

Abstract
Cellular senescence and ferroptosis are the two main, fine-tuned processes in tissue damage restraint; however, they can be overactivated in pathologies such as nonalcoholic fatty liver disease/nonalcoholic steatohepatitis (NAFLD/NASH), becoming dangerous stimuli. Senescence is characterized by a decline in cell division and an abnormal release of reactive oxygen species (ROS), and ferroptosis is represented by iron deposition associated with an excessive accumulation of ROS. ROS and cellular stress pathways are also drivers of NAFLD/NASH development. The etiology of NAFLD/NASH lies in poor diets enriched in fat and sugar. This food regimen leads to liver steatosis, resulting in progressive degeneration of the organ, with a late onset of irreversible fibrosis and cirrhosis. Few studies have investigated the possible connection between senescence and ferroptosis in NAFLD/NASH progression, despite the two events sharing some molecular players. We hypothesized a possible link between senescence and ferroptosis in a NAFLD background. To thoroughly investigate this in the context of “Western-style” diet (WSD) abuse, we used an amylin-modified liver NASH mouse model. The main NASH hallmarks have been confirmed in this model, as well as an increase in apoptosis, and Ki-67 and p53 expression in the liver. Senescent beta-galactosidase-positive cells were elevated, as well as the expression of the related secretory molecules Il-6 and MMP-1. Features of DNA damage and iron-overload were found in the livers of NASH mice. Gpx4 (glutathione peroxidase 4) expression, counteracting ferroptotic cell death, was increased. Notably, an increased number of senescent cells showing overexpression of gpx4 was also found. Our data seem to suggest that senescent cells acquire a gpx4-mediated mechanism of ferroptosis resistance and thus remain in the liver, fostering the deterioration of liver fitness.
Key words: NAFLD/NASH, senescence, ferroptosis, beta-galactosidase, gpx4
Introduction
Senescence is a complex biological process in which cells gradually lose their ability to differentiate and replicate, blocking the cell cycle in the G1/S phase and changing their morphology (becoming flattened and with enlarged nuclei).1,2 In response to harmful events, such as oxidative stress, oncogene activation, or dysregulation of epigenomic control, senescence coupled with apoptosis comprises a physiological defense mechanism aimed to preserve tissue homeostasis and functionality. Senescent cells release a complex secretome (composed of chemokines, interleukins and matrix proteases) known as senescence-associated secretory phenotype (SASP) members. This system warns the tissue microenvironment and the immune system of the onset of the injury, and coordinates the deletion of senescent cells. Finally, damaged cells are discarded by immune cells and the progression of the injury is confined.1,2 On the contrary, when the exposure to the SASP persists, an accumulation of senescent cells resistant to apoptosis occurs,3-7 resulting in several degenerative conditions, including cardiovascular disease,8 obesity,9 and liver fibrosis.10,11 Recently, senescence-associated mechanisms have been highlighted in dysmetabolic syndromes such as nonalcoholic fatty liver disease (NAFLD), a hepatic disorder induced by lipid accumulation.12 nonalcoholic steatohepatitis (NASH) is the late stage of steatotic degeneration, histologically defined by features of steatosis (>5%), hepatocyte ballooning and neutrophil satellitosis with lobular inflammation, with or without fibrosis.13 Almost 20% of NASH patients develop cirrhosis and metabolic syndromes over time, leading to hepatocarcinogenesis and onset of hepatocarcinoma, the major histological type of primary liver cancer, accounting for 70-85% of all liver cancers.14
Genetic predisposition and lifestyle are the major causes of NAFLD/NASH. In particular, alimentary habits are the driving cause of liver steatosis, and abuse of a high-lipid content diet, known as the “Western-style” diet (WSD), is associated with the onset of steatosis and NAFLD/NASH.15,16 Lipid storage in hepatocytes is able to induce an increase in reactive oxygen species (ROS), accelerating apoptosis and senescence, thereby contributing to progressive organ decline and the development of disease.17
Additionally, ROS accumulation is associated with a recentlyidentified form of non-apoptotic cell death, known as ferroptosis. This process is mechanistically and biochemically distinct from apoptosis, necrosis and autophagic cell death.18 Ferroptosis is triggered by dysfunctional iron homeostasis and accumulation, leading to mitochondrial alterations, such as a decrease in mitochondrial volume and reduction of their cristae. Notably, the cellular membrane remains intact and the nucleus size appears normal.19-21 Interestingly, there is a tight correlation between lipid metabolism and ferroptosis, since the cytoplasmic accumulation of ironinduced lipid peroxides is a primary cause of ferroptosis.22
This condition is also biochemically characterized by the exhaustion of intracellular glutathione, which in turn contributes to ROS accumulation, fueling ferroptosis.20 Some homeostatic mechanisms are known to counteract and terminate the ferroptotic pathway, mainly orchestrated by the gpx4 and Xc systems. The presence of redox-active iron and the loss of lipid peroxide repair activity by gpx4 are considered to be typical markers of ferroptosis. Ferroptosis kills damaged and malignant cells, but may also be incorrectly activated in pathologies like neurodegeneration and ischemia.22-25
Several papers emphasize that a high-carbohydrate and highlipid diet promotes NAFLD/NASH disorders, liver fibrosis, hepatocarcinoma, and bowel alteration.26-31 Recent reports described an intimate association between the progression of liver disease and an increase in senescence markers or the onset of ferroptosis in experimental models of NAFLD/NASH induced by several dietary regimens.31,32 However, most of the studies looked at the early phase of the liver injury during NAFLD onset and the related contribution of ferroptosis, while the overt or late stages are poorly investigated. Furthermore, the majority of these studies focused singularly on liver senescence or ferroptosis, while the link between them has not been properly investigated. To date, the cause-effect relationship between senescence and ferroptosis is still under investigation, being hypothesized to trigger a vicious cycle that exacerbates NAFLD/NASH progression.
