/epidemiology/index.html
- 36.Gallo F.N., Enderle A.G., Pardo L.A., Leal E.S., Bollini M. Challenges and perspectives in the discovery of dengue virus entry inhibitors. Curr. Med. Chem. 2022;29:719–740. doi: 10.2174/0929867328666210521213118. [DOI] [PubMed] [Google Scholar]
- 37.Troost B., Smit J.M. Recent advances in antiviral drug development towards dengue virus. Curr. Opin. Virol. 2020;43:9–21. doi: 10.1016/j.coviro.2020.07.009. [DOI] [PubMed] [Google Scholar]
- 38.Watanabe S., Low J.G.-H., Vasudevan S.G. Preclinical antiviral testing for dengue virus infection in mouse models and its association with clinical studies. ACS Infect. Dis. 2018;4:1048–1057. doi: 10.1021/acsinfecdis.8b00054. [DOI] [PubMed] [Google Scholar]
- 39.Lim S.P. Dengue drug discovery: progress, challenges and outlook. Antivir. Res. 2019;163:156–178. doi: 10.1016/j.antiviral.2018.12.016. [DOI] [PubMed] [Google Scholar]
- 40.Diamond M.S., Pierson T.C. Molecular insight into dengue virus pathogenesis and its implications for disease control. Cell. 2015;162:488–492. doi: 10.1016/j.cell.2015.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lai J.H., Lin Y.L., Hsieh S.L. Pharmacological intervention for dengue virus infection. Biochem. Pharmacol. 2017;129:14–25. doi: 10.1016/j.bcp.2017.01.005. [DOI] [PubMed] [Google Scholar]
- 42.Perera R., Kuhn R.J. Structural proteomics of dengue virus. Curr. Opin. Microbiol. 2008;11:369–377. doi: 10.1016/j.mib.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Smith J.L., Sheridan K., Parkins C.J., Frueh L., Jemison A.L., Strode K., Dow G., Nilsen A., Hirsch A.J. Characterization and structure-activity relationship analysis of a class of antiviral compounds that directly bind dengue virus capsid protein and are incorporated into virions. Antivir. Res. 2018;155:12–19. doi: 10.1016/j.antiviral.2018.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Byk L.A., Gamarnik A.V. Properties and functions of the dengue virus capsid protein. Annu. Rev. Virol. 2016;3:263–281. doi: 10.1146/annurev-virology-110615-042334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nemésio H., Palomares-Jerez M.F., Villalaín J. Hydrophobic segment of dengue virus C protein. Interaction with model membranes. Mol. Membr. Biol. 2013;30:273–287. doi: 10.3109/09687688.2013.805835. [DOI] [PubMed] [Google Scholar]
- 46.Markoff L., Falgout B., Chang A. A conserved internal hydrophobic domain mediates the stable membrane integration of the dengue virus capsid protein. Virology. 1997;233:105–117. doi: 10.1006/viro.1997.8608. [DOI] [PubMed] [Google Scholar]
- 47.Samsa M.M., Mondotte J.A., Caramelo J.J., Gamarnik A.V. Uncoupling cis -acting RNA elements from coding sequences revealed a requirement of the N-terminal region of dengue virus capsid protein in virus particle formation. J. Virol. 2012;86:1046–1058. doi: 10.1128/jvi.05431-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hsieh S.C., Wu Y.C., Zou G., Nerurkar V.R., Shi P.Y., Wang W.K. Highly conserved residues in the helical domain of dengue virus type 1 precursor membrane protein are involved in assembly, precursor membrane (prM) protein cleavage, and entry. J. Biol. Chem. 2014;289:33149–33160. doi: 10.1074/jbc.M114.610428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yeo A.S.L., Rathakrishnan A., Wang S.M., Ponnampalavanar S., Manikam R., Sathar J., Kumari Natkunam S., Sekaran S.D. Dengue patients exhibit higher levels of PrM and e antibodies than their asymptomatic counterparts. BioMed Res. Int. 2015 doi: 10.1155/2015/420867. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dias R.S., Teixeira M.D., Xisto M.F., Prates J.W.O., Silva J.D.D., Mello I.O., Silva C.C.D., De Paula S.O. DENV-3 precursor membrane (prM) glycoprotein enhances E protein immunogenicity and confers protection against DENV-2 infections in a murine model. Hum. Vaccines Immunother. 2021;17:1271–1277. doi: 10.1080/21645515.2020.1826798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fahimi H., Mohammadipour M., Haddad Kashani H., Parvini F., Sadeghizadeh M. Dengue viruses and promising envelope protein domain III-based vaccines. Appl. Microbiol. Biotechnol. 2018;102:2977–2996. doi: 10.1007/s00253-018-8822-y. [DOI] [PubMed] [Google Scholar]
- 52.Modis Y., Ogata S., Clements D., Harrison S.C. Structure of the dengue virus envelope protein after membrane fusion. Nature. 2004;427:313–319. doi: 10.1038/nature02165. [DOI] [PubMed] [Google Scholar]
- 53.Lindenbach B.D., Rice C.M. Genetic interaction of flavivirus nonstructural proteins NS1 and NS4A as a determinant of replicase function. J. Virol. 1999;73:4611–4621. doi: 10.1128/jvi.73.6.4611-4621.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Alayli F., Scholle F. Dengue virus NS1 enhances viral replication and pro-inflammatory cytokine production in human dendritic cells. Virology. 2016;496:227–236. doi: 10.1016/j.virol.2016.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Modhiran N., Watterson D., Muller D.A., Panetta A.K., Sester D.P., Liu L., Hume D.A., Stacey K.J., Young P.R. Dengue virus NS1 protein activates cells via Toll-like receptor 4 and disrupts endothelial cell monolayer integrity. Sci. Transl. Med. 2015;7 doi: 10.1126/scitranslmed.aaa3863. [DOI] [PubMed] [Google Scholar]
- 56.Glasner D.R., Puerta-Guardo H., Beatty P.R., Harris E. The good, the bad, and the shocking: the multiple roles of dengue virus nonstructural protein 1 in protection and pathogenesis. Annu. Rev. Virol. 2018;5:227–253. doi: 10.1146/annurev-virology-101416-041848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xie X., Zou J., Zhang X., Zhou Y., Routh A.L., Kang C., Popov V.L., Chen X., Wang Q.Y., Dong H., Shi P.Y. Dengue NS2A protein orchestrates virus assembly. Cell Host Microbe. 2019;26:606–622. doi: 10.1016/j.chom.2019.09.015. e8. [DOI] [PubMed] [Google Scholar]
