

Abstract
A comprehensive analysis of the phosphoproteome is essential for understanding molecular mechanisms of human diseases. However, current tools used to enrich phosphotyrosine (pTyr) are limited in their applicability and scope. Here, we engineered new superbinder Src-Homology 2 (SH2) domains that enrich diverse sets of pTyr-peptides. We used phage display to select a Fes-SH2 domain variant (superFes; sFes1) with high affinity for pTyr and solved its structure bound to a pTyr-peptide. We performed systematic structure–function analyses of the superbinding mechanisms of sFes1 and superSrc-SH2 (sSrc1), another SH2 superbinder. We grafted the superbinder motifs from sFes1 and sSrc1 into 17 additional SH2 domains and confirmed increased binding affinity for specific pTyr-peptides. Using mass spectrometry (MS), we demonstrated that SH2 superbinders have distinct specificity profiles and superior capabilities to enrich pTyr-peptides. Finally, using combinations of SH2 superbinders as affinity purification (AP) tools we showed that unique subsets of pTyr-peptides can be enriched with unparalleled depth and coverage.
Graphical Abstract
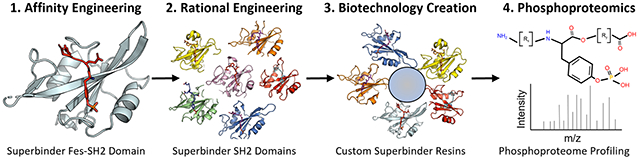
INTRODUCTION
The detection of phosphorylated peptides and their inferred proteins using MS remains challenging due to the low stoichiometry of phosphorylation events. This is particularly true for the analysis of pTyr as its rapid turnover and low cellular abundance relative to phosphorylated serine/threonine residues (pSer/pThr) often result in under-representation of pTyr-sites in proteomes.1 Typically, pTyr-containing proteins (pTyr-proteins) and pTyr-peptides are enriched from tissues and cells after protein digestion, frequently utilizing immobilized metal-affinity chromatography (IMAC) binding pSer/pThr/pTyr-peptides.2 Alternatively, pTyr-specific antibodies can be used, albeit they are expensive, nonspecific, and of low binding efficiency.3 Therefore, efficient analysis of phosphoproteomes necessitates pTyr enrichment tools that are easy to produce and can recover a high percentage of the pTyr-proteome in a cost-effective manner.
SH2 domains naturally bind pTyr-proteins in cells to mediate pTyr-dependent signal transduction networks.4 SH2 domains are comprised of a three-strand antiparallel β-sheet flanked by a pair of α-helices.5 The pTyr-binding pocket is characterized by a highly conserved Arg residue in the βB6 position that co-ordinates the phosphoryl moiety of the pTyr-residue.6 Adjacent to the pTyr-binding pocket are a series of hydrophobic pockets that interact with side chains of amino acids C-terminal to the pTyr-residue. These hydrophobic pockets dictate SH2 ligand specificity and are rendered either accessible, or occluded, by the co-operative action of the EF- and BG-loops.7 Thus, the SH2 domain employs a two-pronged binding mode that depends first on pTyr-binding and second on interactions of the residues flanking pTyr in the polypeptide.8–14
Using phage display, we previously engineered sSrc1 and superFyn-SH2, high-affinity variants of the SH2 domains (SH2 superbinders) of the Src and Fyn tyrosine kinases.8 sSrc1 was used as an affinity purification tool in mass spectrometry (AP-MS) experiments to enrich pTyr-peptides with unprecedented coverage,3,15 whereas superFyn-SH2 variants with altered specificity profiles enabled enrichment of different pTyr-peptides.16 Therefore, SH2 superbinders with complementary specificity profiles could be used to probe the pTyr-proteome with greater depth and coverage than conventional IMAC, anti-pTyr antibodies, or natural SH2 domains that bind pTyr-peptides with modest affinity.17,18
The human genome encodes 122 SH2 domains,19,20 offering a potentially vast palette for engineering superbinders with diverse specificities. Here, we report a general strategy to engineer high-affinity variants of human SH2 domains as superior tools for AP-MS-based pTyr-phosphoproteomics. We used phage display to engineer superbinder variants of the Fes-SH2 domain and subsequently developed a modular method to increase the affinity of SH2 domains by several orders of magnitude. We characterized the structure and function of several different SH2 superbinders using X-ray crystallography and MS. Finally, we demonstrate unprecedented depth and coverage of the pTyr-proteome using combinations of SH2 superbinders as AP-MS tools.
RESULTS AND DISCUSSION
Development of High-Affinity Variants of the Fes-SH2 Domain.
We previously used phage display to develop high-affinity variants of Src-SH2, which belongs to class XII and exhibits specificity for ligands of the type pTyr-X-X-Φ (Φ denotes hydrophobic residues).21 To further expand the range of ligand specificities that could be targeted with SH2 superbinders, we chose to engineer Fes-SH2 that shares only 35% amino acid sequence identity with Src-SH2 and exhibits a different specificity profile (class XVI) that recognizes ligands of the type pTyr-E-X-[V/I].21
To aid library design, we examined the structure of Fes-SH2. However, as there is no structure of ligand-bound Fes-SH2 currently available, we superposed the structure of Fes-SH2 with Src-SH2 in complex with a pTyr-peptide ligand. We then identified Fes-SH2 residues that were in analogous positions with those selected for randomization in the Src-SH2 phage-display library.8 We selected residues that were oriented toward the ligand pTyr-residue and had at least one atom within 10 Å of the pTyr-residue. We excluded the highly conserved Arg residue in the βB6 position, as it was invariant in previous selections for Src-SH2 variants.8 Applying these criteria, we chose a set of 13 residues for diversification, including 2 residues in the αA-helix, 5 residues in the β-sheet, and all 6 residues comprising the BC-loop. (Figure 1a,b). We constructed a phage-display library containing 1.6 × 1010 unique variants, using a soft randomization strategy that favored the wild-type (wt) sequence but allowed for diversity across all 13 positions.22 Phage pools representing the library were cycled through 5 rounds of selections for binding to an immobilized pTyr-peptide (pEZ) derived from the natural Fes-SH2 ligand Ezrin.23 Phage ELISAs were used to identify individual clones that exhibited binding signals for the peptide pEZ that were at least 10-fold higher than those for an unphosphorylated peptide (EZ) with the same primary sequence, and DNA sequencing revealed six unique Fes-SH2 variants, named superFes-SH2-1-6 (sFes,1–6 Figure 1c). The variants exhibited conservation of the wt sequence across all positions in the αA-helix and the β-sheet, except for variants 5 and 6, which contained a single conservative substitution in position βB3 or βD6, respectively. In contrast, all six variants exhibited highly diverse sequences in the BC-loop, which differed greatly from the wt and among each other.
Figure 1.
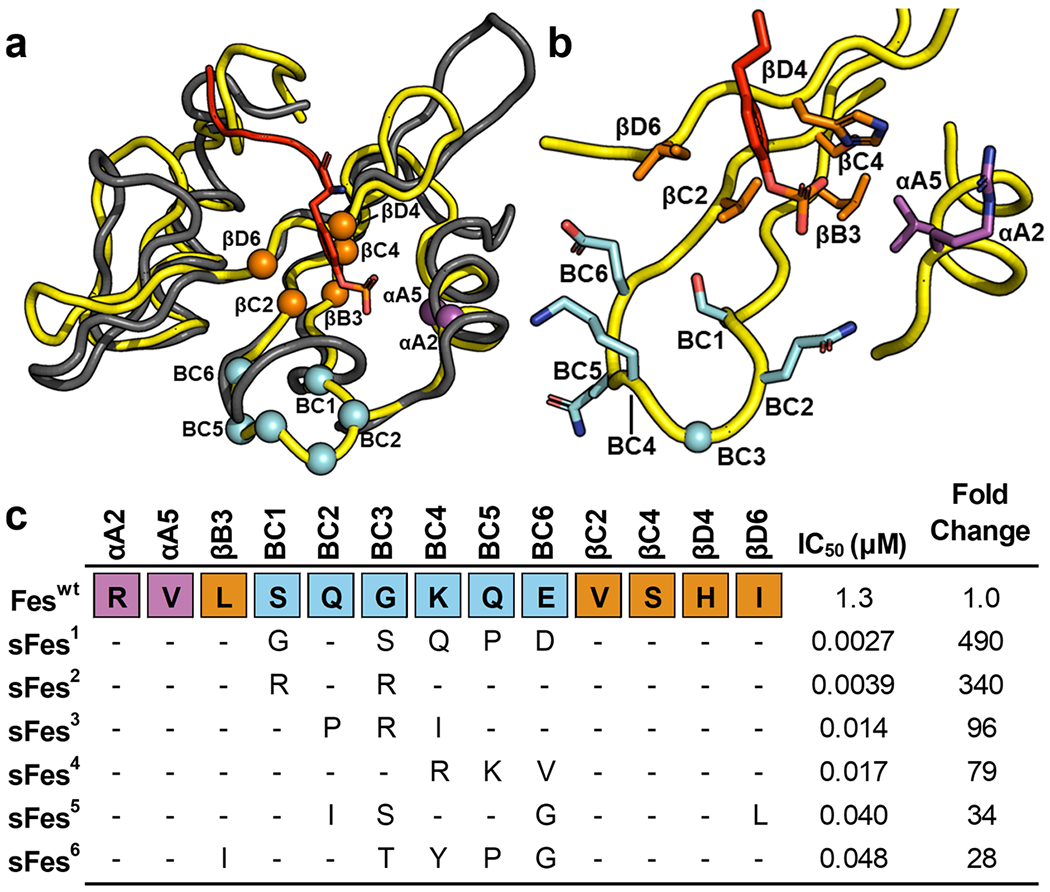
Fes-SH2 library design and selection results. (a) Superposition of the structure of Fes-SH2 (yellow, PDB ID: 1WQU) and Src-SH2 (gray) in complex with a pTyr-peptide (sequence: pTyrEEIE, red, PDB ID: 1HCT). Positions that were diversified in the Fes-SH2 library are shown as spheres colored magenta, orange, or cyan for residues that reside in the αA-helix, the β-sheet, or the BC-loop, respectively. The pTyr side chain of the peptide ligand is shown as sticks. (b) Details of the pTyr-binding pocket. The ligand pTyr side chain and Fes-SH2 side chains at positions that were diversified in the library are shown and colored as in (a). (c) Sequence alignment of Fes-SH2 and variants selected for binding to pTyr-peptide pEZ (PPPVpTyrEPVSYH). The alignment shows only those residues that were diversified in the library and positions that were conserved as the wt sequence are shown as dashes. The IC50 values were determined by fluorescence polarization assays with pTyr-peptide pEZ, and the fold change relative to the Feswt IC50 is also shown for each variant.
