

Abstract
Mitochondrial dysfunction has a role in sepsis-associated acute kidney injury (S-AKI), so the restoration of normal mitochondrial homeostasis may be an effective treatment strategy. Transcription factor nuclear factor erythroid 2 p45-related factor 2 (NRF2) is a main regulator of cell-redox homeostasis, and recent studies reported that NRF2 activation helped to preserve mitochondrial morphology and function under conditions of stress. However, the role of NRF2 in the process of S-AKI is still not well understood. The present study investigated whether NRF2 regulates mitochondrial homeostasis and influences mitochondrial function in S-AKI. We demonstrated activation of NRF2 in an in vitro model: lipopolysaccharide (LPS) challenge of ductal epithelial cells of rat renal tubules (NRK-52e cells), and an in vivo model: cecal ligation and puncture (CLP) of rats. Overexpression of NRF2 attenuated oxidative stress, apoptosis, and the inflammatory response; enhanced mitophagy and mitochondrial biogenesis; and mitigated mitochondrial damage in the in vitro model. In vivo experiments showed that rats treated with an NRF2 agonist had higher adenosine triphosphate (ATP) levels, lower blood urea nitrogen and creatinine levels, fewer renal histopathological changes, and higher expression of mitophagy-related proteins [PTEN-induced putative kinase 1 (PINK1), parkin RBR E3 ubiquitin protein ligase (PRKN), microtubule-associated protein 1 light chain 3 II (LC3 II)] and mitochondrial biogenesis-related proteins [peroxisome proliferator-activated receptor γ coactivator-1 (PGC-1α) and mitochondrial transcription factor A (TFAM)]. Electron microscopy of kidney tissues showed that mitochondrial damage was alleviated by treatment with an NRF2 agonist, and the opposite response occurred upon treatment with an NRF2 antagonist. Overall, our findings suggest that mitochondria have an important role in the pathogenesis of S-AKI, and that NRF2 activation restored mitochondrial homeostasis and function in the presence of this disease. This mitochondrial pathway has the potential to be a novel therapeutic target for the treatment of S-AKI.
Key words: Sepsis, acute kidney injury, transcription factor nuclear factor erythroid 2 p45-related factor 2, mitochondrial, inflammation
Introduction
Acute kidney injury (AKI) is a clinical complication characterized by the sudden loss of renal function, a condition that leads to altered levels of electrolytes, acid-base disorders, and fluid imbalance. 1 Sepsis is the leading cause of AKI, and accounts for approximately 50% of AKI cases among Intensive Care Unit (ICU) patients.2,3 Sepsis-associated acute kidney injury (S-AKI) has a mortality rate approaching 60%.4,5 Under normal physiological conditions, tubular cells consume a large amount of energy to maintain normal function and survival, and this requires a large number of mitochondria to produce adenosine triphosphate (ATP).6 In S-AKI, mitochondrial defects can lead to an insufficient supply of cellular energy.7 In addition, because mitochondria are the main source of reactive oxygen species (ROS), mitochondria function as a hub of innate immune signals and inflammation during sepsis.8,9 Recent studies reported that mitochondria are not just the targets in sepsis, but they also drive the progression of cell damage and organ dysfunction.10 Danger-associated molecular patterns (DAMPs) are endogenous protein and non-protein molecules released from damaged or dysfunctional mitochondria that can further enhance inflammation, trigger apoptosis, and increase oxidative damage.11,12
Maintenance of mitochondrial homeostasis is essential for the recovery of cell and organ function in patients with sepsis.13 This homeostasis relies on the precise regulation of mitochondrial quality- control mechanisms involving mitochondrial biogenesis, mitophagy, and mitochondrial dynamics.14-16 Recent studies found a key link between mitochondrial homeostasis and human diseases, especially inflammatory and neurodegenerative diseases.17,18 Dysregulation of mitochondrial homeostasis was recently considered in models of S-AKI, and this was characterized by deficient mitophagy and inhibition of mitochondrial biogenesis.19,20 Therefore, promotion of mitochondrial homeostasis may be an effective strategy for preventing and treating S-AKI.
Transcription factor nuclear factor erythroid 2 p45-related factor 2 (NRF2) is a major regulator of cell-redox homeostasis, and it functions in the detoxification of xenobiotics, repair and removal of damaged proteins, and inhibition of inflammation.21,22 The NRF2 signaling pathway is one of the body’s endogenous antioxidant defense mechanisms, the main regulator of antioxidant responses, and can resist oxidative stress responses caused by various reasons.23 After activation of this pathway, its downstream target genes can control cellular antioxidant response, the expression of cryoprotection and detoxification gene, and have inhibitory effects on pro-inflammatory cytokines.24 However, there is little known about the role of NRF2 expression in S-AKI. Liu et al.25 administered an endotoxin (lipopolysaccharide, LPS) to experimental animals to establish an in vivo model of S-AKI. They found no obvious changes in total NRF2, but that NRF2 tended to localize in the cytoplasm rather than in the nucleus. However, two other studies reported that total and nuclear NRF2 levels increased following LPS insult.26,27 Recent findings suggested that the increased NRF2 activity helped to preserve mitochondrial morphology and function under conditions of stress. Similarly, Murata et al.28 found that NRF2 promoted the expression of PTEN-induced putative kinase 1 (PINK1) and affected mitochondrial mitophagy in a different line of tumor cells. Piantadosi et al.29 reported a role of NRF2 in the activation of mitochondrial biogenesis. In particular, they found that NRF2 could bind to the NRF1 promoter adenosine and uridine rich elements (AREs), leading to activation of mitochondrial transcription factor A (TFAM), which is directly involved in the mitochondrial DNA (mtDNA) replication.29 However, the relationship of NRF2 with renal mitochondrial function and mitochondrial homeostasis in S-AKI remains unclear.
In this study, we hypothesized that NRF2 could attenuate sepsis- associated kidney dysfunction and help to restore mitochondrial homeostasis.
Materials and Methods
Animals’ treatment and grouped
Thirty-five male Sprague-Dawley (SD) rats (200-250 g) were obtained from the Laboratory Animal Center of Southern Medical University in Guangzhou, China. All animal experiments were approved by the Laboratory Animal Ethics Committee of Zhujiang Hospital (Ethical Approval No. B2019-11-5), Southern Medical University and the Animal Ethical and Welfare Committee of Guangdong Medical Laboratory Animal Center and were in accordance with U.S. National Institutes of Health guidelines for the care and use of laboratory animals.30 Before the experiments, all animals received a standard diet and were maintained at room temperature (20-25°C), at a humidity of 30-70%, and in a 12 h light/dark regimen.
The cecal ligation and puncture (CLP) procedure was used to establish the animal model of sepsis, as previously described.31 Rats were first anesthetized by intraperitoneal injection of 4% chloral hydrate (300 mg/kg). Then a midline laparotomy was performed, and the cecum with adjoining intestine were exposed, with care taken to avoid damage to the blood vessels. Then, a tight ligation of the cecum was applied at the bottom of the ileocecal valve, and it was then punctured twice with a 20-gauge needle. The applied pressure was sufficient to extrude fecal material from the puncture site into the peritoneal cavity. The abdomen was then closed with three sutures. After surgery, the rats were resuscitated by a subcutaneous injection of pre-warmed 0.9% saline solution (3 mL/100 g body weight), and returned to their cages. The dose of NRF2 inhibitor and NRF2 agonist were selected based on previous reports.32,33 The NRF2 inhibitor ML385 (T4360, TargetMol Chemicals Inc., Boston, MA, USA) was administered by intraperitoneal injection (30 mg/kg) at 4 h before the CLP procedure in the CLP 24 h + ML385 group; the NRF2 agonist tert-butylhydroquinone (TBHQ) (HY-100489, MedChemExpress LLC, Princeton, NJ, USA) was administered by intraperitoneal injection (50 mg/kg) at 4 h before CLP in the CLP 24 h + TBHQ group. Rats were sacrificed at different times after treatment by intraperitoneal injection of 4% chloral hydrate (300 mg/kg), followed by cervical dislocation; 5 mL of blood were taken from the abdominal aorta, stored at room temperature for 2 h, centrifuged at 3000 r/min at 4°C for 10 min to separate the upper serum, that was stored at −80 °C for later use. Death was confirmed by cardiac arrest. After washing with a pre-cooled phosphate buffer solution, kidney tissues were quickly stored at −80°C for subsequent analysis.