- 58.Xie X., Gayen S., Kang C., Yuan Z., Shi P.-Y. Membrane topology and function of dengue virus NS2A protein. J. Virol. 2013;87:4609–4622. doi: 10.1128/jvi.02424-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nemésio H., Villalaín J. Membrane interacting regions of dengue virus NS2A protein. J. Phys. Chem. B. 2014;118:10142–10155. doi: 10.1021/jp504911r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li Y., Li Q., Wong Y.L., Liew L.S.Y., Kang C. Membrane topology of NS2B of dengue virus revealed by NMR spectroscopy. Biochim. Biophys. Acta - Biomembr. 2015;1848:2244–2252. doi: 10.1016/j.bbamem.2015.06.010. [DOI] [PubMed] [Google Scholar]
- 61.León-Juárez M., Martínez-Castillo M., Shrivastava G., García-Cordero J., Villegas-Sepulveda N., Mondragón-Castelán M., Mondragón-Flores R., Cedillo-Barrón L. Recombinant Dengue virus protein NS2B alters membrane permeability in different membrane models. Virol. J. 2016;13:1–11. doi: 10.1186/s12985-015-0456-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Leung D., Schroder K., White H., Fang N.X., Stoermer M.J., Abbenante G., Martin J.L., Young P.R., Fairlie D.P. Activity of recombinant dengue 2 virus NS3 protease in the presence of a truncated NS2B Co-factor, small peptide substrates, and inhibitors. J. Biol. Chem. 2001;276:45762–45771. doi: 10.1074/jbc.M107360200. [DOI] [PubMed] [Google Scholar]
- 63.Kim Y.M., Gayen S., Kang C.B., Joy J., Huang Q., Chen A.S., Wee J.L.K., Ang M.J.Y., Lim H.A., Hung A.W., Li R., Noble C.G., Lee L.T., Yip A., Wang Q.Y., Chia C.S.B., Hill J., Shi P.Y., Keller T.H. NMR analysis of a novel enzymatically active unlinked dengue NS2B-NS3 protease complex. J. Biol. Chem. 2013;288:12891–12900. doi: 10.1074/jbc.M112.442723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wu H., Bock S., Snitko M., Berger T., Weidner T., Holloway S., Kanitz M., Diederich W.E., Steuber H., Walter C., Hofmann D., Weißbrich B., Spannaus R., Acosta E.G., Bartenschlager R., Engels B., Schirmeister T., Bodem J. Novel dengue virus NS2B/NS3 protease inhibitors. Antimicrob. Agents Chemother. 2015;59:1100–1109. doi: 10.1128/AAC.03543-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Swarbrick C.M.D., Basavannacharya C., Chan K.W.K., Chan S.A., Singh D., Wei N., Phoo W.W., Luo D., Lescar J., Vasudevan S.G. NS3 helicase from dengue virus specifically recognizes viral RNA sequence to ensure optimal replication. Nucleic Acids Res. 2017;45:12904–12920. doi: 10.1093/nar/gkx1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Silva E.M., Conde J.N., Allonso D., Ventura G.T., Coelho D.R., Carneiro P.H., Silva M.L., Paes M.V., Rabelo K., Weissmuller G., Bisch P.M., Mohana-Borges R. Dengue virus nonstructural 3 protein interacts directly with human glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and reduces its glycolytic activity. Sci. Rep. 2019;9:1–19. doi: 10.1038/s41598-019-39157-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cui T., Sugrue R.J., Xu Q., Lee A.K.W., Chan Y.C., Fu J. Recombinant dengue virus type 1 NS3 protein exhibits specific viral RNA binding and NTPase activity regulated by the NS5 protein. Virology. 1998;246:409–417. doi: 10.1006/viro.1998.9213. [DOI] [PubMed] [Google Scholar]
- 68.Wengler G., Wengler G. The NS 3 nonstructural protein of flaviviruses contains an RNA triphosphatase activity. Virology. 1993;197:265–273. doi: 10.1006/viro.1993.1587. [DOI] [PubMed] [Google Scholar]
- 69.Shiryaev S.A., Chernov A.V., Aleshin A.E., Shiryaeva T.N., Strongin A.Y. NS4A regulates the ATPase activity of the NS3 helicase: a novel cofactor role of the non-structural protein NS4A from West Nile virus. J. Gen. Virol. 2009;90:2081–2085. doi: 10.1099/vir.0.012864-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.He Z., Zhu X., Wen W., Yuan J., Hu Y., Chen J., An S., Dong X., Lin C., Yu J., Wu J., Yang Y., Cai J., Li J., Li M. Dengue virus subverts host innate immunity by targeting adaptor protein MAVS. J. Virol. 2016;90:7219–7230. doi: 10.1128/jvi.00221-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Uno N., Ross T.M. Dengue virus and the host innate immune response. Emerg. Microb. Infect. 2018;7 doi: 10.1038/s41426-018-0168-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Miller S., Kastner S., Krijnse-Locker J., Bühler S., Bartenschlager R. The non-structural protein 4A of dengue virus is an integral membrane protein inducing membrane alterations in a 2K-regulated manner. J. Biol. Chem. 2007;282:8873–8882. doi: 10.1074/jbc.M609919200. [DOI] [PubMed] [Google Scholar]
- 73.Tajima S., Takasaki T., Kurane I. Restoration of replication-defective dengue type 1 virus bearing mutations in the N-terminal cytoplasmic portion of NS4A by additional mutations in NS4B. Arch. Virol. 2011;156:63–69. doi: 10.1007/s00705-010-0816-8. [DOI] [PubMed] [Google Scholar]
- 74.Muñoz-Jordán J.L., Sánchez-Burgos G.G., Laurent-Rolle M., García-Sastre A. Inhibition of interferon signaling by dengue virus. Proc. Natl. Acad. Sci. U.S.A. 2003;100:14333–14338. doi: 10.1073/pnas.2335168100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nemésio H., Palomares-Jerez F., Villalaín J. NS4A and NS4B proteins from dengue virus: membranotropic regions. Biochim. Biophys. Acta - Biomembr. 2012;1818:2818–2830. doi: 10.1016/j.bbamem.2012.06.022. [DOI] [PubMed] [Google Scholar]
- 76.Kapoor M., Zhang L., Ramachandra M., Kusukawa J., Ebner K.E., Padmanabhan R. Association between NS3 and NS5 proteins of dengue virus type 2 in the putative RNA replicase is linked to differential phosphorylation of NS5. J. Biol. Chem. 1995;270:19100–19106. doi: 10.1074/jbc.270.32.19100. [DOI] [PubMed] [Google Scholar]
