

Abstract
Noncoding RNAs are important for the regulation of cardiac hypertrophy. The function of MALAT1 (a long noncoding mRNA), miR-181a, and HMGB2, their contribution to cardiac hypertrophy, and the regulatory relationship between them during this process remain unknown. In the present study, we treated primary cardiomyocytes with angiotensin II (Ang II) to mimic cardiac hypertrophy. MALAT1 expression was significantly downregulated in Ang II-treated cardiomyocytes compared with control cardiomyocytes. Ang II-induced cardiac hypertrophy was suppressed by overexpression of MALAT1 and promoted by genetic knockdown of MALAT1. A dual-luciferase reporter assay demonstrated that MALAT1 acted as a sponge for miR-181a and inhibited its expression during cardiac hypertrophy. Cardiac hypertrophy was suppressed by overexpression of an miR-181a inhibitor and enhanced by overexpression of an miR-181a mimic. HMGB2 was downregulated during cardiac hypertrophy and was identified as a target of miR-181a by bioinformatics analysis and a dual-luciferase reporter assay. miR-181a overexpression decreased the mRNA and protein levels of HMGB2. Rescue experiments indicated that MALAT1 overexpression reversed the effect of miR-181a on HMGB2 expression. In summary, the results of the present study show that MALAT1 acts as a sponge for miR-181a and thereby regulates expression of HMGB2 and development of cardiac hypertrophy. The novel MALAT1/miR-181a/HMGB2 axis might play a crucial role in cardiac hypertrophy and serve as a new therapeutic target.
Key words: Hypertrophy, cardiomyocytes, MALAT1, miR-181a, HMGB2
Introduction
Cardiovascular disease has a high incidence worldwide, and some severe cardiovascular diseases are more dangerous than cancer. 1-3 Cardiac hypertrophy is an important indicator for judging the pathological process and occurs during ischemic heart disease, hypertension, and heart failure.4,5 The pathological changes of cardiac hypertrophy include an increase in the size of cardiomyocytes, myocardial interstitial cell proliferation, and cardiac extracellular matrix remodeling.6 The responses of myocytes to various pathophysiological insults, such as angiotensin II (Ang II), and hypertrophy increase myocardial oxygen consumption and reduce myocardial compliance, leading to heart failure and an increase in the incidence of sudden death.7 Reversal of myocardial hypertrophy has become an important goal for treatment of hypertension and chronic congestive heart failure.8
In mammalian genomes, 98% of transcripts are noncoding RNAs.9 Long non-coding RNAs (lncRNAs) are a type of non-coding RNAs longer than 200 nucleotides and are closely related to cardiovascular diseases. For example, lncRNAs are involved in regulation of endothelial cell function,10 myocardial infarction and heart failure,11 and cardiomyocyte apoptosis.12 They also play an important role in regulation of cardiac hypertrophy.12-14 The lncRNA MALAT1 is upregulated during the regulation of endothelial cell function and vessel growth;15 however, its role in cardiac hypertrophy remains unknown.
miRNAs are small non-coding RNAs containing 20-25 nucleotides that regulate gene expression by inhibiting translation or inducing degradation of mRNAs, mainly by interacting with the 3’ untranslated region (3’UTR) of target genes, including coding and non-coding genes.16 They are involved in various cellular processes, including proliferation, differentiation, survival, growth, and apoptosis,17-19 and in regulation of a variety processes related to heart-related diseases, such as angiogenesis, autophagy, and fibrosis.20,21 Gain- and loss-of-function approaches have shown that miRNAs play a critical role in cardiac hypertrophy and ventricular remodeling.22 miR-181a is involved in cardiac hypertrophy23-25 but its role must be further clarified.
High mobility group protein (HMG) is a nonhistone DNAbinding protein that is widely distributed in nuclei of higher eukaryotes. HMGB can be divided into HMGB1, HMGB2, and HMGB3.26 HMGB2 can bind to a variety of transcription factors, such as steroid hormone receptor, p53, p73, LEF1, and Runx2, and enhance the transcription and recombination activity of its chaperone protein.27,28 It is also involved in the regulation of various differentiation processes, including production of red blood cells, sperm, nerve cells, and muscle cells.29-31 HMGB2 is closely related to cardiac hypertrophy. Quantitative chromatin proteomics showed that HMGB2, but not HMGB1, is a regulator of cardiac hypertrophy. 32 HMGB2 can regulate gene expression by controlling chromosome structure remodeling to mediate cardiac hypertrophy.33 However, it remains to be determined how HMGB2 is modulated by upstream signals, especially during cardiac hypertrophy.
In the current study, we investigated the expression levels of MALAT1, miR-181a, and HMGB2 in an Ang II-induced cell model of cardiac hypertrophy. We explored the regulatory relationship between MALAT1, miR-181a, and HMGB2, and sought to elucidate the important role of the MALAT1/miR-181a/HMGB2 axis in progression of cardiac hypertrophy.
Materials and Methods
Animals and cell culture
All animal procedures were approved by the Animal Research Committee of Ganzhou People’s Hospital [approval No. SJTY(E) 2018-067] and performed according to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH).
Sprague-Dawley rats were purchased from the Laboratory Animal Center of Nanchang University. Cardiomyocytes were isolated from the hearts of newborn Sprague-Dawley rats (1 day old). Briefly, the ventricles were excised and transferred to Hank’s solution (Invitrogen, Thermo Fisher Scientific, Inc., Waltham, MA, USA). Thereafter, cells were isolated and cultured in Dulbecco’s Modified Eagle’s Medium (Gibco, Thermo Fisher Scientific, Inc.) containing 10% fetal bovine serum (Gibco) and 1% penicillinstreptomycin (Gibco) at 37°C with 5% CO2 in humidified air. Cardiomyocytes were cultured under semiconfluent conditions and transferred to 6-well plates at a density of 2 × 105 cells/ml. H9c2 cells, a rat cardiomyoblast cell line, were obtained from the Chinese Academy of Sciences Cell Bank (Shanghai, China) and cultured in Dulbecco’s Modified Eagle’s Medium (Gibco) containing 10% fetal bovine serum (Gibco) and 1% penicillin-streptomycin (Gibco). For experiments, cells were transferred to 6- or 96- well plates at 60–75% confluency and a density of 1 × 105 cells/mL. After 24 h of culture, cells were transfected with the indicated fragments or plasmids using Lipofectamine 3000 (Invitrogen). After 24 h of transfection, cells were treated with 1 μM Ang II (Sigma-Aldrich, CA, USA) for additional 48 h as reported, 34,35 to generate a cell model of cardiac hypertrophy.