Cell culture and transfection
NRK-52E cells derived from renal tubular duct epithelium of Rattus norvegicus (normal proximal tubule epithelial cells with epithelial-like morphology) were cultured in Dulbecco’s modified Eagle’s medium (11995065, Gibco, Thermo Fisher Scientific, Inc., Waltham, MA, USA) containing 10% fetal bovine serum (10270106, Gibco) and 1% penicillin/streptomycin at 37°C in a cell culture incubator with 5% CO2. A recombinant lentivirus carrying an expression construct gene NRF2 (LV-NRF2) and a recombinant lentivirus carrying a negative control (LV-NC) were generated by iGene Biotechnology (Guangzhou, China). NRK-52e cells were infected with a multiplicity of infection (MOI) of 20, according to the manufacturer’s instructions. After 72 h, cells were exposed to lipopolysaccharide (LPS, 50 μg/mL, L-2880, SigmaAldrich, St. Louis, MO, USA) for induction of in vitro sepsis. For NRF2 inhibition in cell culture, NRK-52E cells were pretreated with the NRF2 inhibitor ML385 (T4360, TargetMol) at 5 μM for 12 h before LPS insult.34
Methyl thiazolyl tetrazolium assay
For measurement of cell viability, 5 . 103 cells were inoculated into each well of a 96-well plate. After adhering, the cells were given different treatments, and then 20 μL of methyl thiazolyl tetrazolium (MTT) (500 μg/mL) was added to each well. After incubation for 4 h, the culture was terminated, and the supernatant in each well was carefully aspirated. Then, 150 μL of dimethyl sulphoxide (DMSO) was added, and the mixture was shaken for 10 min to allow the crystals to fully dissolve. The absorption of each well was determined at 490 nm using a microplate reader (CLARIOstar Plus, BMG Labtech, Ortenberg, Germany).
Renal function
The blood samples were collected at the end of the experiment and centrifuged to obtain sera and kept at −20°C for analysis. Renal function in rats was assessed by measurement of blood urea nitrogen (BUN) and serum creatinine levels were estimated as previous studies.16,35 These were determined using a Beckman Coulter AU5800 system (Beckman Coulter, Inc., Brea, CA, USA) according to the manufacturer’s instructions.
Inflammatory cytokines
The concentrations of cytokines in rat serum and cell supernatants were determined using enzyme-linked immunosorbent assay (ELISA) kits (EK306/3-01, EK382/3-96, MultiSciences). Sets of 6 wells were then prepared as standard wells in an ELISA plate, to which 50 μL of the corresponding diluted standard was then added, which contained 40 μL of the sample dilution and tested the 10 μL of sample, uniformly mixed together. Blanks (i.e., no sample, enzyme standard reagent only) were then set, and sealed the plate with a sealing film and incubated in an incubator (37°C, 30 min). The 50 . concentrated washing solution was then diluted into the 1. washing solution, and set aside. After that, the sealing film was removed, and up to 250 μL of washing solution was poured into each well, let stand for 30 s, and then poured out and dried 5 times. 50 μL of enzyme labeling reagent were added to each well. After 30 min, wells were washed 5 times, added 50 μL of coloring solution A, and coloring solution B, and then these were mixed well and placed in an incubator in darkness at 37°C for 15 min. The microplate was removed from the incubator, and 50 μL of the stop solution were added to terminate the reaction. Quantitation was based on absorbance at 450 nm, and was measured using a CLARIOstar Plus microplate reader, according to the manufacturer’s instructions.
SOD activity, MDA, and ATP
The activity of superoxide dismutase (SOD), and the levels of malondialdehyde (MDA) and adenine nucleoside triphosphate (ATP) were measured in rat renal tissue and tubular epithelial cells using commercial kits (S0101S, S0131S, S0027, Beyotime Biotechnology, Haimen, China) according to the manufacturer’s instructions. In this method, at least 100 μL of sample (diluted between 5-100 μg of proteins) must be contained in one assay tube. As standard, bovine serum albumin was used. By adding 2 mg/mL of bovine serum albumin in 1000 μL volume standard was prepared which must be in the range of 200-2000 μg. Then, the mixture containing 4 mL dye reagent was incubated for 10 min. The level of each parameter was expressed relative to total protein concentration.
ROS, mtROS, and mitochondrial transmembrane potential
The 2’,7’-Dichlorofluorescent yellow diacetate (DCFH-DA) kit (S0033S, Beyotime Biotechnology) was used to measure the intracellular production of reactive oxygen species (ROS). Cells were subjected to various treatments, and loaded with 10 μM DCFH-DA according to the manufacturer’s instructions. Then, the cells were washed with PBS three times and visualized by fluorescence microscopy (BX63, Olympus, Tokyo, Japan). Quantitative detection of ROS fluorescence was measured using a CLARIOstar Plus microplate reader (excitation: 488 nm, emission cutoff: 525 nm).
Mitochondrial ROS (mtROS) were detected using a MitoROS™ 580 kit (16052, AAT Bioquest, Inc., Sunnyvale, CA, USA), which selectively targets mitochondria. Briefly, cells were loaded with 1. MitoROS 580 dye in the dark for 30 min at 37°C and then observed by confocal microscopy (FV10i, Olympus). Quantitation of mtROS was determined by measuring fluorescence using a CLARIOstar Plus microplate reader (excitation: 540 nm, emission cutoff: 590 nm). Mitochondrial transmembrane potential (MMP) was determined using a JC-1 kit (C1071M, Beyotime Biotechnology). Briefly, cells were washed with a phosphate buffer solution (PBS) solution. Then, a JC-1 working solution was added, and cells were maintained at 37°C for 20 min in a cell culture incubator. Next, the working solution was removed, cells were washed twice with the JC-1 staining buffer, and then changes in mitochondrial membrane potential were observed using fluorescence microscopy (BX63, Olympus; JC-1 monomer: excitation 480 nm, emission cutoff 525 nm; JC-1 aggregates: excitation 540 nm, emission cutoff 590 nm). The mean fluorescence intensity of was quantified using Image J software (Media Cybernetics, Rockville, MD, USA).
TUNEL assay
Apoptosis was assessed using a terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay kit (C1089, Beyotime Biotechnology), according to the manufacturer’s instructions. The nuclei were then counterstained with a 5 μg/mL solution of 4’,6-diamidino-2-phenylindole (DAPI), and cells were identified by their nuclei. A total of 10 random fields of each sample section were assessed using fluorescence microscopy (Olympus BX63, Tokyo, Japan). After being viewed with the microscope, labeled nuclei were counted to determine the apoptotic index (number of labeled nuclei / total cell nuclei ·100).