Each variant was recombinantly expressed, purified, and fluorescence polarization binding assays revealed that, compared to the wt protein (Feswt, IC50 = 1.3 μM), the variants exhibited 28–490-fold enhancements in apparent affinities (IC50 = 2.7–48 nM). None of the SH2 domains exhibited any detectable binding to unphosphorylated peptide EZ, even at peptide concentrations of 10 μM (Figure S1). For further studies, we focused on sFes1, the variant that exhibited the highest apparent affinity for peptide pEZ.
Site-Directed Mutagenesis Analysis of Determinants of Superbinder Activity.
The Src-SH2 domain was converted into sSrc1 by making only one substitution in the BC-loop, together with two hydrophilic-to-hydrophobic substitutions at positions βC2 and βD6. Notably, the two additional positions cluster together in the three-dimensional structure and form what we refer to as the “backside” of the pTyr-binding pocket. Feswt already contains hydrophobic residues at these two positions. Thus, we hypothesized that enhanced affinity for pTyr is achieved by establishing hydrophobic interactions between the SH2 backside residues and the aromatic ring of the pTyr moiety, together with a particular BC-loop conformation that promotes additional interactions with the pTyr-residue. To test this hypothesis, we designed site-directed mutagenesis studies in sSrc1 and sFes1 to explore whether grafting of substitutions could endow enhanced affinity in the opposing domain.
We assembled a panel of Src-SH2 domain variants and assessed their affinity for a pTyr-peptide derived from the pTyr324 site of the hamster polyomavirus middle T antigen24 (peptide pMT). Compared to Srcwt, we confirmed that sSrc1 exhibited 690-fold enhanced binding (Figures 2a and S2a). Moreover, conversion of the BC-loop or backside sequence of sSrc1 to the wt sequence produced variants (sSrc1a and sSrc1b, respectively) with only 25- or 2.2-fold enhanced affinity, relative to wt. These results showed that the BC-loop and the backside substitutions work co-operatively to enhance the affinity of sSrc1 relative to Srcwt. Next, we grafted the entire sFes1 BC-loop into either Srcwt or sSrc1 (SrcF1 and SrcF2, respectively), and in both cases, ligand binding was almost undetectable (IC50 > 50μM). In sharp contrast, grafting of the BC-loop together with the two backside residues of sFes1 into Src-SH2 resulted in a superbinder (sSrcF) with 510-fold enhanced affinity relative to Srcwt, which was almost as great as that of sSrc1. Taken together, these results showed that the BC-loop and backside residues work co-operatively to greatly enhance the affinity of Src-SH2, but either set of substitutions alone creates a nonfunctional Src-SH2 variant.
Figure 2.
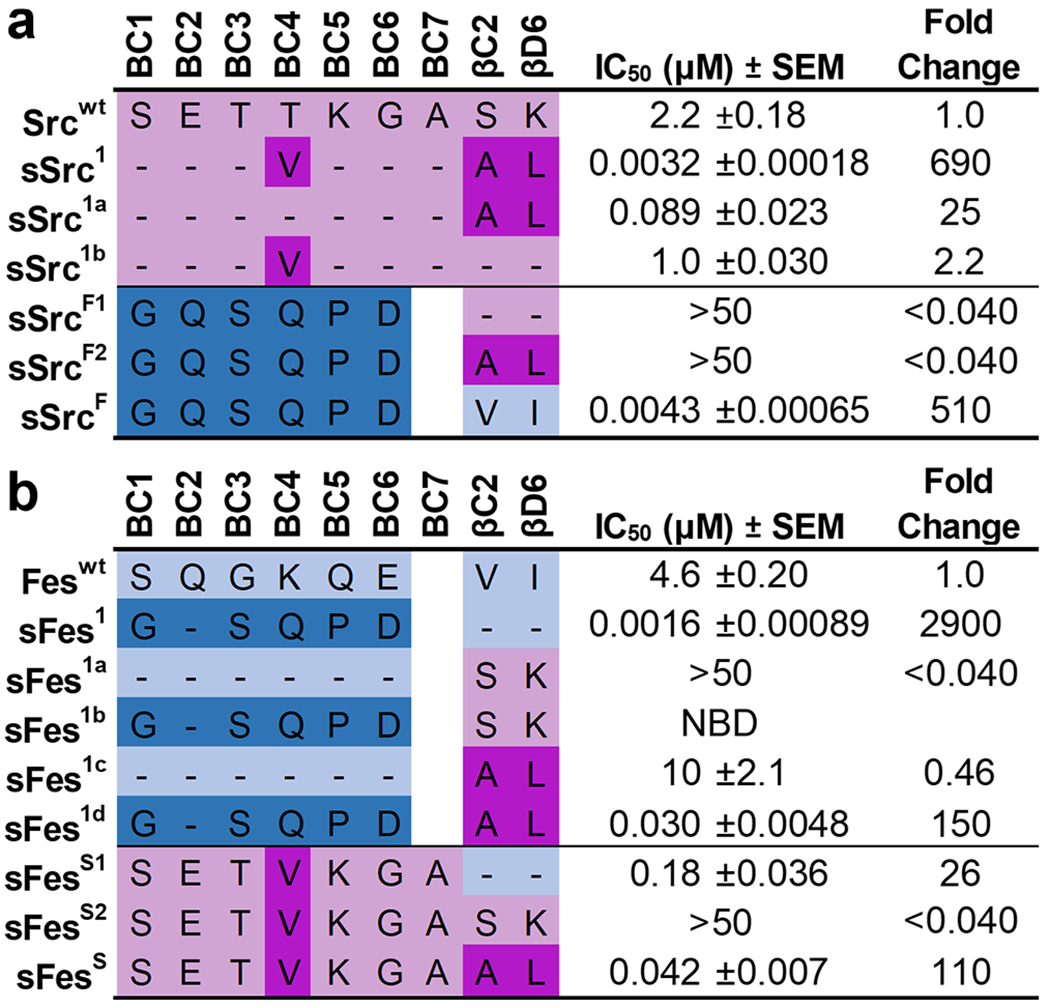
IC50 values for Src and Fes-SH2 variants binding to phosphopeptides. Schema depicting the different Src-derived and Fes-derived sequences grafted into the (a) Src-SH2 domain or (b) Fes-SH2 domain. Binding to pTyr-peptide pMT (sequence: EEPQpTyrEEIPIY) or pTyr-peptide pEZ (sequence: PPPVpTyrEPVSYH) for Src or Fes variants, respectively, was assayed by competitive SH2 phage ELISAs. The IC50 value was derived from the binding curves, and the fold change relative to the IC50 for the wt domain was calculated. Data are an average of 3 to 4 experimental replicates ± SEM. Sequences derived from Src or Fes are colored pink or blue, respectively, and light or dark colors indicate wt or superbinder sequences, respectively. “NDI” indicates “no detectable inhibition”.
Analogous to the study with Src-SH2, we also assembled a panel of Fes-SH2 variants and assessed affinities. Compared to Feswt, sFes1 exhibited 2900-fold enhanced affinity (Figured 2b and S2b). Grafting of the Srcwt backside sequence into Feswt or sFes1 produced nonfunctional domains (Fes1a and Fes1b, respectively) with little to no detectable affinity for the ligand. Conversely, transfer of the sSrc1 backside sequence into Feswt or sFes1 created variants (Fes1c and sFes1d) with two-fold decreased or 150-fold increased affinity, respectively. Moreover, the transfer of the sSrc1 BC-loop into Fes-SH2 in the context of Feswt or sSrc1 backside sequences created variants (sFesS1 and sFesS) with 26- or 110-fold affinity enhancements, respectively, relative to Feswt, whereas binding was barely detectable in the context of the Srcwt backside sequence (sFesS2). These results show that the BC-loop of sFes1 works co-operatively with the hydrophobic Feswt backside to enhance affinity and it can also work well with the hydrophobic backside of sSrc1 but cannot function with the hydrophilic backside of Srcwt. Similarly, the BC-loop of sSrc1 can enhance the affinity of Fes-SH2 in the context of either the Feswt or sSrc1 backsides but cannot function in the context of the hydrophilic Srcwt backside residues.
Elucidation of Structures for Src and Fes Superbinders.
To gain mechanistic insights into the enhanced affinities of SH2 superbinders, we solved three new crystal structures to enable detailed comparisons between wt and superbinder SH2 domains in both the ligand-bound and unbound states (Figure 3 and Table S1). In each structure, the protein adapted the typical SH2 domain fold, with the pTyr-residue bound by the canonical pTyr-binding pocket.
Figure 3.
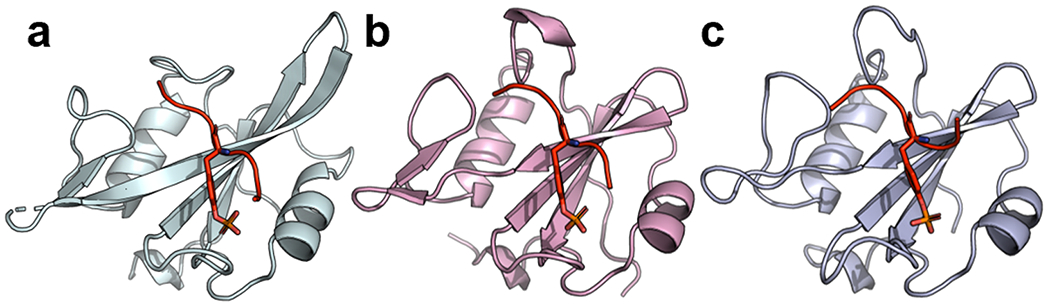
Structures of sSrcF, sFes1, and sFesS in complex with pTyr-peptides. (a) Structure of sSrcF (cyan) in complex with the pTyr-peptide pMT (sequence: EPQpTyrEEI). (b) Structure of sFes1 (pink) and (c) structure of sFess (light blue) both in complex with the pTyr-peptide pEZ (sequence: PPVpTyrEPV). SH2 domains are shown as ribbons and pTyr-peptides are shown in red with the pTyr side chain shown as sticks.
Structural Comparison of Src-SH2 and Its Superbinders.