RT-qPCR
Total RNA was extracted from cardiomyocytes or H9c2 cells using TRIzol reagent (Invitrogen), according to the manufacturer’s instructions. EasyScript cDNA Synthesis SuperMix (TransGen Biotech, Beijing, China) was used to synthesize cDNA from mRNA, and TransScript® miRNA First-Strand cDNA Synthesis SuperMix (TransGen Biotech) was used to synthesize cDNA from miRNA. Quantitative PCR (qPCR) was performed using TransStart Top Green qPCR SuperMix (TransGen Biotech). mRNA levels were normalized to that of actin, and miRNA levels were normalized to that of U6, using a delta-delta-Ct method as previously described.36,37 All experiments were independently performed in triplicate. The following primers were used: 5’-CTCACTAAAGGCACCGAAGG- 3’ and 5’-GGCAGAGAAGTTGCTTGTGG- 3’ (MALAT1); 5’-CTTCGGGGGTAGGATTGAC- 3’ and 5’-CTTGGGATCTTTTGCGATCT-3’ (ANP); 5’-TGATTCTGCTCCTGCTTTTCC-3’ and 5’-TTCTTTTGTAGGGCCTTGGTC- 3’ (BNP); 5’-CGCCTGTCAGCTTGTAAATG- 3’ and 5’-ACAACCCCTACGATTATGCG-3’ (β- MHC); 5’-TCCTGGTAGGCCAACAGGCT-3’ and 5’-AGCTAATGTTGAGCTGCACTTG- 3’ (HMGB2); 5’-CCGCATCTTCTTGTGCAGTG- 3’ and 5’-GAGAAGGCAGCCCTGGTAAC- 3’ (GAPDH); 5’-GGGAACATTCAACGCTGTCG-3’ and 5’-GTGCGTGTCGTGGAGTCG-3’ (miR-181a); and 5’- CTCGCTTCGGCAGCACA-3’ and 5’-AACGCTTCACGAATTTGCGT- 3’ (U6).
RNA interference and overexpression
For the MALAT1 overexpression assay, cDNA encoding MALAT1 was cloned into the pEGFP-C1 vector (BD Biosciences Clontech, USA) and verified by sequencing. For genetic knockdown of MALAT1, the following siRNA fragments were synthesized as previously reported38 by GenePharma (Shanghai, China): siMALAT1, 5’-AAGAAAAAUAAAAGCUUUCCU-3’ and negative control (NC), 5’-ACGUCACACGUUCGGAGAATT-3’. For miR-181a modulation, a rno-miR-181a mimic and inhibitor were synthesized by GenePharma.39 A NC mimic and inhibitor were used as controls. The efficiency was determined by RT-qPCR. All experiments were independently performed in triplicate.
Western blotting
Western blotting was performed as previously reported.40 Samples were harvested and lysed with RIPA lysis buffer (Beyotime, Shanghai, China) containing protease inhibitors. The lysates were separated by 10% SDS-PAGE (Beyotime), and proteins were transferred to a PVDF membrane (Millipore, Burlington, MA, USA). Membranes were blocked with 5% nonfat milk for 1 h and incubated with primary antibodies against ANP (ab180649, 1:500), BNP (ab19645, 1:500), β-MHC (ab170867, 1:500), and HMGB2 (ab172967, 1:1000) (all from Abcam, Cambridge, UK). Membranes were incubated with goat antimouse (1:5000) or antirabbit (1:5000) secondary antibodies (Abclonal Biotechnology, Wuhan, China) for 1 h at room temperature, and protein signals were detected with enhanced chemiluminescence (Beyotime). All experiments were independently performed in triplicate.
Bioinformatics analysis
Targetscan and starBase were used to predict potential binding interactions between MALAT1, miR-181a, and HMGB2. With starBase (http://starbase.sysu.edu.cn/index.php), ‘miRNA-mRNA’ and ‘miRNA-lncRNA’ analysis was performed using MALAT1 and miRNA (miR-181a) as the input and HMGB2 as the target. Significant interactions were found between miR-181a and HMGB2 in multiple databases, such as RNA22, miRmap, PicTar (https://pictar.mdc-berlin.de/), and TargetScan. The results were verified using TargetScan (http://www.targetscan.org/vert_72/) with the human gene symbol ‘HMGB2’ as the input, and the matching sequences were shown.
Luciferase reporter assay
The pGL3-Control vector (Promega, Madison, WI, USA) was used to generate the luciferase reporter construct. A luciferase reporter containing the MALAT1 or HMGB2 3’UTR was inserted into the pGL3-luc vector. The mutant (MUT) nucleotides were synthesized by GenePharm. H9c2 cells were plated in 6-well plates (2.5 × 105 cells/well) and transfected with the miR-181a mimic (50 nM) or NC mimic (50 nM) and the wild-type (WT) or MUT MALAT1 or HMGB2 reporter plasmid using Lipofectamine 3000 (Invitrogen). After 48 h of transfection, luciferase activity was detected using a BioTek Synergy 2 luminometer (BioTek Instruments Inc., Beijing, China), and firefly luciferase activity was normalized against Renilla luciferase activity, according to the manufacturer’s instructions. All experiments were independently performed in triplicate.
