

Abstract
Silencing of tumor-suppressor genes by hypermethylation of promoter CpG islands is well documented in human cancer and may be mediated by methyl-CpG-binding proteins, like MeCP2, that are associated in vivo with chromatin modifiers and transcriptional repressors. However, the exact dynamic between methylation and chromatin structure in the regulation of gene expression is not well understood. In this study, we have analyzed the methylation status and chromatin structure of three CpG islands in the p14(ARF)/p16(INK4A) locus in a series of normal and cancer cell lines using methylation-sensitive digestion, MspI accessibility in intact nuclei and chromatin immunoprecipitation (ChIP) assays. We demonstrate the existence of an altered chromatin structure associated with the silencing of tumor-suppressor genes in human cancer cell lines involving CpG island methylation, chromatin condensation, histone deacetylation and MeCP2 binding. The data showed that MeCP2 could bind to methylated CpG islands in both promoters and exons; MeCP2 does not interfere with transcription when bound at an exon, suggesting a more generalized role for the protein beyond transcriptional repression. In the absence of methylation, it is demonstrated that CpG islands located in promoters versus exons display marked differences in the levels of acetylation of associated histone H3, suggesting that chromatin remodeling can be achieved by methylation-independent processes and perhaps explaining why non-promoter CpG islands are more susceptible to de novo methylation than promoter islands.
INTRODUCTION
Aberrant methylation patterns in CpG islands are a hallmark of human cancers, and the hypermethylation of islands in gene promoters acts as a powerful suppressor of transcriptional activity (1,2). The inverse relationship between methylation levels of promoters and transcriptional activity has been well documented (3,4). It has been suggested that methylation interferes with transcriptional initiation rather than by preventing elongation because only methylation in CpG islands located in promoters has been shown to repress transcription in mammals (5).
Much work has been done to elucidate the mechanism of the methylation-mediated inhibition of transcription. The discovery of methylation-dependent DNA-binding proteins such as methyl-CpG-binding protein 2 (MeCP2) suggests that transcriptional repression by methylation may, in part, be due to the binding of these methyl CpG-binding proteins that prevent the functional binding of transcription factors or may act as transcriptional repressors themselves (6,7). Furthermore, it has been demonstrated that certain members of the methyl-CpG-binding domain protein family (MBDs), like MeCP2, are associated in vivo with other transcriptional modulators, such as co-repressors and chromatin modifiers, like histone deacetylase (HDAC) and Mi-2, a member of the SWI2/SNF2 family of ATPases (8–10). Deacetylation of histones, correlated with decreased gene expression, allows stronger interactions between the DNA backbone and histones, possibly inducing a tight chromatin structure that is inaccessible to the transcription machinery; hyperacetylation of histones, on the other hand, has been associated with increased transcription (11). The data suggest a model in which MBDs act as anchors on methylated DNA, recruiting accessory proteins like HDAC that are able to modulate chromatin structure and the transcriptional activity of the gene.
The purpose of the present study was to characterize the relationship between methylation and the chromatin structure of three CpG islands linked in tandem and with different methylation levels, in a series of normal and cancer cells. The two promoters and shared exon of p14(ARF) and p16(INK4A) were selected for study as the two gene products are both negative regulators of the cell cycle and cellular proliferation. Because of the roles p14 and p16 play in the control of cell cycling, perturbation of their expression has been implicated as a critical event in cell transformation (12–14). We have analyzed the methylation status and chromatin structure at the three CpG islands of the locus using methylation-sensitive restriction digestion, susceptibility of the loci to endonuclease digestion within intact nuclei, and chromatin immunoprecipitation (ChIP) assays in order to study their interplay in the transcriptional regulation of these two genes.
We found that the presence of methylation at discrete CCGG sites correlated well with inaccessibility to digestion by MspI in nuclei. It was also observed that nuclease accessibility at a CpG island was associated with an increased level of acetylated histone H3, and that the degree of histone H3 acetylation at an island was higher in a promoter than in an exon when both were unmethylated. Furthermore, we demonstrate in this report that MeCP2 binds to methylated CpG islands, regardless of whether they are within a promoter or an exon, and does not prevent transcriptional elongation when present at an island downstream of an active promoter. Our data are a clarification of the relationship between methylation, chromatin structure and transcription of cancer-linked genes and represent a step in the understanding of epigenetics in tumorigenesis.
MATERIALS AND METHODS
Tissue culture
The human fetal bladder fibroblast cell line LD419 was maintained in McCoy’s medium with 20% fetal bovine serum and antibiotics (penicillin and streptomycin). The human bladder cancer cell line J82 was maintained in MEM medium with 10% fetal bovine serum, non-essential amino acids, sodium pyruvate, glutamine and antibiotics. The human bladder cancer cell line T24 was maintained in McCoy’s medium with 10% fetal bovine serum and antibiotics. The human colon cancer cell line HCT15 was maintained in RPMI 1640 medium with 10% fetal bovine serum, sodium pyruvate, glutamine and antibiotics. All lines were maintained at 37°C and 5% CO2. LD419 cells were established in our laboratory by Dr Louis Dubeau. J82, T24 and HCT15 cells were obtained from the American Type Culture Collection.