Immunoblot analysis
Nuclear, cytoplasmic and mitochondrial proteins were extracted using commercially available kits (P0027 and C3601, Beyotime Biotechnology). Proteins were extracted from tissues and cells, and the concentration was determined using a bicinchoninic acid (BCA) protein assay kit (P0012S, Beyotime Biotechnology). Protein samples were then separated by SDS polyacrylamide gel electrophoresis (25 μg per lane) and transferred to polyvinylidene fluoride (PVDF) membranes (Millipore, Burlington, MA, USA). Then, the membrane was blocked with a solution containing 5% bovine serum albumin for 60 min at room temperature, and a specific primary antibody was added for incubation at 4°C overnight. Then, the membrane was incubated with a horse radish peroxidase (HRP)-conjugated secondary antibody for 60 min at room temperature. The primary antibodies were against: transcription factor nuclear factor erythroid 2 p45-related factor 2 (NRF2) (1:1000, 16396-1-AP, Proteintech, Rosemont, IL, USA), mitochondrial transcription factor A (TFAM) (1:1000, 22586-1-AP, Proteintech), PTEN-induced putative kinase 1 (PINK1) (1:1000, 23274-1-AP, Proteintech), parkin RBR E3 ubiquitin protein ligase (PRKN) (1:1000, 14060-1-AP, Proteintech), microtubule associated protein 1 light chain 3 II (LC3 II) (1:1000, 14600-1-AP, Proteintech ), peroxisome proliferator-activated receptor γ coactivator-1 (PGC-1α) (1:1000, 66369-1-Ig, Proteintech), cytochrome c (1;1000, 10993-1-AP, Proteintech), cytochrome c oxidase subunit IV isoform 1 (COXIV) (1:1000, 60251-1-Ig, Proteintech), Lamin B (1:1000, WL01775, Wanleibio, Shenyang, China), β-actin (1:2000, 60008-1-Ig, Proteintech), Bcl-2 (1:1000, WL01556, Wanleibio), Bax (1:1000, WL01637, Wanleibio), Erk (1:1000, WL01864, Wanleibio), NF-κB p65 (1:1000, WL01980, Wanleibio), p-NF-κB p65 (1:1000, WL02169, Wanleibio), p38 (1:1000, WL00764, Wanleibio) and p-p38 (1:1000, WLP1576, Wanleibio). The results were visualized using a chemiluminometry or film with electrochemiluminescence (ECL) reagents. Protein levels were standardized to β-actin (internal reference). Image J software (Media Cybernetics) was used for quantitation of band density.
Histological, immunohistochemical, and immunofluorescence assays
Kidney tissue was fixed in 4% paraformaldehyde (Bio-Rad, Hercules, CA, USA) for 24 h, embedded in paraffin, end cut into 4-μm sections. After rehydration with xylene (15 min), and graded ethanol (100%, 95%, 90%, 80%, 70% and 50% for 5 min each), the sections were washed with distilled water three times and stained with hematoxylin (10 min) and eosin (10 s). After dehydration and mounting, the slides were observed and photographed using an MshOT microscope (Guangzhou Mingmei Technology Co., Ltd., Guangzhou, China). For NRF2 immunohistochemistry, the kidney tissues were fixed overnight with 4% paraformaldehyde (Bio-Rad), then cut into 4-μm sections using routine procedures. The dewaxed tissue sections were heat-treated to retrieve epitopes by immersing in a citrate buffer solution at pH 6.0 in an autoclave at 121°C for 1 min. The tissue sections were then incubated with 3% hydrogen peroxide for 1 h, and then with 5% normal goat serum for 30 min. Next, the sections were incubated overnight at 4°C with anti-NRF2 antibodies (1:1000, 16396-1-AP, Proteintech). After washing, an HRP-labeled polymer antibody (GAPDH; 1:1000, Abcam, Cambridge, MA, USA) was used as the secondary antibody and incubated for 1 h at room temperature. The Immunohistochemical reaction was visualized by microscopy (BX51, Olympus). For immunofluorescence, the NRK-52E cells were removed from the culture medium and fixed with 4% paraformaldehyde (Bio-Rad) for 15 min. Then, 0.1% Triton X-100 was added for 5 min at room temperature. The cells were separately incubated with antibodies against LC3 II (1:1000, 14600-1-AP, Proteintech), COXIV (1:1000, 60251-1-Ig, Proteintech), or NRF2 (1:1000, 16396-1-AP, Proteintech) at 4°C overnight. The next day, cells were washed in 1. PBS and incubated with secondary antibodies (GAPDH; 1:1000, Abcam) conjugated with Alexa Fluor for 1 h at 37°C. Finally, they were stained with DAPI (5 μg/mL) for labeling the nuclei for 10 min, washed with 1. PBS for 3 times, and then directly irradiated with fluorescent sheets. Images were visualized using confocal microscopy (FV10i, Olympus).
Transmission electron microscopy
Kidney tissues and cells from different treatment groups were washed with PBS, collected, fixed with 2.5% glutaraldehyde, dehydrated with a gradient of acetone, and then embedded. Thin sections were cut, routinely stained with uranyl acetate and lead citrate, and observed under transmission electron microscopy (TEM; H-7650; Hitachi, Tokyo, Japan).
Statistical analysis
SPSS Statistics ver. 22.0 (IBM SPSS Statistics, Chicago, IL, USA) was used for data analysis. Data are expressed as means ± standard deviations (SDs). Analysis of variance (ANOVA) was used to determine differences among experimental groups and by Student’s t-test for unpaired data, followed by post hoc pairwise comparisons using the least significant difference (LSD) test (homogeneous variance) or Dunnett’s T3 analysis (heteroscedastic variance). A p-value below 0.05 was considered statistically significant.
Results
NRF2 activation in NRK-52e cells following LPS insult
We first performed in vitro studies using LPS-induced NRK- 52e cells to simulate S-AKI. The MTT assay showed that addition of LPS at 24 or 50 μg/mL for 12 or 24 h significantly reduced cell viability (Figure 1 A,B). We thus treated cells with 50 μg/mL LPS for 24 h in subsequent experiments. Immunoblot analysis showed that LPS led to increased expression of NRF2 over time (Figure 1 C,D). Further, immunofluorescence staining showed that LPS treatment increased the level of NRF2 in the nucleus (Figure 1E). These results suggest that NRF2 is activated in this in vitro model of AKI.
NRF2 inhibition aggravated LPS-induced injury and apoptosis in NRK-52e cells
Apoptosis contributes to and promotes the progression of AKI.36 Thus, we used the specific NRF2 inhibitor ML385 to examine the effect of NRF2 on apoptosis in NRK-52e cells under LPSinsult. The results of the TUNEL assay showed more apoptotic cells in the LPS+ML385 group than in the LPS+Vehicle group (Figure 2A). Furthermore, Figure 2B shows that cell viability decreased remarkably in the LPS-treated cells compared with control cells, and treatment with ML385 and LPS further decreased the cell viability. Immunoblot analysis showed that ML385 significantly reduced the LPS-mediated increase of total NRF2 and nuclear NRF2 (Figure 2 C,D,H). NRF2 inhibition exacerbated the LPS-induced alterations in the expression of apoptosis-related proteins, in that it further increased the level of Bax and further decreased the level of Bcl-2 (Figure 2 C,E,F). In addition, LPS significantly decreased the level of cytochrome c in the mitochondria, but increased its level in the cytoplasm, and NRF2 inhibition exacerbated this effect (Figure 2 C,G,I). These results indicated that upregulation of NRF2 mitigated LPS-induced cell injury and apoptosis.