Immunofluorescence
Cardiomyocytes were cultured on coverslips, treated with or without Ang II for 48 h, fixed with 4% paraformaldehyde (Sigma- Aldrich, St. Louis, MO, USA) for 40 min at 4°C, permeabilized with phosphate-buffered saline containing 0.1% Triton X-100, blocked with 3% appropriate serum, incubated with primary antibodies against actin (Abcam, ab137346, 1:1000) overnight at 4°C, and then labeled with secondary antibodies conjugated with Alexa Fluor 488 (Thermo Fisher Scientific, Inc.) or Alexa Fluor 555 (Thermo Fisher Scientific, Inc.) at room temperature for 2 h. Cells incubated with no primary antibodies were used as negative control Cells were mounted in Fluro-Gel II containing DAPI (Electron Microscopy Sciences, Hatfield, PA, USA), and images were captured with a Carl Zeiss LSM 780 confocal microscope (Zeiss, Jean, Germany). The surface area was calculated using ImageJ software (NIH, v. 1.43; http://rsb.info.nih.gov/ij/).
Statistical analysis
All data are presented as the mean ± standard deviation. Statistical analyses were performed with SPSS software (version 17.0; SPSS Inc., Chicago, IL, USA). More than two groups were compared using an analysis of variance followed by Tukey’s posthoc test, and two groups were compared using Student’s t-test; p<0.05 was considered statistically significant. All experiments were independently performed in triplicate.
Results
MALAT1 is involved in regulation of cardiac hypertrophy
To elucidate the detailed molecular mechanism underlying cardiac hypertrophy, an in vitro model was established by treating primary cardiomyocytes with Ang II. Immunofluorescence staining of actin indicated that Ang II treatment significantly increased the size of cardiomyocytes (Figure 1A). This was confirmed by measurement of cell surface areas (Figure 1B). The mRNA expression levels of ANP, BNP, and β-MHC determined by RT-qPCR, and their protein expression levels determined by Western blotting, were markedly upregulated in hypertrophic cardiomyocytes compared with control cells (Figure 1 C,D). These data suggest that a cell model of cardiac hypertrophy was successfully established. RT-qPCR demonstrated that MALAT1 was significantly downregulated upon Ang II treatment (Figure 2A). To further elucidate the effect of MALAT1 during cardiac hypertrophy, we constructed a MALAT1-encoding plasmid and synthesized siRNA fragments against MALAT1. The level of MALAT1 in cardiomyocytes was sharply increased upon overexpression of MALAT1 and decreased upon genetic knockdown of MALAT1 (Figure 2B). Ang II treatment markedly increased the cell surface area, but this effect was abolished by overexpression of MALAT1 (Figure 2 C,E) and enhanced by knockdown of MALAT1 (Figure 2 D,F). These data indicate that MALAT1 protects against cardiac hypertrophy.
miR-181a promotes development of cardiac hypertrophy
MALAT1 reportedly regulates the downstream target miR- 181a in a variety of diseases and physiological processes, such as M1 polarization of macrophages,41 liver fibrosis,42 proliferation of myeloma cells,43 and gastric adenocarcinoma.44 However, the relationship between MALAT1 and miR-181a during cardiac hypertrophy remains unknown. To investigate this, we first determined expression of miR-181a in our model. RT-qPCR showed that miR- 181a was significantly upregulated in Ang II-treated cells compared with control cells (Figure 3A). We synthesized an miR-181a mimic and inhibitor. A NC mimic and inhibitor were used as controls. RT-qPCR showed that the level of miR-181a was significantly upregulated by transfection of the miR-181a mimic and downregulated by transfection of the miR-181a inhibitor compared with the NC mimic or inhibitor (Figure 3B). Transfection of the miR- 181a inhibitor markedly suppressed Ang II-induced hypertrophy and significantly attenuated the increase in the cell surface area (Figure 3 C,E). Transfection of the miR-181a mimic further increased the cell surface area upon Ang II treatment (Figure 3 D,F). These data suggest that miR-181a promotes cardiac hypertrophy, as previously reported.23
MALAT1 acts as a sponge for miR-181a to regulate cardiac hypertrophy
We next investigated whether MALAT1 regulated miR-181a during cardiac hypertrophy. According to bioinformatics analysis, there is a matching sequence with rno-miR-181a in the 3’UTR region of MALAT1 (Figure 4A). We thus constructed luciferase reporter plasmids containing the WT and MUT 3’UTR of MALAT1. These constructs were transfected into H9c2 cells together with the miR-181a or NC mimic. The miR-181a mimic significantly suppressed the luciferase signals from MALAT1 WT, but not those from MALAT1 MUT (Figure 4B). Furthermore, expression of MALAT1 was investigated in cultured primary rat cardiomyocytes transfected with the miR-181a inhibitor or mimic. Expression of MALAT1 was significantly upregulated by transfection of the miR-181a inhibitor and downregulated by transfection of the miR-181a mimic (Figure 4C), indicating that MALAT1 acts as a sponge for miR-181a in cardiac cells. Moreover, cells transfected with MALAT1 were cotransfected with or without the miR- 181a mimic and treated with Ang II to induce hypertrophy. MALAT1 overexpression prevented Ang II-induced hypertrophy, but this effect was reversed by cotransfection of the miR-181a mimic (Figure 4D). These data indicate that MALAT1 acts as a sponge for and thereby inhibits miR-181a, and thus regulates cardiac hypertrophy.
HMGB2 is a target of miR-181a during cardiac hypertrophy
Next, we explored the underlying effector of MALAT1/miR- 181a during cardiac hypertrophy. Using bioinformatics tools, we screened all the reported and potential downstream targets, and HMGB2 attracted our attention because of its role during cardiac hypertrophy. Protein expression of HMGB2 was significantly downregulated in Ang II-treated cells (Figure 5 A,B). Bioinformatics analysis showed there is a matching sequence for miR-181a in the 3’UTR of HMGB2 (Figure 5C). The luciferase assay showed that transfection of miR-181a markedly suppressed signals of HMGB2 WT, but not of HMGB2 MUT, in H9c2 cells (Figure 5B). Furthermore, RT-qPCR showed that the mRNA and protein levels of HMGB2 significantly decreased by transfection of the miR-181a mimic and increased by transfection of the miR- 181a inhibitor (Figure 5 E,F). These data indicate that HMGB2 is a direct target of miR-181a during cardiac hypertrophy.