Given these bases, the purpose of the study is to investigate the contribution of the senescence and ferroptosis mechanisms in the liver parenchyma in a mouse model of NASH upon prolonged administration of a high fat/high-carbohydrate diet (WSD) compared with the standard diet (SD). To this aim, we used a NAFLD/NASH phenotype mouse model provided by Taconic, known as the Amylin (AMLN) liver NASH model, induced by a diet containing 40 Kcal% fat (of which around 18% trans-fat), 20 Kcal% fructose, and 2% cholesterol, also known as the WSD.33-37
Materials and Methods
Mice, diets and experimental design
Eight livers from diet-induced male C57BL/6N NASH mice were purchased from Taconic Biosciences (Taconic Biosciences A/S, Lille Skensved, Denmark) for use in this study. Ethical review and approval did not apply to this study because it relies exclusively on the use of commercial biospecimens from Taconic Biosciences. The company maintained mice on a modified Amylin liver NASH (AMLN) diet, also known as a “Western-style diet” (#D09100310i Research Diets), characterized by high fat and carbohydrate content.
Eight livers from C57BL/6N control mice fed with a standard diet (NIH #31M Research Diets) were used as controls. The sample size of the experimental groups was determined using G-Power software, v. 3.1.9.7 (Heinrich-Heine-Universitat Dusseldorf, Germany). Table 1 shows the full description of the main nutrients of the two diets. All mice were put on the different diets at 6 weeks of age and housed at reduced density. According to the aim of our study, and the information provided by the company, we purchased the samples after 23 weeks of the diet. The company performed routine zoometric and biochemical laboratory tests for NASH features before delivering the samples (Table 1). At this timepoint, the AMLN diet led to development of obesity, fatty liver, liver inflammation, and early signs of fibrosis. The livers were explanted and perfused with 10% neutral buffered formalin. The murine biospecimens for NASH and control animals used in the study were purchased preserved in 10% neutral buffered formalin.
Histological, immunohistochemical and immunofluorescence analysis
Liver fragments were washed and embedded in low-temperature fusion paraffin. Sections of 3 μm thickness were stained with Mayer’s hematoxylin and eosin (H&E) (Bio Optica, Milan, Italy) to evaluate the degree of steatosis and inflammation. Masson’s trichrome (Bio Optica) staining was conducted to detect the deposition of connective tissue and fibrosis.
The stained sections were then observed under an Olympus BX51 Light Microscope (Olympus Optical Co. Ltd, Tokyo, Japan). Features of NAFLD/NASH (steatosis, inflammation and fibrosis) were scored based on the parameters proposed by Kleiner et al. and Bedossa et al.38,39 Steatosis was graded as 0 (<5%), 1 (5-33%), 2 (33-66%), or 3 (>66%). For each specimen, four fields were evaluated (10× magnification). Inflammation was defined as a foci of two or more inflammatory cells within the lobule. A focus was defined as an aggregate of ≥50 mononuclear (predominantly lymphocytic) inflammatory cells. All foci were counted at 20× magnification (0: none; 1: ≤2 foci; 2: >2 foci). Stage of fibrosis was assigned a score of 0 (none), 1 (perisinusoidal zone or periportal fibrosis), 2 (perisinusoidal and periportal fibrosis without bridging), 3 (bridging fibrosis), or 4 (cirrhosis).38,39 All morphological and morphometrical data were assessed by two blinded morphologists.
Prussian Blue solution was prepared by mixing equal parts of 20% (vol/vol) hydrochloric acid and 10% (g/vol) potassium ferrocyanide (Sigma-Aldrich/Merck KGaA, Darmstadt, Germany; code H1758) solution just before use. The samples were immersed in this solution for 20 min. The nuclei were counterstained with nuclear fast red (code N3020; (Sigma-Aldrich/Merck KGaA) for 5 min. Iron deposition (number and surface of blue deposits) was counted across the whole section for each sample (40× magnification) using ImageJ software with a scale set (8.97 pixels/μm) and normalized per mm2 of section. The data obtained were plotted as histograms. For the immunostaining analysis, the samples were incubated for 40 min in methanol and then in 3% hydrogen peroxide solution for 5 min. The specimens were incubated overnight at 4°C with the specific antibodies: β-gal (galactosidase), Ki-67; pH2AX (phosphorylate H2A histone family member X); and IL-6 (interleukin-6), dilutions 1:50; MMP1 (matrix metalloproteinase-1), dilution 1:100; p53 (tumor protein p53); gpx4 (phospholipid hydroperoxide glutathione peroxidase4); COX2 (cyclooxygenase2), CD11c; CD80; dilutions 1:50; and IgG-HRP (immunoglobulin-horseradish peroxidase); IgG-FITC (immunoglobulin-fluorescein isothiocyanate); IgG-PE (immunoglobulin-phycoerythrin), dilutions 1:200. All antibodies were purchased from Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA.