NRF2 upregulation alleviated LPS-induced oxidative stress and inflammatory responses in NRK-52e cells
The NF-κB and MAPK signaling pathways are important in regulating inflammation and sepsis-associated organ damage.37,38 Thus, we used lentivirus transfection to establish NRK-52e cells with overexpression of NRF2. Immunoblot analysis verified efficient transfection (Figure 3A). Our results indicated that LPS treatment of NRK-52e cells led to up-regulation of p-p65, p-p38, and erk, and down-regulation of IKB-α, and that NRF2 over-expression reversed these effects (Figure 3 B,C). Furthermore, LPS led to increased levels of TNF-α and IL-6 in the cell culture supernatant, and NRF2 over-expression reversed this effect (Figure 3 D,E). Analysis of oxidative stress indicated that LPS increased the level of MDA and decreased SOD activity, and NRF2 also reversed these effects (Figure 3 F,G). Measurement of intracellular ROS production indicated much lower levels of total ROS in the LPS+LV-NRF2 group than in the LPS group (Figure 3 H,I). These results showed that NRF2 over-expression alleviated the LPSinduced oxidative stress and inflammatory response.
NRF2 upregulation alleviated LPS-induced mitochondrial defects in NRK-52e cells
The disruption of mitochondrial morphology and function is a key feature of S-AKI.39 Therefore, maintenance of mitochondrial homeostasis and function may promote the physiological function and improve the survival of renal tubular cells. Thus, we examined the effect of NRF2 upregulation on LPS-induced mitochondrial defects of NRK-52e cells. TEM of the mitochondrial ultrastructure indicated that LPS led to widespread swelling, rupture of the cristae, and disintegration and fragmentation of the mitochondria, and that NRF2 over-expression alleviated all of these effects (Figure 4F). Our mtROS, ATP, and MMP confirmed that LPS led to mitochondrial dysfunction, in that it increased the level of mtROS, and decreased the levels of MMP and ATP. NRF2 overexpression alleviated all of these effects (Figure 4 A-E).
Mitochondrial homeostasis is maintained by a tight regulation of mitochondrial biogenesis and mitophagy. PGC-1α and TFAM function in mitochondrial biogenesis and PINK1, PRKN, and LC3 II function in mitophagy. Our immunoblotting results indicated that LPS significantly decreased the levels of PGC-1α and TFAM in NRK-52e cells and that NRF2 alleviated these effects (Figure 5 A,B,E). LPS also significantly increased the levels of PINK1, PRKN, LC3 II, and NRF2 enhanced this effect (Figure 5 A-D,F). To verify the activation of mitophagy, we performed double staining of LC3 II and COXIV. The results indicated minimal co-localization of LC3 II with COXIV in the control group, significant colocalization in the LPS group, and even more co-localization in the LPS+LV-NRF2 group (Figure 5G). Collectively, these results suggest that NRF2 over-expression reduced the damage of mitochondrial function and morphology induced by LPS and also restored mitochondrial homeostasis.
Rats with CLP have upregulated renal NRF2
We also used an in vivo model to investigate the role of NRF2 in S-AKI. Thus, we first examined the expression of NRF2 in the kidney tissues of rats after CLP. The expression of NRF2 was increased in the CLP group, and the level was greatest after 12 h (Figure 6 A,B). Immunohistochemistry confirmed upregulation of NRF2 in the kidneys, and that NRF2 was mainly expressed in the renal tubular epithelium (Figure 6C). These results are consistent with our in vitro experiments and suggest that NRF2 may function in S-AKI.
NRF2 activation reduced oxidative stress, inflammatory response, and kidney injury in rats with CLP
We investigated the potentially protective effect of NRF2 in SAKI by administering an intraperitoneal injection of an NRF2 agonist (TBHQ) or inhibitor (ML385) to rats subjected to CLP. The effects of TBHQ and ML385 in our rat model of S-AKI (Figure 7 A,C) are consistent with previous reports. In particular, the protein level of NRF2 was greater in the CLP group, ML385 treatment partially reversed this effect, and TBHQ treatment increased this effect (Figure 7 B,D,E). Next, we examined the effects of these two agents on oxidative stress and the inflammatory response. CLP decreased the activity of SOD, M385 treatment increased this effect, and TBHQ partially reversed this effect (Figure 7F). CLP treatment also increased the levels of MDA, M385 increased this effect, and TBHQ partially reversed this effect (Figure 7G). Measurements of TNF-α, IL-6, Scr, and BUN indicated the different treatment groups had responses that were similar to those for MDA (Figure 7 H-K). Histological examination of kidney tissues indicated intraepithelial vacuolar degeneration and interstitial inflammation in the CLP group, TBHQ treatment attenuated this effect, and ML385 exacerbated this effect (Figure 7L). These data showed that activation of NRF2 signaling attenuated kidney damage, oxidative stress, and the inflammatory response, and that antagonism of NRF2 signaling had the opposite effects.
Figure 1.
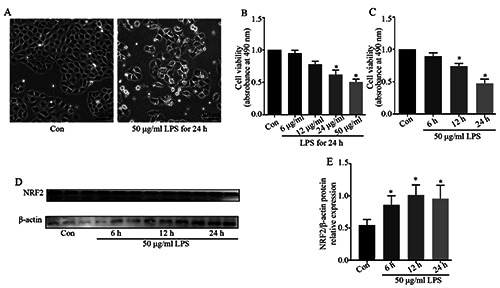
Effect of LPS on NRF-2 activation in NRK-52e cells. A) Cell viability after addition of 6–50 μg/mL LPS for 24 h (n=5). B) Cell viability after addition of 50 μg/mL LPS for 6–24 h (n=5). C) Western blots showing NRF2 expression in cells at 6, 12, and 24 h after addition of LPS (50 μg/mL). D) Quantitative analysis of the western blot data, with expression relative to β-actin (n=3). E) Immunofluorescence detection of the expression and distribution of NRF2 following LPS treatment. Data are presented as means ± SD; *p<0.05 vs control.
NRF2 activation decreased CLP-induced mitochondrial damage, enhanced mitophagy, and restored impaired mitochondrial biogenesis
Next, we assessed the effect of NRF2 signaling on mitochondrial damage. TEM of renal mitochondria indicated they had a normal structure in the sham group, but they were deformed, disintegrated, and swollen, with vacuolation and fragmentation, in the CLP group; these changes were alleviated in the CLP+TBHQ group, but exacerbated in the CLP+ML385 group (Figure 8H). To evaluate mitochondrial function, we measured the ATP levels in kidney tissues. The results show that CLP treatment reduced the ATP level, the NRF2 agonist increased the ATP level and the NRF2 antagonist exacerbated the effect of CLP (Figure 8G).
We examined the possible mechanism of NRF2 in regulating mitochondrial morphology and function by use of western blotting to measure the levels of multiple proteins that function in mitochondrial biogenesis and mitophagy (Figure 8A). The results show that CLP increased the expression of PINK1, PRKN, and LC3 II; ML385 partially reversed the effects of CLP; and THBQ increased the effects of CLP (Figure 8 C,D,F). CLP also decreased the levels of PGC1-α and TFAM; ML385 partially reversed the effects of CLP; and TBHQ increased the effects of CLP (Figure 7 B,E). These data demonstrated that NRF2 activation helped to maintain mitochondrial homeostasis and alleviate mitochondrial injury in this in vivo model of S-AKI.
Figure 2.