- 77.Ashour J., Laurent-Rolle M., Shi P.-Y., García-Sastre A. NS5 of dengue virus mediates STAT2 binding and degradation. J. Virol. 2009;83:5408–5418. doi: 10.1128/jvi.02188-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.De Maio F.A., Risso G., Iglesias N.G., Shah P., Pozzi B., Gebhard L.G., Mammi P., Mancini E., Yanovsky M.J., Andino R., Krogan N., Srebrow A., Gamarnik A.V. The dengue virus NS5 protein intrudes in the cellular spliceosome and modulates splicing. PLoS Pathog. 2016;12:1–29. doi: 10.1371/journal.ppat.1005841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Aromí G., Barrios L.A., Roubeau O., Gamez P. Triazoles and tetrazoles: prime ligands to generate remarkable coordination materials. Coord. Chem. Rev. 2011;255:485–546. doi: 10.1016/j.ccr.2010.10.038. [DOI] [Google Scholar]
- 80.Al-Masoudi Ia A.M.N., Al-Soud Y.A., Al-Salihi N.J. 1, 2, 4-Triazoles: synthetic approaches and pharmacological importance. Chem. Heterocycl. Compd. 2006;42:1377–1403. [Google Scholar]
- 81.De Nino A., Maiuolo L., Costanzo P., Algieri V., Jiritano A., Olivito F., Tallarida M.A. Recent progress in catalytic synthesis of 1,2,3-triazoles. Catalysts. 2021;11:1–48. doi: 10.3390/catal11091120. [DOI] [Google Scholar]
- 82.Neto J.S.S., Zeni G. A decade of advances in the reaction of nitrogen sources and alkynes for the synthesis of triazoles. Coord. Chem. Rev. 2020;409 doi: 10.1016/j.ccr.2020.213217. [DOI] [Google Scholar]
- 83.Sahu J.K., Ganguly S., Kaushik A. Triazoles: a valuable insight into recent developments and biological activities. Chin. J. Nat. Med. 2013;11:456–465. doi: 10.1016/S1875-5364(13)60084-9. [DOI] [PubMed] [Google Scholar]
- 84.Duan J.R., Liu H.B., Jeyakkumar P., Gopala L., Li S., Geng R.X., Zhou C.H. Design, synthesis and biological evaluation of novel Schiff base-bridged tetrahydroprotoberberine triazoles as a new type of potential antimicrobial agents. Medchemcomm. 2017;8:907–916. doi: 10.1039/c6md00688d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Naidu K.M., Srinivasarao S., Agnieszka N., Ewa A.K., Kumar M.M.K., Chandra Sekhar K.V.G. Seeking potent anti-tubercular agents: design, synthesis, anti-tubercular activity and docking study of various ((triazoles/indole)-piperazin-1-yl/1,4-diazepan-1-yl)benzo[d]isoxazole derivatives. Bioorg. Med. Chem. Lett. 2016;26:2245–2250. doi: 10.1016/j.bmcl.2016.03.059. [DOI] [PubMed] [Google Scholar]
- 86.Kumbhare R.M., Kosurkar U.B., Janaki Ramaiah M., Dadmal T.L., Pushpavalli S.N.C.V.L., Pal-Bhadra M. Synthesis and biological evaluation of novel triazoles and isoxazoles linked 2-phenyl benzothiazole as potential anticancer agents. Bioorg. Med. Chem. Lett. 2012;22:5424–5427. doi: 10.1016/j.bmcl.2012.07.041. [DOI] [PubMed] [Google Scholar]
- 87.Asif M. A mini review on antimalarial activities of biologically active substituted triazoles derivatives. Int. J. Adv. Res. Comput. Sci. 2014;1:2349–2403. www.arcjournals.org [Google Scholar]
- 88.Shaikh M.H., Subhedar D.D., Shingate B.B., Kalam Khan F.A., Sangshetti J.N., Khedkar V.M., Nawale L., Sarkar D., Navale G.R., Shinde S.S. Synthesis, biological evaluation and molecular docking of novel coumarin incorporated triazoles as antitubercular, antioxidant and antimicrobial agents. Med. Chem. Res. 2016;25:790–804. doi: 10.1007/s00044-016-1519-9. [DOI] [Google Scholar]
- 89.Ayati A., Emami S., Foroumadi A. The importance of triazole scaffold in the development of anticonvulsant agents. Eur. J. Med. Chem. 2016;109:380–392. doi: 10.1016/j.ejmech.2016.01.009. [DOI] [PubMed] [Google Scholar]
- 90.Rizwana F., Muthu S., Prasana J.C., Abraham C.S., Raja M. Spectroscopic (FT-IR, FT-Raman) investigation, topology (ESP, ELF, LOL) analyses, charge transfer excitation and molecular docking (dengue, HCV) studies on ribavirin. Chem. Data Collect. 2018;17–18:236–250. doi: 10.1016/j.cdc.2018.09.003. [DOI] [Google Scholar]
- 91.Vernekar S.K.V., Qiu L., Zhang J., Kankanala J., Li H., Geraghty R.J., Wang Z. 5′-silylated 3′-1,2,3-triazolyl thymidine analogues as inhibitors of West Nile Virus and Dengue virus. J. Med. Chem. 2015;58:4016–4028. doi: 10.1021/acs.jmedchem.5b00327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Vishvakarma V.K., Shukla N., Reetu, Kumari K., Patel R., Singh P. A model to study the inhibition of nsP2B-nsP3 protease of dengue virus with imidazole, oxazole, triazole thiadiazole, and thiazolidine based scaffolds. Heliyon. 2019;5 doi: 10.1016/j.heliyon.2019.e02124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chauhan A., Sharma P.K., Kaushik N. Pyrazole: a versatile moiety. Int. J. ChemTech Res. 2011;3:11–17. [Google Scholar]
- 94.Ansari A., Ali A., Asif M. Shamsuzzaman, Review: biologically active pyrazole derivatives. New J. Chem. 2016;41:16–41. doi: 10.1039/c6nj03181a. [DOI] [Google Scholar]
- 95.Naim Mj A.P., Alam O., Farah Nawaz M., Alam J. Current status of pyrazole and its biological activities. J. Pharm. BioAllied Sci. 2016;8:2–17. doi: 10.4103/0975-7406.171694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Karrouchi K., Radi S., Ramli Y., Taoufik J., Mabkhot Y.N., Al-Aizari F.A., Ansar M. 2018. Synthesis and Pharmacological Activities of Pyrazole Derivatives: A Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Keter F.K., Darkwa J. Perspective: the potential of pyrazole-based compounds in medicine. Biometals. 2012;25:9–21. doi: 10.1007/s10534-011-9496-4. [DOI] [PubMed] [Google Scholar]
- 98.Kumar V., Kaur K., Gupta G.K., Sharma A.K. Pyrazole containing natural products: synthetic preview and biological significance. Eur. J. Med. Chem. 2013;69:735–753. doi: 10.1016/j.ejmech.2013.08.053. [DOI] [PubMed] [Google Scholar]