MALAT1/miR-181a axis regulates cardiac hypertrophy by modulating expression of HMGB2
We next investigated whether the MALAT1/miR-181a axis regulates HMGB2 expression. Transfection of the miR-181a mimic significantly downregulated luciferase activity of HMGB2, but this effect was reversed by co-transfection of MALAT1 (Figure 6A). Analysis of the mRNA (Figure 6B) and protein (Figure 6C) levels of HMGB2 yielded similar findings. MALAT1 rescued suppression of HMGB2 expression by miR-181a. Rescue experiments were performed to elucidate the role of the MALAT1/miR-181a axis in cardiac hypertrophy. When cells were treated with Ang II, transfection of miR-181a significantly enhanced the cell surface area, but this effect was abrogated by overexpression of MALAT1 (Figure 6D). Taken together, these data suggest that the MALAT1/miR-181a axis regulates expression of HMGB2 and thereby modulates cardiac hypertrophy.
Figure 1.
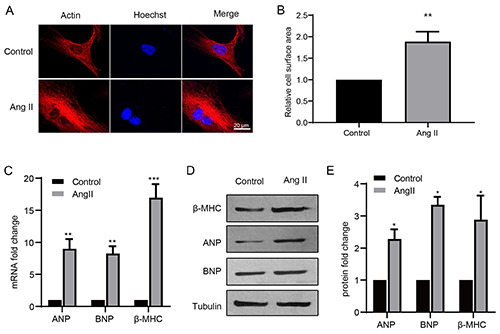
The establishment of cardiac hypertrophy model. A) Cultured primary cardiomyocytes were treated with Ang II or control, then the cells were subjected to immunocytochemistry with actin antibody. The images captured by LSM 780 confocal microscope and were analyzed and the relative cell surface area was measured to indicate hypertrophy response (B). The cells were subjected to RT-qPCR to detect the mRNA level (C) and the protein levels of ANP, BNP and β-MHC. *p<0.05; **p<0.01; ***p<0.001; repeated three times; scale bar: 20 μm.
Discussion
In this study, we demonstrated that the expression levels of MALAT1 and HMGB2 were downregulated in Ang II-treated cardiomyocytes, while that of miR-181a was upregulated. Ang IIinduced cardiac hypertrophy was suppressed by overexpression of MALAT1 and was enhanced by genetic knockdown of MALAT1. Conversely, Ang II-induced cardiac hypertrophy was enhanced by overexpression of miR-181a and was suppressed by inhibition of miR-181a. MALAT1 was identified as a sponge for miR-181a, and HMGB2 was identified as a direct target of miR-181a. MALAT1/miR-181a regulates cardiac hypertrophy by modulating HMGB2 expression. This novel molecular mechanism by which MALAT1/miR-181a/HMGB2 regulate cardiac hypertrophy is summarized in Figure 7.
Figure 2.
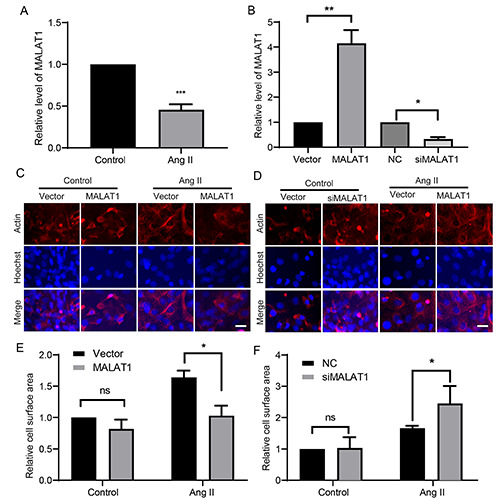
MALAT1 is important for cardiac hypertrophy. A) Cells were treated as in figure 1. The cells were detected with RT-qPCR for the level of MALAT1. B) The levels of MALAT1 were verified. Cell were transfected with MALAT1 overexpression plasmids (C-E) or genetic knockdown siRNA fragments (D-F), or the controls, then the representative images were shown and the relative cell surface area was determined. *p<0.05; **p<0.01; ***p<0.001; ns, no significance; repeated three times; scale bar: 20 μm.
LncRNAs play an important role in transcription and post-transcriptional regulation of gene expression, and are a key node for regulating transcriptional activity and mRNA expression of target genes. MALAT1 is reportedly involved in regulation of various heart functions.45 It regulates the function of endothelial cells, and expression of MALAT1, MEG3, and TUG1 increases upon hypoxia stimulation.46 Inhibition of MALAT1 suppresses proliferation but increases basal sprouting and migration of endothelial cells in vitro and in vivo.15 Furthermore, inhibition of MALAT1 significantly changes the expression of a variety of critical genes related to cell cycle regulation, with downregulated expression of CCNA2, CCNB1, CCNB2, and CDK-115 MALAT1 also regulates proliferation of fibroblasts, and inhibition of MALAT1 induces splicing of the cancer-related transcription factor B-Myb.47 However, the role of MALAT1 during hypertrophy remains unclear. MALAT1 is reportedly a dispensable regulator of cardiac hypertrophy and failure during cardiac pressure overload induced by thoracic aortic constriction.48 In diabetic cardiomyopathy, MALAT1 recruits the histone methyltransferase EZH2 to suppress expression of miR-22, and thereby mediates myocardial damage and cardiomyocyte apoptosis.49 In the current study, we investigated the role of MALAT1 in Ang II-induced cardiomyocyte hypertrophy. The expression level of MALAT1 was significantly downregulated in our model. Cardiac hypertrophy was promoted by knockdown of MALAT1 and suppressed by overexpression of MALAT1. MALAT1 acted as a sponge for miR-181a, and thereby suppressed its expression and finally regulated the level of HMGB2 to execute its function.
Figure 3.