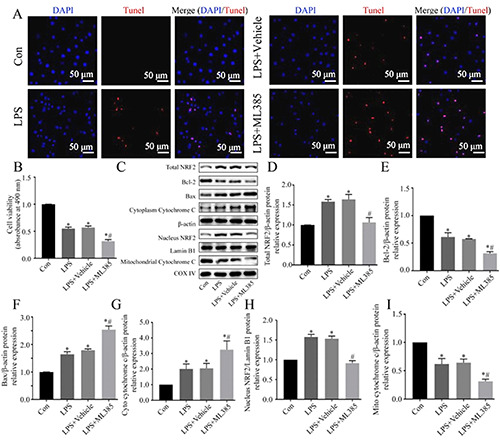
Effect of NRF2 inhibition on viability and apoptosis in LPS-induced NRK-52e cells. A) Representative images of apoptotic cells measured by the TUNEL assay. B) Cell viability, determined by the MTT assay (n=5). (C) Western blotting of Bcl-2, Bax, cytochrome c, and NRF2. D-I) Quantitative analysis of the Western blotting data, with expression relative to β-actin, Lamin BI, or COXIV (n=3). Data are presented as the means ± SD; *p<0.05 vs control, #p<0.05 vs LPS.
Discussion
Sepsis is the leading cause of AKI in ICU patients and increases the short-term mortality of patients with AKI, and some surviving patients progress to chronic kidney disease after discharge.37,38 Mitochondrial dysfunction appears to play a critical role.20,39,40 In addition, mitochondrial dysfunction is closely related to oxidative stress, increased apoptosis, and increased inflammation during sepsis, and these all contribute to the increased risk of mortality. 40,41 Therefore, restoration of mitochondrial homeostasis may provide an effective treatment for sepsis-induced organ injury. Recent research reported that NRF2 may be associated with mitophagy, and that the function of NRF2 in mitophagy may involve the PINK1/PRKN pathway.42 In the present study, we identified NRF2 activation in vitro model of S-AKI. Our in vitro studies showed that NRF2 inhibition of LPS-treated cells further exacerbated cell injury. Moreover, NRF2 activation attenuated oxidative stress, apoptosis, and the inflammatory response; enhanced mitophagy and mitochondrial biogenesis; and mitigated mitochondrial damage.
Figure 3.
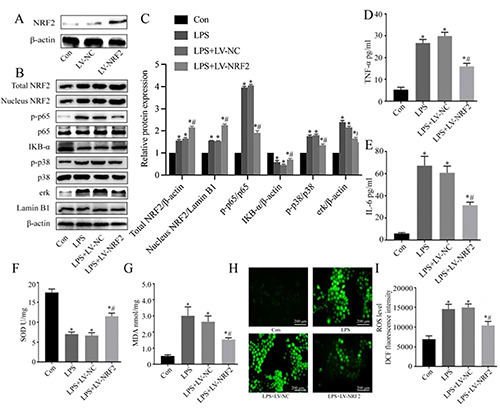
Effect of NRF2 on the inflammatory response and oxidative stress in LPS-induced injury of NRK-52e cells. A) Immunoblotting of NRF2 following transfection with LV-NRF2. B) Western blotting of NRF2, Nucleus NRF2, p-p65, IKB-α, p-p38 and erk. C) Quantitative analysis of the Western blotting data, with expression relative to β-actin, p65, p38 or Lamin B1 (n=3). D,E) ELISA of TNF-α and IL-6 in cell culture supernatants (n=5). (F) SOD activity (n=5). G) MDA level (n=5). H) Representative fluorescence microscopy images of ROS production (DCFH-DA probe). I) Quantitative analysis of the ROS data (n=5). Data are presented as means ± SD; *p<0.05 vs control, #p<0.05 vs LPS.
Mitochondrial dysfunction is the main cause of sepsis-induced organ failure and is closely related to patient prognosis.43 Mitochondrial damage occurs during S-AKI, thus mitochondrial protection strategies may be key to the prevention and treatment of S-AKI.35 A previous study reported that NRF2 activity is closely related to mitochondrial function, and that NRF2 deficiency led to lower basal mitochondrial membrane potential (ΔΨm) and decreased efficiency of oxidative phosphorylation and ATP production. 44 We showed here that NRF2 over-expression alleviated the decline of ATP, alterations in ΔΨm, and the increased mtROS in NRK-52e cells following LPS insult. Our in vivo studies also found that a NRF2 agonist (TBHQ) alleviated the decline in renal ATP caused by CLP, but a NRF2 inhibitor (ML385) had the opposite effect. These results suggested that NRF2 activation helped to maintain mitochondrial function in the presence of experimentally induced sepsis.
Figure 4.
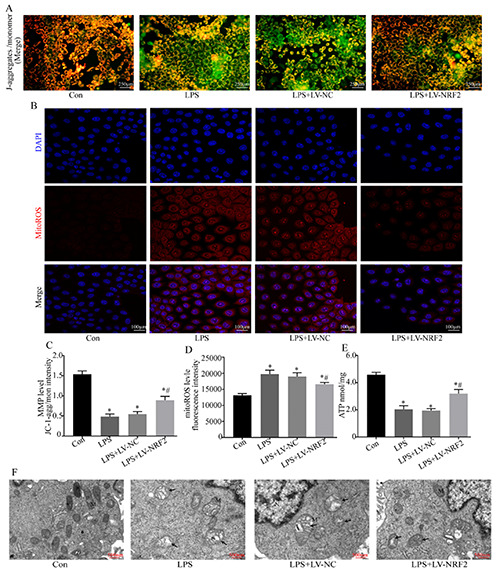
Effect of NRF2 inhibition on viability and apoptosis in LPS-induced NRK-52e cells. A) Representative images of apoptotic cells measured by the TUNEL assay. B) Cell viability, determined by the MTT assay (n=5). (C) Western blotting of Bcl-2, Bax, cytochrome c, and NRF2. D-I) Quantitative analysis of the Western blotting data, with expression relative to β-actin, Lamin BI, or COXIV (n=3). Data are presented as the means ± SD; *p<0.05 vs control, #p<0.05 vs LPS.
Figure 5.
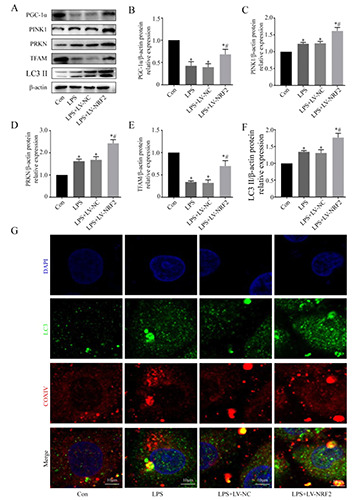
Effect of NRF2 on mitochondrial homeostasis of LPS-induced NRK-52e cells. A) Western blotting of proteins related to mitophagy and mitochondrial-biogenesis. B-F) Quantitative analysis of the Western blotting data, with expression relative to β-actin (n=3). G) Representative fluorescence microscopy images showing co-localization of anti-LC3 II (green)/anti-COXIV (red) antibodies. Data are presented as means ± SD; *p<0.05 vs control, #p<0.05 vs LPS.