- 99.Li M., Zhao B.X. Progress of the synthesis of condensed pyrazole derivatives (from 2010 to mid-2013) Eur. J. Med. Chem. 2014;85:311–340. doi: 10.1016/j.ejmech.2014.07.102. [DOI] [PubMed] [Google Scholar]
- 100.Faria J.V., Vegi P.F., Miguita A.G.C., dos Santos M.S., Boechat N., Bernardino A.M.R. Recently reported biological activities of pyrazole compounds. Bioorg. Med. Chem. 2017;25:5891–5903. doi: 10.1016/j.bmc.2017.09.035. [DOI] [PubMed] [Google Scholar]
- 101.Saudi M., Zmurko J., Kaptein S., Rozenski J., Gadakh B., Chaltin P., Marchand A., Neyts J., Van Aerschot A. Synthetic strategy and antiviral evaluation of diamide containing heterocycles targeting dengue and yellow fever virus. Eur. J. Med. Chem. 2016;121:158–168. doi: 10.1016/j.ejmech.2016.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lee J.C., Tseng C.K., Lin C.K., Tseng C.H. Discovery of novel diarylpyrazolylquinoline derivatives as potent anti-dengue virus agents. Eur. J. Med. Chem. 2017;141:282–292. doi: 10.1016/j.ejmech.2017.10.001. [DOI] [PubMed] [Google Scholar]
- 103.Zamri A I.I., Teruna H.Y., Wulansari S., Herfindo N., Frimayanti N. Vol. 4. 2019. pp. 1–8. (3-(3,4-Dimethoxyphenyl)-5-(2-Fluorophenyl)-1-Phenyl- 4,5-Dihydro-1H-Pyrazole). Short note. [DOI] [Google Scholar]
- 104.Bhatnagar N., Sharma A., Kumar P.K. A review on " imidazoles " : their chemistry and pharmacological potentials. Int. J. PharmaTech Res. 2011;3:268–282. [Google Scholar]
- 105.Dmitrii A. Shabalin and Jason E. Camp, Recent advances in the synthesis of imidazoles. Org. Biomol. Chem. 2020;18:3950–3964. doi: 10.1039/d0ob00350f. [DOI] [PubMed] [Google Scholar]
- 106.Sisko J., Mellinger M. Development of a general process for the synthesis of highly substituted imidazoles. Pure Appl. Chem. 2002;74:1349–1357. [Google Scholar]
- 107.Tahlan S., Kumar S., Narasimhan B. Pharmacological significance of heterocyclic 1H-benzimidazole scaffolds: a review. BMC Chem. 2019;13:1–21. doi: 10.1186/s13065-019-0625-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ahmad N., Azad M.I., Khan A.R., Azad I. Benzimidazole as a promising antiviral heterocyclic scaffold: a review. J. Sci. Arts. 2021;21:273–284. doi: 10.46939/j.sci.arts-21.1-b05. [DOI] [Google Scholar]
- 109.Swain S.P., Mohanty S. Imidazolidinones and imidazolidine-2,4-diones as antiviral agents. ChemMedChem. 2019;14:291–302. doi: 10.1002/cmdc.201800686. [DOI] [PubMed] [Google Scholar]
- 110.Ali I., Lone M.N., Aboul-Enein H.Y. Imidazoles as potential anticancer agents. Medchemcomm. 2017 doi: 10.1039/C7MD00067G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sucipto T.H., Churrotin S., Setyawati H., Martak F., Mulyatno K.C., Amarullah I.H., Kotaki T., Kameoka M., Yotopranoto S., Soegijanto S. A new copper (II)-imidazole derivative effectively inhibits replication of denv-2 in vero cell. African J. Infect. Dis. 2018;12:116–119. doi: 10.2101/Ajid.12v1S.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sucipto F.M. Teguh H. Inhibition of dengue virus serotype 2 in Vero cells with [Cu(2,4,5-triphenyl-1H-imidazole) 2(H2O)2].Cl2. Infect. Dis. Rep. 2020;12:93–97. doi: 10.4081/idr.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sumarsih S., Pratiwi M.D., Ainni I.N., Sinatriya H.R., Soegijanto S., Sucipto T.H., Setyawati H. The influence of metal on the performance of 2,4,5-triphenylimidazole as an inhibitor of dengue virus replication, Asia-Pacific. J. Mol. Biol. Biotechnol. 2020;28:113–121. doi: 10.35118/apjmbb.2020.028.3.11. [DOI] [Google Scholar]
- 114.Wibrianto A., Martak F., Sucipto T.H., Churrotin S., Amarullah I.H. 2020. Eff Ect of Zinc(II)-2 , 4 , 5-triphenyl-1 H -imidazole Complex against Replication DENV-2 in Vero Cell Chemicals and Media. [Google Scholar]
- 115.Okano Y., Saito-Tarashima N., Kurosawa M., Iwabu A., Ota M., Watanabe T., Kato F., Hishiki T., Fujimuro M., Minakawa N. Synthesis and biological evaluation of novel imidazole nucleosides as potential anti-dengue virus agents. Bioorg. Med. Chem. 2019;27:2181–2186. doi: 10.1016/j.bmc.2019.04.015. [DOI] [PubMed] [Google Scholar]
- 116.Altaf A.A., Shahzad A., Gul Z., Rasool N., Badshah A., Lal B., Khan E. A review on the medicinal importance of pyridine derivatives. J. Drug Des. Med. Chem. 2015;1:1–11. doi: 10.11648/j.jddmc.20150101.11. [DOI] [Google Scholar]
- 117.Salim R., Ech-chihbi E., Oudda H., El Hajjaji F., Taleb M., Jodeh S. A review on the assessment of imidazo[1,2-a]pyridines as corrosion inhibitor of metals. J. Bio- Tribo-Corrosion. 2019;5 doi: 10.1007/s40735-018-0207-3. [DOI] [Google Scholar]
- 118.Chaban T.I., Ogurtsov V.V., Matiychuk V.S., Chaban I.G., Demchuk I.L., Nektegayev I.A. Synthesis, anti-inflammatory and antioxidant activities of novel 3H-thiazolo[4,5-b]pyridines. Acta Chim. Slov. 2019;66:103–111. doi: 10.17344/acsi.2018.4570. [DOI] [PubMed] [Google Scholar]
- 119.Henary M., Kananda C., Rotolo L., Savino B., Owens E.A., Cravotto G. Benefits and applications of microwave-assisted synthesis of nitrogen containing heterocycles in medicinal chemistry. RSC Adv. 2020;10:14170–14197. doi: 10.1039/d0ra01378a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hong-Tao Xu, Colby-Germinario Susan P., Hassounah Said, Quashie Peter K., Han Yingshan, Oliveira Maureen, Stranix Brent R., Wainberg M.A. Identification of a pyridoxine-derived small-molecule inhibitor, antimicrob. Agents Chemother. 2016;60:600–608. doi: 10.1128/AAC.02203-15.Address. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Pu S., Wouters R., Schor S., Rozenski J., Barouch-bentov R., Prugar L.I., Brien C.M.O., Brannan J.M., Dye J.M., Herdewijn P., De Jonghe S., Einav S. Optimization of isothiazolo[4,3-b]pyridine-based inhibitors of cyclin G associated kinase (GAK) with broad-spectrum antiviral activity. J. Med. Chem. 2018;61:6178–6192. doi: 10.1021/acs.jmedchem.8b00613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Belen Martinez-Gualda D.S., Saul Sirle, Froeyen Mathy, Piet Herdewijn S.D.J., Einav Shirit. Discovery of 3-phenyl- and 3- N -piperidinyl-isothiazolo [ 4 , 3- b ] pyridines as highly potent inhibitors of cyclin G-associated kinase. Eur. J. Med. Chem. 2021;213 doi: 10.1016/j.ejmech.2021.113158. [DOI] [PubMed] [Google Scholar]