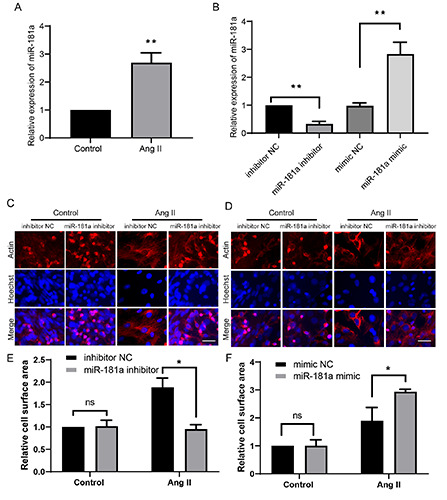
The effect of miR-181a in cardiac hypertrophy. A) Cells were treated as in figure 1. The miR-181a level was detected by RTqPCR. (B) The efficiency of miR-181a inhibitor or mimic was determined by RT-qPCR in H9c2 cells. Cells were transfected with miR- 181a inhibitor (C-E) or mimic (D-F), then the representative images were shown and the relative cell surface area was measured. *p<0.05; **p<0.01; ns, no significance; repeated three times.
miR-181a reportedly plays a role in cardiac hypertrophy. It is highly expressed in bone marrow-derived mesenchymal stem cells and regulates glucagon secretion via the PTEN/AKT signaling pathway.50 rno-miR-181a is downregulated in exosomes of induced pluripotent stem cell-derived cardiomyocytes, and this attenuates myocardial fibrosis and hypertrophy.51 In a diabetesinduced cardiac hypertrophy model, miR-30a and miR-181a synergistically function via modulating the p53-p21 pathway. The expression level of miR-181a is decreased in diabetic patients, rats with diabetic cardiomyopathy, and high glucose-treated cardiomyocytes, and overexpression of miR-181a decreases p53, p21, and ANP expression levels and apoptosis in high glucose-induced cardiomyocytes.52 In rats with myocardial hypertrophy, expression of miR-181a is downregulated to regulate autophagy. Moreover, expression of MALAT1 is increased and miR-181a downregulated during high glucose-induced cardiomyocyte apoptosis, and MALAT1 functions by acting as a sponge for miR-181a.25 Using cultured primary rat cardiac cardiomyocytes, we found that miR- 181a was markedly upregulated in Ang II-treated cells, and inhibition of miR-181a suppressed Ang II-induced hypertrophy. MALAT1 was identified as a sponge for miR-181a and thereby regulated cardiac hypertrophy. Our results differ from those of the aforementioned reports. This may be because different models (e.g., high glucose and Ang II treatment) or sources of cells were used. An in vivo model in rats or even human samples should be used to further elucidate the detailed mechanism.
Figure 4.
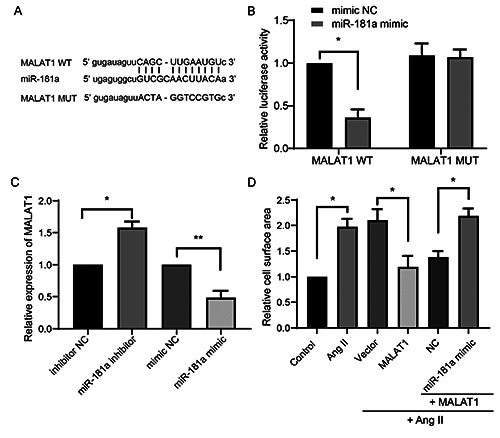
MALAT1 targets miR-181a in cardiac hypertrophy. A) The bioinformatics analysis between the 3’ UTR of MALAT1 against miR-181a and the construction of MALAT1 MUT was shown. B) H9c2 cells were transfected with MALAT1 WT or MUT luciferase reporter plasmids, with or without miR-181a mimic. Then the relative luciferase activity was determined. C) The levels of MALT1 in primary cardiomyocytes were determined upon the transfection of miR-181a inhibitor or mimic. D) Cardiomyocytes were transfected with the indicated plasmids or fragments, then were subjected with Ang II treatment. The relative cell surface area was determined. *p<0.05; **p<0.01; repeated three times.
HMGB is present in almost every cell type and is essential for development. Indeed, HMGB1-deficient mice die soon after birth.53 Cardiac hypertrophy is the gradual thickening of the myocardium caused by hypertrophy of myocardial cells, and HMGB proteins play an important role in this process. In animal models of cardiac hypertrophy induced by transverse aortic constriction, HMGB1 is upregulated in infiltrating cells and cardiomyocytes. 54,55 Maintenance of stable intracellular HMGB1 levels can prevent cardiac hypertrophy.56 Extracellular recombinant HMGB1 mediates pressure overload-induced cardiac hypertrophy and heart failure. Thus, HMGB proteins have dual functions in regulation of cardiac hypertrophy. HMGB2 is involved in cardiac hypertrophy. Quantitative chromatin proteomics found that HMGB2, but not HMGB1, acts as a regulator of myocardial hypertrophy.57 HMGB2 regulates gene expression by modulating chromosome remodeling to mediate cardiac hypertrophy.33 In HMGB2-knockout mice (Hmgb2-/-), the AKT and endoplasmic reticulum calcium-ATP regulatory pathways are inhibited, which results in severe myocardial dysfunction, indicating that HMGB2 has an important role in maintaining normal heart function.58 Our results showed that HMGB2 was downregulated during cardiac hypertrophy. HMGB2 was a downstream target of MALAT1/miR-181a, and regulation of its expression level played a critical role in modulating cardiac hypertrophy.
Figure 5.