We compared the unbound and bound structures of Srcwt and sSrc1 to see whether changes induced upon ligand binding may contribute to affinity differences. Thus, we compared structures for (1) unbound v-Src, (2) Srcwt bound to pTyr-peptide pTyrEEIE, (3) unbound sSrc1, and (4) sSrc1 bound to pTyr. Although the v-Src sequence differs at three positions compared to Srcwt, these differences are all far from the pTyr-binding pocket, and v-Src is the closest Srcwt homologue with a solved structure that does not contain any co-ordinating solvent molecules in the pTyr-binding pocket.
To compare main chain conformations, we superposed the four structures using Cα co-ordinates (Figure 4a; left). For unbound and bound sSrc1 structures, the main chains of residues around the pTyr-residue were nearly identical. In contrast, structures of unbound v-Src and bound Srcwt showed significant differences, with the main chains of the BC-loop and αA-helix of unbound v-Src positioned further away or closer to the pTyr-residue, respectively. Consequently, a comparison of the main chains of the bound Srcwt and sSrc1 structures shows that the BC-loops are nearly identical, but that the αA-helix of Srcwt is farther from the pTyr-residue.
Figure 4.
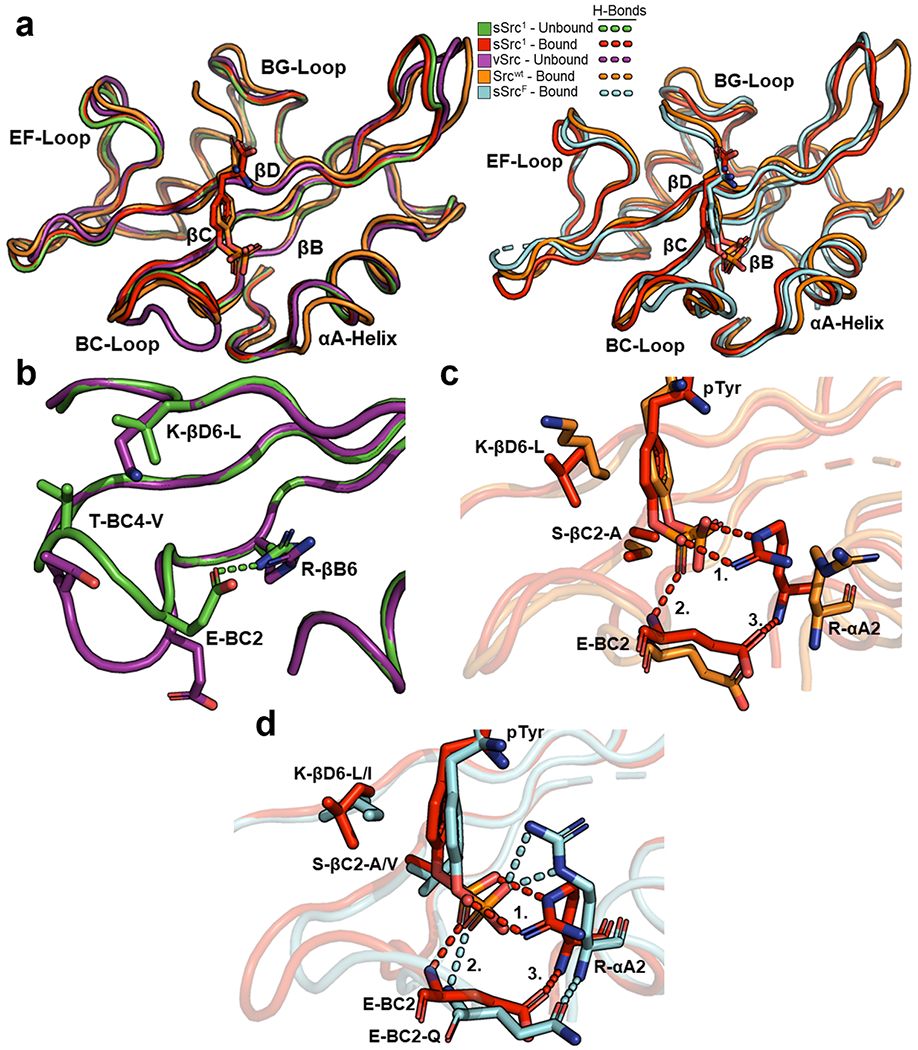
Structural comparison of Src-SH2 and its superbinders. (a) Superposition of Src-SH2 and its variants. The left panel depicts the following structures: unbound v-Src (purple, PDB ID: 1BKL), unbound sSrc1 (green, PDB ID: 4F59), bound Srcwt (orange, PDB ID: 1HCT), and bound sSrc1 (red, PDB ID: 4F5B). The right panel depicts the following structures: bound Srcwt (orange), bound sSrc1 (red), and bound sSrcF (cyan). Structures were aligned based on Cα co-ordinates using the ALIGN function in PyMol. (b) Superposition of the pTyr-binding pockets of unbound v-Src (purple) and unbound sSrc1 (green). (c) Superposition of the pTyr-binding pockets of bound Srcwt (orange) and bound sSrc1 (red). Hydrogen bonds are shown as dashed lines and numbers refer to interactions described in the main text. (d) Superposition of the pTyr-binding pockets of bound sSrc1 (red) and bound sSrcF (cyan).
Inspection of intramolecular side-chain interactions in the unbound structures revealed several differences that may contribute to the different conformations of the sSrc1 BC-loop compared to that of the wt loop of v-Src (Figure 4b). In particular, the sSrc1 loop contained a hydrogen bond between the Glu-BC2 side chain and the Arg-βB6 side chain, whereas in the wt loop, these side chains point away from each other and do not interact. Moreover, two hydrophobic substitutions in sSrc1 compared to Srcwt (Thr-BC4-Val and Lys-βD6-Leu) make hydrophobic interactions in sSrc1 but not in Srcwt. Consequently, the BC-loop of unbound sSrc1 is more compact and more closely resembles the loop in the bound state than does the unbound wt loop.
Furthermore, we analyzed intermolecular side-chain interactions between the SH2 domains and the pTyr ligand residues (Figure 4c). Notably, the two hydrophobic substitutions in the backside of the pTyr-binding pocket (Ser-βC2-Ala and Lys-βD6-Leu) result in hydrophobic packing interactions with the aromatic ring of the pTyr side chain in the case of sSrc1 compared to Srcwt. Additionally, sSrc1 makes a series of hydrogen bonds involving (1) the Arg-αA2 side chain and the pTyr phosphoryl group, (2) the main chain amide of BC2 and the pTyr phosphoryl group, and (3) the Glu-BC2 side chain and the main chain amide of αA2. This hydrogen-bond network is not formed in Srcwt. Thus, it appears that the enhanced affinity of sSrc1 for pTyr arises from (1) a BC-loop that is pre-organized in the unbound state to resemble the bound conformation, (2) a hydrophobic backside that interacts with the pTyr aromatic ring, and (3) closer proximity of the αA-helix that facilitates hydrogen-bond formation between Arg-αA2 and the pTyr phosphoryl group.
We next superposed the structure of the superbinder sSrcF bound to the pTyr-peptide EPQpTyrEEI with the pTyr-bound structures of sSrc1 and Srcwt (Figure 4a; right). The primary sequence of the sSrcF BC-loop is very different from those of sSrc1 and Srcwt, and unsurprisingly, it superposed poorly with them. Nonetheless, the position of the αA-helix of sSrcF resembled that of sSrc1 and was closer to the pTyr-residue than was that of Srcwt. Moreover, both domains form similar hydrogen-bond networks with the pTyr phosphoryl group, despite the different rotamer conformations of the Arg-αA2 side chains (Figure 4d). Finally, the wt backside of sSrcF (Val-βC2 and Ile-βD6) resembles that of sSrc1 in that both contain hydrophobic side chains that pack with the pTyr aromatic ring. Thus, despite having very different BC-loops, sSrc1 and sSrcF use similar mechanisms to enhance the affinity for pTyr side chains, and these include hydrophobic interactions with the pTyr aromatic ring and hydrogen bonds with the phosphoryl moiety.
Structural Comparison of Fes-SH2 and Its Superbinders.
We also examined the structures of sFes1 and sFesS bound to pTyr-peptides and compared these to a previous structure of Feswt bound to a sulfate ion (no structures of Feswt bound to pTyr are available but we reasoned that the sulfate ion is a good mimic).
Superposition of the main chains of the three structures showed significant differences in the BC-loops, and small differences in the N-terminal end of the αA-helix, which was positioned closer to the pTyr in the two superbinder structures (Figure 5a). Analysis of intermolecular side-chain interactions of Feswt and the sulfate ion showed that there are four residues whose side chains establish hydrogen bonds with the sulfate ion: (1) Arg-αA2, (2) Arg-βB6, (3) Ser-BC1, and (4) Lys-BC4 (Figure 5b). In the sFes1 structure, there are also four residues whose side chains form hydrogen bonds with the pTyr phosphoryl moiety: (1) Arg-αA2, (2) Arg-βB6, (3) Ser-BC3, and (4) Gln-BC4 (Figure 5c). However, a crucial difference compared to Feswt is that, instead of Ser-BC1, sFes1 has (5) Ser-BC3 forming a hydrogen bond with pTyr with additional contacts being made with the main chain amides of Gln-BC2 and Ser-BC3. These additional interactions between the pTyr moiety and the BC-loop appear to be supported by the formation of (6) an intramolecular hydrogen bond between the side chain of Gln-BC2 and the main chain amide of Arg-αA2, which is also present in the Src superbinder motifs. Finally, the two pTyr-binding pocket backside residues, Val-βC2 and Ile-βD6, pack against the phenyl ring of pTyr and are in a similar position to the same residues in Feswt.
Figure 5.
