

Graphical abstract
Keywords: Protein-protein interaction network, Neighborhood, Diffusion, Context-specific network
Abstract
Protein-protein interaction network (PPIN) analysis is a widely used method to study the contextual role of proteins of interest, to predict novel disease genes, disease or functional modules, and to identify novel drug targets. PPIN-based analysis uses both generic and context-specific networks. Multiple contextualization methodologies have been described, such as shortest-path algorithms, neighborhood-based methods, and diffusion/propagation algorithms. This review discusses these methods, provides intuitive representations of PPIN contextualization, and also examines how the quality of such context-specific networks could be improved by considering additional sources of evidence. As a heuristic, we observe that tasks such as identifying disease genes, drug targets, and protein complexes should consider local neighborhoods, while uncovering disease mechanisms and discovering disease-pathways would gain from diffusion-based construction.
1. Introduction
The emergence and growth of network medicine in the last decade has been facilitated by the growth of publicly-accessible molecular datasets [1]. Various kinds of interactions such as protein-protein interactions (PPIs), transcription factor-gene regulatory interactions, perturbation effects of drugs and small molecules on gene expression, etc., have been systematically documented [2], [3], [4]. These interactions can be represented as networks or graphs, which are composed of a set of nodes, also called vertices, representing the discrete interacting entities (whose nature depends on the type of data under study), and a set of links, also called edges, representing physical or functional interactions between nodes. Networks consisting of different data types have been leveraged for a variety of tasks, such as the identification of novel disease proteins and drug targets, predictions of drug side-effects and toxicity, or the discovery of functional and disease modules, among others [5], [6]. Thereby, different types of biological networks have been explored, such as gene-regulatory networks, metabolic networks, protein–protein interaction networks (PPIN), drug-target networks, etc. [7]. Amongst the PPINs, a variety of applications have been previously described. Table 1 illustrates some of the different application areas of PPINs.
Table 1.
Some applications of PPINs mentioned in literature.
| Authors | Type | Application | Comments | Ref |
|---|---|---|---|---|
| Tomkins and Manzoni | Review | PPIN analysis of Parkinson's disease | Illustrates the different uses of PPIN analysis, including exploration of the neighbourhood of a single gene, disease genes prioritization, exploration of novel functions, disease candidates and pathways, and for comparative studies with other neurodegenerative diseases | [8] |
| Vinayagam et al. | Method | Predicted novel cancer-associated genes | Applied concepts from control theory to PPIN analysis | [9] |
| Cheng et al. | Method | Predicted hundreds of drug-disease associations | Method based on network proximity of disease proteins and drug targets in a PPIN | [10] |
| Cheng et al. | Method | Predicted drug combinations | Network proximity applied to prediction of drug combinations. Approach validated for a combination of anti-hypertensives | [11] |
| Chautard E et al. | Review | Identifying drug targets | Analysis of drug targets in a PPIN identifies characteristics of drug targets, can guide drug design | [5] |
| Choobdar S et al. | Review | Identify various protein communities, functional and disease modules | DREAM challenge exhibits various approaches for identification of modules based on topology of networks, including PPINs | [12] |
| Maron et al. | Method | Patient specific subnetwork identification and disease sub-typing | Differences in types of cardiomyopathies (hypertrophic and dilated) detected based on patient-specific networks | [13] |
| Vavouraki et al. | Method | Disease stratification and exploring molecular mechanism | PPIN based study of Hereditary Spastic Paraplegia | [14] |
Some applications call for analysis of generic PPINs. For instance, a study of human disease symptoms showed that shared symptoms are linked to shared protein interactions. This study was based on analysis of a PPIN combined from 5 different databases [15]. However, in order to investigate a smaller system for specific tissues, or localized perturbations, a relevant context-specific network needs to be designed. As an example, a disease-specific network was constructed by mapping Parkinson's disease (PD) associated genes to 3 different generic PPINs [16]. The resulting consensus PD-specific network was analysed to identify novel candidates and drug targets. Approaches to obtain context-specific networks can be broadly divided into local ones (such as neighbourhood methods), and more global ones (such as diffusion-based methods) [17]. Each approach may lead to different context-specific networks, in terms of size and structure. Thus, any analysis performed using such a contextualized network is dependent on the choice of the method used for the construction. However, it might not always be clear what an optimal approach to network building would be or when a certain method should be preferred.
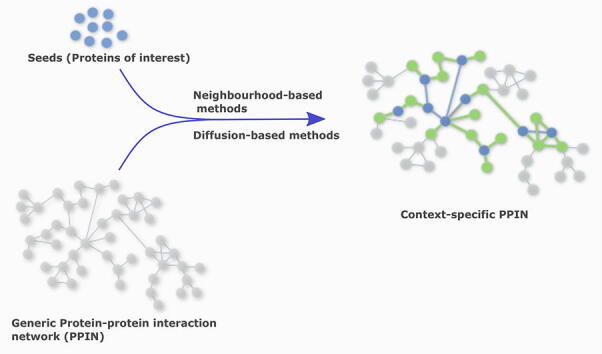
This review presents an overview of the network contextualization process (Fig. 1). To begin with, a description of generic PPINs is presented, together with some of the main public databases of protein-protein interactions. We then discuss the two approaches – neighbourhood and diffusion-based – in detail, drawing examples from various studies mentioned in the literature. Further, we explore methods of refining networks obtained using these approaches. We also examine the suitability of each approach in different contexts. The review concludes with a summary of our observations and suggestions, highlighting the application-specific and contextual nature of the PPIN construction process.
Fig. 1.

Flowchart of the major steps discussed in the manuscript to obtain a context-specific network. Inputs are taken to be proteins representing a specific context and a generic PPIN. Two main methods of contextualization – neighbourhood-based and diffusion based- are elaborated. We also discuss additional options for curation, using different data sources. Such a contextualized network can then be subject to further analysis such as identification of important nodes, and clustering.