- 123.Wouters R., Pu S., Froeyen M., Lescrinier E., Einav S., Herdewijn P., De Jonghe S. European Journal of Medicinal Chemistry Cyclin G-associated kinase (GAK) af fi nity and antiviral activity studies of a series of 3- C -substituted isothiazolo [4 , 3- b] pyridines. Eur. J. Med. Chem. 2019;163:256–265. doi: 10.1016/j.ejmech.2018.11.065. [DOI] [PubMed] [Google Scholar]
- 124.Jiahong Li S., Kovackova Sona, Pu Szuyuan, Rozenski Jef, De Jonghe Steven, Einav P.H. Isothiazolo[4,3-b]pyridines as inhibitors of cyclin G associated kinase : synthesis, structure-activity relationship studies and antiviral activity. Medchemcomm. 2015;6:1666–1672. doi: 10.1039/c5md00229j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Verdonck S., Pu S.Y., Sorrell F.J., Elkins J.M., Froeyen M., Gao L.J., Prugar L.I., Dorosky D.E., Brannan J.M., Barouch-Bentov R., Knapp S., Dye J.M., Herdewijn P., Einav S., De Jonghe S. Synthesis and structure-activity relationships of 3,5-Disubstituted-pyrrolo[2,3- b]pyridines as inhibitors of adaptor-associated kinase 1 with antiviral activity. J. Med. Chem. 2019;62:5810–5831. doi: 10.1021/acs.jmedchem.9b00136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Shi B., Conner S.D., Liu J. Dysfunction of endocytic kinase AAK1 in ALS. Int. J. Mol. Sci. 2014;15:22918–22932. doi: 10.3390/ijms151222918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Martinez-gualda B., Pu S., Froeyen M., Herdewijn P., Einav S., De Jonghe S. Bioorganic & Medicinal Chemistry Structure-activity relationship study of the pyridine moiety of isothiazolo [ 4 , 3- b ] pyridines as antiviral agents targeting cyclin G-associated kinase. Bioorg. Med. Chem. 2020;28 doi: 10.1016/j.bmc.2019.115188. [DOI] [PubMed] [Google Scholar]
- 128.Dolezal M., Zitko J. Pyrazine derivatives: a patent review (June 2012-present) Expert Opin. Ther. Pat. 2015;25:33–47. doi: 10.1517/13543776.2014.982533. [DOI] [PubMed] [Google Scholar]
- 129.Zaki R.M., Abdul-Malik M.A., Saber S.H., Radwan S.M., El-Dean A.M.K. A convenient synthesis, reactions and biological evaluation of novel pyrazolo[3,4-b]selenolo[3,2-e]pyrazine heterocycles as potential anticancer and antimicrobial agents. Med. Chem. Res. 2020;29:2130–2145. doi: 10.1007/s00044-020-02635-z. [DOI] [Google Scholar]
- 130.MVessally Esmail, Hosseinian Akram, Edjlali Ladan, Bekhradnia Ahmadreza, Esrafili D. New page to access pyrazines and their ring fused analogues: synthesis from N-propargylamines. Curr. Org. Synth. 2017;14:557–567. [Google Scholar]
- 131.Kim J., Park M., Choi J., Singh D.K., Kwon H.J., Kim S.H., Kim I. Design, synthesis, and biological evaluation of novel pyrrolo[1,2-a]pyrazine derivatives. Bioorg. Med. Chem. Lett. 2019;29:1350–1356. doi: 10.1016/j.bmcl.2019.03.044. [DOI] [PubMed] [Google Scholar]
- 132.Tantawy E.S., Amer A.M., Mohamed E.K., Abd Alla M.M., Nafie M.S. Synthesis, characterization of some pyrazine derivatives as anti-cancer agents: in vitro and in Silico approaches. J. Mol. Struct. 2020;1210 doi: 10.1016/j.molstruc.2020.128013. [DOI] [Google Scholar]
- 133.Miniyar P., Murumkar P., Patil P., Barmade M., Bothara K. Unequivocal role of pyrazine ring in medicinally important compounds: a review. Mini-Rev. Med. Chem. 2015;13:1607–1625. doi: 10.2174/1389557511313110007. [DOI] [PubMed] [Google Scholar]
- 134.Boschi D., Pippione A.C., Sainas S., Lolli M.L. European Journal of Medicinal Chemistry Dihydroorotate dehydrogenase inhibitors in anti-infective drug research. Eur. J. Med. Chem. 2019;183 doi: 10.1016/j.ejmech.2019.111681. [DOI] [PubMed] [Google Scholar]
- 135.Sharma V., Chitranshi N., Agarwal A.K. 2014. Significance and Biological Importance of Pyrimidine in the Microbial World. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Mishra I., Tomar R. Pyrimidine: the molecule of diverse biological and medicinal importance. Int. J. Pharma Sci. Res. 2011;2:758–771. [Google Scholar]
- 137.Sahu M., Siddiqui N. A review on biological importance of pyrimidines in the new era. Int. J. Pharm. Pharmaceut. Sci. 2016;8:8–21. [Google Scholar]
- 138.Theivendren Panneer Selvam S.K.V., Richa James Caiado, Vijaysarathy Dniandev Phadte. A mini review of pyrimidine and fused pyrimidine marketed drugs. Res. Pharm. 2012;2:1–9. [Google Scholar]
- 139.Clark M.J., Miduturu C., Schmidt A.G., Zhu X., Pitts J.D., Wang J., Potisopon S., Zhang J., Wojciechowski A., Hann Chu J.J., Gray N.S., Yang P.L. GNF-2 Inhibits Dengue Virus by Targeting Abl Kinases and the Viral e Protein. Cell Chem. Biol. 2016;23:443–452. doi: 10.1016/j.chembiol.2016.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.McGuigan C., Serpi M., Slusarczyk M., Ferrari V., Pertusati F., Meneghesso S., Derudas M., Farleigh L., Zanetta P., Bugert J. Anti-flavivirus activity of different tritylated pyrimidine and purine nucleoside analogues. ChemistryOpen. 2016;5:227–235. doi: 10.1002/open.201500216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Leal E.S., Adler N.S., Fernández G.A., Gebhard L.G., Battini L., Aucar M.G., Videla M., Monge M.E., Hernández de los Ríos A., Acosta Dávila J.A., Morell M.L., Cordo S.M., García C.C., Gamarnik A.V., Cavasotto C.N., Bollini M. De novo design approaches targeting an envelope protein pocket to identify small molecules against dengue virus. Eur. J. Med. Chem. 2019;182 doi: 10.1016/j.ejmech.2019.111628. [DOI] [PubMed] [Google Scholar]