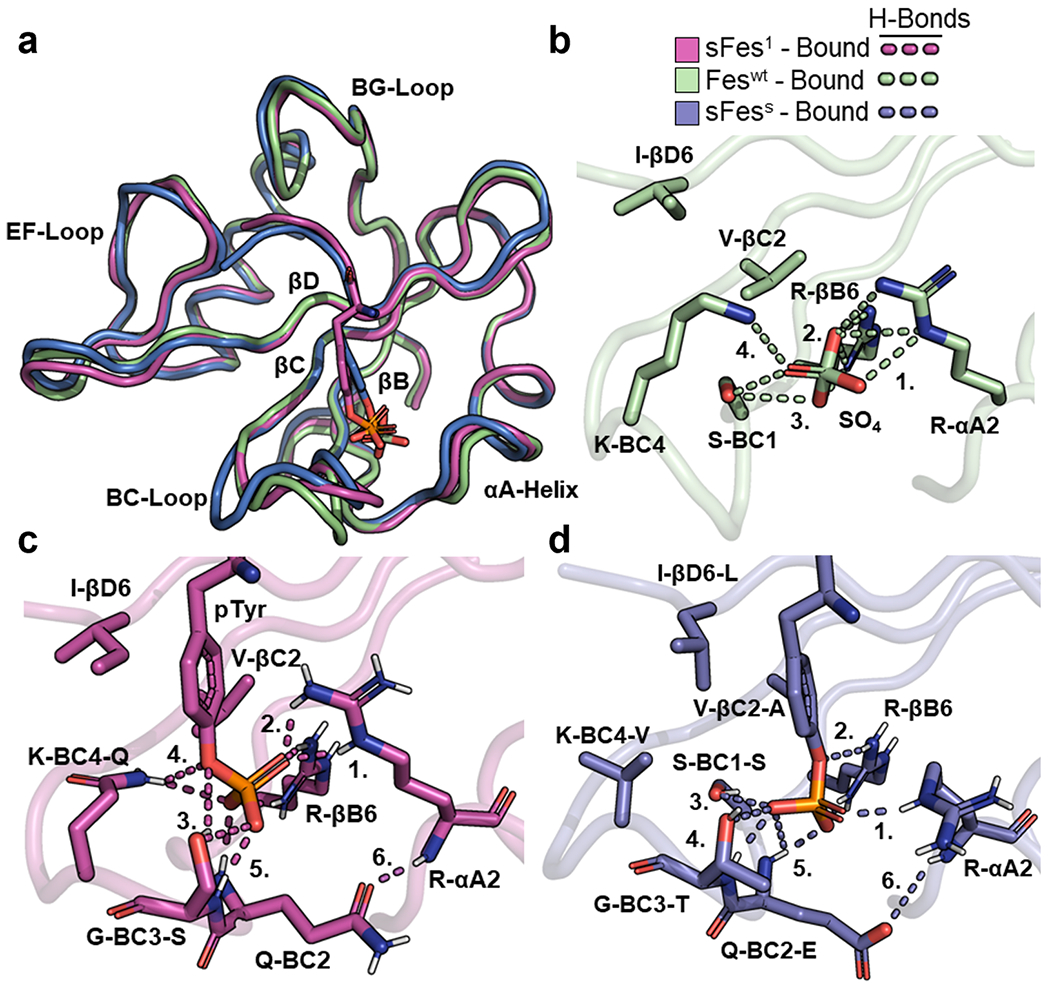
Structural comparison of Fes-SH2 and its variants. (a) Superposition of Fes-SH2 and its variants. The following bound structures are depicted: Feswt (green, PDB ID: 3BKB), sFes1 (magenta), and sFesS (blue). Structures were aligned based on Cα co-ordinates using the ALIGN function in PyMol. (b) pTyr-binding pocket of Feswt (bound). Hydrogen bonds are shown as dashed lines and numbers refer to interactions described in the main text. (c) pTyr-binding pocket of sFes1 (bound). Hydrogen bonds are shown as dashed lines and numbers refer to interactions described in the main text. (d) pTyr-binding pocket of sFesS (bound). Hydrogen bonds are shown as dashed lines and numbers refer to interactions described in the main text.
We also analyzed the pTyr-binding pocket of sFesS. Like sFes1, there are four side chains that form hydrogen bonds with pTyr (Figure 5d): (1) Arg-αA2, (2) Arg-βB6, (3) Ser-BC1, and (4) Thr-BC3. There are also (5) hydrogen bonds between pTyr and the main chain amides of Glu-BC2 and Thr-BC3. As for sFes1, the BC-loop contains (6) an intramolecular hydrogen bond between the side chain of Glu-BC2 and the main chain amide of Arg-αA2. The two residues comprising the backside of the pTyr-binding pocket, Ala-βC2 and Leu-βD6, also pack against the phenyl ring of pTyr with additional hydrophobic interactions between Val-BC4 and Leu-βD6 contributing to this interaction.
Therefore, the Fes superbinders maintain an extensive hydrogen-bond network, crucially, with a residue in the BC3 position that also allows additional hydrogen bonds with the main chain amides of other residues in the BC-loop. One notable consequence is the formation of hydrogen bonds between the BC-loop and the αA-helix, which likely act to further stabilize contacts between Arg-αA2 and pTyr. These contacts are not made in Feswt.
Structural Comparison of Src and Fes Superbinders.
To complete our structural analysis, we compared the pTyr-binding pockets of pairs of Src and Fes superbinders with matching substitutions and performed alanine scanning of essential hydrogen-bond-forming residues (Figure S3). These analyses demonstrate that the sSrc1 and sFes1 superbinder motifs can reconstitute the pTyr-binding pocket in different SH2 domains.
Grafting of sSrc1 and sFes1 Superbinder Motifs into Diverse SH2 Domains.
Having established the structural basis for the enhanced affinities of sSrc1 and sFes1, we explored whether the superbinder sequence motifs of these domains could be transferred into a diverse set of SH2 domains. We performed a sequence alignment of all 122 human SH2 domains and assembled the alignment into an unrooted phylogenetic tree (Figure S4). To identify suitable, diverse domains for engineering, we focused on domains that (1) could be recombinantly expressed in bacteria,25 (2) had a reported structure, and (3) had a defined specificity profile.21,26–28 This process yielded a panel of 17 new SH2 domains that were spread out across the phylogenetic tree, indicating high diversity among the sequences.
For each of the 17 new domains, we engineered variants containing the BC-loop and the pTyr-binding pocket backside sequences from either sSrc1 or sFes1, thus obtaining a panel of 51 SH2 domains consisting of the 17 wt domains and two potential superbinders for each SH2. We purified each domain, and for each related trio, we estimated affinities by competitive ELISAs (Figures 6 and S5–6). For 13 or 8 of the 17 domains, grafting of either the sSrc1 or sFes1 superbinder sequence motif, or sometimes both, increased affinity for the phosphopeptide by >10-fold or >50-fold, respectively, relative to the wt.
Figure 6.
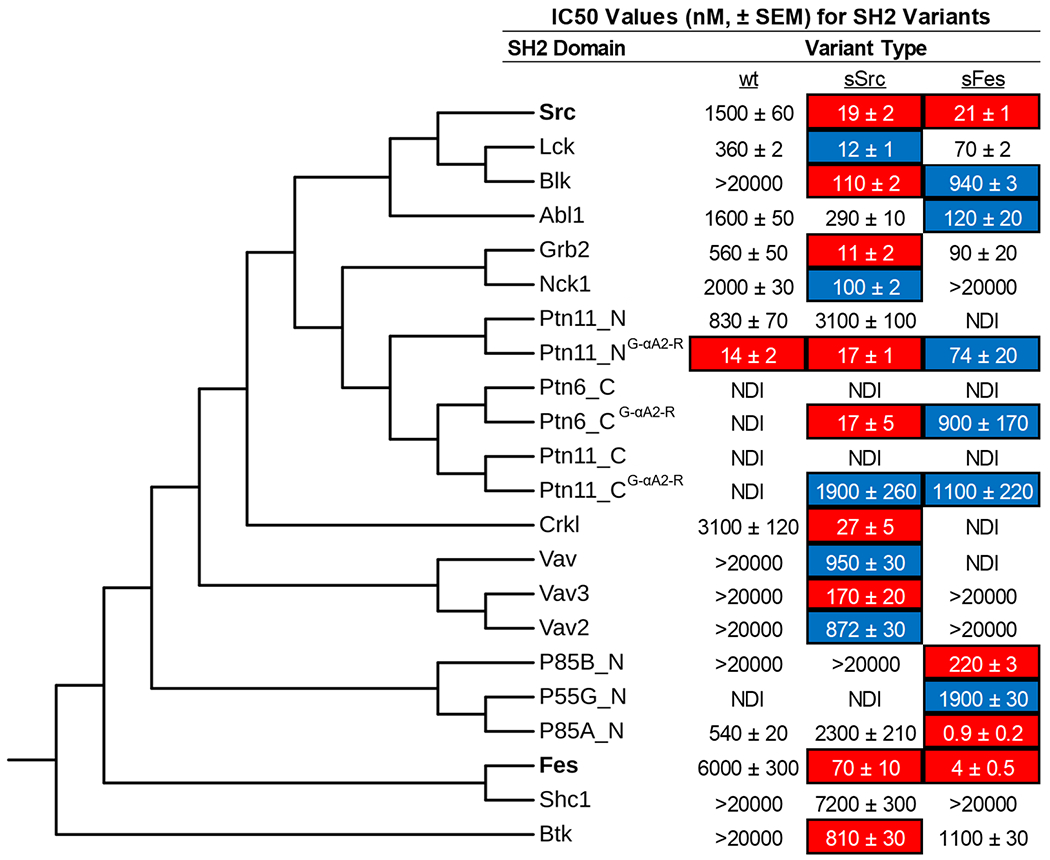
Grafting of sSrc1 and sFes1 superbinder motifs into diverse SH2 domains. A panel of 17 different SH2 domains was selected to receive the sSrc1 or sFes1 superbinder graft. The amino acid sequences of these domains, in addition to Src and Fes-SH2 domains, were aligned using COBALT and assembled into an unrooted phylogenetic tree, using fast minimum evolution with a maximum sequence dissimilarity cutoff of 0.9. We performed competitive ELISA to determine IC50 values for each trio of domains with a unique pTyr-peptide that the domain was reported to bind in the literature (Figure S8). Data are an average of 3–4 experiments ± SEM. IC50 values for which curve fitting could not be performed are listed as >20,000 nM. SH2 variants with >10-fold or >50-fold reduction in IC50 compared to wt are highlighted in blue or red, respectively. NDI indicates no detectable inhibition.
Among the four domains for which neither superbinder motif improved affinity substantially, Shc1-SH2 clustered close to Fes-SH2, indicating high sequence homology, and it is not clear why the grafting failed to produce a high-affinity variant. Notably, the other failed domains (Ptn11_N–SH2, Ptn11_C-SH2, Ptn6_C-SH2) clustered together and away from both Src-SH2 and Fes-SH2. Notably, these three domains all contained a glycine at position αA2, whereas the other domains in the panel contained an arginine at this position (Figure S7). In the sSrc1 and sFes1 structures, Arg-αA2 forms a hydrogen bond with the pTyr phosphoryl group, and we hypothesized that the absence of this side chain may explain the failure of the grafting strategy. To test this hypothesis, we substituted Gly for Arg at position αA2 and for Ptn11_N–SH2, this single substitution was sufficient to improve the affinity ~60-fold. For Ptn11_C-SH2 and Ptn6_C-SH2, affinity was not improved by the single substitution, but in both cases, affinity was substantially improved by the grafting of the superbinder motifs in this context.