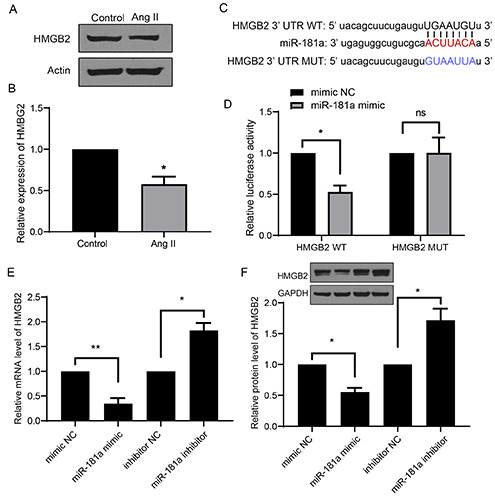
HMGB2 is a target of miR-181a in cardiac hypertrophy. A,B) The expression level of HMGB2 was show in cardiac hypertrophy model. C) The bioinformatics analysis between the 3’ UTR of HMGB2 and miR-181a was shown. D) The luciferase activity from HMGB2 WT or MUT transfected with or without miR-181a mimic was shown. E,F) The mRNA and protein levels of HMGB2 were shown. *p<0.05; **p<0.01; repeated three times.
Figure 6.
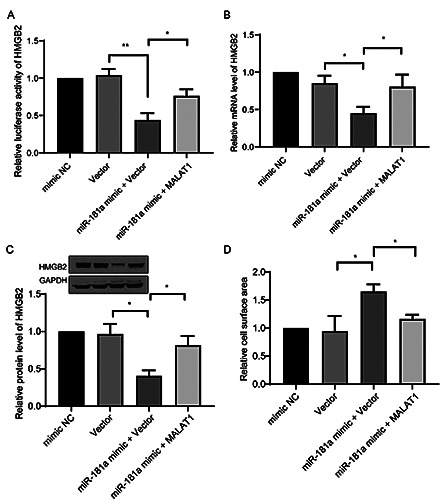
MALAT1/miR-181a axis regulate HMGB2 in cardiac hypertrophy. A) The luciferase reporter of HMGB2 WT were co-transfected with indicated fragments or MALT1 encoding plasmids; then the relative luciferase activity of HMGB2 was determined. The mRNA level (B) and the protein level (C) of HMGB2, and the relative cell surface area (D) were determined in cells same treated in (A). *p<0.05; **p<0.01; repeated three times.
Figure 7.
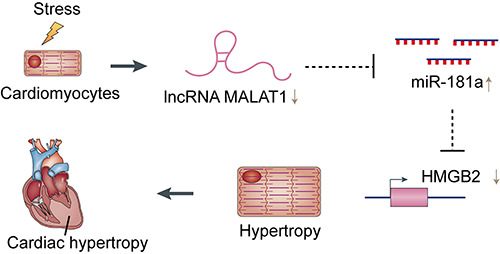
The schematic diagram of regulatory mechanism of MALAT1/miR-181a/HMGB2 in cardiac hypertrophy.
In summary, MALAT1 participates in regulation of cardiac cell function by acting as a sponge for miR-181a, leading to downregulation of miR-181a and upregulation of HMGB2. This study identifies a novel regulatory mechanism by which MALAT1/miR- 181a/HMGB2 regulate cardiac hypertrophy. These findings could provide new ideas and intervention targets for clinical treatment and prevention of cardiac hypertrophy.
Funding Statement
Funding: This work was supported by the Science and Technology Project of Jiangxi Province (No. 20203BBGL73188) and the Natural Science Foundation of Jiangxi, China (No. 20212ACB206031).
References
- 1.Yurista SR, Chong CR, Badimon JJ, Kelly DP, de Boer RA, Westenbrink BD. Therapeutic potential of ketone bodies for patients with cardiovascular disease: JACC state-of-the-art review. J Am Coll Cardiol 2021;77:1660-9. [DOI] [PubMed] [Google Scholar]
- 2.Tada H, Fujino N, Hayashi K, Kawashiri MA, Takamura M. Human genetics and its impact on cardiovascular disease. J Cardiol 2022;79:233-9. [DOI] [PubMed] [Google Scholar]
- 3.Zhang Y, Xin L, Xiang M, Shang C, Wang Y, Wang Y, et al. The molecular mechanisms of ferroptosis and its role in cardiovascular disease. Biomed Pharmacother 2022;145:112423. [DOI] [PubMed] [Google Scholar]
- 4.Mishra S, Kass DA. Cellular and molecular pathobiology of heart failure with preserved ejection fraction. Nat Rev Cardiol 2021;18:735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lei H, Hu J, Sun K, Xu D. The role and molecular mechanism of epigenetics in cardiac hypertrophy. Heart Fail Rev 2021;26:1505-14. [DOI] [PubMed] [Google Scholar]
- 6.Ramachandra CJA, Cong S, Chan X, Yap EP, Yu F, Hausenloy DJ. Oxidative stress in cardiac hypertrophy: From molecular mechanisms to novel therapeutic targets. Free Radic Biol Med 2021;166:297-312. [DOI] [PubMed] [Google Scholar]
- 7.Liu CF, Tang WHW. Epigenetics in Cardiac hypertrophy and heart failure. JACC Basic Transl Sci 2019;4:976-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhu L, Li C, Liu Q, Xu W, Zhou X. Molecular biomarkers in cardiac hypertrophy. J Cell Mol Med 2019;23:1671-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.He J, Luo Y, Song J, Tan T, Zhu H. Non-coding RNAs and pathological cardiac hypertrophy. Adv Exp Med Biol 2020;1229:231-45. [DOI] [PubMed] [Google Scholar]
- 10.Qiu GZ, Tian W, Fu HT, Li CP, Liu B. Long noncoding RNAMEG3 is involved in diabetes mellitus-related microvascular dysfunction. Biochem Biophys Res Commun 2016;471:135-41. [DOI] [PubMed] [Google Scholar]
- 11.Li Y, Liang Y, Zhu Y, Zhang Y, Bei Y. Noncoding RNAs in cardiac hypertrophy. J Cardiovasc Transl Res 2018;11:439-49. [DOI] [PubMed] [Google Scholar]
- 12.Greco CM, Condorelli G. Epigenetic modifications and noncoding RNAs in cardiac hypertrophy and failure. Nat Rev Cardiol 2015;12:488-97. [DOI] [PubMed] [Google Scholar]
- 13.Sun J, Wang C. Long non-coding RNAs in cardiac hypertrophy. Heart Fail Rev 2020;25:1037-45. [DOI] [PubMed] [Google Scholar]
- 14.Zhang J, Feng C, Song C, Ai B, Bai X, Liu Y, et al. Identification and analysis of a key long non-coding RNAs (lncRNAs)-associated module reveal functional lncRNAs in cardiac hypertrophy. J Cell Mol Med 2018;22:892-903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Michalik KM, You X, Manavski Y, Doddaballapur A, Zornig M, Braun T, et al. Long noncoding RNA MALAT1 regulates endothelial cell function and vessel growth. Circ Res 2014;114:1389-97. [DOI] [PubMed] [Google Scholar]
- 16.Scolari FL, Faganello LS, Garbin HI, Piva EMB, Biolo A. A systematic review of microRNAs in patients with hypertrophic cardiomyopathy. Int J Cardiol 2021;327:146-54. [DOI] [PubMed] [Google Scholar]