Finally, the specimens were incubated for 1 h at room temperature with the appropriate secondary antibody, horseradish (HRP) [EnVisionR+ Dual Link System-HRP (DAB+); Agilent, Santa Clara, CA, USA] or a fluorophore-conjugated antibody. Finally, for immunohistochemistry (IHC), the sections were counterstained with Mayer’s hematoxylin and mounted with Eukitt medium, while for immunofluorescence (IF), the sections were mounted with Fluorlast with 4′,6-diamidino-2-phenylindole (DAPI) medium for nuclear counterstaining (Biovision, Milpitas, CA, USA). To control the specificity of the immunostaining, all reactions included negative controls (sections were incubated omitting the primary antibody). The specimens were observed under an Olympus BX51 light microscope equipped with a laser source (Olympus, Optical Co. Ltd.).
Senescence assay
The presence of active endogenous acidic beta-galactosidase was revealed using the CellEvent™ Senescence Green Assay Kit (Thermo Fisher Scientific, Waltham, MA USA), according to the manufacturer’s instructions.
TUNEL assay
Apoptosis was investigated using the ApopTagR Peroxidase in Situ Apoptosis Detection Kit, (Merckmillipore, Merck KGaA, Darmstadt, Germany), according to the manufacturer’s instructions.
Semi-quantitative digital image analysis of immunohistochemical staining
Semi-quantitative comparison of IHC staining was performed using the ImageJ IHC profiler software plugin, as previously described.15 Immunopositivity was expressed as a percentage of the total software-classified areas, and the data obtained were plotted as histograms.
Statistical analyses
Results were expressed as mean ± standdard deviation and unpaired non-parametric Mann-Whitney tests were used for statistical comparisons between experimental groups; a p-value <0.05 was considered statistically significant.
Results
NASH-related alterations in the liver of WSD-fed mice
The main zoometric and biochemical NAFLD/NASH-related parameters were confirmed to be changed as expected (Table 1). Histological liver analyses by H&E staining in WSD-fed mice showed evident signs of steatosis compared to control mice, as confirmed by steatosis score (Figure 1 A,B). Additionally, mild/moderate features of inflammation were found in the parenchyma of WSD-fed mice, while only scattered inflammatory cells were detected in the control group, as confirmed by inflammation scores (Figure 1C). To assess signs of fibrosis, Masson’s Trichrome staining was conducted (Figure 1D). WSD-fed mice showed mild perisinusoidal or portal fibrosis, while no collagen deposition was noted in the control group (Figure 1D). These data were confirmed by fibrotic scores, which were close to zero in SD mice (Figure 1E). Accordingly, an increase in apoptosis, commonly occurring during liver injury, was revealed by the TUNEL assay (Figure 1 F,G), as well as increased hepatocellular proliferation, a typical hallmark of steatosis, evaluated by Ki-67 (Figure 1 H,I) in the liver parenchyma of the WSD-fed group, as expected.
Table 1.
Detailed description of the main nutrients of the diets used in the experimental procedures, and zoometric and biochemical parameters of mice undergoing dietary regimens used in this study. Values represent means ± standard deviation (n=8 for each group). Statistics were performed using the Mann-Whitney test.
| Diet ingredients | SD | WSD | |
|---|---|---|---|
| Protein | Total 21 kcal% | Total 20 kcal% | |
| Casein | 800 kcal | ||
| L-cystine | 12 kcal | ||
| Carbohydrate | Total 65 kcal% | Total 40 kcal% | |
| Maltodextrin 10 | 400 kcal | ||
| Fructose | 800 kcal | ||
| Sucrose | 384 kcal | ||
| Fat | Total 14 kcal% | Total 40 kcal% | |
| Soybean oil | 225 kcal | ||
| Lard | 180 kcal | ||
| Palm oil | 1215 kcal | ||
| Parameters | SD | WSD | p |
| Body weight (g) | 32.69±3.53 | 44.67±2.02 | *** |
| Liver weight (g) | 1.469±0.15 | 4.027±0.57 | *** |
| % Liver/body weight | 4.51±0.22 | 8.99±0.99 | *** |
| Serum ALT (U/L) | 41.96±5.85 | 621.2±322.3 | ** |
| Liver triglycerides (mg/g) | 26.35±5.29 | 232.4±39.29 | ** |
SD, standard diet; WSD, Western-style diet; U/L, units/liter of serum. **p<0.01; ***p<0.001.
Figure 1.