MspI nuclear accessibility assays
At least 108 cells from each line were harvested and washed twice with cold phosphate-buffered saline (PBS). The cell pellets were resuspended in 9 ml of RSB buffer [10 mM Tris–HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2 and 1 mM phenylmethylsulfonyl fluoride (PMSF)] and allowed to stand on ice for 10 min. After, 1 ml of 10% NP-40 was added to the cells which were then homogenized with a Dounce homogenizer using the tight plunger for 10 strokes. The resulting nuclei were pelleted and washed once in RSB buffer plus 10% NP-40 and then twice in RSB buffer without detergent. The nuclei were then washed once in MspI buffer (10 mM Tris–HCl pH 7.5, 10 mM dithiothreitol, 10 mM MgCl2 and 0.3 M sucrose). Finally, the nuclei were resuspended in MspI buffer to a concentration of 7.5 × 107 nuclei per ml.
Digestion by MspI endonuclease (Roche Molecular Biochemicals) was carried out in 1.5 ml microfuge tubes containing 1.5 × 107 nuclei each. Nuclei from each cell line were digested in a series of increasing MspI concentrations (0, 10, 50, 100 and 250 U/ml) at 37°C for 1 h. The reactions were then halted by addition of an equal volume of stop solution (20 mM Tris–HCl pH 7.5, 0.6 M NaCl, 1% SDS, 10 mM EDTA and 400 µg/ml proteinase K). Samples were incubated in stop solution at 37°C for 2 h. DNA was purified from these samples with phenol/chloroform extractions and ethanol precipitation.
Twenty micrograms of DNA purified from the MspI nuclear digestions were then digested by BstXI and PstI in order to liberate the CpG islands of interest. The DNA was digested with 4 U/µg DNA of BstXI for 16 h at 55°C followed by an additional 1 U/µg for a further 6 h. The DNA was then purified and subjected to a similar time-course of digestion with PstI at 37°C. The digested DNA was separated by electrophoresis on a 1.5% agarose gel and then Southern blotted. Because all CpG islands of interest (p14 promoter, p16 promoter and p14/p16 exon 2) were liberated by double digestion with BstXI and PstI, we were able to utilize the same blot for analysis of all islands. The blot was hybridized overnight at 42°C with a probe in or near the CpG island of interest. The blot was washed twice in 2× SSC and 0.1% SDS and twice in 1× SSC and 0.1% SDS at 50°C. Visualization of the bands was achieved by exposure of the blot to film at –80°C for a period of 3 days.
Probes specific for each of the three CpG islands of interest were PCR amplified from genomic DNA using the following sets of primers: p14 promoter (5′ region) sense, 5′-GAGTTTGAGCATGTGCAATGTTAGG-3′ and antisense, 5′-GGGATTATTACTCCTGTTTTACAGGTG-3′; p14 promoter (3′ region) sense, 5′-ATCTTGGAGGTCCGGGTGGGAGT-3′ and antisense, 5′-GGGCCTTTCCTACCTGTCTTCTAGGA-3′; p16 promoter sense, 5′-CTCCAAAGCATTTTCTTTATATGCCTCAAAACAAG-3′ and antisense, 5′-AATACGGACGGGGGAGAATTCTG-3′; p14/p16 exon 2 sense, 5′-GAAGTTCAACATTCCCAGAAGCTAAGTG-3′ and antisense, 5′-AGAGAGAACAGAATGGTCAGAGCC-3′. Probes were labeled with [α-32P]dCTP using a random-primed labeling kit (Roche Molecular Biochemicals), and then purified using S-400 HR microspin columns (Amersham Pharmacia Biotech).
HpaII methylation analysis of CpG islands
To analyze the methylation of discrete CCGG sites whose accessibility was assayed with MspI digestion, we extracted DNA from each of the four cell lines as previously described (15). Twenty micrograms of DNA was subjected to a double digest with BstXI and PstI to liberate fragments containing the CpG islands of interest as described for the MspI accessibility assay. The DNA was subsequently digested with 4 U/µg DNA of HpaII endonuclease for 16 h followed by an additional 1 U/µg for a further 6 h. The digested DNA was electrophoresed on a 1.5% agarose gel and Southern blotted. The blot was hybridized by the same CpG-island-specific probes used in our MspI nuclear accessibility assay. After an overnight hybridization at 65°C and washes, the blot was exposed to film at –80°C for 3 days.