As the main regulator of cell redox homeostasis, NRF2 promotes cell adaptive responses in the presence of stress.45 Under normal physiological conditions, NRF2 is sequestered in the cytoplasm and maintained at a low level due to ubiquitination by Kelch-like ECH-related protein 1 (KEAP1).46 However, under stress conditions, modification of certain cysteine residues in KEAP1 disrupts its ubiquitination and degradation of NRF2, so that NRF2 accumulates and activates the expression of target genes by binding with antioxidant elements, thus exerting anti-oxidant and protective effects.47 Our results verified activation of NRF2 in an in vitro and in vivo model of S-AKI. In previous studies, the authors used i.p. injection of LPS at a dosage of 5 to 7.5 mg/kg body weight;26,27,48 Liu et al.25 used i.p. injection of LPS at a dosage of 10 mg/kg body weight. We used the CLP-induced sepsis model in our experiments and was the first to report activation of NRF2 in the CLP model of S-AKI, and the value of NRF2 as a possible therapeutic target for S-AKI was also examined. It may hypothesize that initiation of inflammation and stress activates NRF2, and this exerts an endogenous cytoprotective effect,49 but that this protective mechanism is insufficient when damage continues. Our in vivo studies showed that CLP induced mitochondrial damage, oxidative stress, an inflammatory response, and kidney injury, and that these were aggravated by NRF2 inhibition. Furthermore, mitophagy and mitochondrial biogenesis were inhibited in CLP+ML385 group compared with CLP group. Based on these results, we suggested that activation of NRF2 during sepsis was a cellular protective response to mitochondrial defects, oxidative stress, and inflammation. A healthy mitochondrial network is essential for maintaining cell function, and mitochondrial “quality control” processes regulate the mitochondrial network by assuring appropriate mitochondrial biogenesis, mitophagy, and mitochondrial dynamics.9 Our TEM analysis indicated that mitochondrial structural damage (swelling, vacuolation, and rupture) was less severe in rats that received an NRF2 agonist, similar to the results of our in vitro model in which there was upregulation of NRF2. The NRF2-associated improvement of mitochondrial structure and function in S-AKI may be related its regulation of mitochondrial “quality control”. In septic conditions, damaged mitochondria release cytochrome c, mtDNA, and calcium, and these are considered to be DAMPs that can induce apoptosis, inflammation, and cell damage.50,51 In addition, damaged mitochondria have disrupted electron transport chains, and this promotes the production of ROS.52 Timely removal of damaged mitochondria through mitophagy in stressful conditions is important for maintaining cell homeostasis.39 Previous studies demonstrated that insufficient mitophagy aggravated cell damage, and mitophagy activation attenuated myocardial, kidney, and lung injury in animal models of sepsis.19,53-55 A recent study reported the protective effects of MitoQ in a model of diabetic kidney disease via NRF2/PINKI activation. 56 In particular, PINK1 functions as a serine/threonine kinase that acts as a molecular sensor for mitochondria.28 When there is mitochondrial damage or a decrease of the ΔΨm, PINK1 aggregates form on the outer membrane of the mitochondria, and they recruit PRKN.57 This leads to the ubiquitination of mitochondrial outer membrane proteins, recruitment of autophagy receptors, and binding to LC3 II to form autophagosomes18,58 In this study, we showed that NRF2 overexpression promoted the expression of mitophagy-associated proteins, and increased the co-localization of LC3 II and COXIV in cells subjected to LPS treatment. These data supported the role of NRF2 in promoting mitophagy. However, uncontrolled mitophagy may led to a depletion of mitochondria, which can cause energy deficiency.25 To maintain a healthy mitochondrial population, an appropriate balance between mitophagy and mitochondria biogenesis is necessary.42 Other studies also reported a relationship between NRF2 and mitochondrial biogenesis.59,60 In addition to NRF1, these studies found that NRF2 was also involved in the regulation of PGC1-a in maintaining mitochondrial biogenesis, which becomes predominant under conditions of stress.59,60 Here, we showed that up-regulation or activation of NRF2 attenuated the decrease of mitochondrial biogenesisrelated proteins PGC-1α and TFAM in a model of S-AKI.
Figure 6.
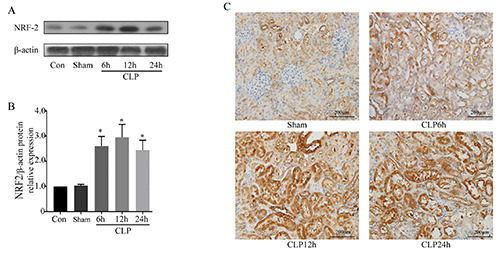
Effect of CLP of rats on renal expression of NRF2. A) Western blotting of NRF2 in kidney tissues. B) Quantitative analysis of the Western blotting data, with expression relative to β-actin (n=5). C) Representative immunohistochemical images showing NRF2 expression. Data are presented as means ±SD; *p<0.05 vs control.
Figure 7.
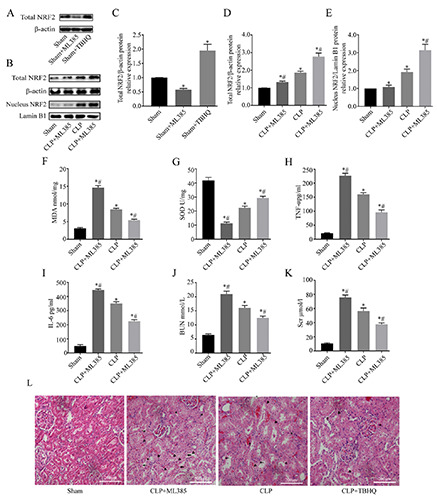
Effect of NRF2 activation on oxidative stress, inflammatory responses, and renal function in rats that received CLP. A,B) Western blotting of NRF2 in kidney tissues. C-E) Quantitative analysis Western blotting data, with expression relative to β-actin or Lamin B1 (n=5). F) MDA levels in kidney tissues (n=8). G) SOD activity in kidney tissues (n=8). H,I) ELISA of serum levels TNF-α and IL-6 (n=8). J,K) Serum creatinine and BUN (n=8). L) Representative hematoxylin and eosin staining images of renal tissues. Data are presented as means ± SDs; asterisks, edema; triangle, vacuolization; arrow, loss of brush border; *p<0.05 vs sham; #p<0.05 vs CLP.
Figure 8.
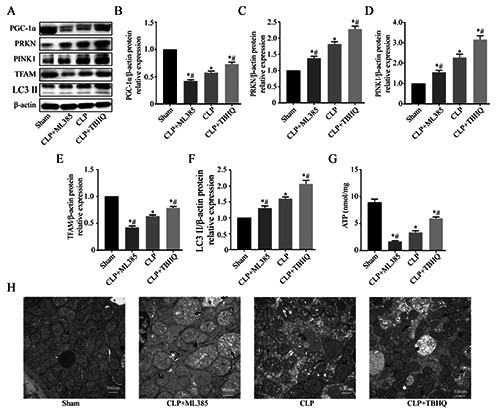
Effect of NRF2 on mitochondrial damage and mitochondrial homeostasis in rats that received CLP. A) Western blotting of kidney proteins related to mitophagy and mitochondrial biogenesis. B-F) Quantitative analysis of the western blotting data with expression relative to β-actin (n=5; data are presented as means ± SD; *p<0.05 vs sham, #p<0.05 vs CLP). G) ATP level in kidney tissues (n=8). H) Representative transmission electron micrographs of renal mitochondria. M, mitochondrion; N, nucleus; CLP, the arrow shows a mitochondrion with swelling and cristae fracture; CLP+ML385, the arrow shows a mitochondrion with swelling, cristae fracture, and rupture of membranes; CLP+TBHQ, the arrow shows a mitochondrion with mild swelling and cristae fracture.