- 142.hong Wan Y., yu Wu W., xin Guo S., jun He S., dong Tang X., yun Wu X., Nandakumar K.S., Zou M., Li L., guang Chen X., wen Liu S., gang Yao X. [1,2,4]Triazolo[1,5-a]pyrimidine derivative (Mol-5) is a new NS5-RdRp inhibitor of DENV2 proliferation and DENV2-induced inflammation. Acta Pharmacol. Sin. 2020;41:706–718. doi: 10.1038/s41401-019-0316-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kaushik N.K., Kaushik N., Attri P., Kumar N., Kim C.H., Verma A.K., Choi E.H. Biomedical importance of indoles. Molecules. 2013;18:6620–6662. doi: 10.3390/molecules18066620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kumar S., Ritika A brief review of the biological potential of indole derivatives. Futur. J. Pharm. Sci. 2020;6:1–19. [Google Scholar]
- 145.Inman M., Moody C.J. Indole synthesis – something old, something new. Chem. Sci. 2013;4:29–41. doi: 10.1039/c2sc21185h. [DOI] [Google Scholar]
- 146.Taber D.F., Tirunahari P.K. Indole synthesis: a review and proposed classification. Tetrahedron. 2011;67:7195–7210. doi: 10.1016/j.tet.2011.06.040.Indole. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Gobi G.V.T.K.T. Evaluation of larvicidal efficacy of amino acids Schiff base against Aedes aegypti mosquito vectors. Int. J. Res. Anal. Rev. 2018;5:799–804. [Google Scholar]
- 148.De S., Jain A., Barman P. Recent advances in the catalytic applications of chiral schiff-base ligands and metal complexes in asymmetric organic transformations. 2022. 7,1-24. [DOI]
- 149.Savir S., Wei Z.J., Liew J.W.K., Vythilingam I., Lim Y.A.L., Saad H.M., Sim K.S., Tan K.W. Synthesis, cytotoxicity and antimalarial activities of thiosemicarbazones and their nickel (II) complexes. J. Mol. Struct. 2020;1211 doi: 10.1016/j.molstruc.2020.128090. [DOI] [Google Scholar]
- 150.Jain A., De S., Barman P. Microwave-assisted synthesis and notable applications of Schiff-base and metal complexes: a comparative study. Res. Chem. Intermed. 2022;48:2199–2251. doi: 10.1007/s11164-022-04708-7. [DOI] [Google Scholar]
- 151.T.A. de Janaina R. Sousa, Fábio A. Silva, Sabrina K. Targanski, Bruno R. Fazolo, Oliveira, Jessica, M. Souza, Mateus, G. Campos, Lucas, C. C. Vieira, M.A. Soares Mendes, Synthesis and larvicidal activity of indole derivatives against Aedes aegypti (Diptera : Culicidae), J. Appl. Entomol. 143 (2019) 1–10. 10.1111/jen.12685. [DOI]
- 152.Dorothée Bardiot A.M., Mohamed Koukni, Smets Wim, Gunter Carlens, McNaughton Michael, Kaptein Suzanne, Dallmeier Kai, Chaltin Patrick, Neyts Johan. Discovery of indole derivatives as novel and potent dengue virus inhibitors. J. Med. Chem. 2018;61:8390–8401. doi: 10.1021/acs.jmedchem.8b00913. [DOI] [PubMed] [Google Scholar]
- 153.Qian W., Xue J.X., Xu J., Li F., Zhou G.F., Wang F., Luo R.H., Liu J., Zheng Y.T., Zhou G.C. Design, synthesis, discovery and SAR of the fused tricyclic derivatives of indoline and imidazolidinone against DENV replication and infection. Bioorg. Chem. 2022;120 doi: 10.1016/j.bioorg.2022.105639. [DOI] [PubMed] [Google Scholar]
- 154.Nie S., Zhao J., Wu X., Yao Y., Wu F., Lin Y., Li X., Kneubehl A.R., Vogt M.B., Rico-hesse R., Song Y. Synthesis , structure-activity relationship and antiviral activity of indole-containing inhibitors of Flavivirus NS2B-NS3 protease. Eur. J. Med. Chem. 2021;225 doi: 10.1016/j.ejmech.2021.113767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Narwal S., Kumar S., Kumar P. Synthesis and therapeutic potential of quinoline derivatives. Res. Chem. Intermed. 2017;43:2765–2798. doi: 10.1007/s11164-016-2794-2. [DOI] [Google Scholar]
- 156.Suliphuldevara B., Pattanashettar R., Yernale G. A comprehensive review on the biological interest of quinoline and its derivatives. Bioorg. Med. Chem. 2021;32 doi: 10.1016/j.bmc.2020.115973. [DOI] [PubMed] [Google Scholar]
- 157.Marella A., Tanwar O.P., Saha R., Ali M.R., Srivastava S., Akhter M., Shaquiquzzaman M. Quinoline : a versatile heterocyclic. Saudi Pharmaceut. J. 2013;21:1–12. doi: 10.1016/j.jsps.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Weyesa A., Mulugeta E. Recent advances in the synthesis of biologically and pharmaceutically active quinoline and its analogues : a review. RSC Adv. 2020;10 doi: 10.1039/d0ra03763j. 20784–20793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Afzal O., Kumar S., Ali R., Kumar R., Jaggi M., Bawa S. A review on anticancer potential of bioactive heterocycle quinoline. Eur. J. Med. Chem. 2015;97:871–910. doi: 10.1016/j.ejmech.2014.07.044. [DOI] [PubMed] [Google Scholar]
- 160.Pinheiro L.C.S., Feitosa L.M., Gandi M.O., Silveira F.F., Boechat N. The development of novel compounds against malaria: quinolines, triazolpyridines, pyrazolopyridines and pyrazolopyrimidines. Molecules. 2019;24:1–20. doi: 10.3390/molecules24224095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Dhiman A.K., Thakur A., Kumar R., Sharma U. Rhodium-catalyzed selective C−H bond functionalization of quinolines. Asian J. Org. Chem. 2020;9:1502–1518. doi: 10.1002/ajoc.202000341. [DOI] [Google Scholar]
- 162.Huang P., Saul S., Einav S., Asquith C.R.M. Optimization of 4-anilinoquinolines as dengue virus inhibitors. Molecules. 2021;26:1–12. doi: 10.3390/molecules26237338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Selvam T.P., Kumar P.V. Quinazoline marketed drugs – a review. Res. Pharm. 2011;1:1–21. www.researchinpharmacy.com [Google Scholar]