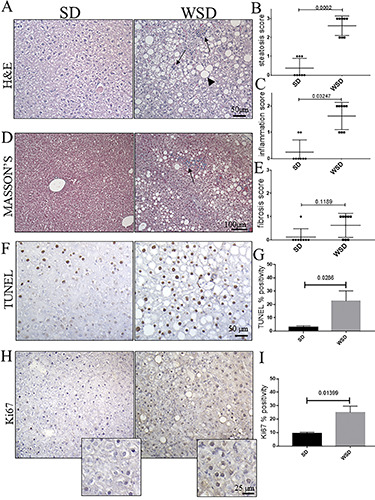
A) Hematoxylin and eosin; the liver parenchyma of WSD-fed mice showed severe signs of steatosis (arrowhead), compared with SD-fed mice; rare and isolated inflammation foci are detected in SD mice compared with the WSD group (arrows); original magnification 20×, scale bar: 50 μm. B,C) Steatosis and inflammation scores differed significantly between WSD and SD mice. D) Masson’s trichrome staining; no collagen depositions are detected in SD mice, while mild fibrosis is highlighted (arrows) in WSD animals; original magnification 10×, scale bar: 100 μm. E) Fibrosis scores were not significantly different in WSD vs SD mice. F) The TUNEL assay and (H) the immunohistochemical labeling for Ki-67 showed an increase in Tunel-positive cells in WSD compared to SD mice; original magnification 20×, scale bar: 50 μm. G,I) Semi-quantitative analyses confirmed a significant increase of apoptotic pathways, and Ki-67 expression. These images are representative of n=8 SD, and n=8 WSD mice. Values represent means ± standard deviation. Data were analyzed using the Mann-Whitney test; SD, standard diet; WSD, Western-style diet.
Senescent cell phenotype induced by WSD
In order to evaluate whether NAFLD/NASH features induced by prolonged WSD were accompanied by cellular senescence, IHC for the main senescence-associate marker, acidic beta-galactosidase, was performed; this was found to be increased in the NASH group compared to the control (Figure 2 A,B). These cells were not undergoing the apoptotic program, being negative for TUNEL (Figure 2C). This molecular profile, strictly linked to cell cycle arrest, was also enriched in the expression of the key markers of DNA damage, p53 (Figure 3A) and p-H2AX (Figure 3C). The expression of all of these molecules was increased in WSD-fed mice compared to the control diet group (Figure 3 B,D). Additionally, we tested the SASP markers IL-6 and MMP-1, and found a noticeable increase in these indirect indicators of senescence in WSD-fed mice compared to control (Figure 3 E,G), validating the presence of senescent cells associated with inflammation and fibrosis (Figure 3 F,H).
Ferroptotic environment induced by the WSD
Since p53 induction is also a feature of iron-induced cell stress and ferroptosis, we investigated the iron accumulation in the liver parenchyma. We found an increase in iron deposits in WSD-fed mice compared to controls (Figure 4 A,B). Furthermore, in order to confirm the triggering of ferroptosis-related pathways, we assessed cyclooxygenase 2 (Cox2), one of the main enzymes that contributes to the production of ROS and promotion of ferroptosis (Figure 4C). The IHC highlighted increased expression of Cox2 in WSD-fed mice compared to controls (Figure 4D). Finally, we measured glutathione peroxidase (Gpx4), a key inhibitor of ferroptotic cell death, which is supposed to be downregulated upon ferroptosis activation (Figure 4 E,F). Surprisingly, an increase in Gpx4 was seen in the WSD group compared to the SD-fed mice (Figure 4 E,F). This result prompts us to conclude that, under our experimental conditions, the liver parenchyma, at least in part, is able to react to iron overload and oxidative stress, and counteract the triggering of ferroptosis. In order to deeply dissect the molecular profile of the liver, we looked at the relationship between senescence and antioxidant activity occurring in hepatic cells. Interestingly, multiplex IF staining revealed a significant increase in the number of β- gal positive senescent cells expressing Gpx4 in the WSD-fed group compared to the SD group (Figure 5). Finally, we investigated whether the accumulation of β-gal/Gpx4 double-positive cells induced the activation of the immune response, looking at the monocyte/macrophage lineage. We found a significant increase of phagocytic CD11c-positive and costimulatory CD80-positive macrophages in WSD-fed mice compared to controls (Figure 6).
Figure 2.
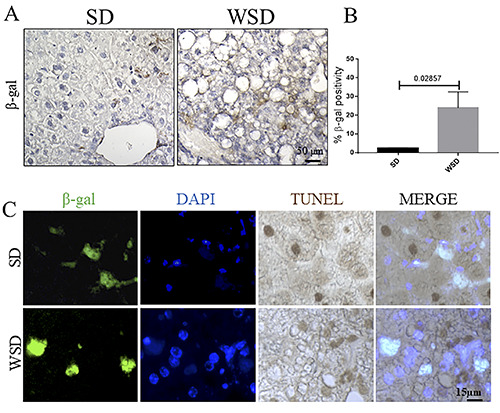
A) Immunohistochemistry for β-gal; original magnification 40×, scale bar: 25 μm. B) Semi-quantitative analyses showed a significant increase in β-gal in WSD-fed mice compared to the SD group. C) Immunostaining for β-gal (IF, green) and Tunel (IHC, brown) in WSD-fed mice compared to the SD group; the staining confirmed the activation of the apoptosis-resistant senescent pathway; nuclei are counterstained with DAPI (IF, blue); original magnification 60×, scale bar 15 μm. These images are representative of n=8 SD, and n=8 WSD mice. Data were analyzed using the Mann-Whitney test; SD, standard diet; WSD, Western-style diet.
Altogether, our results revealed that senescence and oxidative stress with iron accumulation are the main features of the overt stage of NAFLD in mice upon prolonged WSD. In this context, accumulation of senescent cells with a high level of the ferroptosis- protective enzyme Gpx4 occurred, contributing to liver degeneration.
Figure 3.