RT–PCR analysis of p14 and p16 expression
RNA was extracted from each of the four cell lines as described previously (16). Reverse transcription was carried out with random hexamers (Pharmacia), M-MuLV reverse transcriptase (Gibco Life Sciences) and 2.5 µg of RNA as described previously (16). Subsequent PCR reactions were carried out with 100 ng of the cDNA product, dNTPs (Roche Molecular Biochemicals) and Taq DNA polymerase (Sigma) as described previously (17). The following primers were used: p14 sense, 5′-CATGGTGCGCAGGTTCTTG-3′; p14 antisense, 5′-TTCCCGAGGTTTCTCAGAG-3′; p16 sense, 5′-AGCCTTCGGCTGACTGGCTGG-3′; p16 antisense, 5′-CTGCCCAT-CATCATGACCTGGA-3′; GAPDH sense, 5′-CAGCCGAGCCACATCGCTCAGACT-3′; GAPDH antisense, 5′-TGAGGCTGTTGTCATACTTCTC-3′. Reaction conditions for each PCR were carried out as described previously (16). PCR products were resolved on 2% agarose gels, transferred to Zetaprobe nylon membrane (Bio-Rad), and probed with a γ-32P-end-labeled internal oligonucleotide.
Western blot analysis of MeCP2 protein levels
Cells in 150 mm dishes were rinsed with 2 vol of ice-cold PBS. RIPA buffer (1× PBS, 1% SDS, 0.5% NP-40 and 0.5% sodium deoxycholate) was then added, and cells were scraped off the dish and placed on ice for 30 min. The mixture was then centrifuged at 13 000 r.p.m. for 30 min at 4°C. The supernatant was removed and used for western analysis. Thirty micrograms of total protein were loaded onto 4–15% Tris–HCl gels (Bio-Rad), electrophoresed in Tris–glycine–SDS running buffer, and transferred to a PVDF membrane in Tris–glycine buffer overnight at 4°C. The membranes were hybridized with antibody against MeCP2 (1:500 dilution; Upstate Biotechnology) in TBS-T buffer with 5% non-fat dry milk for 1 h at room temperature. The membranes were subsequently washed five times with TBS-T at room temperature. The membranes were then incubated with secondary anti-rabbit-IgG-HRP antibody (1:1000 dilution) for 1 h at room temperature. The proteins were detected with the enhanced chemiluminescent detection kit (Amersham-Pharmacia) and exposed to Kodak X-OMAT AR film.
ChIP assays
Chromatin proteins of interest were cross-linked to DNA by addition of formaldehyde directly to the culture medium to a final concentration of 1%. After a 10 min incubation at room temperature, the cells were washed and scraped off the dishes in ice-cold PBS containing 1 µg/ml pepstatin A and 1× protease inhibitor cocktail (Roche Molecular Biochemicals). Cells were pelleted and then resuspended in SDS lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris–HCl pH 8.1, 1× protease inhibitor cocktail and 1 µg/ml pepstatin A) for 10 min on ice. The resulting lysate was subjected to sonication to reduce the size of DNA to 300–1000 bp. The sample was centrifuged to remove cell debris and diluted 10-fold in ChIP dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris–HCl pH 8.1, 167 mM NaCl, 1× protease inhibitor cocktail and 1 µg/ml pepstatin A). The chromatin solution was pre-cleared with 80 µl of a mixture of salmon sperm DNA–protein A agarose slurry (Upstate Biotechnology) to reduce non-specific background. After the pre-clearing, the solution was centrifuged, and the supernatant was collected. Either 5 µl of anti-acetylated histone H3 or anti-MeCP2 antibody (Upstate Biotechnology) was added to the chromatin solution and incubated overnight at 4°C with agitation; a no-antibody control was also performed for each ChIP assay. After the overnight antibody incubation, resulting immune complexes were collected by addition of 60 µl of salmon sperm DNA–protein A agarose slurry and incubated at 4°C with agitation for 2 h. The beads were washed several times, and the attached immune complexes were eluted with a buffer containing 1% SDS and 0.1 M NaHCO3. Cross-links were reversed by addition of 5 M NaCl and incubation at 65°C for 4 h. The samples were then treated with proteinase K for 1 h, and DNA was purified by phenol/chloroform extraction and ethanol precipitation.