2. Data availability and quality
PPIs considered here are physical interactions between proteins that lead to downstream biological changes in an organism [18]. (Fig. 2 (a)) Such interactions are responsible, for example, for signalling cascades involved in all biological processes. There are several generic and tissue-specific PPI databases available which offer experimentally verified and computationally predicted interactions. These databases document interactions available in the literature, such as those detected experimentally using methods such as yeast two-hybrid screening, affinity purification, etc., as well as those predicted by computational algorithms. Some of the most widely used databases include HPRD, APID, BioGRID, HINT, HIPPIE, STRING, and IntAct [19], [20], [21], [22], [23], [24], [25] Some databases provide confidence scores, which can be used to construct weighted networks, in which an interaction’s weight corresponds to the degree of certainty that the interaction is, indeed, a part of the real network. These generic PPIs contain interactions collected across multiple cell/tissue types, occurring at different times, in multiple biological contexts. However, not all interactions occur in all entities simultaneously, as each cell/tissue type has a characteristic protein expression profile. Contextualizing generic PPINs into tissue-specific networks is based on tissue-specific expression data. For example, in their study, Magger et al., identified 60 tissue-specific PPINs based on gene expression data and a generic PPIN [26]. Such tissue-specific networks are used to study localized conditions, or to identify specific drug targets. A database of cell-line specific networks, BioPLEX, is also under construction [27]. However, gene expression might not always be a good predictor of protein expression, and several studies have reported conflicting results in this regard [28], [29], [30]. Several mechanisms exist, such as transcriptional regulation, ribosomal competition, post-translational modifications, ubiquitin-dependent degradation etc., which regulate the protein levels, especially after the mRNA has been transcribed [31]. Hence, caution must be exercised when using gene expression as a proxy for protein expression. Background on various experimental techniques, an overview of how PPI databases are built, and a recent list of PPI databases can be found here [3], [32]. Some examples of PPI databases are shown in Table 2.
Fig. 2.

Different components of contextualized PPIN construction.
Table 2.
Some PPIN databases.
| Database | Size (Human)* | Type | Organisms | Comments | Website | Ref. |
|---|---|---|---|---|---|---|
| HPRD | 41,327 | Primary | 1 (h. sapiens) | Manually curated from literature. Last updated in 2010 | https://www.hprd.org/ | [19] |
| APID | 667,805 | Secondary | >400 | Experimentally validated interactions. Last updated in 2021. Collection of interactions from IntAct, HPRD, BioGRID, DIP and BioPlex | https://cicblade.dep.usal.es:8080/APID/init.action | [20] |
| BioGRID | 841,206+ 15,642 ++ | Primary | 81 | Lists physical and genetic interactions for various organisms. Contains a ‘muti-validated’ dataset with high confidence interactions, based on presence of multiple evidences of a given interaction. Updated monthly | https://thebiogrid.org/ | [21] |
| IntAct | 3,62,712** | Primary | 16 | Experimentally obtained data, curated data from literature | https://www.ebi.ac.uk/intact/home | [25] |
| BioPlex | ∼120,000 (HEK293T) ∼ 71,000 (HCT116) | Primary | 2 human Cell lines | Experimentally obtained Affinity-Purification Mass Spectrometry (AP-MS) data | https://bioplex.hms.harvard.edu/ | [27] |
| STRING | 1,19,38,498# | Secondary/Predictive | 14,094 | Physical and functional interactions obtained from experiments, computational predictions, text-mining and other databases. Provides confidence scores associated with each interaction. | https://string-db.org/ | [24] |
| HIPPIE | 7,83,182 | Secondary | 1 (h. sapiens) | Provides confidence scores and functional annotation for experimentally verified interactions. Last updated April 2022 | https://cbdm-01.zdv.uni-mainz.de/~mschaefer/hippie/index.php | [23] |
| HINT | 119,526 | Secondary | 12 | Manually curated high-throughput experimental data, curated from 8 different databases | https://hint.yulab.org/ | [22] |
| GeneMANIA | 1,17,49,785^ | Secondary | 9 | Physical and functional interactions. Can be used as a curation tool, e.g for adding missing members in a network; as a tool for functional annotation and interpretation | https://genemania.org/ | [33] |
*As obtained from the database website, May 2022.
** For Human species, only intra-species physical interactions between proteins considered here.
^All human interactions.
Non-Redundant-Physical, ++Non-Redundant – Genetic.
Physical and functional interactions.
3. Methods for constructing and contextualizing PPINs
3.1. Overview
Generic PPINs, as described above, catalogue interactions between different proteins. A contextualized network refers to a specific subset of such a generic PPIN, with a defined biological context. Biological contexts of interest include, for example, tissue-specific sets of interactions, the network of all PPIs in a specific cell type, or the network of all the proteins associated and suspected to be involved in, say, Alzheimer’s disease. The construction of context-specific PPINs from a generic PPIN involves the creation of a relevant network around specific proteins, or seeds. We define seeds as the proteins of interest that form a specific biological context. Seeds can be differentially expressed proteins obtained from a proteomics or transcriptomic experiment, or prior knowledge from literature on disease-associated proteins. Creating a contextualized network (graph) of these seeds involves mapping these seeds onto a generic PPIN, thus identifying the connections between the seeds, and adding new nodes from the neighbourhood of the seeds. Curation of obtained network can be done based on additional databases. Such a contextualized network can then be subjected to various ways of network analysis such as centrality calculations and clustering.
Interactions between proteins can be either transient or permanent. Description of changes in network topology over time is possible as a sequence of static networks, which are snapshots of the effective interactions at specific timepoints, therefore enabling the study of the network over time. Some methods, explicitly considering the protein levels as states of the model, require specific construction methods, kinetic parameters, which draw from a variety of sources and are highly dependent of the mathematical approach. In this work, we consider only static contextualized networks in detail, leaving aside dynamic networks, where the structural changes of the network are studied over time.
The first step of static PPIN contextualization is the mapping of the seeds (Fig. 2 (b)). Often, not all of the seeds map to the generic PPINs. This could either be because the protein is missing from the database, or due to protein annotation discrepancies. Hence, network construction may begin with a reduced number of seeds (Fig. 3 (a)). This initial seed mapping allows one to already obtain the first impressions about their topology, connectivity, helps visualize their placement in the network, and indicate their importance. Following the initial seed mapping, one can explore their contextual, topological and biological characteristics further via connecting the seeds based on existing direct edges (Fig. 2(c)) and/or expanding the network (Fig. 2(d), (e), (f)). The two broad approaches to contextualization are the neighbourhood-based methods – which include shortest-path and k-step neighbour methods – and the diffusion-based methods.
Fig. 3.