- 164.Wang D., Gao F. Quinazoline derivatives: synthesis and bioactivities. Chem. Cent. J. 2013;7 doi: 10.1186/1752-153X-7-95. 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Jafari E., Khajouei M.R., Hassanzadeh F., Hakimelahi G.H., Khodarahmi G.A. Quinazolinone and quinazoline derivatives: recent structures with potent antimicrobial and cytotoxic activities. Res. Pharm. Sci. 2016;11:1–14. [PMC free article] [PubMed] [Google Scholar]
- 166.Das R., Mehta D.K., Dhanawat M. Bestowal of quinazoline scaffold in anticancer drug discovery. Anti Cancer Agents Med. Chem. 2020;21:1350–1368. doi: 10.2174/1871520620666200627205321. [DOI] [PubMed] [Google Scholar]
- 167.Í Kune J., Jaroslav B., Pour M., Waisser K., Iosárek M., Í Janota J. Quinazoline derivatives with antitubercular activity. Farmaco. 2000;55:725–729. doi: 10.1016/S0014-827X(00)00100-2. [DOI] [PubMed] [Google Scholar]
- 168.Venugopala K.N., Nayak S.K., Gleiser R.M., Sanchez-Borzone M.E., Garcia D.A., Odhav B. Synthesis, polymorphism, and insecticidal activity of methyl 4-(4-chlorophenyl)-8-iodo-2-methyl-6-oxo-1,6-dihydro-4H-pyrimido[2,1-b]quinazoline-3-Carboxylate against Anopheles arabiensis mosquito. Chem. Biol. Drug Des. 2016:88–96. doi: 10.1111/cbdd.12736. [DOI] [PubMed] [Google Scholar]
- 169.Venkatesham A., Saudi M., Kaptein S., Neyts J., Rozenski J., Froeyen M., Van Aerschot A. Aminopurine and aminoquinazoline scaffolds for development of potential dengue virus inhibitors. Eur. J. Med. Chem. 2017;126:101–109. doi: 10.1016/j.ejmech.2016.10.008. [DOI] [PubMed] [Google Scholar]
- 170.Saul S., Pu S.Y., Zuercher W.J., Einav S., Asquith C.R.M. Potent antiviral activity of novel multi-substituted 4-anilinoquin(az)olines. Bioorg. Med. Chem. Lett. 2020;30 doi: 10.1016/j.bmcl.2020.127284. [DOI] [PubMed] [Google Scholar]
- 171.Saul S., Huang P.T., Einav S., Asquith C.R.M. Identification and evaluation of 4-anilinoquin(az)olines as potent inhibitors of both dengue virus (DENV) and Venezuelan equine encephalitis virus (VEEV) Bioorg. Med. Chem. Lett. 2021;52 doi: 10.1016/j.bmcl.2021.128407. [DOI] [PubMed] [Google Scholar]
- 172.Mishra S., Pandey A., Manvati S. Coumarin: an emerging antiviral agent. Heliyon. 2020;6 doi: 10.1016/j.heliyon.2020.e03217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Tataringa G., Sathyamurthy B., Sandu I., Zbancioc A.M. In silico docking study of some coumarin derivatives as potential inhibitors on different dengue viral proteins. Rev. Chim. 2019;70:3387–3391. doi: 10.37358/rc.19.9.7555. [DOI] [Google Scholar]
- 174.Li Z., Kong D., Liu Y., Li M. Pharmacological perspectives and molecular mechanisms of coumarin derivatives against virus disease. Genes Dis. 2022;9:80–94. doi: 10.1016/j.gendis.2021.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Gómez-calderón C., Mesa-castro C., Robledo S., Gómez S., Bolivar-avila S. Antiviral effect of compounds derived from the seeds of Mammea americana and Tabernaemontana cymosa on Dengue and Chikungunya virus infections. BMC Compl. Alternative Med. 2017;17:1–12. doi: 10.1186/s12906-017-1562-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Rouf A., Tanyeli C. Bioactive thiazole and benzothiazole derivatives. Eur. J. Med. Chem. 2015;97:911–927. doi: 10.1016/j.ejmech.2014.10.058. [DOI] [PubMed] [Google Scholar]
- 177.Kumar G., Singh N.P. Synthesis , anti-inflammatory and analgesic evaluation of thiazole/oxazole substituted benzothiazole derivatives. Bioorg. Chem. 2021;107:1–12. doi: 10.1016/j.bioorg.2020.104608. [DOI] [PubMed] [Google Scholar]
- 178.Asiri Y.I., Alsayari A., Muhsinah A.B., Mabkhot Y.N., Hassan M.Z. Benzothiazoles as potential antiviral agents. J. Pharm. Pharmacol. 2020;72:1459–1480. doi: 10.1111/jphp.13331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Naaz F., Srivastava R., Singh A., Singh N., Verma R., Singh V.K., Singh R.K. Molecular modeling , synthesis , antibacterial and cytotoxicity evaluation of sulfonamide derivatives of benzimidazole , indazole , benzothiazole and thiazole. Bioorg. Med. Chem. 2018;26:3414–3428. doi: 10.1016/j.bmc.2018.05.015. [DOI] [PubMed] [Google Scholar]
- 180.Nguyen W., Lee E.F., Evangelista M., Lee M., Harris J., Colman P.M., Smith N.A., Williams L.B., Jarman K.E., Lowes K.N., Haeberli C., Keiser J., Smith B.J., Fairlie W.D., Sleebs B.E. Optimization of benzothiazole and thiazole hydrazones as inhibitors of schistosome BCL - 2. ACS Infect. Dis. 2021;7:1143–1163. doi: 10.1021/acsinfecdis.0c00700. [DOI] [PubMed] [Google Scholar]
- 181.Ghonim A.E., Ligresti A., Rabbito A., Mokhtar A., Di V., Osman N.A., Abadi A.H. Structure-activity relationships of thiazole and benzothiazole derivatives as selective cannabinoid CB2 agonists with in vivo anti- in fl ammatory properties. Eur. J. Med. Chem. 2019;180:154–170. doi: 10.1016/j.ejmech.2019.07.002. [DOI] [PubMed] [Google Scholar]
- 182.Jadav S.S., Kaptein S., Timiri A., De Burghgraeve T., Badavath V.N., Ganesan R., Sinha B.N., Neyts J., Leyssen P., Jayaprakash V. Design, synthesis, optimization and antiviral activity of a class of hybrid dengue virus e protein inhibitors. Bioorg. Med. Chem. Lett. 2015;25:1747–1752. doi: 10.1016/j.bmcl.2015.02.059. [DOI] [PubMed] [Google Scholar]
- 183.Batool F., Saeed M., Saleem H.N., Kirschner L., Bodem J. Facile synthesis and in vitro activity of n-substituted 1,2-benzisothiazol-3(2H)-ones against dengue virus NS2BNS3 protease. Pathogens. 2021;10:1–17. doi: 10.3390/pathogens10040464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Maus H., Barthels F., Hammerschmidt S.J., Kopp K., Millies B., Gellert A., Ruggieri A., Schirmeister T. SAR of novel benzothiazoles targeting an allosteric pocket of DENV and ZIKV NS2B/NS3 proteases. Bioorg. Med. Chem. 2021;47:1–23. doi: 10.1016/j.bmc.2021.116392. [DOI] [PubMed] [Google Scholar]