PCR analysis of immunoprecipitated DNA
Amplification was achieved using hot-started reactions with Expand DNA polymerase (Roche Molecular Biochemicals) and 5 µl of either immunoprecipitated DNA, a no-antibody control or a 1:100 dilution of input chromatin. Experimental reactions were performed to determine optimal PCR conditions so that the yield of PCR products was dependent on the amount of input DNA (data not shown). The conditions for all reactions were as follows: 95°C for 3 min, 28 cycles at 95°C for 1 min, 62°C for 1 min (64°C for p16 promoter), 72°C for 75 s and 72°C for 5 min. The primers used are as follows: p14 promoter sense, 5′-GAAGAATGGAAGACTTTCGACGAGG-3′; p14 promoter antisense, 5′-ACCTCCAAGATCTCGGAACGG-3′; p16 promoter sense, 5′-GAAAGATACCGCGGTCCCTC-3′; p16 promoter antisense, 5′-ACCGTAACTATTCGGTGCGTTGG-3′; p14/p16 exon 2 sense, 5′-GGGCTCTACACAAGCTTCCTTTC-3′; p14/p16 exon 2 antisense, 5′-TATCTATGCGGGCATGGTTACTGC-3′. PCR products were electrophoresed on 2% agarose gels and quantitated with the Bio-Rad Gel Doc 1000 system and Molecular Analyst software. Fold enrichment in each immunoprecipitation was determined by quantifying the intensities of the PCR product in immunoprecipitated DNA versus input DNA (total chromatin).
RESULTS
Analysis of chromatin structure at the three CpG islands of the p14/p16 locus by MspI accessibility assay
The p14/p16 locus on chromosome 9p21 is a unique model system in which to study the interplay of chromatin structure, DNA methylation and gene transcription. The p14 and p16 genes initiate transcription at different first exons that are then spliced to a common second exon, albeit in different reading frames (Fig. 1) (18–20). Moreover, the promoters of both transcripts, as well as the second exon, fulfill the criteria for being considered CpG islands. This afforded us the unique opportunity to study three linked CpG islands in which one, the promoter of p16, serves as both a promoter and a downstream island and in which the islands are variably methylated in different cell lines (16,17).
Figure 1.

Map of the p14/p16 locus. Located on chromosome 9p21, this locus contains two distinct promoters that initiate transcripts with unique first exons that are then spliced, in different reading frames, onto a common second exon.
Antequera et al. (21) demonstrated that methylated CCGG sites located within CpG islands are resistant to cutting by MspI when in their intact chromatin domains. We therefore prepared nuclei from the four cell lines (LD419, J82, T24 and HCT15), digested them with increasing concentrations of MspI, and analyzed the products on Southern blots hybridized with probes in or near the islands (Fig. 2). Because of the large size of the promoter region of p14, a second probe located in the 3′-end of the promoter was also used to fully characterize the accessibility profile of the entire locus (radiographic data not shown; summarized in Fig. 3)
Figure 2.

Southern blot depicting results of an MspI nuclear accessibility assay. Nuclei from each cell line were subjected to a series of digestions with increasing concentrations of MspI: 0, 10, 50, 100 and 250 U/ml. For each CpG island analyzed, the presence of lower molecular weight bands running below the uncut fragment indicated accessibility of a discrete CCGG site to MspI digestion. The last lane in each panel shows a positive control of naked DNA from LD419 that was digested to completion by 4 U/µg of MspI. The arrows to the right of each panel indicate the positions of every possible band that would result if MspI were able to access every CCGG site within the island studied.
Figure 3.

Summary of the methylation, nuclear accessibility and expression data for the p14/p16 locus in all cell lines. The circles above the line represent the methylation status of individual CCGG sites as assayed by HpaII digest (16,17). A black circle represents complete methylation; a white circle is indicative of no methylation; and a gray circle represents intermediate methylation of a particular site within a population of cells. The arrows underneath the line indicate cutting by MspI at discrete CCGG sites. Black boxes indicate the position of the DNA probes used in the hybridizations, and hatched boxes indicate the location of coding regions.
The p14 promoter was quite accessible to increasing concentrations of MspI in all cell lines with susceptibility to cleavage at almost all sites observed in J82 and T24 cells, whereas LD419 and HCT15 cells demonstrated more limited accessibility to endonuclease (Fig. 2, top panel). A detailed map of the cleavage sites is shown in Figure 3, which shows the methylation data from HpaII restriction digests published previously (16,17) and additional data not shown (available upon request), and also the expression of each transcript. Interestingly, there was not a complete correlation between the accessibility to MspI and the level of expression; the p14 promoter in LD419 cell nuclei showed incomplete accessibility when compared with J82 even though the cell lines expressed the p14 transcript at approximately equal levels. Moreover, the non-expressing p14 promoter of HCT15 demonstrated limited accessibility similar to LD419. However, p14 expression did correlate well with the absence of methylation at the promoter (Fig. 3, top panel). The p14 promoter showed either partial or no methylation at the HpaII sites in LD419, whereas the J82 and T24 cell lines were unmethylated at all sites assayed; all three cell lines were positive for p14 expression. HCT15 displayed incomplete to complete methylation at all sites assayed and did not express the transcript.