Our results showed that, in most cases (15 of 19, including Src-SH2 and Fes-SH2), the SH2 domain affinity for pTyr-peptides was substantially improved by grafting of superbinder motifs from sSrc1 or sFes1. In three of the four cases where this approached failed, an additional change from Gly to Arg at position αA2 resulted in success, presumably by enabling the favorable interactions observed in the structures of sSrc1 and sFes1. Thus, despite significant sequence diversity at the global level, it appears that the pTyr-binding pockets of most SH2 domains are structurally and functionally conserved such that similar mechanisms can be used to optimize affinity across the SH2 family.
Specificity Profiling of SH2 Superbinders by Mass Spectrometry.
Having developed a panel of SH2 domain variants with an increased affinity for pTyr-peptides, we assessed their utility as AP-MS tools to enrich pTyr-peptides from cell lysates. Thirty SH2 variants (superbinder and wt versions) from 12 different SH2 domains were selected, if the family contained a superbinder variant with a reasonably high affinity for a pTyr-peptide (IC50 < 200 nM, Figure 7). We fused each SH2 domain to a N-terminal AviTag for site-specific biotinylation and determined the biotinylation extent via the band-shift assay29 (Figure S8). All domains were well expressed, soluble, and stable with an average biotinylation of >90%. To diversify the type and number of available pTyr-peptides, we prepared cell lysates from unstimulated and stimulated K562 cells. To enable sample characterization and increased throughput utilizing SH2 superbinders in phosphoproteomic studies, we established an automated enrichment process that included the binding of biotinylated SH2 domains to streptavidin-coated magnetic beads and incubation of immobilized SH2 domains with the cell digest. While phosphoproteomic studies often rely on large amounts of the starting material (mg range), we demonstrated here the successful affinity isolation of pTyr-peptides from 200 μg of the unfractionated K562 protein digest. We compared the high-affinity SH2 domains to their wt counterparts and the Ti/Zr-IMAC microparticle beads. Biotinylated-GST served as the negative control.
Figure 7.
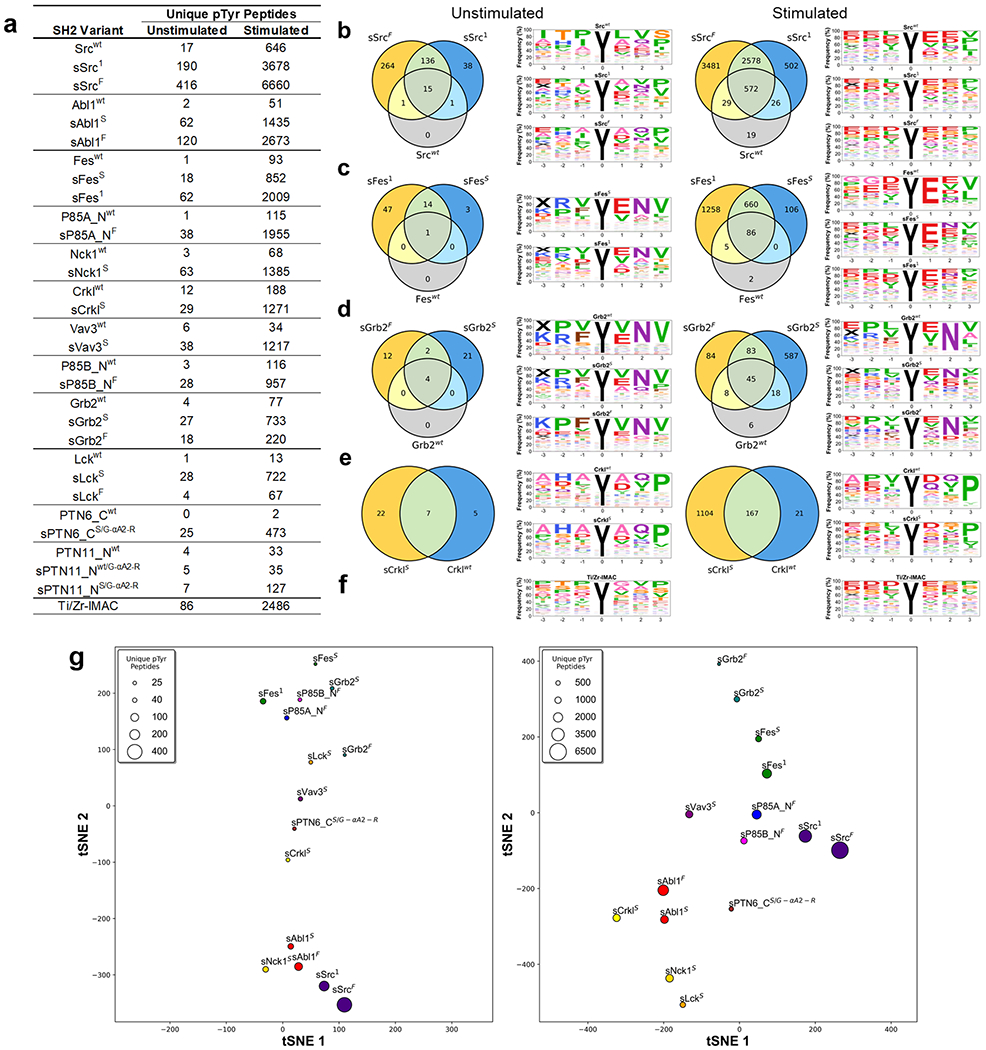
SH2 variant specificity profiling using AP-MS. (a) Number of unique pTyr-peptides enriched by SH2 variants in AP-MS experiments. SH2 domains are rank-ordered from most unique pTyr-peptides enriched (top) to least (bottom). (b–e) Venn diagrams depicting the number of unique pTyr-peptides and sequence motifs depicting profiles of pTyr-peptides enriched from unstimulated and stimulated K562 cell lysates by (b) Src, (c) Fes, (d) Grb2, or (e) Crkl-SH2 domain variants. (f) Sequence motifs of Ti/Zr-IMAC pTyr-peptide enrichments. (g) t-distributed stochastic neighbor embedding (tSNE) analysis of pTyr-peptide enrichment profiles for superbinders from unstimulated (left) or stimulated (right) K562 cell lysates. Relationships in the plot are qualitative with respect to pTyr-peptide enrichment similarities. The marker size is indicative of the number of unique pTyr-peptides enriched.
Figure 7a depicts the number of unique pTyr-peptides enriched from SH2 variants (see Tables S2 and S3 for mass spectrometry results in greater detail including the number of pTyr-sites). For each SH2 variant tested, the sSrc1 or sFes1 superbinder grafts increased the number of identified unique pTyr-peptides from both unstimulated and stimulated cell lysates. Each tested SH2 domain family had at least one superbinder variant that enriched >10-fold the number of unique pTyr-peptides compared to its wt counterpart. The most dramatic of these were sSrcF, sAbl1F, sFes1, sP85A_NF, sNck1S, sCrklS, and sVav3S, each of which enriched >1000 unique pTyr-peptides from stimulated cells. Unsurprisingly, stimulated K562 cells correlated with greater pTyr-peptide enrichment, as previously reported,30 but we observed a very similar trend for unstimulated cells, highlighting the direct correlation between the SH2 domain binding affinity and the pTyr-peptide isolation performance. Remarkably, sSrcF, sSrc1, and sAbl1F outperformed Ti/Zr-IMAC by isolating more unique pTyr-peptides in both unstimulated and stimulated cells. Previous studies have used the Src-SH2 superbinder to enrich pTyr-peptides from nine different cell lines,3 and crucially, the Src superbinder significantly outperformed anti-pTyr antibodies for phosphoproteome analysis. Also, we previously showed that SH2 domains with diverse binding specificities and enhanced affinities enable efficient and differential coverage of the human phosphoproteome.16
Next, we assessed the binding specificity of SH2 variants by analyzing how similar the enriched pTyr-peptide profiles were for superbinders compared to their wt counterpart. We computed the Pearson correlation coefficient of pTyr-peptides enriched by each SH2 variant relative to one another and plotted the data as a heatmap for both the unstimulated and stimulated conditions (Figure S9). Overall, grafting superbinder motifs into SH2 domains did not significantly affect the binding specificity, as SH2 superbinders recognized similar peptides to their wt counterparts. However, the overall degree of relatedness both within a type of the SH2 domain and between different SH2 domains decreased in the stimulated condition, in agreement with the biological effect of different phosphatase inhibitor treatments on phospho-signaling.31 To investigate these relationships further, we generated sequence motifs and Venn diagrams depicting peptide enrichment similarities for each type of the SH2 domain. We highlight our results for the Src (class XII), Fes (class XVI), Grb2 (class XIII), and Crkl (class VI) SH2 domains that have well-defined specificity profiles21 (Figure 7b–e). Despite the superbinders of these domains enriching many more unique pTyr-peptides than their wt counterparts, in both the unstimulated and stimulated conditions, the obtained sequence motifs of peptides enriched were similar to one another within a condition. This was particularly evident in the +1 to +3 positions for Src (E-X-[V/I]), Fes (E-X-Φ, Φ denotes hydrophobic residues), Grb2 (X-N-X), and Crkl (X-X-P) in the stimulated condition. These binding preferences were maintained for Grb2 and Crkl but slightly changed for Src and Fes in the unstimulated condition. The binding specificity of wt domains was maintained in all SH2 superbinders (Figure S10), except for sVav3s, for which the binding preference was noticeably different from that of the wt domain (Figure S10h). The sequence motif of Ti/Zr-IMAC (Figure 7f) shows a nonsequence specific enrichment pattern that also changes between unstimulated and stimulated conditions.
We next assessed how superbinder enrichment profiles compared to one another. We focused on the best 15 superbinders that enriched >25 or >450 unique pTyr-peptides in the unstimulated or stimulated conditions, respectively, and represent unique specificity classes. To assess the similarity of pTyr-peptides enriched by different high-affinity superbinders, we used t-distributed stochastic neighbor embedding (tSNE)32 to display the unique pTyr-peptide enrichment relationships between the superbinders in either the unstimulated (left) or stimulated (right) conditions (Figure 7g). Two superbinder versions of a given SH2 domain (if both were used) were found closer together compared to superbinders derived from other SH2 domains. Overall, different superbinders recognized diverse pTyr-peptides in both stimulated and untreated K562 cells, and such preferences were independent of the number of pTyr-peptides enriched but reflected the intrinsic binding specificity of each parental SH2 domain.