- 17.Gebert LFR, MacRae IJ. Regulation of microRNA function in animals. Nat Rev Mol Cell Biol 2019;20:21-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mens MMJ, Ghanbari M. Cell cycle regulation of stem cells by MicroRNAs. Stem Cell Rev Rep 2018;14:309-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Y, Blelloch R. Cell cycle regulation by MicroRNAs in embryonic stem cells. Cancer Res 2009;69:4093-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fazmin IT, Achercouk Z, Edling CE, Said A, Jeevaratnam K. Circulating microRNA as a biomarker for coronary artery disease. Biomolecules 2020;10:1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Omidkhoda N, Wallace Hayes A, Reiter RJ, Karimi G. The role of MicroRNAs on endoplasmic reticulum stress in myocardial ischemia and cardiac hypertrophy. Pharmacol Res 2019;150:104516. [DOI] [PubMed] [Google Scholar]
- 22.Ooi JY, Bernardo BC, McMullen JR. The therapeutic potential of miRNAs regulated in settings of physiological cardiac hypertrophy. Future Med Chem 2014;6:205-22. [DOI] [PubMed] [Google Scholar]
- 23.Li AL, Lv JB, Gao L. MiR-181a mediates Ang II-induced myocardial hypertrophy by mediating autophagy. Eur Rev Med Pharmacol Sci 2017;21:5462-70. [DOI] [PubMed] [Google Scholar]
- 24.Garg A, Foinquinos A, Jung M, Janssen-Peters H, Biss S, Bauersachs J, et al. MiRNA-181a is a novel regulator of aldosterone- mineralocorticoid receptor-mediated cardiac remodelling. Eur J Heart Fail 2020;22:1366-77. [DOI] [PubMed] [Google Scholar]
- 25.Cheng Y, Li J, Wang C, Yang H, Wang Y, Zhan T, et al. Inhibition of long non-coding RNA metastasis-associated lung adenocarcinoma transcript 1 attenuates high glucose-induced cardiomyocyte apoptosis via regulation of miR-181a-5p. Exp Anim 2020;69:34-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Niu L, Yang W, Duan L, Wang X, Li Y, Xu C, et al. Biological functions and theranostic potential of HMGB family members in human cancers. Ther Adv Med Oncol 2020;12:1758835920970850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stros M, Ozaki T, Bacikova A, Kageyama H, Nakagawara A. HMGB1 and HMGB2 cell-specifically down-regulate the p53- and p73-dependent sequence-specific transactivation from the human Bax gene promoter. J Biol Chem 2002;277:7157-64. [DOI] [PubMed] [Google Scholar]
- 28.Taniguchi N, Carames B, Kawakami Y, Amendt BA, Komiya S, Lotz M. Chromatin protein HMGB2 regulates articular cartilage surface maintenance via beta-catenin pathway. Proc Natl Acad Sci U S A 2009;106:16817-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Laurent B, Randrianarison-Huetz V, Marechal V, Mayeux P, Dusanter-Fourt I, Dumenil D. High-mobility group protein HMGB2 regulates human erythroid differentiation through trans-activation of GFI1B transcription. Blood 2010;115:687-95. [DOI] [PubMed] [Google Scholar]
- 30.Ronfani L, Ferraguti M, Croci L, Ovitt CE, Scholer HR, Consalez GG, et al. Reduced fertility and spermatogenesis defects in mice lacking chromosomal protein Hmgb2. Development 2001;128:1265-73. [DOI] [PubMed] [Google Scholar]
- 31.Zhou X, Li M, Huang H, Chen K, Yuan Z, Zhang Y, et al. HMGB2 regulates satellite-cell-mediated skeletal muscle regeneration through IGF2BP2. J Cell Sci 2016;129:4305-16. [DOI] [PubMed] [Google Scholar]
- 32.Franklin S, Chen H, Mitchell-Jordan S, Ren S, Wang Y, Vondriska TM. Quantitative analysis of the chromatin proteome in disease reveals remodeling principles and identifies high mobility group protein B2 as a regulator of hypertrophic growth. Mol Cell Proteomics 2012;11:M111.014258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Monte E, Rosa-Garrido M, Karbassi E, Chen H, Lopez R, Rau CD, et al. reciprocal regulation of the cardiac epigenome by chromatin structural proteins Hmgb and Ctcf: Implications for transcriptional regulation. J Biol Chem 2016;291:15428-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nie X, Fan J, Li H, Yin Z, Zhao Y, Dai B, et al. miR-217 promotes cardiac hypertrophy and dysfunction by targeting PTEN. Mol Ther Nucleic Acids 2018;12:254-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gao Y, Zhao D, Xie WZ, Meng T, Xu C, Liu Y, et al. Rap1GAP mediates angiotensin II-induced cardiomyocyte hypertrophy by inhibiting autophagy and increasing oxidative stress. Oxid Med Cell Longev 2021;2021:7848027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Poltronieri C, Maccatrozzo L, Simontacchi C, Bertotto D, Funkenstein B, Patruno M, et al. Quantitative RT-PCR analysis and immunohistochemical localization of HSP70 in sea bass Dicentrarchus labrax exposed to transport stress. Eur J Histochem 2007;51:125-35. [PubMed] [Google Scholar]