The p14 promoter data showed that complete methylation at a CCGG site mostly precluded accessibility of that site to MspI digestion in nuclei (Fig. 3, top panel). Conversely, the absence of methylation at a CCGG site was consistently associated with MspI accessibility, although there are a few instances in which the data do not conform to this generalization (the last CCGG site in the 3′-end of the promoter of p16 and the first CCGG site in the 5′-end of the shared exon 2). Incomplete methylation at discrete CCGG sites demonstrated no clear correlation with MspI accessibility; in HCT15, some CCGG sites displaying incomplete methylation were accessible to MspI digestion whereas others were resistant.
The p16 promoter showed limited accessibility to MspI digestion in LD419 as demonstrated by the weakness of the digested bands even at the highest concentration of MspI (Fig. 2, middle panel). The p16 promoter of J82, on the other hand, showed substantial digestion by MspI at all possible CCGG sites. T24 and HCT15 showed complete resistance to MspI digestion. In this case, nuclease accessibility and expression level correlated very well; J82, which was highly accessible to MspI digestion, showed a considerably higher level of p16 expression than LD419 (Fig. 3, middle panel). Both LD419 and J82 were unmethylated at all CCGG sites analyzed, whereas T24 and HCT15 showed substantial methylation (Fig. 3, middle panel). Methylation at the p16 promoter corresponded to inaccessibility to MspI digestion and absence of gene expression.
The shared exon 2 of the p14/p16 locus proved to be the least accessible to MspI digestion of the three CpG islands (Fig. 2, bottom panel). Nuclei from LD419 cells were the only nuclei examined to show limited digestion by the enzyme; however, the uncut band persisted at the highest concentration of MspI, and the bands corresponding to accessible MspI sites were very weak. LD419 was unmethylated at all CCGG sites. J82 and T24 were completely methylated, whereas HCT15 displayed either partial or complete methylation at all sites (Fig. 3, bottom panel). The presence of methylation at exon 2 correlated with a closed chromatin configuration inaccessible to MspI digestion.
Furthermore, the data demonstrate that a closed chromatin configuration inaccessible to MspI is resistant to transcriptional initiation but amenable to transcriptional elongation from an active upstream promoter. This was demonstrated in the case of the T24 cell line in which the methylated and inaccessible p16 promoter was transcriptionally inactive but did not stop the elongation and strong expression of the transcript from the active upstream promoter of p14 (see Fig. 3).
Characterization of chromatin composition at each CpG island using the ChIP assay
ChIP is a powerful technique to test for the presence of certain DNA-binding proteins that might modulate chromatin structure and/or transcriptional characteristics of the specific region of DNA with which they are associated. We made use of polyclonal antibodies generated against MeCP2 as well as acetylated histone H3, both of which are proteins that have been linked to chromatin modification and regulation of transcription. Both sets of ChIPs were repeated three times to confirm the reproducibility of the PCR results.
The results show that the three CpG islands analyzed were occupied by acetylated histone H3 in all cell lines, regardless of methylation or accessibility of a particular island to nuclease digestion (Fig. 4A). However, higher levels of acetylated H3 were found on promoter versus non-promoter islands that were equally unmethylated as observed in LD419 (Fig. 4B). In addition, higher levels of acetylated H3 were generally found on unmethylated rather than methylated islands; the one exception to this is the greater level of acetylated H3 found on the methylated exon 2 of J82 compared with the unmethylated exon 2 in LD419. Levels of acetylated histone H3 at a particular island also tended to correlate with how accessible it was to MspI digestion in nuclei. For example, in the T24 cell line, only the p14 promoter was accessible to MspI digestion (Fig. 2); this corresponded well with the higher level of acetylated histone H3 found on the p14 promoter when compared with the inaccessible p16 promoter and shared exon 2 (Fig. 4B).
Figure 4.

(A) Results of ChIP assays performed with antibody against acetylated histone H3. IP, immunoprecipitated DNA; NAC, no-antibody control; Input, sample representing amplification from a 1:100 dilution of total input chromatin from each ChIP experiment. Experimental reactions were performed to determine optimal PCR conditions so that the yield of PCR products was dependent on the amount of input DNA (data not shown). The lengths of the PCR products corresponding to the p14 promoter, p16 promoter and shared exon 2 are 383, 312 and 363 bp, respectively. Each ChIP experiment was repeated three times to confirm reproducibility of results. (B) UV quantitation of acetylated histone H3 ChIP PCR results. Relative differences in the levels of immunoprecipitated DNA (which are reflective of the levels of the chromatin protein of interest occupying a particular island) between different islands and cell lines were quantified by dividing UV intensity of immunoprecipitated bands by the intensity of the input band, which is representative of the level of total input chromatin in a particular ChIP assay. (C) Results of ChIP assay performed with antibody against MeCP2. (D) UV quantitation of MeCP2 ChIP PCR results.