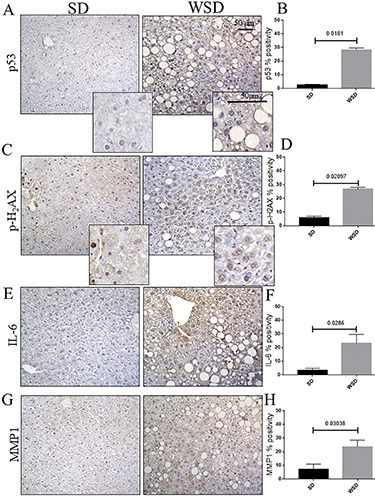
A,C) Immunohistochemistry for p53, and p-H2AX; upper, original magnification 20×, scale bar: 50 μm; inset, scale bar: 50 μm. B,D) The expression of p53 and p-H2AX was significantly increased in WSD mice compared to the SD group, as shown by semiquantitative analyses. E G) Immunohistochemistry for IL-6 and MMP1; original magnification 20×, scale bar: 50 μm. F,H) Semi-quantitative analyses showed a significant increase in both IL-6 and MMP1 in WSD-fed mice compared to the SD group. Values represent means ± standard deviation. Data were analyzed using the Mann-Whitney test; SD, standard diet; WSD, Western-style diet.
Figure 4.
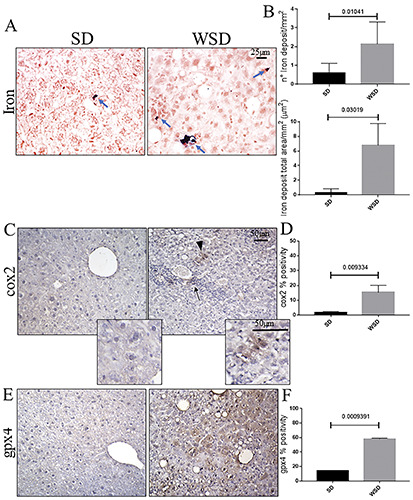
A) Prussian blue staining showed increased iron deposits (arrows) in WSD mice compared to the SD group; original magnification 40×, scale bar: 25 μm. B) Quantification of iron deposits, in both number and area, showed a significant increase in iron content in WSD-fed mice compared to the SD group. C) Immunohistochemistry for Cox2 showed an increase in immunopositivity in WSD compared to SD mice; original magnification 20×, scale bar 50 μm; inset, scale bar: 50 μm; arrowhead, epithelial-like Cox2-positive cells; arrow, Cox2-positive inflammation focus. D) Semi-quantitative analyses confirmed a significant increase in Cox2 expression. E) Immunohistochemistry of Gpx4 showed increased positivity in WSD mice compared to the SD group; original magnification 20×, scale bar: 50 μm. E) The semi-quantitative evaluation highlighted a significant increase of expression in WSD-fed mice compared to the SD group. These images are representative of n=8 SD, and n=8 WSD mice. Values represent means ± standard deviation. Data were analyzed using the Mann-Whitney test; SD, standard diet; WSD, Western-style diet.
Discussion
Senescence is commonly described as a physiological process characterized by an irreversible arrest of the cellular cycle. However, the accumulation of senescent cells in organs has been shown to be a risk for many chronic disorders. In this context, it is increasingly accepted that senescence is associated with other events, including ferroptosis, a newly-discovered form of irondependent programmed cell death. Although it is well-known that both senescence and ferroptosis are involved in the progression of aging or age-related pathologies, including NAFLD/NASH,2,17,32,40 the true relationship between these two processes is still poorly characterized, especially in the overt stage of liver degeneration and NASH. In this paper, we describe the ferroptotic-resistant phenotype of the senescent cells in the NAFLD/NASH background, thus refuting the hypothesis of ferroptosis activation, at least in the context of the overt NAFLD/NASH stage.
Figure 5.
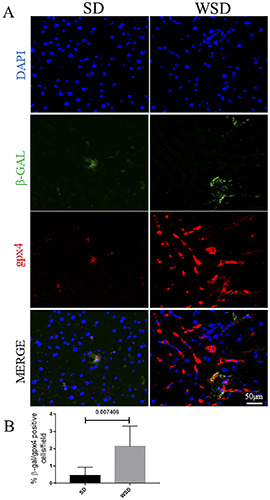
A) Immunofluorescence for β-gal (green) and Gpx4 (red) showed an increase in β-gal/Gpx4-positive cells in WSD-fed mice compared to the SD group; Nuclei are counterstained with DAPI (blue); original magnification 20×, scale bar: 50 μm. B) Quantification of A. These images are representative of n=8 SD, and n=8 WSD mice. Data were analyzed using the Mann-Whitney test; SD, standard diet; WSD, Western-style diet.
The WSD regimen is responsible for the occurrence of the typical hallmarks of the NAFLD/NASH, following the canonical timeframe of liver degeneration: steatosis (16 weeks of diet), inflammation (20 weeks of diet), and fibrosis (>20 weeks of diet). We first confirmed the senescent phenotype of the livers upon extended WSD exposure, highlithing a consistent increase in senescence-associated beta-galactosidase and SASP members. Furthermore, the p53 protein, a player shared between senescence and ferroptosis, was increased in mice under the WSD regimen. Therefore, we planned to investigate the ferroptosis pathway activation, looking at other features of iron-induced stress. Consistent with this, increased iron deposition, as well as Cox2 expression (another marker in common between senescence and ferroptosis), was detected in the WSD-fed group.