Illustration of network building methods: In terms of number of nodes and edges, simply connecting seed nodes (a) after mapping them to a generic PPIN (which could result in the loss of number of seed nodes) will keep the number of nodes constant, but will lead to a modest increase in the number of edges. Connecting nodes via shortest paths (b) would increase the number of nodes as well as edges. Here, the choice of whether one or more shortest paths are considered will indeed affect the size of the final network. A steep increase may be expected in the size of the network in terms of both number of nodes and edges when the neighbourhood/diffusion-based network building is applied (c), especially if hub nodes are present in the seed genes. The largest size of a network is the entire generic PPIN used in the process. We can illustrate the network building process, as a bottom-up construction method (d) or a top-down contextualization process (e). In the bottom-up construction, starting from nodes of interest, one can build up the connections between the nodes based on available evidence in generic PPIN, or add new nodes and edges to the starting nodes to understand how they influence and are influenced by their interaction partners. On the other hand, one can start with a generic PPIN, and trim away nodes and edges that may not be expressed in specific tissues or cell types, or in certain disease contexts. In a constructed network or in contextualization of a network, multiple criteria can be used to reach a final network. A constructed/contextualized network can further be appended or pruned (f). For example, for a network constructed by connecting the seeds via shortest paths, one would need to consider interactions among the newly added seeds, thus increasing the number of edges of the network while keeping the number of nodes constant. Alternatively, given evidence of expression, one may consider including additional nodes, and thus additional edges to the network. On the other hand, one may prune the network based on additional criteria, such as removing peripheral nodes (reducing number of nodes and edges), or simply removing some of the edges that may not have experimental support. The loss of an edge may or may not reduce the number of nodes. While these steps may seem trivial, they affect the size and topology of the network, and have a major effect on the predictions and conclusions of network-based analyses.
The size of the contextualized network – measured in terms of nodes and edges of the network – varies, based on the contextualization process followed. In case only the existing edges between the seeds are considered, the number of nodes remains constant, after the initial loss of seeds during mapping, while the edges corresponding to their connectivity are added (Fig. 3 (a)). Connecting seeds via shortest paths usually results in the inclusion of additional, non-seed nodes (Fig. 2(d) and Fig. 3 (b)). Network expansion around seeds via either neighbourhood approaches or diffusion-based approaches generally leads to much larger networks, compared with the starting number of seeds, especially if some seeds are hub proteins (interacting with a large number of proteins) (Fig. 2(e), (f) and Fig. 3 (c)).
The final size and structure of the resulting network depends on documented links between the proteins – thus, the state of completion of the database-, the approach, and any additional curation steps. For neighbourhood approaches, one criterion could be a confidence scores threshold for the inclusion of interactions. When using diffusion algorithms, the size of the obtained network is dependent on the cut-off values considered for inclusion of nodes.
Conceptually, PPIN construction can be viewed either as a bottom-up process: starting from a set of known proteins of interest, one can construct a network based on physical interactions between these known nodes, and expand the network (Fig. 3 (d)), or it can also be thought of as a top-down approach (Fig. 3 (e)): starting from a generic database, one then prunes nodes and edges to give a more specific network.
In the next section, variants of two broad construction/contextualization approaches: local neighbourhood-based, and diffusion-based, are discussed.
3.2. Connecting proteins and neighbourhood-based methods
The simplest approach to obtaining a context-specific PPIN is to connect every pair of seeds when such interactions are documented in a database (Fig. 2 (c)). The resulting network would have the same number of seeds, with an increased number of edges. (Fig. 3 (a)) This could lead to either a dense, single-component network, or several disconnected components, depending on the number of seeds and of relevant interactions present in the database. In the case of disconnected components, while these small components may help understand the interactions and functions in which the seeds are involved, the lack of a single network may limit the scope of analysis.
Another approach is to connect the seeds via the shortest paths (Fig. 2 (d)): for every pair of seed node, if there exist a path in the generic network which is shorter and not yet part of the contextualized one, include all nodes and interactions along this shortest path. This method of network construction was the idea behind some tools such as Lists2Networks and POINeT [34], [35]. These tools use different generic PPIs, and are designed to contextualize them, based on user-provided seeds. They also have users choose the maximum number of intermediary nodes on such shortest paths. POINeT has other options to refine networks such as removal of peripheral (degree 1) nodes, setting filters on the number of references for interactions, and adding confidence to interactions if two interactors of one of the interactions were both present in query list, if two interactors shared a GO term, or interologs (conserved interactions across species, such that interactions between two proteins in one organism are conserved in another organism’s orthologs) are present [36]. These tools are no longer maintained, however. Wang and Loscalzo proposed the Seed Connector algorithm (SCA) to connect disease-associated proteins scattered in a PPI network by adding as few additional linking nodes as possible [37]. This is done by iteratively adding neighbours of seeds, and identifying additional nodes that yield the largest connected component containing the maximum number of the seeds. The authors showed that the resulting modules are coherent and correspond to disease mechanisms and pathways, and are also enriched in known targets of existing drugs.
When multiple shortest paths exist between seeds, the user must choose a criterion, such as including nodes on all of the shortest paths, or only including paths and nodes that are shared among different seeds. In a method proposed by Garcia-Vaquero et al., aiming to identify proteins involved in multiple conditions, betweenness centrality is used to ascertain proteins that appear most frequently in the shortest paths between different disease modules [38]. A similar approach was used to define a ‘flow centrality score’ based on betweenness centrality considering paths between the disease modules of asthma and COPD [39]. These choices underlie the trade-off between the size and quality of the context-specific network: if one prioritizes a minimal number of additions, then algorithms designed to arrive at an optimal network may be resource intensive. On the other hand, allowing for all possible paths would considerably increase the size of the network, increase computation time, and may add a high number of false positives. Approximations can be used to save computational time, but running such optimizations for large sets of networks may still be resource-intensive. For weighted networks, when the choice of the shortest path is linked to minimizing or maximizing the edge weights, Dijkstra’s algorithm provides an exact solution [40]. For example, in PPINs that provide edge weights based on the quantity of evidence available for a given interaction, prioritizing paths with highest edge weights would be equivalent to ensuring only the most certain interactions are included in the network.
The direct neighbourhood approach to network construction is based on the “guilt-by-association” principle – interacting proteins may belong to similar functional modules, and thus contribute towards the same biological processes (Fig. 2 (e)) [41]. In the case of protein complexes, each of the members of a complex would be a first neighbour of at least one other member of the complex. Proteins playing a role in the etiology or progression of a certain diseases may have other nodes in their neighbourhood, which relay external stimuli, or are interaction partners involved in the same signalling pathway, and may be involved in the disease state as well. These neighbourhood nodes should be identified and functionally annotated.