Phosphoproteome Profiling Using Combinations of SH2 Superbinders.
We reasoned that combinations of SH2 superbinders with different binding specificities could be used to isolate unique subsets of pTyr-peptides from cell lysates. We selected five combinations of SH2 superbinders (C1–C5) based on the total number of unique pTyr-peptides isolated and the degree of relatedness to one another in terms of the types of unique pTyr-peptides enriched. For example, C1 was a combination of sSrcF, sSrc1, and sAbl1F, the top three superbinders that bound the greatest numbers of unique pTyr-peptides from K562 cell lysates, whereas C3 was a combination of sAbl1F, sP85A_NF, sFes1, sNck1S, and sCrklS, five superbinders that bound different subsets of pTyr-peptides. We used the five combinations to enrich pTyr-peptides from unstimulated and stimulated K562 cell lysates and demonstrated that different combinations of superbinders were able to isolate more unique pTyr-peptides compared to Ti/Zr-IMAC (Figure 8a). Although the combinations were not able to enrich more unique pTyr-peptides than sSrcF alone, in both the unstimulated and stimulated conditions, they were all able to isolate distinct subsets of pTyr-peptides. Like individual superbinders, the sequence motifs of the combinations were different when enriching pTyr-peptides from unstimulated (Figure S11a) or stimulated samples (Figure S11b). The sequence motifs between enrichment tools were consistent with one another. However, when compared to sSrcF and Ti/Zr-IMAC in terms of unique pTyr-peptides enriched, each combination enriched a substantial number of unique pTyr-peptides from both unstimulated (top) and stimulated (bottom) cell lysates (Figure 8b).
Figure 8.
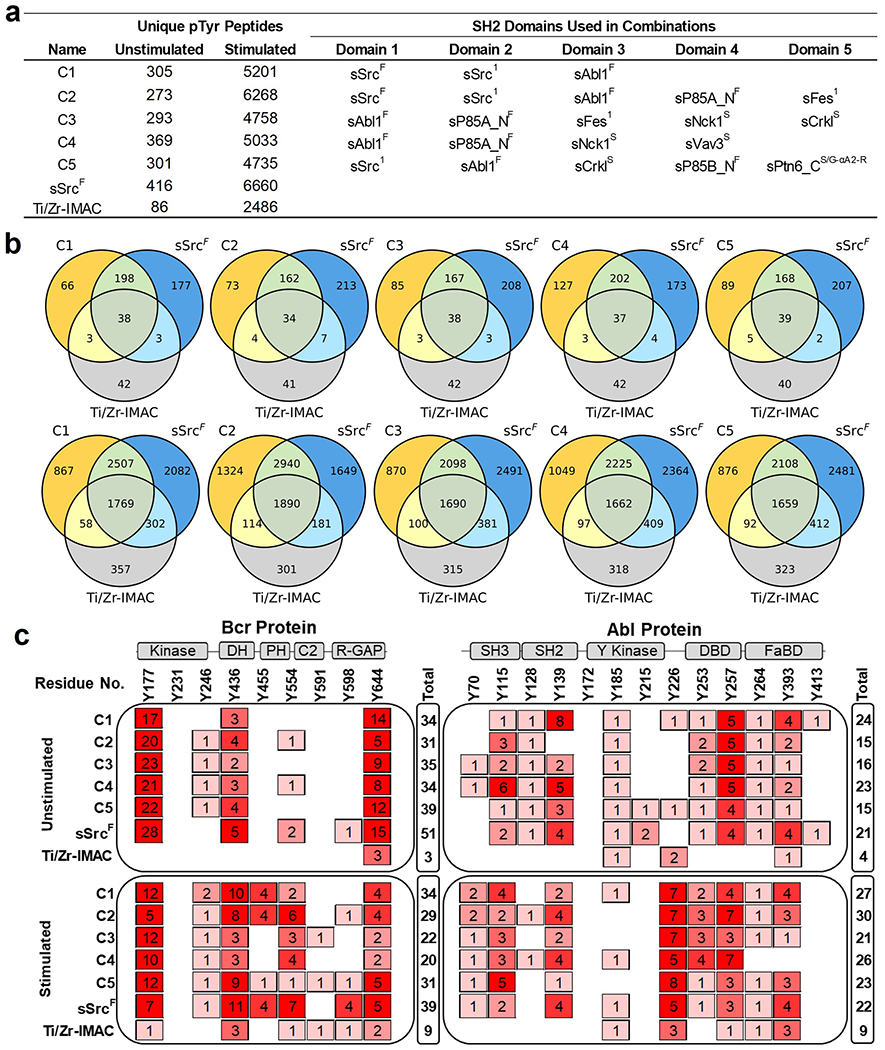
Phosphoproteome profiling using combinations of SH2 superbinders. (a) Number of pTyr-peptides enriched from combinations of SH2 superbinders used in AP-MS experiments. (b) Venn diagrams of pTyr-peptide enrichment profiles for each superbinder combination, sSrcF and Ti/Zr-IMAC from unstimulated (top) and stimulated (bottom) K562 cell lysates. (c) pTyr-site mapping for Bcr (P11274) and Abl1 (P00519) human proteins. Columns represent reported pTyr-sites of Bcr and Abl proteins in public resources including Uniprot, NextProt, and PeptideAtlas (Human phosphoproteome build 2017). Rows represent observed spectra for the pTyr-site using superbinder combinations (C1–C5), sSrcF, or Ti/Zr-IMAC from unstimulated (top) or stimulated (bottom) K562 cell lysates. “Total” represents the sum of observed spectra for pTyr-sites detected for Bcr and Abl proteins. Cartoon representations of secondary structures of Bcr and Abl proteins are shown above the plot. Abbreviations: Kinase; Serine/Threonine Kinase, DH; Dbl Homology, PH; Pleckstrin Homology, R-GAP; Rho-GTPase Activating Protein, SH3; Src-Homology 3, SH2; Src-Homology 2, Y Kinase; Tyrosine Kinase, DBD; DNA Binding Domain, and FaBD; F-actin Binding Domain.
K562 cells express Bcr (P11274) and Abl1 (P00519) or the Bcr–Abl fusion protein, which drives oncogenesis. We assessed the ability of superbinder combinations to detect pTyr-sites of Bcr and Abl1 phosphopeptides previously reported in UniProt,33 NeXtProt,34 and PeptideAtlas.35,36 We mapped pTyr-sites of the MS-detected pTyr-peptides from the superbinder combinations, sSrcF and Ti/Zr-IMAC in both unstimulated and stimulated cell lysates to known pTyr-sites on Bcr and Abl1 (Figure 8c). These resources report up to 9 and 13 pTyr-sites for Bcr and Abl1, respectively. We detected most of these pTyr-sites with the superbinder combinations and sSrcF, specifically 8 of 9 pTyr-sites in Bcr and 12 of 13 pTyr-sites in Abl1 in addition to other pTyr-sites in these two proteins. Overall, we observed that a larger number of unique pTyr-peptides and pTyr-sites were detected with superbinder combinations and sSrcF in comparison to Ti/Zr-IMAC. Superbinder combinations outperformed Ti/Zr-IMAC, especially in unstimulated cells, and all pTyr-sites enriched with Ti/Zr-IMAC were also detected with one of our superbinder combination or sSrcF. These results demonstrate that superbinder combinations are superior enrichment tools that have the capability to bind a broad range of pTyr-peptides.
Summary.
We established a general strategy to enhance the affinities of a diverse set of SH2 domains. Furthermore, we demonstrated that our new panel of superbinders offers a novel, scalable, and superior set of tools to enrich pTyr-peptides. SH2 superbinders can be used alone or in combinations to access distinct subsets of pTyr-peptides from various, complex biological samples.
We benchmarked our SH2 superbinder AP-MS workflow against a sequential Ti-IMAC and Zr-IMAC enrichment. Three of our SH2 superbinders and the five tested combinations of SH2 superbinders enriched substantially more unique pTyr-peptides from 200 μg of unstimulated or stimulated K562 cell lysates than currently used IMAC methods. As demonstrated, IMAC enrichment provides little unique pTyr-peptides (less than 5% for stimulated and 10% for unstimulated cell lysates) in comparison to the enhanced SH2 domains. Moreover, our newly engineered sSrcF significantly outperformed sSrc1.
These results highlight that much of the pTyr-proteome is inaccessible by conventional pTyr enrichment technologies. Our newly engineered set of superbinders is an important addition to the suite of tools available for pTyr-proteome analysis that can be used in basic research, diagnostic, or drug discovery workflows.
METHODS
pTyr-Peptide Enrichment with SH2 Domain Superbinders.
Phosphopeptide enrichment with superbinders was performed on a KingFisher robot. Streptavidin MagBeads (GenScript) were tested for binding capacity using the AVIDITY method.37 One hundred microliters of streptavidin MagBeads (45 nmol mL−1 capacity) was added to 400 μL of Buffer 1 (10 mM sodium phosphate, 150 mM NaCl, pH 7.5), mixed for 10 s, collected, and washed three times in 500 μL of Buffer 1 (3 min, slow speed, beads collected at each step). Next, streptavidin beads and 100 μg of superbinder in 600 μL of Buffer 1 were mixed for 1 h at a slow speed. Beads were collected and washed in 600 μL of Buffer 1 (3 min, slow speed). The digest was resuspended in 200 μL of 0.1 M Tris 8.5, 150 mM NaCl and mixed with collected superbinder beads for 1 h at a slow speed. Streptavidin MagBeads with bound superbinder were collected and washed three times in 600 μL of Buffer 1 (3 min, slow speed) and once in 600 μL of H2O (3 min, slow speed), with beads collected at each step. Peptides were eluted in 0.15% trifluoracetic acid (TFA) (15 min, medium speed). The eluate was immediately applied to a 10 mg Atlas Cerex column, which had been washed in 100% ACN and then pre-equilibrated with 0.1% TFA/H2O. The bound peptides were washed with 1.5 mL of 0.1% TFA and eluted in three steps with 250 μL of 15% ACN/0.1% TFA, 30% ACN/0.1% TFA, and 70% ACN/0.1% TFA. Samples were dried by SpeedVac, resuspended in 20 μL of H2O/0.1% FA, loaded on Evotips (Evosep) following the vendor’s protocol, and subjected to LC-MS analysis. To test combinations of superbinders, different engineered SH2 domains were mixed in equal amounts for a total of 100 μg and the mixture was processed as described above for individual superbinders.
LC-MS Analysis.