- 185.Cascioferro S., Parrino B., Carbone D., Schillaci D., Giovannetti E., Cirrincione G., Diana P. Thiazoles , their benzofused systems , and thiazolidinone derivatives : versatile and promising tools to combat antibiotic resistance. J. Med. Chem. 2020;63:7923–7956. doi: 10.1021/acs.jmedchem.9b01245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.de Siqueira A.C.L., de Moraes Gomes L.R.P., de Lima Ferreira P.A.T., de Melo Rêgo L.P., Leite M.J.B. Multi-target compounds acting in cancer progression : focus on thiosemicarbazone , thiazole and thiazolidinone analogues. Eur. J. Med. Chem. 2019;170:237–260. doi: 10.1016/j.ejmech.2019.03.024. [DOI] [PubMed] [Google Scholar]
- 187.Kumar A., Vaidya A., Ravichandran V., Kumar S., Kishore R. Bioorganic & Medicinal Chemistry Recent developments and biological activities of thiazolidinone derivatives : a review. Bioorg. Med. Chem. 2012;20:3378–3395. doi: 10.1016/j.bmc.2012.03.069. [DOI] [PubMed] [Google Scholar]
- 188.Omar K., Geronikaki A., Zoumpoulakis P., Camoutsis C., Ana C., Glamoc J., Sokovic M. Novel 4-thiazolidinone derivatives as potential antifungal and antibacterial drugs. Bioorg. Med. Chem. 2010;18:426–432. doi: 10.1016/j.bmc.2009.10.041. [DOI] [PubMed] [Google Scholar]
- 189.Nitsche C., Schreier V.N., Behnam M.A.M., Kumar A., Bartenschlager R., Klein C.D. Thiazolidinone-peptide hybrids as dengue virus protease inhibitors with antiviral activity in cell culture. J. Med. Chem. 2013;56:8389–8403. doi: 10.1021/jm400828u. [DOI] [PubMed] [Google Scholar]
- 190.Manvar D., Küçükgüzel I., Erensoy G., Tatar E., Deryabaşoʇullari G., Reddy H., Talele T.T., Cevik O., Kaushik-Basu N. Discovery of conjugated thiazolidinone-thiadiazole scaffold as anti-dengue virus polymerase inhibitors. Biochem. Biophys. Res. Commun. 2016;469:743–747. doi: 10.1016/j.bbrc.2015.12.042. [DOI] [PubMed] [Google Scholar]
- 191.Gautam N., Garg A., Gautam D.C. Synthesis , spectral characterization , and pharmacological importance of new 4 H -1 , 4- benzothiazines , their sulfone analogues , and ribofuranosides, nucleosides. Nucleotides Nucleic Acids ISSN. 2015;34:40–55. doi: 10.1080/15257770.2014.955194. [DOI] [PubMed] [Google Scholar]
- 192.Maheshwari M., Goyal A. A review : synthesis and medicinal importance of 1 , 4-benzothiazine analogs. Asian J. Pharmaceut. Clin. Res. 2019;8:41–46. [Google Scholar]
- 193.Aman F., Asiri Abdullah M., Siddiqui Waseeq A., Nadeem Arshad Muhammad, Ashraf Adnan. Multilevel topological description of molecular packings in 1, 2-benzothiazines. CrystEngComm. 2014;16 doi: 10.1039/c3ce42218f. 1963–1970. [DOI] [Google Scholar]
- 194.Lal Badshah Syed, Abdul Naeem Bioactive thiazine and benzothiazine derivatives : green synthesis methods and their medicinal importance. Molecules. 2016;22:1–20. doi: 10.3390/molecules21081054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Cannalire R., Tarantino D., Astolfi A., Barreca M.L., Sabatini S., Massari S., Tabarrini O., Milani M., Querat G., Mastrangelo E., Manfroni G., Cecchetti V. Functionalized 2,1-benzothiazine 2,2-dioxides as new inhibitors of Dengue NS5 RNA-dependent RNA polymerase. Eur. J. Med. Chem. 2018;143:1667–1676. doi: 10.1016/j.ejmech.2017.10.064. [DOI] [PubMed] [Google Scholar]
- 196.Somani R.R., Shirodkar P.Y. Oxadiazole: a biologically important heterocycle. Der Pharma Chem. 2009;1:130–140. [Google Scholar]
- 197.Frost J.R., Scully C.C.G., Yudin A.K. Oxadiazole grafts in peptide macrocycles. Nat. Chem. 2016;8:1105–1111. doi: 10.1038/nchem.2636. [DOI] [PubMed] [Google Scholar]
- 198.Spink E., Ding D., Peng Z., Boudreau M.A., Leemans E., Lastochkin E., Song W., Lichtenwalter K., O'Daniel P.I., Testero S.A., Pi H., Schroeder V.A., Wolter W.R., Antunes N.T., Suckow M.A., Vakulenko S., Chang M., Mobashery S. Structure-activity relationship for the oxadiazole class of antibiotics. J. Med. Chem. 2015;58:1380–1389. doi: 10.1021/jm501661f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Bakht M.A., Yar M.S., Abdel-Hamid S.G., Al Qasoumi S.I., Samad A. Molecular properties prediction, synthesis and antimicrobial activity of some newer oxadiazole derivatives. Eur. J. Med. Chem. 2010;45:5862–5869. doi: 10.1016/j.ejmech.2010.07.069. [DOI] [PubMed] [Google Scholar]
- 200.Janardhanan J., Chang M., Mobashery S. The oxadiazole antibacterials. Curr. Opin. Microbiol. 2016;33:13–17. doi: 10.1016/j.mib.2016.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Verma S.K., Verma R., Verma S., Vaishnav Y., Tiwari S.P., Rakesh K.P. Anti-tuberculosis activity and its structure-activity relationship (SAR) studies of oxadiazole derivatives: a key review. Eur. J. Med. Chem. 2021;209 doi: 10.1016/j.ejmech.2020.112886. [DOI] [PubMed] [Google Scholar]
- 202.Tok F., Kocyigit-Kaymakcioglu B., Tabanca N., Estep A.S., Gross A.D., Geldenhuys W.J., Becnel J.J., Bloomquist J.R. Synthesis and structure–activity relationships of carbohydrazides and 1,3,4-oxadiazole derivatives bearing an imidazolidine moiety against the yellow fever and dengue vector, Aedes aegypti. Pest Manag. Sci. 2018;74:413–421. doi: 10.1002/ps.4722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Hamdani S.S., Khan B.A., Hameed S., Batool F., Saleem H.N., Mughal E.U., Saeed M. Synthesis and evaluation of novel S-benzyl- and S-alkylphthalimide- oxadiazole -benzenesulfonamide hybrids as inhibitors of dengue virus protease. Bioorg. Chem. 2020;96:1–11. doi: 10.1016/j.bioorg.2020.103567. [DOI] [PubMed] [Google Scholar]
- 204.Adawara S.N., Shallangwa G.A., Mamza P.A., Ibrahim A. In silico studies of oxadiazole derivatives as potent dengue virus inhibitors. Chem. Africa. 2021;4:861–868. doi: 10.1007/s42250-021-00255-7. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No data was used for the research described in the article.