The ChIP results for MeCP2 showed that the protein bound to methylated CpG islands but not to unmethylated islands (Fig. 4C). No MeCP2 binding was demonstrated at any of the unmethylated islands in LD419. J82 showed binding of MeCP2 only to the methylated exon 2. MeCP2 occupied the methylated p16 promoter and shared exon 2 in T24 as well as all three methylated islands in HCT15. Expression of MeCP2 in the four cell lines studied was confirmed by western blot (Fig. 5).
Figure 5.
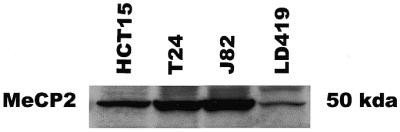
Western blot confirming expression of MeCP2 in HCT15, T24, J82 and LD419 cell lines.
Comparison of the quantitated levels of MeCP2 and acetylated histone H3 at each of the three CpG islands demonstrated an inverse relationship between the two proteins. Islands occupied by MeCP2 were significantly deacetylated whereas islands unbound by MeCP2 demonstrated higher levels of histone H3 acetylation (Fig. 4B and D). One exception to this observation was seen when comparing the acetylation levels of the shared exon 2 between LD419 and J82. Even though the exon 2 of J82 was bound by MeCP2 (and LD419 was not), the exon demonstrated a higher level of acetylation in J82.
Interestingly, MeCP2 was observed to bind to a methylated exon 2 regardless of the methylation and transcriptional status of the upstream promoter islands. The presence of MeCP2 at a methylated p14/p16 exon 2 did not block transcriptional elongation through the region, as in the cases of J82 (with active p14 and p16 promoters) and T24 (with an active p14 promoter).
DISCUSSION
Many human cancers display alterations in the methylation patterns of promoter CpG islands that may perturb the expression of genes critical to the regulation of cell proliferation. Hypermethylation of the promoter regions of genes, such as p16(INK4a) or p14(ARF), has been correlated with downregulation of their expression, suggesting that aberrant methylation interferes with transcriptional initiation (16,17,22). Methyl-CpG-binding proteins, like MeCP2, recruit accessory proteins to methylated DNA that may contribute to transcriptional inhibition at promoter islands (8,9). Such proteins include transcriptional co-repressors as well as chromatin remodeling proteins, like Mi-2 and HDAC (10). Although many of the molecular components of this methylation-dependent chromatin remodeling system have been characterized, the exact dynamic between methylation and chromatin within the context of abnormal gene silencing in cancer has not been well delineated.
Using the MspI nuclear accessibility assay in conjunction with the ChIP assay, we demonstrated that accessibility of discrete CpG sites (within the context of the CCGG recognition sequence) within an island correlated well with the unmethylated state as well as with an increased level of acetylated histone H3 present at the island. All of the active promoters in the p14/p16 locus in the four cell lines studied fulfilled these three criteria. However, inactive promoters were found to be less accessible to digestion, highly methylated and generally deacetylated (when compared with active promoters). Interestingly, we found that, in the absence of methylation (LD419), promoters and exons displayed inherent differences in their acetylation profile, with promoter islands displaying higher levels of acetylated histone H3. This phenomenon of differential acetylation in the absence of methylation differences (and of methylation itself) indicates that chromatin remodeling at CpG islands can also be mediated through methylation-independent processes.
Our data show that MeCP2 can bind to a methylated CpG island regardless of whether it is located within a promoter or an exon, and that binding of MeCP2 is associated with deacetylation. The one exception to the latter observation is in the shared exon 2, which has more acetylated H3 in J82 than LD419 even though it is methylated and bound by MeCP2 in J82. This may be due to the greater transcriptional activity of the p14/p16 locus in J82 versus LD419 (Fig. 3); increased transcription through exon 2 in J82 may account for the higher level of acetylation despite the presence of MeCP2. Moreover, the presence of MeCP2 at a methylated CpG island does not inhibit transcriptional elongation from an active upstream promoter. The discovery of MeCP2 binding to a non-promoter CpG island, whose methylation and accessibility has no known effect on upstream promoter activity, is particularly fascinating in light of the in vivo associations of MeCP2 and its proposed function as a transcriptional repressor (8,9). Our finding of MeCP2 binding to the shared exon 2 of the p14/p16 locus in J82, in which both upstream promoters are active and unmethylated, argues for a more generalized function for the protein and against the current model that it is exclusively a mediator of transcriptional repression. While it is entirely possible that MeCP2 is responsible for the accessibility and acetylation profiles we observed at a methylated exon 2, such chromatin remodeling has no known effect on the level of transcription of the gene.
Our MeCP2 data are in contrast to a previous study in which it was demonstrated that the methylated promoters of p14 and p16 in cancer cells were occupied by MBD2, another methyl-CpG-binding protein, but not by MeCP2 (23). Such a discrepancy could be due to different antibodies with different affinities for MeCP2 used in the immunoprecipitation. Our data are self-consistent in that they are internally controlled, e.g. the methylated exon 2 of J82 was occupied by MeCP2, whereas the unmethylated p14 and p16 promoters of the same cell line were unbound by MeCP2. We therefore are unable to fully explain this discrepancy at the present time.