Samples were analyzed with an Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific) equipped with an EvoSep One LC system and an Evo Easy-Spray adapter (Evosep). Peptides were eluted at low pressure from Evotips and separated on an analytical column (25 × 0.15 mm ID, 1,9 μm dp, C18 (PepSep)) connected to a stainless-steel emitter (Evosep) using a pre-programmed 88 min gradient (extended method 15 SPD) with 0.1% FA in Milli-Q water (mobile phase A) and 0.1% FA in ACN (mobile phase B). Survey full-scan MS spectra were acquired in the mass range 375–1550 m/z at 120,000 resolution, the automatic gain control (AGC) target set to 3e6, and maximum ion injection time (IT) at 20 ms. Peptides were fragmented above a threshold of 2.5e4 by higher energy collisional dissociation (HCD) at a resolution of 30,000, AGC target 1e6, maximum IT 60 ms, TopN of 15, an isolation width of 1.6 m/z, and a normalized collision energy of 28%. Charge state z = 1, unassigned charges and z ≥ 6 were rejected; dynamic exclusion was set to 30 s. A spray voltage of 1900 V in positive mode and an RF lens at 30% were used.
Data Analysis.
Thermo RAW files were converted to mzML38 using msconvert (ProteoWizard, version 3.0.21068)39 with “peakPicking true 1-” and “zeroSamplesremoveExtra” filters. mzML files were searched with Comet40 (version 2021.01 rev. 0) against the reviewed UniProt/SwissProt Homo sapiens proteome containing 20,386 proteins (downloaded October 12, 2021), appended with the common Repository of Adventitious Proteins (cRAP),41 and randomized decoys. The search was performed with a precursor mass tolerance of 20 ppm, a fragment bin tolerance of 0.02 m/z, and up to two missed cleavages allowed. Carbamidomethyl (+57.021464 Da) on cysteine was used as fixed modification, oxidation (+15.9949 Da) on methionine and tryptophan, and phosphorylation (+79.966331 Da) on serine, threonine, and tyrosine were set as variable modification. The search results were processed and statistically validated with the Trans-Proteomic-Pipeline (v6.0.0 OmegaBlock)42 including PeptideProphet,43 iProphet,44 and PTMProphet.45 PeptideProphet was run with accurate mass binning, Expect Score as discriminant, and one f-value distribution for all charge states. Decoy hits were used to pin down the negative distribution and the nonparametric model was enabled. PeptideProphet results were further processed with iProphet to refine peptide-spectrum match (PSM) probabilities. Correct localization of modified residues was determined with PTMProphet45 considering phosphorylation on serine, threonine, and tyrosine as well as oxidation on methionine and tryptophan, a minimum PSM probability for computation of 0.9 and 20 ppm MS2 peak tolerance. A minimum iProphet probability of 0.9 was applied in each experiment corresponding to an error rate of <0.01, and only peptides with a minimum PTMProphet mean best probability of 0.75 were considered for further analysis. pTyr-sites were determined using the reference proteome and an in-house Perl script.
Data Availability.
The data and results supporting this study are available within this manuscript and supplementary data files. The X-ray crystallographic data for newly solved structures have been deposited to the Protein Data Bank and can be accessed using the following IDs: sSrcF; 7T1U, sFes1; 7T1K, sFesS; and 7T1L. The MS phosphoproteomics data have been deposited to the ProteomeX-change Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE46 partner repository with the dataset identifier PXD030038. Additional data or materials used in this study, such as plasmids, for protein expression are available upon reasonable request.
Extended Methods.
A detailed description of the methods used in the study is available in the “Extended Methods” section of the Supporting Information.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank A. de Marco (University of Nova Gorica) for Erv1p and DsbC plasmids. This work was partially funded by the National Institutes of Health, National Institute of General Medical Sciences grant R01GM087221 (R.L.M), the Office of the Director S10OD026936 (R.L.M), the National Institute of Allergy and Infectious Diseases grant R21AI142302 (R.L.M), the National Institute on Aging rant U19AG023122 (R.L.M), and the National Science Foundation Award 1920268 (R.L.M). X-ray diffraction experiments were conducted at the Northeastern Collaborative Access Team beamlines at the Advanced Photon Source (APS), which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P30 GM124165). The APS is a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by the Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Funding was provided by the Canadian Institutes of Health Research grant MOP-93684 (S.S.S) and FDN-143277 (FS).
ABBREVIATIONS USED
- ELISA
enzyme linked immunosorbent assay
- MS
mass spectrometry
- pSer
phosphoserine
- pThr
phosphothreonine
- pTyr
phosphotyrosine
- PTM
post-translational modification
- SH2
Src-Homology 2
- Fes
Feline sarcoma
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/acschembio.2c00051
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschembio.2c00051.
Data collection and refinement statistics for newly solved structures (XLSX)
Detailed MS data (XLSX)
A table of human SH2 domains with associated sequences and structures (XLSX)
Supporting work can be presented as “Supporting Figures” (Figures S1–11); A detailed description of the methods, including automated AP-MS protocols, can be found in the “Extended Methods” section (PDF)
The authors declare the following competing financial interest(s): D.G.M, G.V. B.P.Y. and S.S.S. are inventors on a US patent application No. 63/214,946 which applies to the development and use of SH2 domain superbinders for the analysis of proteomes and synthetic biology.
Contributor Information
Gregory D. Martyn, Donnelly Centre for Cellular and Biomolecular Research, University of Toronto, Toronto, Ontario M5S3E1, Canada; Department of Molecular Genetics, University of Toronto, Toronto, Ontario M5S 1A8, Canada.
Gianluca Veggiani, Donnelly Centre for Cellular and Biomolecular Research, University of Toronto, Toronto, Ontario M5S3E1, Canada.
Ulrike Kusebauch, Institute for Systems Biology, Seattle, Washington 98109, United States.
Seamus R. Morrone, Institute for Systems Biology, Seattle, Washington 98109, United States
Bradley P. Yates, Donnelly Centre for Cellular and Biomolecular Research, University of Toronto, Toronto, Ontario M5S3E1, Canada
Alex U. Singer, Donnelly Centre for Cellular and Biomolecular Research, University of Toronto, Toronto, Ontario M5S3E1, Canada
Jiefei Tong, Program in Cell biology, Hospital for Sick Children, Toronto M5G 0A4, Canada.
Noah Manczyk, Department of Biochemistry, University of Toronto, Toronto, Ontario M5S 1A8, Canada; Lunenfeld-Tanenbaum Research Institute, Mount Sinai Hospital, Toronto, Ontario M5G 1X5, Canada.
Gerald Gish, Lunenfeld-Tanenbaum Research Institute, Mount Sinai Hospital, Toronto, Ontario M5G 1X5, Canada.
Zhi Sun, Institute for Systems Biology, Seattle, Washington 98109, United States.
Igor Kurinov, Department of Chemistry and Chemical Biology, Cornell University, NE-CAT, Argonne, Illinois 60439, United States.
Frank Sicheri, Department of Molecular Genetics and Department of Biochemistry, University of Toronto, Toronto, Ontario M5S 1A8, Canada; Lunenfeld-Tanenbaum Research Institute, Mount Sinai Hospital, Toronto, Ontario M5G 1X5, Canada.
Michael F. Moran, Department of Molecular Genetics, University of Toronto, Toronto, Ontario M5S 1A8, Canada; Program in Cell biology, Hospital for Sick Children, Toronto M5G 0A4, Canada; The Hospital for Sick Children, SPARC Biocentre, Toronto, Ontario M5G 0A4, Canada
Robert L. Moritz, Institute for Systems Biology, Seattle, Washington 98109, United States.
Sachdev S. Sidhu, Donnelly Centre for Cellular and Biomolecular Research, University of Toronto, Toronto, Ontario M5S3E1, Canada; Department of Molecular Genetics, University of Toronto, Toronto, Ontario M5S 1A8, Canada.