Surprisingly, further evaluation of the ferroptosis hallmark failed to confirm the activation of the ferroptosis pathway. In fact, the antioxidant enzyme and master protector from ferroptosis initiation, Gpx4, has been found to increase in the WSD group. Under our experimental conditions, a deeper investigation at the cellular level of the ferroptotic and senescent markers revealed a complex mixed phenotype. In fact, besides senescent beta-galactosidase-positive cells and iron-induced Gpx4-positive cells, a population of Gpx4- expressing senescent cells has been observed. Thus, we demonstrate that a subpopulation of the senescent cells in the liver presented high expression of Gpx4, being presumably protected from ferroptosis and contributing to the alterations of the liver parenchyma undergoing steatohepatitis. This complex phenotype can contribute to the accumulation and progression of tissue damage and dysfunction in the steatotic liver, since two major molecular programs aimed to oppose the elimination of injured cells are activated. These cells are hard to eradicate from the liver parenchyma, being protected from both apoptosis and ferroptosis, and continuing to release SASP members, which spread damaging stimuli into the surroundings.
The parenchyma degeneration in NAFLD/NASH is accompanied by senescence. This is a biological process involved in tissue homeostasis, which is able to prevent the degeneration of the cell toward oncogenic transformation upon replicative or stressinduced injuries.41 However, the accumulation of senescent cells in a tissue is associated with the onset and progression of many degenerative and chronic diseases, such as Parkinson’s disease, atherosclerosis, and osteoporosis.5,12,17 In line with this data, the use of senolytic drugs is a promising therapeutic strategy to treat these pathologies.42 Senescence is induced by endogenous and exogenous stimuli that cause DNA damage, culminating in irreversible cell cycle arrest and senescence initiation. These cells undergo chromatin remodeling, present the SASP, change in morphology, and gain other characteristics of the fully-fledged senescence phenotype, and become resistant to apoptotic cell death. The SASP is constituted by a plethora of molecules, among which are cytokines, MMPs (i.e., IL-6 and MMP1) and extracellular matrix, and insoluble factors (collagens). It attracts immune cells and induces senescent reprogramming in the neighboring cells.41
Figure 6.
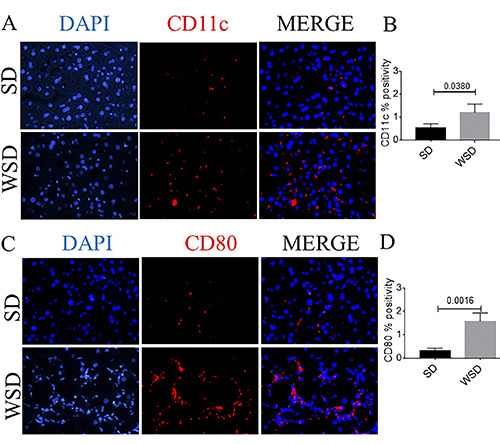
A) Immunofluorescence analysis of CD11c (red) showed an increase in CD11c-positive cells in WSD-fed mice compared to the SD group.(B) Quantification of A. C) Immunofluorescence analysis of CD80 (red) showed an increase in CD80-positive cells in WSD-fed mice compared to the SD group; nuclei are counterstained with DAPI (blue); original magnification 40×, scale bar: 50 μm. (D) Quantification of C. These images are representative of n=8 SD, n=8 WSD mice. Values represent means ± standard deviation. Data were analyzed using the Mann-Whitney test; SD, standard diet; WSD, Western-style diet.
The key involvement of senescence in NAFLD/NASH has been described in the liver parenchyma in both rodents and humans, with steatosis being successfully reverted by the genetic or pharmacological elimination of senescent hepatocytes.12 In line with this evidence, our analyses revealed a cellular switch toward a senescent phenotype. Several reports have described the contribution of ferroptosis to NAFLD/NASH.32,40 Notably, senescent and ferroptotic pathways show some resemblances: senescence is accompanied by iron accumulation and glutathione exhaustion, as well as p53 induction, features in common with ferroptosis.3,20 Accordingly with this body of literature, in WSD-fed mice, we described some key members of the ferroptotic pathway, such as iron deposition and Cox2 overexpression.
Surprisingly, Gpx4 activation was found, indicating that ferroptosis is not a pivotal player in steatotic degeneration, at least in the context of the overt NAFLD/NASH stage. In particular, senescent cells presented antioxidant activity through the expression of Gpx4. This molecular profile renders senescent cells possibly prone to remain and accumulate in the liver parenchyma. We are conscious that this study has mainly provided a descriptive molecular profile of liver cells, and further evaluations will be necessary to uncover the mechanistic events underlying the senescence-iron overload link. However, to our knowledge, we have provided the first evidence of Gpx4 overexpression in the context of a senescent steatotic background, probably due to the experimental model and diet regimen used in the present study.
In conclusion, we have demonstrated a tight link between senescence and ferroptosis resistance in the context of overt NAFLD/NASH (Figure 7), highlighting the complexity and plasticity of the molecular profile during steatohepatitis degeneration.
Figure 7.