The methylation and presence of MeCP2 at the shared exon 2 of p14 and p16, regardless of upstream promoter methylation status, in the three cancer cell lines studied are consistent with previous work carried out in our laboratory in which it was demonstrated that non-promoter CpG islands are more susceptible to de novo methylation and may be foci for the seeding of de novo methylation which can then spread to adjacent islands (24). Recent work has shown that certain MBD-containing proteins interact in vivo with DNMT1, which itself has been found in complexes with HDAC (25–28). Although such an interaction with MeCP2 has not been found, it is still possible that it could interact with DNMT1 directly or indirectly through HDAC recruitment. It is conceivable that MeCP2, bound to an aberrantly methylated patch in an exon, might recruit methyltransferase activity to the island either directly or indirectly and mediate the spread of aberrant methylation through an island.
Moreover, such a model might explain why non-promoter islands seem to be more vulnerable than promoter islands to de novo methylation. Our ChIP data demonstrate that unmethylated exonic islands are less acetylated then unmethylated promoter islands. Such data might imply that higher levels of HDAC would be found occupying the exon, possibly leading to recruitment of DNMT1, seeding of de novo methylation, binding by MeCP2 and subsequent amplification of the methylation signal. However, the mechanism by which an unmethylated exon is rendered less accessible and acetylated than an unmethylated promoter is still unclear. Perhaps steric hindrance by components of the transcription initiation complex at a promoter prevents access by HDAC or proteins that recruit HDAC activity; such steric hindrance would not exist at an exon.
Further studies are required to fully identify the interplay between DNA methylation and chromatin structure in the silencing of gene expression in cancer cells. Whether DNA methylation directly induces the assembly of closed chromatin or whether a closed chromatin structure might recruit methyltransferase activity is still a question to be answered. Extensive characterization of the chromatin proteins occupying promoter versus non-promoter islands may help in clarifying the differential patterns of accessibility, acetylation and methylation between different islands we have observed in this study.
Acknowledgments
ACKNOWLEDGEMENT
This work was supported by the United States Public Health Service Grant R35 CA49758 from the National Cancer Institute.
REFERENCES
- 1.Jones P.A. and Laird,P.W. (1999) Cancer epigenetics comes of age. Nature Genet., 21, 163–167. [DOI] [PubMed] [Google Scholar]
- 2.Baylin S.B. and Herman,J.G. (2000) DNA hypermethylation in tumorigenesis: epigenetics joins genetics. Trends Genet., 16, 168–174. [DOI] [PubMed] [Google Scholar]
- 3.Kass S.U., Pruss,D. and Wolffe,A.P. (1997) How does DNA methylation repress transcription? Trends Genet., 13, 444–449. [DOI] [PubMed] [Google Scholar]
- 4.Razin A. and Cedar,H. (1991) DNA methylation and gene expression. Microbiol. Rev., 55, 451–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jones P.A. (1999) The DNA methylation paradox. Trends Genet., 15, 34–37. [DOI] [PubMed] [Google Scholar]
- 6.Zhang X.Y., Ehrlich,K.C., Wang,R.Y. and Ehrlich,M. (1986) Effect of site-specific DNA methylation and mutagenesis on recognition by methylated DNA-binding protein from human placenta. Nucleic Acids Res., 14, 8387–8397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nan X., Campoy,F.J. and Bird,A. (1997) MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell, 88, 471–481. [DOI] [PubMed] [Google Scholar]
- 8.Jones P.L., Veenstra,G.J., Wade,P.A., Vermaak,D., Kass,S.U., Landsberger,N., Strouboulis,J. and Wolffe,A.P. (1998) Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nature Genet., 19, 187–191. [DOI] [PubMed] [Google Scholar]
- 9.Nan X., Ng,H.H., Johnson,C.A., Laherty,C.D., Turner,B.M., Eisenman,R.N. and Bird,A. (1998) Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature, 393, 386–389. [DOI] [PubMed] [Google Scholar]