REFERENCES
- (1).Sharma K; D’Souza RCJ; Tyanova S; Schaab C; Wiśniewski J; Cox J; Mann M Ultradeep Human Phosphoproteome Reveals a Distinct Regulatory Nature of Tyr and Ser/Thr-Based Signaling. Cell Rep. 2014, 8, 1583–1594. [DOI] [PubMed] [Google Scholar]
- (2).Humphrey SJ; Karayel O; James DE; Mann M High-throughput and high-sensitivity phosphoproteomics with the Easy-Phos platform. Nat. Protoc 2018, 13, 1897–1916. [DOI] [PubMed] [Google Scholar]
- (3).Bian Y; Li L; Dong M; Liu X; Kaneko T; Cheng K; Liu H; Voss C; Cao X; Wang Y; et al. Ultra-deep tyrosine phosphoproteomics enabled by a phosphotyrosine superbinder. Nat. Chem. Biol 2016, 12, 959–966. [DOI] [PubMed] [Google Scholar]
- (4).Moran MF; Koch CA; Anderson D; Ellis C; England L; Martin GS; Pawson T Src homology region 2 domains direct protein-protein interactions in signal transduction. Proc. Natl. Acad. Sci. U.S.A 1990, 87, 8622–8626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Waksman G; Kominos D; Robertson SC; Pant N; Baltimore D; Birge RB; Cowburn D; Hanafusa H; Mayer BJ; Overduin M; et al. Crystal structure of the phosphotyrosine recognition domain of SH2 of v-src complexed with tyrosine-phosphorylated peptides. Nature 1992, 358, 646–653. [DOI] [PubMed] [Google Scholar]
- (6).Bradshaw JM; Waksman G Calorimetric examination of high-affinity Src SH2 domain-tyrosyl phosphopeptide binding: Dissection of the phosphopeptide sequence specificity and coupling energetics. Biochemistry 1999, 38, 5147–5154. [DOI] [PubMed] [Google Scholar]
- (7).Kaneko T; Huang H; Zhao B; Li L; Liu H; Voss CK; Wu C; Schiller MR; Li SS-C Loops govern SH2 domain specificity by controlling access to binding pockets. Sci. Signal 2010, 3, ra34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Kaneko T; Huang H; Cao X; Li X; Li C; Voss C; Sidhu SS; Li SSC Superbinder SH2 domains act as antagonists of cell signaling. Sci. Signal 2012, 5, ra68. [DOI] [PubMed] [Google Scholar]
- (9).Akishiba M; Takeuchi T; Kawaguchi Y; Sakamoto K; Yu HH; Nakase I; Takatani-Nakase T; Madani F; Gräslund A; Futaki S Cytosolic antibody delivery by lipid-sensitive endosomolytic peptide. Nat. Chem 2017, 9, 751–761. [DOI] [PubMed] [Google Scholar]
- (10).Malabarba MG; Milia E; Faretta M; Zamponi R; Pelicci PG; Di Fiore PP A repertoire library that allows the selection of synthetic SH2s with altered binding specificities. Oncogene 2001, 20, 5186–5194. [DOI] [PubMed] [Google Scholar]
- (11).Virdee S; Macmillan D; Waksman G Semisynthetic Src SH2 Domains Demonstrate Altered Phosphopeptide Specificity Induced by Incorporation of Unnatural Lysine Derivatives. Chem. Biol 2010, 17, 274–284. [DOI] [PubMed] [Google Scholar]
- (12).Marengere LEM; Songyang Z; Gish GD; Schaller MD; Parsons JT; Stern MJ; Cantley LC; Pawson T SH2 domain specificity and activity modified by a single residue. Nature 1994, 369, 502–505. [DOI] [PubMed] [Google Scholar]
- (13).Kaneko T; Sidhu SS; Li SSC Evolving specificity from variability for protein interaction domains. Trends Biochem. Sci 2011, 36, 183–190. [DOI] [PubMed] [Google Scholar]
- (14).Tiruthani K; Mischler A; Ahmed S; Mahinthakumar J; Haugh JM; Rao BM Design and evaluation of engineered protein biosensors for live-cell imaging of EGFR phosphorylation. Sci. Signal 2019, 12, No. aap7584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Tong J; Cao B; Martyn GD; Krieger JR; Taylor P; Yates B; Sidhu SS; Li SSC; Mao X; Moran MF Protein-phosphotyrosine proteome profiling by superbinder-SH2 domain affinity purification mass spectrometry, sSH2-AP-MS. Proteomics 2017, 17, No. 1600360. [DOI] [PubMed] [Google Scholar]
- (16).Veggiani G; Huang H; Yates BP; Tong J; Kaneko T; Joshi R; Li SSC; Moran MF; Gish G; Sidhu SS Engineered SH2 domains with tailored specificities and enhanced affinities for phosphoproteome analysis. Protein Sci. 2019, 28, 403–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Ladbury JE; Arold ST Energetics of Src homology domain interactions in receptor tyrosine kinase-mediated signaling. In Methods Enzymol, 1st ed.; Elsevier Inc: New York., 2011; pp 147–183. [DOI] [PubMed] [Google Scholar]
- (18).Ladbury JE; Lemmon MA; Zhou M; Green J; Botfield MC; Schlessinger J Measurement of the binding of tyrosyl phosphopeptides to SH2 domains: a reappraisal. Proc. Natl. Acad. Sci. U.S.A 1995, 92, 3199–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Liu BA; Jablonowski K; Raina M; Pawson T; Nash PD; Arce M The Human and Mouse Complement Resource of SH2 Domain Proteins - Establishing the Boundaries of Phosphotyrosine Signaling. Mol. Cell 2006, 22, 851–868. [DOI] [PubMed] [Google Scholar]
- (20).Liu BA; Shah E; Jablonowski K; Stergachis A; Engelmann B; Nash PD The SH2 Domain-Containing Proteins in 21 Species Establish the Provenance and Scope of Phosphotyrosine Signaling in Eukaryotes. Sci. Signal 2011, 4, ra83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Tinti M; Kiemer L; Costa S; Miller M; Sacco F; Jesper V; Carducci M; Paoluzi S; Langone F; Christopher T; et al. The SH2 domain interaction landscape. Cell Rep. 2013, 3, 1293–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Sidhu SS; Lowman HB; Cunningham BC; Wells JA Phage Display for Selection of Novel Binding Peptides. Methods Enzymol. 2000, 328, 333–363. [DOI] [PubMed] [Google Scholar]
- (23).Filippakopoulos P; Kofler M; Hantschel O; Gish GD; Grebien F; Salah E; Neudecker P; Kay LE; Turk BE; Superti-Furga G; et al. Structural Coupling of SH2-Kinase Domains Links Fes and Abl Substrate Recognition and Kinase Activation. Cell 2008, 134, 793–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Dunant NM; Messerschmitt AS; Ballmer-Hofer K Functional interaction between the SH2 domain of Fyn and tyrosine 324 of hamster polyomavirus middle-T antigen. J. Virol 1997, 71, 199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Machida K; Thompson CM; Dierck K; Jablonowski K; Kärkkäinen S; Liu B; Zhang H; Nash PD; Newman DK; Nollau P; et al. High-Throughput Phosphotyrosine Profiling Using SH2 Domains. Mol. Cell 2007, 26, 899–915. [DOI] [PubMed] [Google Scholar]
- (26).Jones RB; Gordus A; Krall JA; MacBeath G A quantitative protein interaction network for the ErbB receptors using protein microarrays. Nature 2006, 439, 168–174. [DOI] [PubMed] [Google Scholar]
- (27).Hause RJ; Leung KK; Barkinge JL; Ciaccio MF; Chuu C. pin.; Jones RB Comprehensive Binary Interaction Mapping of SH2 Domains via Fluorescence Polarization Reveals Novel Functional Diversification of ErbB Receptors. PLoS One 2012, 7, No. e44471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Huang H; Li L; Wu C; Schibli D; Colwill K; Ma S; Li C; Roy P; Ho K; Songyang Z; et al. Defining the Specificity Space of the Human Src Homology 2 Domain. Mol. Cell. Proteomics 2008, 7, 768–784. [DOI] [PubMed] [Google Scholar]
- (29).Fairhead M; Howarth M Site-specific biotinylation of purified proteins using BirA. In Site-Specific Protein Labeling: Methods and Protocols, Gautier A; Hinner MJ, Eds.; Springer: New York, New York, NY., 2015; pp 171–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Chua XY; Mensah T; Aballo T; Mackintosh SG; Edmondson RD; Salomon AR Tandem mass tag approach utilizing pervanadate BOOST channels delivers deeper quantitative characterization of the tyrosine phosphoproteome. Mol. Cell. Proteomics 2020, 19, 730–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Kauko O; Imanishi SY; Kulesskiy E; Yetukuri L; Laajala TD; Sharma M; Pavic K; Aakula A; Rupp C; Jumppanen M; et al. Phosphoproteome and drug-response effects mediated by the three protein phosphatase 2A inhibitor proteins CIP2A, SET, and PME-1. J. Biol. Chem 2020, 295, 4194–4211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Van Der Maaten L; Hinton G Visualizing data using t-SNE. J. Mach. Learn. Res 2008, 9, 2579–2625. [Google Scholar]
- (33).Schneider M; Lane L; Boutet E; Lieberherr D; Tognolli M; Bougueleret L; Bairoch A The UniprotKB/Swiss-Prot knowledgebase and its Plant Proteome Annotation Program. J. Proteomics 2009, 72, 567–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Lane L; Argoud-Puy G; Britan A; Cusin I; Duek PD; Evalet O; Gateau A; Gaudet P; Gleizes A; Masselot A; et al. NeXtProt: A knowledge platform for human proteins. Nucleic Acids Res. 2012, 40, 76–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Desiere F; Deutsch EW; Nesvizhskii AI; Mallick P; King NL; Eng JK; Aderem A; Boyle R; Brunner E; Donohoe S; et al. Integration with the human genome of peptide sequences obtained by high-throughput mass spectrometry. Genome Biol. 2004, 6, No. R9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Deutsch EW; Sun Z; Campbell D; Kusebauch U; Chu CS; Shteynberg D; Omenn GS; Moritz RL The State of the Human Proteome in 2014/2015 as viewed through PeptideAtlas: enhancing accuracy and coverage through AtlasProphet. J. Proteome Res 2015, 14, 3461–3473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Berg Luecke L; Gundry RL Assessment of Streptavidin Bead Binding Capacity to Improve Quality of Streptavidin-based Enrichment Studies. J. Proteome Res 2021, 20, 1153–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Martens L; Chambers M; Sturm M; Kessner D; Levander F; Shofstahl J; Tang WH; Römpp A; Neumann S; Pizarro AD; et al. mzML - A community standard for mass spectrometry data. Mol. Cell. Proteomics 2011, 10, R110.000133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Kessner D; Chambers M; Burke R; Agus D; Mallick P ProteoWizard: Open source software for rapid proteomics tools development. Bioinformatics 2008, 24, 2534–2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Eng JK; Jahan TA; Hoopmann MR Comet: An open-source MS/MS sequence database search tool. Proteomics 2013, 13, 22–24. [DOI] [PubMed] [Google Scholar]
- (41).Mellacheruvu D; Wright Z; Couzens AL; Lambert JP; St-Denis NA; Li T; Miteva YV; Hauri S; Sardiu ME; Low TY; et al. The CRAPome: A contaminant repository for affinity purification-mass spectrometry data. Nat. Methods 2013, 10, 730–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Deutsch EW; Mendoza L; Shteynberg D; Slagel J; Sun Z; Moritz RL Trans-Proteomic Pipeline, a standardized data processing pipeline for large-scale reproducible proteomics informatics. Proteomics: Clin. Appl 2015, 9, 745–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Keller AD; Nesvizhskii AI; Kolker E; Aebersold R Empirical statistical model to estimate the accuracy of protein identifications made by MS/MS and database search. Anal. Chem 2002, 74, 37–38. [DOI] [PubMed] [Google Scholar]
- (44).Shteynberg D; Deutsch EW; Lam H; Eng JK; Sun Z; Tasman N; Mendoza L; Moritz RL; Aebersold R; Nesvizhskii AI iProphet: Multi-level integrative analysis of shotgun proteomic data improves peptide and protein identification rates and error estimates. Mol. Cell. Proteomics 2011, 10, No. M111.007690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Shteynberg DD; Deutsch EW; Campbell DS; Hoopmann MR; Kusebauch U; Lee D; Mendoza L; Midha MK; Sun Z; Whetton AD; Moritz RL PTMProphet: Fast and Accurate Mass Modification Localization for the Trans-Proteomic Pipeline. J. Proteome Res 2019, 18, 4262–4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Perez-Riverol Y; Csordas A; Bai J; Bernal-Llinares M; Hewapathirana S; Kundu DJ; Inuganti A; Griss J; Mayer G; Eisenacher M; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data and results supporting this study are available within this manuscript and supplementary data files. The X-ray crystallographic data for newly solved structures have been deposited to the Protein Data Bank and can be accessed using the following IDs: sSrcF; 7T1U, sFes1; 7T1K, sFesS; and 7T1L. The MS phosphoproteomics data have been deposited to the ProteomeX-change Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE46 partner repository with the dataset identifier PXD030038. Additional data or materials used in this study, such as plasmids, for protein expression are available upon reasonable request.