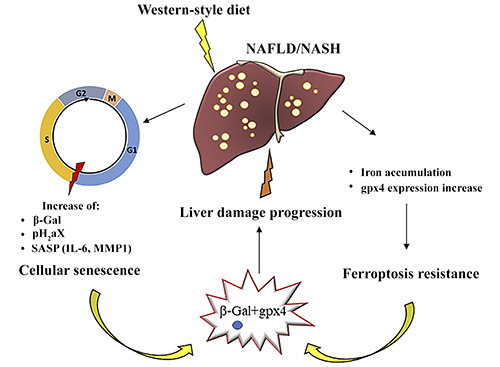
Western-style diet assumption in NAFLD/NASH was accompanied by cellular senescence, characterized by the arrest of the cell cycle in the G1/S phase, and by an increase in levels of several molecules (β-Gal, pH2aX, and the SASP: IL-6, MMP1). NAFLD/NASH is also associated with iron deposition in the liver parenchyma and concomitantl increased Gpx4 expression, leading to ferroptosis resistance. These mechanisms can co-occur in the same cell, cooperating to exacerbate NAFLD/NASH progression.
Acknowledgements
The authors would like to thank Dompè farmaceutici S.p.A., L’Aquila, Italy, for providing Taconic’s samples.
Funding Statement
Funding: This research received no external funding.
References
- 1.Aravinthan AD, Alexander GJM. Senescence in chronic liver disease: Is the future in aging? J Hepatol 2016;65:825–34. [DOI] [PubMed] [Google Scholar]
- 2.Papatheodoridi AM, Chrysavgis L, Koutsilieris M, Chatzigeorgiou A. The role of senescence in the development of nonalcoholic fatty liver disease and progression to nonalcoholic steatohepatitis. Hepatology 2020;71:363-74. [DOI] [PubMed] [Google Scholar]
- 3.Campisi J. Senescent cells, tumor suppression, and organismal aging: Good citizens, bad neighbors. Cell 2005;120:513-22. [DOI] [PubMed] [Google Scholar]
- 4.Hampel B, Wagner M, Teis D, Zwerschke W, Huber LA, Jansen-Durr P. Apoptosis resistance of senescent human fibroblasts is correlated with the absence of nuclear IGFBP-3. Aging Cell 2005;4:325–30. [DOI] [PubMed] [Google Scholar]
- 5.Herbig U, Ferreira M, Condel L, Carey D, Sedivy JM. Cellular senescence in aging primates. Science 2006;311:1257. [DOI] [PubMed] [Google Scholar]
- 6.Hoenicke L, Zender L. Immune surveillance of senescent cells-biological significance in cancer-and non-cancer pathologies. Carcinogenesis 2012;33:1123–6. [DOI] [PubMed] [Google Scholar]
- 7.Marcotte R, Lacelle C, Wang E. Senescent fibroblasts resist apoptosis by downregulating caspase-3. Mech Ageing Dev 2004;125:S777–83. [DOI] [PubMed] [Google Scholar]
- 8.Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J, Van Deursen JM. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science 2016;354:472-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Minamino T, Orimo M, Shimizu I, Kunieda T, Yokoyama M, Ito T, et al. A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nat Med 2009;15:1082-7. [DOI] [PubMed] [Google Scholar]
- 10.Moncsek A, Al-Suraih MS, Trussoni CE, O’Hara SP, Splinter PL, Zuber C, et al. Targeting senescent cholangiocytes and activated fibroblasts with B-cell lymphoma-extra large inhibitors ameliorates fibrosis in multidrug resistance 2 gene knockout (Mdr2−/−) mice. Hepatology 2018;67:247–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pompili S, Sferra R, Gaudio E, Viscido A, Frieri G, Vetuschi A, et al. Can Nrf2 modulate the development of intestinal fibrosis and cancer in inflammatory bowel disease? Int J Mol Sci 2019;20:4061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ogrodnik M, Miwa S, Tchkonia T, Tiniakos D, Wilson CL, Lahat A, et al. Cellular senescence drives age-dependent hepatic steatosis. Nat Commun 2017;8:15691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sheka AC, Adeyi O, Thompson J, Hameed B, Crawford PA, Ikramuddin S. Nonalcoholic Steatohepatitis. JAMA 2020;323:1175. [DOI] [PubMed] [Google Scholar]
- 14.Anstee QM, Reeves HL, Kotsiliti E, Govaere O, Heikenwalder M. From NASH to HCC: current concepts and future challenges. Nat Rev Gastroenterol Hepatol 2019;16:411–28. [DOI] [PubMed] [Google Scholar]
- 15.Pompili S, Vetuschi A, Gaudio E, Tessitore A, Capelli R, Alesse E, et al. Long-term abuse of a high-carbohydrate diet is as harmful as a high-fat diet for development and progression of liver injury in a mouse model of NAFLD/NASH. Nutrition 2020;75-76:10782. [DOI] [PubMed] [Google Scholar]
- 16.Tessitore A, Mastroiaco V, Vetuschi A, Sferra R, Pompili S, Cicciarelli G, et al. Development of hepatocellular cancer induced by long term low fat-high carbohydrate diet in a NAFLD/NASH mouse model. Oncotarget 2017;8:53482–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schade M, Sanabria JJ, Aguilar R, Modarresi M, Gillon B, Hunter Z, et al. Cellular senescence and their role in liver metabolism in health and disease: Overview and future directions. World J Surg Surg Res 2018;1:1035-51. [Google Scholar]
- 18.Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012;149:1060-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cillero-Pastor B, Carames B, Lires-Dean M, Vaamonde- Garcia C, Blanco FJ, Lopez-Armada MJ. Mitochondrial dysfunction activates cyclooxygenase 2 expression in cultured normal human chondrocytes. Arthritis Rheum 2008;58:2409-19. [DOI] [PubMed] [Google Scholar]