To contextualize a network exploring the neighbourhood of known disease proteins, the number of neighbours (1-step, 2-step, k-step) to be included depends on the interest and context, such as whether the aim is to identify drug targets close to the known disease protein, or whether the interest is elucidating the disease mechanism and pathways involved. Multiple tools exist that allow for expanding seed nodes and obtaining subnetworks via k-step interactors, for example STRING and BIANA (Biologic Interactions and Network Analysis), which is available as a part of Galaxy InteractoMIX [24], [42], [43].
Generally, in the literature, only first (direct) and sometimes second-step neighbours are included. Liu et al. used an epidemiological network model to evaluate the effect of neighbourhood on spreading efficacy, using the SIR epidemiological model for simulating the spread of information through 6 different types of network [44]. While they proposed a novel measure –‘neighbourhood centrality’ – they state that 2-step neighbourhood returns higher rankings of nodes they refer to as ‘influential spreaders’. However, as the networks tested in this study were not biological networks, the applicability of this conclusion for biomedical research needs to be verified. Using signalling and PPI networks, Módos et al., showed that first neighbours of cancer-related proteins are important in pathogenesis and can be effective drug targets [45]. They also showed that several of the approved compounds target first neighbours.
However, one problem with this k-step neighbourhood approach is that it can yield a network where the number of nodes is considerably greater than the number of seeds, and could include several unrelated interactors. Indeed, some of the best-known proteins have many hundreds of known interactors both on account of their ubiquitous physiological roles – for example p53 and ubiquitin – and due to the fact that these are very well-studied proteins. Some of these interactions could be spurious, while some others may be context-specific, and thus, not found under all the conditions. The selection of such hub proteins may lead to very large networks encompassing many pathways and systems, in a way that is less dependent on the specific seed proteins, making these networks less useful for the study of specific processes or diseases.
3.3. Diffusion-based algorithms
Because neighbourhood and shortest-path approaches are local approaches, these may not capture perturbations from peripheral connections. A seed may have a limited role in a neighbourhood but might be connected to a larger component via only a few links. For example, a receptor may be specific to a ligand and have a specific downstream interactor, but at the same time, may be a part of a large signalling cascade via this single interactor. Some of the connecting members of a pathway may be missing, thus, may not be found as direct neighbours of disease-associated seeds. Also, when multiple, equally short paths exist between seed nodes, the approach becomes subjective. Alternative approaches have been developed to overcome these shortcomings based on the idea of information propagation, along the lines of ‘heat/fluid’ diffusion, where seed nodes are treated as ‘hot-spots’ and this heat diffuses through the network structure to identify other, potentially ‘hot’ candidates. The process is referred to as network diffusion or network propagation. Since this is an iterative process where each node is exchanging information with its neighbour, at convergence, each node will have been affected by the entire topology of the background network, and will be attributed a quantitative value based on its proximity to different seed nodes and the topology of the network (Fig. 2(f)). This approach considers all possible paths, and thus overcomes the limitations of the shortest path methods [46].
Propagation over unweighted networks leads to equal flow to the neighbours, while in weighted networks, flows are functions of edge weights. Variants of the method include random walk and random walk with restart (RWR). Details on the algorithms can be found in the review by Cowen et al. [46].
Propagation algorithms such as PRINCE, for example, require an input of disease-disease similarity measure and a background PPIN to identify disease-associated genes and complexes [47]. Other well-known examples of network propagation are the algorithms HotNet and HotNet2, that identify differentially mutated sub-networks in cancer networks, thus pointing to novel candidates [48], [49]. MUFFINN, another tool designed for cancer gene prediction, uses PPINs such as STRING and HumanNet, along with mutation data as seeds for network propagation [50]. It uses three different diffusion algorithms (Gaussian smoothing, RWR and iterative ranking). An application of network contextualization used to assess drug combinations in cancer is SynGeNet, which uses the belief propagation algorithm for subgraph identification [51]. Belief propagation finds the minimum Steiner tree, or an optimal subgraph using edge weights (based on the reliability of presence of the edge), and node weights (expression p-values) [52]. A recent review summarizes the different methods belonging to this class of algorithms and their application in integrating multiple layers of omics data [53].
Some limitations of the method include the need for user-defined parameters. For example, in case of RWR, the restart parameter (the probability at each step to restart the random walk from the initial node) needs to be specified, which ensures that the diffusion process penalizes longer walks from the seed nodes. One also needs a significance threshold to define the network of interest, and these choices are often case-specific. Hence, the application of network propagation algorithms needs to be optimized in the light of the data used and the application context.
Since networks are studied across different fields, some of these methods are discussed under different labels. An example is the PageRank algorithm, on which the Google search engine is based, which describes prioritization based on information diffusion through directed networks – a variant of random walk with restart [54]. Indeed, network building and analysis are tightly linked. Sometimes the boundaries between these steps may be diffuse, hence discretization of these steps can be tricky. For example, clustering or community detection, which is employed for network analysis, can also be seen as a way of network contextualization. As an example, applying a clustering algorithm contextualized the yeast interactome, resulting in clusters based on cellular functions [55]. Thus, clustering can identify specific subsets of proteins, corresponding to specific functional contexts. This approach can help functional annotation of proteins with previously unknown functions. Several of the methods underlying clustering include variants of neighbourhood-based and random-walk based approaches, along with those based on network topology. Several reviews discuss the many approaches and applications of clustering [56], [57].
3.4. Other methods
Another class of methods, called representation learning, is now being used extensively for PPIN analysis. These methods take as inputs entire networks and learn a vectorized representation (embedding) of their topology, which is then used for tasks such as label prediction, edge prediction, etc. [58]. However, underlying these methods is a combination of learning from neighbours and diffusion of information in the network. As mentioned before, studies and methods mentioned above focus on static PPINs. However, most of the PPIs are transient. Dynamic PPINs account for these time-dependent interactions. They are e.g. constructed from temporal correlation data, based on the strength of correlations between expression of different genes, or by combining static, generic PPINs with condition-specific temporal data. Several computational approaches also exist that combine experimental data with modelling approaches e.g. Boolean methods, for constructing dynamic networks, including protein networks [59], [60]. These dynamical networks present more accurate pictures of evolution of interactions, and are able to present, for example, the response of the organism to external stimuli. More discussions on the construction of dynamic networks can be found here [61].