In conclusion, our findings showed that NRF2 enhanced mitophagy in kidney tissues in vivo and in renal tubular cells in vitro, and also restored mitochondrial biogenesis under conditions of experimentally induced sepsis. Thus, NRF2 increased the number of functional mitochondria and maintained a healthier mitochondria network, and this led to attenuation of the inflammatory response, oxidative stress, and apoptosis. These new insights into the function of NRF2 suggest its possible use as a novel target for the treatment of S-AKI.
Funding Statement
Funding: This work was supported by the National Natural Science Foundation of China (81601664), a Youth Research Project of Sichuan Medical Association (2019Q19017), and a Scientific Research Project of Guangdong Provincial Bureau of traditional Chinese Medicine (20201035).
References
- 1.Kellum JA, Romagnani P, Ashuntantang G, Ronco C, Zarbock A, Anders HJ. Acute kidney injury. Nat Rev Dis Primers 2021;7:52. [DOI] [PubMed] [Google Scholar]
- 2.Liu N, Zhang Z, Hong Y, Li B, Cai H, Zhao H, et al. Protocol for a prospective observational study on the association of variables obtained by contrast-enhanced ultrasonography and sepsis-associated acute kidney injury. BMJ Open 2019;9:e023981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Peters E, Antonelli M, Wittebole X, Nanchal R, Francois B, Sakr Y, et al. A worldwide multicentre evaluation of the influence of deterioration or improvement of acute kidney injury on clinical outcome in critically ill patients with and without sepsis at ICU admission: results from The Intensive Care Over Nations audit. Crit Care 2018;22:188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dedionisio A. Establishing a new model of endotoxemia-associated acute kidney injury in zebrafish. University of Pittsburgh; 2020. Available from: http://d-scholarship.pitt.edu/39642/ [Google Scholar]
- 5.Koyner JL, Chawla LS, Bihorac A, Gunnerson KJ, Schroeder R, Demirjian S, et al. Performance of a standardized clinical assay for urinary CC motif chemokine ligand 14 (CCL14) for persistent severe acute kidney injury. Kidney 360 2022:3:1-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bhatia D, Capili A, Choi ME. Mitochondrial dysfunction in kidney injury, inflammation, and disease: Potential therapeutic approaches. Kidney Res Clin Pract 2020;39:244-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li Y, Nourbakhsh N, Pham H, Tham R, Zuckerman JE, Singh P. Evolution of altered tubular metabolism and mitochondrial function in sepsis-associated acute kidney injury. Am J Physiol Renal Physiol 2020;319:F229-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gkikas I, Palikaras K, Tavernarakis N. The role of mitophagy in innate immunity. Front Immunol 2018;9:1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mohanty A, Tiwari-Pandey R, Pandey NR. Mitochondria: the indispensable players in innate immunity and guardians of the inflammatory response. J Cell Commun Signal 2019;13:303-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gomez H, Kellum JA. Sepsis-induced acute kidney injury. Curr Opin Crit Care 2016;22:546-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thiagarajan D. Phospholipid related antigens and protective mechanisms: Implications for cardiovascular diseases, human autoimmunity and inflammation [D]. PhD Thesis, Karolinska Institutet; 2019. Available from: https://openarchive.ki.se/xmlui/handle/10616/46633 [Google Scholar]
- 12.Karkossa I, Raps S, von Bergen M, Schubert K. Systematic review of multi-omics approaches to investigate toxicological effects in macrophages. Int J Mol Sci 2020;21:9371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kraft BD, Chen L, Suliman HB, Piantadosi CA, Welty-Wolf KE. Peripheral blood mononuclear cells demonstrate mitochondrial damage clearance during sepsis. Crit Care Med 2019;47:651-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hara H, Kuwano K, Araya J. Mitochondrial quality control in COPD and IPF. Cells 2018;7:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sedlackova L, Korolchuk VI. Mitochondrial quality control as a key determinant of cell survival. Biochim Biophys Acta Mol Cell Res 2019;1866:575-87. [DOI] [PubMed] [Google Scholar]
- 16.Tang C, Cai J, Yin XM, Weinberg JM, Venkatachalam MA, Dong Z. Mitochondrial quality control in kidney injury and repair. Nat Rev Nephrol 2021;17:299-318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Franco-Iborra S, Vila M, Perier C. Mitochondrial quality control in neurodegenerative diseases: Focus on Parkinson's disease and Huntington's disease. Front Neurosci 2018;12:342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu Y, Yao YM, Lu ZQ. Mitochondrial quality control mechanisms as potential therapeutic targets in sepsis-induced multiple organ failure. J Mol Med (Berl) 2019;97:451-62. [DOI] [PubMed] [Google Scholar]
- 19.Zhu J, Zhang S, Geng Y, Song Y. Transient receptor potential ankyrin 1 protects against sepsis-induced kidney injury by modulating mitochondrial biogenesis and mitophagy. Am J Transl Res 2018;10:4163-72. [PMC free article] [PubMed] [Google Scholar]
- 20.Liu JX, Yang C, Zhang WH, Su HY, Liu ZJ, Pan Q, et al. Disturbance of mitochondrial dynamics and mitophagy in sepsis- induced acute kidney injury. Life Sci 2019;235:116828. [DOI] [PubMed] [Google Scholar]
- 21.Abrescia P, Treppiccione L, Rossi M, Bergamo P. Modulatory role of dietary polyunsaturated fatty acids in Nrf2-mediated redox homeostasis. Prog Lipid Res 2020;80:101066. [DOI] [PubMed] [Google Scholar]
- 22.Dai X, Yan X, Wintergerst KA, Cai L, Keller BB, Tan Y. Nrf2: Redox and metabolic regulator of stem cell state and function. Trends Mol Med 2020;26:185-200. [DOI] [PubMed] [Google Scholar]
- 23.Surai PF, Kochish II, Fisinin VI, Kidd MT. Antioxidant defence systems and oxidative stress in poultry biology: An update. Antioxidants (Basel) 2019;8:235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu Q, Zhang F, Zhang X, Cheng R, Ma JX, Yi J, et al. Fenofibrate ameliorates diabetic retinopathy by modulating Nrf2 signaling and NLRP3 inflammasome activation. Mol Cell Biochem 2018;445:105-15. [DOI] [PubMed] [Google Scholar]
- 25.Liu JX, Yang C, Liu ZJ, Su HY, Zhang WH, Pan Q, et al. Protection of procyanidin B2 on mitochondrial dynamics in sepsis associated acute kidney injury via promoting Nrf2 nuclear translocation. Aging (Albany NY) 2020;12:15638-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang Y, Zhou F, Shen C, Wang H, Xiao Y. LBP reduces theinflammatory injuryof kidney in septic rat and regulates the Keap1-Nrf2∕ARE signaling pathway1. Acta Cir Bras 2019;34:e20190010000003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu Q, Liu LT, Wang XY, Lang ZF, Meng XH, Guo SF, et al. Lycium barbarum polysaccharides attenuate kidney injury in septic rats by regulating Keap1-Nrf2/ARE pathway. Life Sci 2020;242:117240. [DOI] [PubMed] [Google Scholar]