- 10.Wade P.A., Gegonne,A., Jones,P.L., Ballestar,E., Aubry,F. and Wolffe,A.P. (1999) Mi-2 complex couples DNA methylation to chromatin remodelling and histone deacetylation. Nature Genet., 23, 62–66. [DOI] [PubMed] [Google Scholar]
- 11.Kuo M.H. and Allis,C.D. (1998) Roles of histone acetyltransferases and deacetylases in gene regulation. Bioessays, 20, 615–626. [DOI] [PubMed] [Google Scholar]
- 12.Duro D., Bernard,O., Della Valle,V., Berger,R. and Larsen,C.J. (1995) A new type of p16INK4/MTS1 gene transcript expressed in B-cell malignancies. Oncogene, 11, 21–29. [PubMed] [Google Scholar]
- 13.Gardie B., Cayuela,J.M., Martini,S. and Sigaux,F. (1998) Genomic alterations of the p19ARF encoding exons in T-cell acute lymphoblastic leukemia. Blood, 91, 1016–1020. [PubMed] [Google Scholar]
- 14.Nuovo G.J., Plaia,T.W., Belinsky,S.A., Baylin,S.B. and Herman,J.G. (1999) In situ detection of the hypermethylation-induced inactivation of the p16 gene as an early event in oncogenesis. Proc. Natl Acad. Sci. USA, 96, 12754–12759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bell G.I., Karam,J.H. and Rutter,W.J. (1981) Polymorphic DNA region adjacent to the 5′ end of the human insulin gene. Proc. Natl Acad. Sci. USA, 78, 5759–5763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gonzalgo M.L., Hayashida,T., Bender,C.M., Pao,M.M., Tsai,Y.C., Gonzales,F.A., Nguyen,H.D., Nguyen,T.T. and Jones,P.A. (1998) The role of DNA methylation in expression of the p19/p16 locus in human bladder cancer cell lines. Cancer Res., 58, 1245–1252. [PubMed] [Google Scholar]
- 17.Robertson K.D. and Jones,P.A. (1998) The human ARF cell cycle regulatory gene promoter is a CpG island which can be silenced by DNA methylation and down-regulated by wild-type p53. Mol. Cell. Biol., 18, 6457–6473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Quelle D.E., Zindy,F., Ashmun,R.A. and Sherr,C.J. (1995) Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle. Cell, 83, 993–1000. [DOI] [PubMed] [Google Scholar]
- 19.Stone S., Jiang,P., Dayananth,P., Tavtigian,S.V., Katcher,H., Parry,D., Peters,G. and Kamb,A. (1995) Complex structure and regulation of the P16 (MTS1) locus. Cancer Res., 55, 2988–2994. [PubMed] [Google Scholar]
- 20.Liggett W.H. Jr, Sewell,D.A., Rocco,J., Ahrendt,S.A., Koch,W. and Sidransky,D. (1996) p16 and p16 beta are potent growth suppressors of head and neck squamous carcinoma cells in vitro. Cancer Res., 56, 4119–4123. [PubMed] [Google Scholar]
- 21.Antequera F., Macleod,D. and Bird,A.P. (1989) Specific protection of methylated CpGs in mammalian nuclei. Cell, 58, 509–517. [DOI] [PubMed] [Google Scholar]
- 22.Esteller M., Tortola,S., Toyota,M., Capella,G., Peinado,M.A., Baylin,S.B. and Herman,J.G. (2000) Hypermethylation-associated inactivation of p14(ARF) is independent of p16(INK4a) methylation and p53 mutational status. Cancer Res., 60, 129–133. [PubMed] [Google Scholar]
- 23.Magdinier F. and Wolffe,A.P. (2001) Selective association of the methyl-CpG binding protein MBD2 with the silent p14/p16 locus in human neoplasia. Proc. Natl Acad. Sci. USA, 98, 4990–4995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nguyen C., Liang,G., Nguyen,T.T., Tsao-Wei,D., Groshen,S., Lübbert,M., Zhou,J., Benedict,W.F. and Jones,P.A (2001) Susceptibility of nonpromoter CpG islands to de novo methylation in normal and neoplastic cells. J. Natl Cancer Inst., 93, 1465–1472. [DOI] [PubMed] [Google Scholar]
- 25.Tatematsu K.I., Yamazaki,T. and Ishikawa,F. (2000) MBD2-MBD3 complex binds to hemi-methylated DNA and forms a complex containing DNMT1 at the replication foci in late S phase. Genes Cells, 5, 677–688. [DOI] [PubMed] [Google Scholar]
- 26.Fuks F., Burgers,W.A., Brehm,A., Hughes-Davies,L. and Kouzarides,T. (2000) DNA methyltrasnferase Dnmt1 associates with histone deacetylase activity. Nature Genet., 24, 88–91. [DOI] [PubMed] [Google Scholar]
- 27.Robertson K.D., Ait-Si-Ali,S., Yokochi,T., Wade,P.A., Jones,P.L. and Wolffe,A.P. (2000) DNMT1 forms a complex with Rb, E2F1 and HDAC1 and represses transcription from E2F-responsive promoters. Nature Genet., 25, 338–342. [DOI] [PubMed] [Google Scholar]
- 28.Rountree M.R., Bachman,K.E. and Baylin,S.B. (2000) DNMT1 binds HDAC2 and a new co-repressor, DMAP1, to form a complex at replication foci. Nature Genet., 25, 269–277. [DOI] [PubMed] [Google Scholar]