3.5. Use of additional types of ‘interactions’ and databases
Constructed context-specific PPINs based on the above-mentioned approaches can be refined further by ‘appending’ or ‘pruning’ (Fig. 3 (f)) based on additional evidence, from other sources such as gene expression, or proteomics data, protein structure and protein complex databases, tissue expression catalogues, etc. Both appending and pruning may be at node or edge level. For example, if a cluster of proteins in the network forms a part of a known protein complex, the other members of the complex could be added (both nodes and edges). Interactions between proteins can be further filtered based on the number of occurrences of the interactions in databases, or by using confidence scores. For example, several studies use the confidence scores of 0.9 while using the STRING database [62], [63]. Stringent cut-offs will result in small networks, while relaxing the condition allows for more candidates. Appropriate cut-offs ideally limit the number of false positive interactions.
Several of these databases are manually curated, thus reducing the risk of artefacts from text-mining. However, databases still contain noisy data. For example, data observed in experiments such as affinity purification, may not occur in vivo. Certain interactions may only occur following specific perturbations, and may not exist in normal physiological conditions. An interesting observation reported is that some of the tissue-specific proteins interact with proteins which are a part of the core machinery in different cells, while some of the ubiquitously present ‘house-keeping’ proteins may have tissue-specific interactions [64]. Thus, determination of tissue-specific, true positives is challenging. Evidence for interactions may also vary based on the popularity of a protein: it is known that well-studied proteins contribute to a large percentage of the literature, leading to imbalanced data availability [65]. Databases constructed based on literature thus reflect this bias in terms of network connectivity of the well-known genes. On the other hand, the picture of the interactome is incomplete, as many interactions have probably not been evidenced yet, or might only exist in biological conditions that have not yet been investigated [66]. Because of these limitations, there is a need for reliable PPI prediction methods, to reduce the exponential number of experiments that would be required to complete the interactome. Hence, a prudent strategy of accurate predictions followed by experimental validation is needed.
Many databases such as STRING and GeneMANIA offer different levels of connections between an input protein and its known interactors such as co-expression, co-localization, pathway connectivity, and shared protein domains [24], [33]. Such information can be combined with physical interactions. Other databases such as protein complex information, protein structures, and pathway databases can complement information in PPINs and help append/prune interactions. For example, Cheng et al. constructed a ‘structurally resolved PPIN’ based on the availability of experimental or predicted protein structures [67]. Guala et al. provide a comprehensive view of different data types available that can be integrated to build high-confidence networks [68].
However, low overlap between different databases may lead to use of limited data. This is a vicious cycle, that well-studied proteins will have a large body of evidence to support observed interactions, which would then be selected for further study. Thus, while choosing interactions with strong evidence may be preferred, it might impede the discovery of lesser known candidates. Hence, obtaining high-quality basal networks, with sufficient support for inclusion of interactions to ensure new knowledge can be uncovered, while reducing false positives, is crucial for generating superior analytical results.
Using multiple databases and multiple versions of the same database can also be a strategy to reduce database dependency of the results. Since different PPI databases show limited overlap in their listed interactions, several studies have designed strategies to overcome this bias [69]. One is to use separate databases, and collate independently obtained results or identify common ones. Another is to use different databases for prediction and validation. Indeed, different versions of the same database show differences. The study by Cecchini et al., combined the strategies of using multiple datasets, and multiple versions of the same dataset [70]. They used two different PPINs -BioGRID and HPRD – for training and prediction of novel metabolic disease genes, and validated the predictions using a subsequent version of the Comparative Toxicogenomics Database, which led to the validation of 123 new candidates, that were absent in the previous version of the database. The May 2022 version of BioGRID has 841,206 non-redundant human protein interactions listed, while the January 2022 version contained 794,900. Changes in number of interactions affect the number of edges, may also affect the number of nodes, and hence the structure and topology of the network, which has direct implications on the network analysis and results. The scale free nature of biological networks ensures that the core, highly connected nodes retain their importance, but analyses that includes low-degree, peripheral nodes may be affected. Thus, results based on a specific release may need to be revisited over time to ascertain that the relevance and applicability of the analysis are retained.
4. Discussion
Choice of network building approach
In this review, we covered then main methodologies for contextualizing a generic PPI, given the available data and the scope of the study. A key question, then, is how does one choose the most suitable approach? Multiple factors can render the different methods more or less relevant depending on the scientific problem and the specific case. Among some of the constraints to be considered are the availability of data and ease of application.
The applicability of the different methods mentioned previously depends on the availability of specific data. Well-studied diseases with multiple -omics datasets allow for building context-specific networks based on multiple sources of a large quantity of information, while in the case of rare diseases, limited data availability may limit this process. For example, there exist abundant cancer datasets of different -omics types, with a large number of samples. In contrast, rare diseases are limited in the number of patients from which such data can be generated. Some of the algorithms require data such as mutation frequency, copy number variation, etc, which is much more readily available for cancer, than for other diseases.
Accessibility of different algorithms also poses a challenge. Some of these algorithms require coding skills for data handling and integration from multiple sources, and have been implemented in a variety of programming languages, thus requiring specialized training. Some of these methods are available as online tools, and apps for platforms such as Cytoscape [71]. However, many of these apps and online tools are no longer maintained and eventually become deprecated or lose compatibility over time.
For neighbourhood approaches, an appropriate value for the number of steps in k-step methods needs to be identified. Using a low value will fail to include the low-impact peripheral effectors, while a high value will make the contextualized network eventually converge towards the generic PPIN. In the latter case, the influence of the starting seeds would decrease, and the networks would lose their ‘context-specificity’. Diffusion algorithms require user-defined parameters like the rate or extent of diffusion. Thus, these methods may require either arbitrary inputs, or a detailed evaluation of the impact of different parameter values on the outcome.
Depending on the application, a focus on the local neighbourhood would be promising from a therapeutic perspective. Drugs affecting disease proteins or disrupting an interaction such as ligand-receptor binding typically tend to affect the immediate neighbourhoods. As mentioned, protein complexes involve direct-interactors. In such cases, the immediate neighbourhood of seeds would be of interest, rather than a large network, obtained from higher value of k in a k-step expansion or diffusion, which may not necessarily lead to useable predictions but could increase computational time and the required resources.
Few studies have directly compared the different methods of network contextualization. Shim et al. presented an interesting study [17] in which they compared the direct neighbourhood approach to the diffusion-based approach on human and worm functional gene networks for prediction of disease-associated genes. Using the area under the receiver-operating characteristic curve as an evaluation metric, they showed that the direct neighbourhood approach recovers more true positives when only the top predictions are considered. They argue that while it is generally shown that network propagation algorithms perform better in terms of gene prioritization, this outcome is based on the overall ranking of all genes under study. When only the top predictions are of interest (which is the case when experimental validation is the goal), neighbourhood approaches might be preferred.