- 37.Fede C, Albertin G, Petrelli L, Sfriso MM, Biz C, De Caro R, et al. Expression of the endocannabinoid receptors in human fascial tissue. Eur J Histochem 2016;60:2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu Z, Liu J, Wei Y, Xu J, Wang Z, Wang P, et al. LncRNA MALAT1 prevents the protective effects of miR-125b-5p against acute myocardial infarction through positive regulation of NLRC5. Exp Ther Med 2020;19:990-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao H, Zhang X, Zheng Y, Li Y, Wang X, Hu N, et al. Propofol protects rat cardiomyocytes from anthracyclineinduced apoptosis by regulating microRNA-181a in vitro and in vivo. Oxid Med Cell Longev 2018;2018:2109216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jiang C, Liu F, Xiao S, He L, Wu W, Zhao Q. miR-29a-3p enhances the radiosensitivity of oral squamous cell carcinoma cells by inhibiting ADAM12. Eur J Histochem 2021;65:3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu J, Niu Z, Zhang R, Peng Z, Wang L, Liu Z, et al. MALAT1 shuttled by extracellular vesicles promotes M1 polarization of macrophages to induce acute pancreatitis via miR-181a- 5p/HMGB1 axis. J Cell Mol Med 2021;25:9241-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang Y, Mou Q, Zhu Z, Zhao L, Zhu L. MALAT1 promotes liver fibrosis by sponging miR181a and activating TLR4NFkappaB signaling. Int J Mol Med 2021;48:215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sun Y, Jiang T, Jia Y, Zou J, Wang X, Gu W. LncRNA MALAT1/miR-181a-5p affects the proliferation and adhesion of myeloma cells via regulation of Hippo-YAP signaling pathway. Cell Cycle 2019;18:2509-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lu Z, Luo T, Pang T, Du Z, Yin X, Cui H, et al. MALAT1 promotes gastric adenocarcinoma through the MALAT1/miR- 181a-5p/AKT3 axis. Open Biol 2019;9:190095. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 45.Puthanveetil P, Gutschner T, Lorenzen J. MALAT1: a therapeutic candidate for a broad spectrum of vascular and cardiorenal complications. Hypertens Res 2020;43:372-9. [DOI] [PubMed] [Google Scholar]
- 46.Zhang X, Li DY, Reilly MP. Long intergenic noncoding RNAs in cardiovascular diseases: Challenges and strategies for physiological studies and translation. Atherosclerosis 2019;281:180-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tripathi V, Shen Z, Chakraborty A, Giri S, Freier SM, Wu X, et al. Long noncoding RNA MALAT1 controls cell cycle progression by regulating the expression of oncogenic transcription factor B-MYB. PLoS Genet 2013;9:e1003368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Peters T, Hermans-Beijnsberger S, Beqqali A, Bitsch N, Nakagawa S, Prasanth KV, et al. Long non-coding RNA Malat- 1 is dispensable during pressure overload-induced cardiac remodeling and failure in mice. PLoS One 2016;11:e0150236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang C, Liu G, Yang H, Guo S, Wang H, Dong Z, et al. MALAT1-mediated recruitment of the histone methyltransferase EZH2 to the microRNA-22 promoter leads to cardiomyocyte apoptosis in diabetic cardiomyopathy. Sci Total Environ 2021;766:142191. [DOI] [PubMed] [Google Scholar]
- 50.Song J, He Q, Guo X, Wang L, Wang J, Cui C, et al. Mesenchymal stem cell-conditioned medium alleviates high fat-induced hyperglucagonemia via miR-181a-5p and its target PTEN/AKT signaling. Mol Cell Endocrinol 2021;537:111445. [DOI] [PubMed] [Google Scholar]
- 51.Vaskova E, Ikeda G, Tada Y, Wahlquist C, Mercola M, Yang PC. Sacubitril/valsartan improves cardiac function and decreases myocardial fibrosis via downregulation of exosomal miR-181a in a rodent chronic myocardial infarction model. J Am Heart Assoc 2020;9:e015640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Raut SK, Singh GB, Rastogi B, Saikia UN, Mittal A, Dogra N, et al. miR-30c and miR-181a synergistically modulate p53-p21 pathway in diabetes induced cardiac hypertrophy. Mol Cell Biochem 2016;417:191-203. [DOI] [PubMed] [Google Scholar]
- 53.Calogero S, Grassi F, Aguzzi A, Voigtländer T, Ferrier P, Ferrari S, et al. The lack of chromosomal protein Hmg1 does not disrupt cell growth but causes lethal hypoglycaemia in newborn mice. Nat Genet 1999;22:276-80. [DOI] [PubMed] [Google Scholar]
- 54.Zhang L, Liu M, Jiang H, Yu Y, Yu P, Tong R, et al. Extracellular high-mobility group box 1 mediates pressure overload-induced cardiac hypertrophy and heart failure. J Cell Mol Med 2016;20:459-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jia Z, Xue R, Liu G, Li L, Yang J, Pi G, et al. HMGB1 Is involved in the protective effect of the PPAR alpha agonist fenofibrate against cardiac hypertrophy. PPAR Res 2014;2014:541394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Funayama A, Shishido T, Netsu S, Narumi T, Kadowaki S, Takahashi H, et al. Cardiac nuclear high mobility group box 1 prevents the development of cardiac hypertrophy and heart failure. Cardiovasc Res 2013;99:657-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Franklin S, Chen HD, Mitchell-Jordan S, Ren SX, Wang YB, Vondriska TM. Quantitative analysis of the chromatin proteome in disease reveals remodeling principles and identifies high mobility group protein B2 as a regulator of hypertrophic growth. Mol Cell Proteomics 2012;11:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sato M, Miyata K, Tian Z, Kadomatsu T, Ujihara Y, Morinaga J, et al. Loss of endogenous HMGB2 promotes cardiac dysfunction and pressure overload-induced heart failure in mice. Circ J 2019;83:368-78. [DOI] [PubMed] [Google Scholar]