- 20.Lei P, Bai T, Sun Y. Mechanisms of ferroptosis and relations with regulated cell death: A review. Front Physiol 2019;10:139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li J, Cao F, Yin H liang, Huang Z jian, Lin Z tao, Mao N, et al. Ferroptosis: past, present and future. Cell Death Dis 2020;11:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang WS, Stockwell BR. Ferroptosis: Death by lipid peroxidation. Trends Cell Biol 2016;26:165-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tuo Q, Lei P, Jackman KA, Li X, Xiong H, Li X, et al. Taumediated iron export prevents ferroptotic damage after ischemic stroke. Mol Psychiatry 2017;22:1520-30. [DOI] [PubMed] [Google Scholar]
- 24.Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell 2017;171:273-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 2017;547:453-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abid A, Taha O, Nseir W, Farah R, Grosovski M, Assy N. Soft drink consumption is associated with fatty liver disease independent of metabolic syndrome. J Hepatol 2009;51:918–24. [DOI] [PubMed] [Google Scholar]
- 27.Assy N, Nasser G, Kamayse I, Nseir W, Beniashvili Z, Djibre A, et al. Soft drink consumption linked with fatty liver in the absence of traditional risk factors. Can J Gastroenterol 2008;22:811-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Neuschwander-Tetri BA. Carbohydrate intake and nonalcoholic fatty liver disease. Curr Opin Clin Nutr Metabol Care 2013;16: 446–52. [DOI] [PubMed] [Google Scholar]
- 29.Radhakrishnan S, Ke J-Y, Pellizzon MA. Targeted nutrient modifications in purified diets differentially affect nonalcoholic fatty liver disease and metabolic disease development in rodent models. Curr Dev Nutr 2020;4:nzaa078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sferra R, Pompili S, Cappariello A, Gaudio E, Latella G, Vetuschi A. Prolonged chronic consumption of a high fat with sucrose diet alters the morphology of the small intestine. Int J Mol Sci 2021;22:7280–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Velazquez KT, Enos RT, Bader JE, Sougiannis AT, Carson MS, Chatzistamou I, et al. Prolonged high-fat-diet feeding promotes non-alcoholic fatty liver disease and alters gut microbiota in mice. World J Hepatol 2019;11:619–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tsurusaki S, Tsuchiya Y, Koumura T, Nakasone M, Sakamoto T, Matsuoka M, et al. Hepatic ferroptosis plays an important role as the trigger for initiating inflammation in nonalcoholic steatohepatitis. Cell Death Dis 2019;10:1-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Clapper JR, Hendricks MD, Gu G, Wittmer C, Dolman CS, Herich J, et al. Diet-induced mouse model of fatty liver disease and nonalcoholic steatohepatitis reflecting clinical disease progression and methods of assessment. Am J Physiol Gastrointest Liver Physiol 2013;305:483–95. [DOI] [PubMed] [Google Scholar]
- 34.Hansen HH, Feigh M, Veidal SS, Rigbolt KT, Vrang N, Fosgerau K. Mouse models of nonalcoholic steatohepatitis in preclinical drug development. Drug Discov Today 2017;22:1707-18. [DOI] [PubMed] [Google Scholar]
- 35.Kristiansen MNB, Veidal SS, Rigbolt KTG, Tolbol KS, Roth JD, Jelsing J, et al. Obese diet-induced mouse models of nonalcoholic steatohepatitis-tracking disease by liver biopsy. World J Hepatol 2016;8:673–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mells JE, Fu PP, Kumar P, Smith T, Karpen SJ, Anania FA. Saturated fat and cholesterol are critical to inducing murine metabolic syndrome with robust nonalcoholic steatohepatitis. J Nutr Biochem 2015;26:285-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tetri LH, Basaranoglu M, Brunt EM, Yerian LM, Neuschwander-Tetri BA. Severe NAFLD with hepatic necroinflammatory changes in mice fed trans fats and a highfructose corn syrup equivalent. Am J Physiol Gastrointest Liver Physiol 2008;295:987-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kleiner DE, Brunt EM, Natta M Van, Behling C, Contos MJ, Cummings OW, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005;41:1313–21. [DOI] [PubMed] [Google Scholar]
- 39.Bedossa P, Poitou C, Veyrie N, Bouillot J-LL, Basdevant A, Paradis V, et al. Histopathological algorithm and scoring system for evaluation of liver lesions in morbidly obese patients. Hepatology 2012;56:1751–9. [DOI] [PubMed] [Google Scholar]
- 40.Gorgoulis V, Adams PD, Alimonti A, Bennett DC, Bischof O, Bishop C, et al. Cellular senescence: Defining a path forward. Cell 2019;179:813-27. [DOI] [PubMed] [Google Scholar]
- 41.Kirkland JL, Tchkonia T. Senolytic drugs: from discovery to translation. J Intern Med 2020;288:518-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Qi J, Kim JW, Zhou Z, Lim CW, Kim B. Ferroptosis affects the progression of nonalcoholic steatohepatitis via the modulation of lipid peroxidation-mediated cell death in mice. Am J Pathol 2020;190:68-81. [DOI] [PubMed] [Google Scholar]