They, however, added the caveat that the neighbourhood approach may not work if genes belonging to the same pathways are disconnected in the network. This could happen, for example, if the seeds are distant members of a pathway and the intermediate members of the pathway are missing, in which case diffusion algorithms would be preferred. This observation was highlighted in a study by Agrawal et al. [72]. They showed that neighbourhood methods perform poorly in discovering disease-pathways, as compared to random-walk based methods, which showed better performance. Across 519 diseases, the recall of the random-walk method was 0.356, but only 0.242 for the neighbourhood-based method. The Mean Reciprocal Rank of the predictions (proportional to prediction performance) was 0.061 compared to 0.029, indicating a lower (better) rank of the true positives for random-walk based methods. Their analysis also included embedding methods. Diffusion-based network construction can capture perturbation effects from peripheral interactors, and identify components of a pathway spread out across the generic PPIN. Hence, this approach could be useful in uncovering details of disease mechanisms, involving large signalling cascades and disease-pathways spread across the network. However, the authors also noted that performances of different methods varied for different diseases.
In the case of protein function prediction, Cao et al. argue that while hub proteins have a large number of interactors which might be involved in the same biological process, these interactors are not likely to have the same function [73]. They used the example of a chaperone as a hub protein, which interacts with thousands of functionally distinct proteins. They propose a new metric, based on graph embedding, called diffusion state distance (DSD) and show that it outperforms function prediction methods based on the shortest-path approach.
Task-specific benchmarking of different methods will be crucial to evaluate which of the network construction approaches would yield reliable predictions in the different practical applications. In reviewing the performance of different algorithms: network neighbourhood based, diffusion-based and algorithms based on Random walk with restart, Guala and Sonnhammer designed a benchmarking strategy based on Gene Ontology (GO) terms [74]. The benchmarking study re-iterates observations made by Shim et al., that while diffusion-based methods show an overall better performance, neighbourhood methods show a superior performance when only the top part of the output is taken into consideration. The comparison was made based on measures such as Median Rank Ratio of True Positives and partial Area Under the ROC Curve (pAUC). This pAUC was consistently higher for the MaxLink method, using neighbourhood-based approach, than for the methods using the PageRank or random-walk algorithms. Some of the benchmarking efforts have been cancer-specific e.g. this study which reviewed 12 methods of predicting cancer driver genes, based on eight benchmarking datasets [75]. Fine et al. proposed a ‘leave-one chromosome out’ cross-validation, applying it to 20 different gene prioritization methods [76]. Their evaluation method is based on stratified linkage-disequilibrium (LD) regression. The proposed method aims to avoid the pitfalls of relying on ‘gold-standard’ genes for validation, as such datasets do not include as-yet undiscovered disease-gene associations, and may not be useful for validating novel candidates. Other benchmarking approaches for PPIN-based analysis include [12] for assessment of module identification algorithms on different kinds of networks including PPINs. A survey of subnetwork identification was recently undertaken by Nguyen et al. [77], in which they tested 22 different methods. As PPINs are used in several contexts, each application will call for such benchmarking efforts.
Overall, these benchmarking efforts show that different data, network structure and assumptions underlie various methods, which may yield different but complementary insights. Thus, no method may be inherently superior, but could be more or less informative for a given context or application. Benchmarking efforts could help outline strengths, limitations and the range of applicability of different approaches.
5. Summary and outlook
PPINs yield several insights – from highlighting novel disease genes to prioritizing drug targets, and can be useful in diverse applications aiding translational medicine. However, reliable generic and context-specific networks are necessary to obtain reliable and robust predictions. Context-specific networks for disease-specific applications have been obtained using two main approaches: one based on local neighbourhood of seeds and the second being diffusion-based information transfer. In reviewing these approaches, the following observations can be made:
-
•
Several variants of neighbourhood-based and diffusion-based methods exist, and a variety of applications of these methods can be found in the literature. However, there is no standard method for a given application. The choice of the method remains dependent on the kind and availability of input data and the purpose of the analysis.
-
•
Local neighbourhood methods could be more useful for tasks such as identifying disease genes, drug targets, and protein complexes, while diffusion-based methods could yield better insights on protein function prediction, studying disease mechanisms and discovering disease-pathways.
-
•
Standard tools based on different algorithms can be very useful, especially to improve the accessibility of the algorithms. These tools should also be maintained over time which is often not the case.
-
•
Benchmarking of methods is necessary. In practice, it is difficult to compare different studies due to the diversity in available data for different diseases, the heterogeneity of networks, and the use of different datasets. Hence, devising standardized measures of performance of various methods, and studies performing comprehensive evaluation of different techniques are needed.
-
•
The quality of the databases has a direct impact on network-based analysis. Noisy data, incomplete coverage of interactions, and low overlap between databases necessitates several data pre-processing and integration steps during context-specific network construction.
The use of different approaches for PPI network construction makes it difficult to draw conclusions about the superiority of some methods across studies. As distinct approaches towards such network-based analysis might shed a different light on the same problem, the combination of multiple methods could yield complementary insights about the same dataset. However, follow-up experiments validating computational predictions are needed, and findings need to be fed back into the literature, in a continuous benchmarking effort, to ensure that such predictive methods remain useful and relevant. As the number of studies based on PPINs grows, a unifying framework for integrating insights based on different methods and data would be critical for obtaining a comprehensive view of biological systems.