- 28.Murata H, Takamatsu H, Liu S, Kataoka K, Huh NH, Sakaguchi M. NRF2 regulates PINK1 expression under oxidative stress conditions. PLoS One 2015;10:e0142438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Piantadosi CA, Carraway MS, Babiker A, Suliman HB. Heme oxygenase-1 regulates cardiac mitochondrial biogenesis via Nrf2-mediated transcriptional control of nuclear respiratory factor-1. Circ Res 2008;103:1232-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Committee for the Update of the Guide for the Care and Use of Laboratory Animals Institute for Laboratory Animal Research Division on Earth and Life Studies. Guide for the Care and Use of Laboratory Animals. 8th ed. Washington: the National Academies Press; 2011. Available from: https://grants.nih.gov/grants/olaw/guide-for-the-care-and-useof-laboratory-animals.pdf [Google Scholar]
- 31.Toscano MG, Ganea D, Gamero AM. Cecal ligation puncture procedure. J Vis Exp 2011;:2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sukumari-Ramesh S, Alleyne CH. Post-Injury administration of tert-butylhydroquinone attenuates acute neurological injury after intracerebral hemorrhage in mice. J Mol Neurosci 2016;58:525-31. [DOI] [PubMed] [Google Scholar]
- 33.Xian P, Hei Y, Wang R, Wang T, Yang J, Li J, et al. Mesenchymal stem cell-derived exosomes as a nanotherapeutic agent for amelioration of inflammation-induced astrocyte alterations in mice. Theranostics 2019;9:5956-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang J, Tong W, Sun H, Jiang M, Shen Y, Liu Y, et al. Nrf2- mediated neuroprotection by MANF against 6-OHDA-induced cell damage via PI3K/AKT/GSK3β pathway. Exp Gerontol 2017;100:77-86. [DOI] [PubMed] [Google Scholar]
- 35.Peerapornratana S, Manrique-Caballero CL, Gomez H, Kellum JA. Acute kidney injury from sepsis: current concepts, epidemiology, pathophysiology, prevention and treatment. Kidney Int 2019;96:1083-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jacobs R, Honore PM, Joannes-Boyau O, Boer W, De Regt J, De Waele E, et al. Septic acute kidney injury: the culprit is inflammatory apoptosis rather than ischemic necrosis. Blood Purif 2011;32:262-5. [DOI] [PubMed] [Google Scholar]
- 37.Neyra JA, Li X, Canepa-Escaro F, Adams-Huet B, Toto RD, Yee J, et al. Cumulative fluid balance and mortality in septic patients with or without acute kidney injury and chronic kidney disease. Crit Care Med 2016;44:1891-900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chua HR, Wong WK, Ong VH, Agrawal D, Vathsala A, Tay HM, et al. Extended mortality and chronic kidney disease after septic acute kidney injury. J Intensive Care Med 2020;35:527-35. [DOI] [PubMed] [Google Scholar]
- 39.Arulkumaran N, Pollen S, Greco E, Courtneidge H, Hall AM, Duchen MR, et al. Renal Tubular cell mitochondrial dysfunction occurs despite preserved renal oxygen delivery in experimental septic acute kidney injury. Crit Care Med 2018;46:e318-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.West AP, Brodsky IE, Rahner C, Woo DK, Erdjument-Bromage H, Tempst P, et al. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 2011;472:476-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev 2007;87:99-163. [DOI] [PubMed] [Google Scholar]
- 42.Qiu YH, Zhang TS, Wang XW, Wang MY, Zhao WX, Zhou HM, et al. Mitochondria autophagy: a potential target for cancer therapy. J Drug Target 2021;29:576-91. [DOI] [PubMed] [Google Scholar]
- 43.Brealey D, Karyampudi S, Jacques TS, Novelli M, Stidwill R, Taylor V, et al. Mitochondrial dysfunction in a long-term rodent model of sepsis and organ failure. Am J Physiol Regul Integr Comp Physiol 2004;286:R491-7. [DOI] [PubMed] [Google Scholar]
- 44.Holmstrom KM, Baird L, Zhang Y, Hargreaves I, Chalasani A, Land JM, et al. Nrf2 impacts cellular bioenergetics by controlling substrate availability for mitochondrial respiration. Biol Open 2013;2:761-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fetoni AR, Paciello F, Rolesi R, Paludetti G, Troiani D. Targeting dysregulation of redox homeostasis in noise-induced hearing loss: Oxidative stress and ROS signaling. Free Radic Biol Med 2019;135:46-59. [DOI] [PubMed] [Google Scholar]
- 46.Lee DY, Song MY, Kim EH. Role of oxidative stress and Nrf2/KEAP1 signaling in colorectal cancer: Mechanisms and Therapeutic perspectives with phytochemicals. Antioxidants (Basel) 2021;10:743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nguyen T, Nioi P, Pickett CB. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J Biol Chem 2009;284:13291-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gui Y, Yang Y, Xu D, Tao S, Li J. Schisantherin A attenuates sepsis-induced acute kidney injury by suppressing inflammation via regulating the NRF2 pathway. Life Sci 2020;258:118161. [DOI] [PubMed] [Google Scholar]
- 49.Mendonca P, Soliman KFA. Flavonoids activation of the transcription factor Nrf2 as a hypothesis approach for the prevention and modulation of SARS-CoV-2 infection severity. Antioxidants (Basel) 2020;9:659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Scorrano L. Opening the doors to cytochrome c: changes in mitochondrial shape and apoptosis. Int J Biochem Cell Biol 2009;41:1875-83. [DOI] [PubMed] [Google Scholar]
- 51.McArthur K, Whitehead LW, Heddleston JM, Li L, Padman BS, Oorschot V, et al. BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science 2018;359:eaao6047. [DOI] [PubMed] [Google Scholar]
- 52.Kuznetsov AV, Kehrer I, Kozlov AV, Haller M, Redl H, Hermann M, et al. Mitochondrial ROS production under cellular stress: comparison of different detection methods. Anal Bioanal Chem 2011;400:2383-90. [DOI] [PubMed] [Google Scholar]
- 53.Dai XG, Xu W, Li T, Lu JY, Yang Y, Li Q, et al. Involvement of phosphatase and tensin homolog-induced putative kinase 1- Parkin-mediated mitophagy in septic acute kidney injury. Chin Med J (Engl) 2019;132:2340-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gao Y, Dai X, Li Y, Li G, Lin X, Ai C, et al. Role of Parkinmediated mitophagy in the protective effect of polydatin in sepsis-induced acute kidney injury. J Transl Med 2020;18:114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang Z, Chen Z, Liu R, Liang Q, Peng Z, Yin S, et al. Bcl-2 Proteins regulate mitophagy in lipopolysaccharide-induced acute lung injury via PINK1/parkin signaling pathway. Oxid Med Cell Longev 2020;2020:6579696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xiao L, Xu X, Zhang F, Wang M, Xu Y, Tang D, et al. The mitochondria-targeted antioxidant MitoQ ameliorated tubular injury mediated by mitophagy in diabetic kidney disease via Nrf2/PINK1. Redox Biol 2017;11:297-311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yan C, Gong L, Chen L, Xu M, Abou-Hamdan H, Tang M, et al. PHB2 (prohibitin 2) promotes PINK1-PRKN/Parkindependent mitophagy by the PARL-PGAM5-PINK1 axis. Autophagy 2020;16:419-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tanaka K. The PINK1-parkin axis: An overview. Neurosci Res 2020;159:9-15. [DOI] [PubMed] [Google Scholar]
- 59.Piantadosi CA, Withers CM, Bartz RR, MacGarvey NC, Fu P, Sweeney TE, et al. Heme oxygenase-1 couples activation of mitochondrial biogenesis to anti-inflammatory cytokine expression. J Biol Chem 2011;286:16374-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Athale J, Ulrich A, MacGarvey NC, Bartz RR, Welty-Wolf KE, Suliman HB, et al. Nrf2 promotes alveolar mitochondrial biogenesis and resolution of lung injury in Staphylococcus aureus pneumonia in mice. Free Radic Biol Med 2012;53:1584-94. [DOI] [PMC free article] [PubMed] [Google Scholar]