CRediT authorship contribution statement
Apurva Badkas: Conceptualization, Writing – original draft, Writing – review & editing. Sébastien De Landtsheer: Conceptualization, Writing – original draft, Writing – review & editing. Thomas Sauter: Conceptualization, Writing – original draft, Writing – review & editing, Supervision.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- 1.Lee L.-Y.-H., Loscalzo J. Network Medicine in Pathobiology. Am J Pathol. 2019;189(7):1311–1326. doi: 10.1016/j.ajpath.2019.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Silverman E.K., et al. Molecular networks in Network Medicine: Development and applications. WIREs Syst Biol Med. Nov. 2020;12(6):e1489. doi: 10.1002/wsbm.1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rigden D.J., Fernández X.M. The 2022 Nucleic Acids Research database issue and the online molecular biology database collection. Nucleic Acids Res. Jan. 2022;50(D1):D1–D10. doi: 10.1093/nar/gkab1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barabási A.-L., Oltvai Z.N. Network biology: understanding the cell’s functional organization. Nat Rev Genet. 2004;5(2):101–113. doi: 10.1038/nrg1272. [DOI] [PubMed] [Google Scholar]
- 5.Chautard E., Thierry-Mieg N., Ricard-Blum S. Interaction networks: From protein functions to drug discovery. A review. Pathol Biol. 2009;57(4):324–333. doi: 10.1016/j.patbio.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 6.Pujol A., Mosca R., Farrés J., Aloy P. Unveiling the role of network and systems biology in drug discovery. Trends Pharmacol Sci. 2010;31(3):115–123. doi: 10.1016/j.tips.2009.11.006. [DOI] [PubMed] [Google Scholar]
- 7.Zhu X., Gerstein M., Snyder M. Getting connected: analysis and principles of biological networks. Genes Dev. May 2007;21(9):1010–1024. doi: 10.1101/gad.1528707. [DOI] [PubMed] [Google Scholar]
- 8.Tomkins J.E., Manzoni C. Advances in protein-protein interaction network analysis for Parkinson’s disease. Neurobiol Dis. 2021;155 doi: 10.1016/j.nbd.2021.105395. [DOI] [PubMed] [Google Scholar]
- 9.Arunachalam V., et al. Controllability analysis of the directed human protein interaction network identifies disease genes and drug targets. Proc Natl Acad Sci. May 2016;113(18):4976–4981. doi: 10.1073/pnas.1603992113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheng F., et al. Network-based approach to prediction and population-based validation of in silico drug repurposing. Nat Commun. 2018;9(1):2691. doi: 10.1038/s41467-018-05116-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheng F., Kovács I.A., Barabási A.-L. Network-based prediction of drug combinations. Nat Commun. 2019;10(1):1197. doi: 10.1038/s41467-019-09186-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Choobdar S., et al. Assessment of network module identification across complex diseases. Nat Methods. 2019;16(9):843–852. doi: 10.1038/s41592-019-0509-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maron B.A., et al. Individualized interactomes for network-based precision medicine in hypertrophic cardiomyopathy with implications for other clinical pathophenotypes. Nat Commun. 2021;12(1):873. doi: 10.1038/s41467-021-21146-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vavouraki N., et al. Integrating protein networks and machine learning for disease stratification in the Hereditary Spastic Paraplegias. iScience. 2021;24(5) doi: 10.1016/j.isci.2021.102484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou X., Menche J., Barabási A.-L., Sharma A. Human symptoms–disease network. Nat Commun. 2014;5(1):4212. doi: 10.1038/ncomms5212. [DOI] [PubMed] [Google Scholar]
- 16.Quan P., et al. Integrated network analysis identifying potential novel drug candidates and targets for Parkinson’s disease. Sci Rep. 2021;11(1):13154. doi: 10.1038/s41598-021-92701-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shim J.E., Hwang S., Lee I. Pathway-Dependent Effectiveness of Network Algorithms for Gene Prioritization. PLoS One. 2015;10(6) doi: 10.1371/journal.pone.0130589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jubb H.C., Pandurangan A.P., Turner M.A., Ochoa-Montaño B., Blundell T.L., Ascher D.B. Mutations at protein-protein interfaces: Small changes over big surfaces have large impacts on human health. Prog Biophys Mol Biol. 2017;128:3–13. doi: 10.1016/j.pbiomolbio.2016.10.002. [DOI] [PubMed] [Google Scholar]
- 19.Keshava Prasad T.S., et al. Human Protein Reference Database–2009 update. Nucleic Acids Res. 2009;37, no. Database:D767–D772. doi: 10.1093/nar/gkn892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alonso-López D., et al. APID database: redefining protein–protein interaction experimental evidences and binary interactomes. Database. 2019;2019:baz005. doi: 10.1093/database/baz005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oughtred R., et al. The BioGRID interaction database: 2019 update. Nucleic Acids Res. Jan. 2019;47(D1):D529–D541. doi: 10.1093/nar/gky1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Das J., Yu H. HINT: High-quality protein interactomes and their applications in understanding human disease. BMC Syst Biol. 2012;6(1):92. doi: 10.1186/1752-0509-6-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alanis-Lobato G., Andrade-Navarro M.A., Schaefer M.H. HIPPIE v2.0: enhancing meaningfulness and reliability of protein–protein interaction networks. Nucleic Acids Res. Jan. 2017;45(D1):D408–D414. doi: 10.1093/nar/gkw985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Szklarczyk D., et al. Correction to ‘The STRING database in 2021: customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets’. Nucleic Acids Res. Oct. 2021;49(18):10800. doi: 10.1093/nar/gkab835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.del Toro N., et al. The IntAct database: efficient access to fine-grained molecular interaction data. Nucleic Acids Res. Jan. 2022;50(D1):D648–D653. doi: 10.1093/nar/gkab1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Magger O., Waldman Y.Y., Ruppin E., Sharan R. Enhancing the Prioritization of Disease-Causing Genes through Tissue Specific Protein Interaction Networks. PLOS Comput. Biol. 2012;8(9) doi: 10.1371/journal.pcbi.1002690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huttlin E.L., et al. The BioPlex Network: A Systematic Exploration of the Human Interactome. Cell. 2015;162(2):425–440. doi: 10.1016/j.cell.2015.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu Y., Beyer A., Aebersold R. On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell. 2016;165(3):535–550. doi: 10.1016/j.cell.2016.03.014. [DOI] [PubMed] [Google Scholar]
- 29.Fortelny N., Overall C.M., Pavlidis P., Freue G.V.C. Can we predict protein from mRNA levels? Nature. 2017;547(7664):E19–E20. doi: 10.1038/nature22293. [DOI] [PubMed] [Google Scholar]
- 30.Li G.-H., et al. System-level metabolic modeling facilitates unveiling metabolic signature in exceptional longevity. Aging Cell. Apr. 2022;21(4):e13595. doi: 10.1111/acel.13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Buccitelli C., Selbach M. mRNAs, proteins and the emerging principles of gene expression control. Nat Rev Genet. 2020;21(10):630–644. doi: 10.1038/s41576-020-0258-4. [DOI] [PubMed] [Google Scholar]
- 32.Iacobucci I., Monaco V., Cozzolino F., Monti M. From classical to new generation approaches: An excursus of -omics methods for investigation of protein-protein interaction networks. J Proteomics. 2021;230 doi: 10.1016/j.jprot.2020.103990. [DOI] [PubMed] [Google Scholar]
- 33.Franz M., et al. GeneMANIA update 2018. Nucleic Acids Res. Jul. 2018;46(W1):W60–W64. doi: 10.1093/nar/gky311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lachmann A., Ma’ayan A. Lists2Networks: Integrated analysis of gene/protein lists. BMC Bioinf. 2010;11(1):87. doi: 10.1186/1471-2105-11-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee S.-A., et al. POINeT: protein interactome with sub-network analysis and hub prioritization. BMC Bioinf. 2009;10(1):114. doi: 10.1186/1471-2105-10-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yu H., et al. Annotation transfer between genomes: protein-protein interologs and protein-DNA regulogs. Genome Res. Jun. 2004;14(6):1107–1118. doi: 10.1101/gr.1774904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang R.-S., Loscalzo J. Network-Based Disease Module Discovery by a Novel Seed Connector Algorithm with Pathobiological Implications. J. Mol. Biol. 2018;430(18, Part A):2939–2950. doi: 10.1016/j.jmb.2018.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Garcia-Vaquero M.L., Gama-Carvalho M., Las Rivas J.D., Pinto F.R. Searching the overlap between network modules with specific betweeness (S2B) and its application to cross-disease analysis. Sci Rep. 2018;8(1):11555. doi: 10.1038/s41598-018-29990-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maiorino E., et al. Discovering the genes mediating the interactions between chronic respiratory diseases in the human interactome. Nat Commun. 2020;11(1):811. doi: 10.1038/s41467-020-14600-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dijkstra E.W. A note on two problems in connexion with graphs. Numer Math. 1959;1(1):269–271. doi: 10.1007/BF01386390. [DOI] [Google Scholar]
- 41.Schwikowski B., Uetz P., Fields S. A network of protein–protein interactions in yeast. Nat Biotechnol. 2000;18(12):1257–1261. doi: 10.1038/82360. [DOI] [PubMed] [Google Scholar]
- 42.Garcia-Garcia J., Guney E., Aragues R., Planas-Iglesias J., Oliva B. Biana: a software framework for compiling biological interactions and analyzing networks. BMC Bioinf. 2010;11(1):56. doi: 10.1186/1471-2105-11-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mirela-Bota P., et al. Galaxy InteractoMIX: An Integrated Computational Platform for the Study of Protein-Protein Interaction Data. J Mol Biol. 2021;433(11) doi: 10.1016/j.jmb.2020.09.015. [DOI] [PubMed] [Google Scholar]
- 44.Liu Y., Tang M., Zhou T., Do Y. Identify influential spreaders in complex networks, the role of neighborhood. Phys A Stat Mech its Appl. 2016;452:289–298. doi: 10.1016/j.physa.2016.02.028. [DOI] [Google Scholar]
- 45.Módos D., et al. “Neighbours of cancer-related proteins have key influence on pathogenesis and could increase the drug target space for anticancer therapies. NPJ Syst Biol Appl. 2017;3(1):2. doi: 10.1038/s41540-017-0003-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cowen L., Ideker T., Raphael B.J., Sharan R. Network propagation: a universal amplifier of genetic associations. Nat Rev Genet. 2017;18(9):551–562. doi: 10.1038/nrg.2017.38. [DOI] [PubMed] [Google Scholar]
- 47.Vanunu O., Magger O., Ruppin E., Shlomi T., Sharan R. Associating Genes and Protein Complexes with Disease via Network Propagation. PLOS Comput. Biol. 2010;6(1) doi: 10.1371/journal.pcbi.1000641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vandin F., Upfal E., Raphael B.J. Algorithms for Detecting Significantly Mutated Pathways in Cancer. J Comput Biol. Mar. 2011;18(3):507–522. doi: 10.1089/cmb.2010.0265. [DOI] [PubMed] [Google Scholar]
- 49.Leiserson M.D.M., et al. Pan-cancer network analysis identifies combinations of rare somatic mutations across pathways and protein complexes. Nat Genet. 2015;47(2):106–114. doi: 10.1038/ng.3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cho A., Shim J.E., Kim E., Supek F., Lehner B., Lee I. MUFFINN: cancer gene discovery via network analysis of somatic mutation data. Genome Biol. 2016;17(1):129. doi: 10.1186/s13059-016-0989-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Regan-Fendt K.E., et al. “Synergy from gene expression and network mining (SynGeNet) method predicts synergistic drug combinations for diverse melanoma genomic subtypes”. NPJ Syst Biol Appl. 2019;5(1):6. doi: 10.1038/s41540-019-0085-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bailly-Bechet M., et al. Finding undetected protein associations in cell signaling by belief propagation. Proc. Natl. Acad. Sci. 2011;108(2):882–887. doi: 10.1073/pnas.1004751108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Di Nanni N., Bersanelli M., Milanesi L., Mosca E. Network Diffusion Promotes the Integrative Analysis of Multiple Omics. Front Genet. 2020;11:106. doi: 10.3389/fgene.2020.00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Page L., Brin S., Motwani R., Winograd T. The PageRank citation ranking: Bringing order to the web. Stanford InfoLab. 1999 [Google Scholar]
- 55.Brun C., Chevenet F., Martin D., Wojcik J., Guénoche A., Jacq B. Functional classification of proteins for the prediction of cellular function from a protein-protein interaction network. Genome Biol. 2003;5(1):R6. doi: 10.1186/gb-2003-5-1-r6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bhowmick S.S., Seah B.S. Clustering and Summarizing Protein-Protein Interaction Networks: A Survey. IEEE Trans Knowl Data Eng. 2016;28(3):638–658. doi: 10.1109/TKDE.2015.2492559. [DOI] [Google Scholar]
- 57.Rasti S., Vogiatzis C. A survey of computational methods in protein–protein interaction networks. Ann Oper Res. 2019;276(1):35–87. doi: 10.1007/s10479-018-2956-2. [DOI] [Google Scholar]
- 58.Nelson W., Zitnik M., Wang B., Leskovec J., Goldenberg A., Sharan R. To Embed or Not: Network Embedding as a Paradigm in Computational Biology. Front Genet. 2019;10 doi: 10.3389/fgene.2019.00381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Razzaq M., Paulevé L., Siegel A., Saez-Rodriguez J., Bourdon J., Guziolowski C. Computational discovery of dynamic cell line specific Boolean networks from multiplex time-course data. PLOS Comput. Biol. 2018;14(10) doi: 10.1371/journal.pcbi.1006538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rodriguez A., Crespo I., Androsova G., del Sol A. Discrete Logic Modelling Optimization to Contextualize Prior Knowledge Networks Using PRUNET. PLoS One. 2015;10(6) doi: 10.1371/journal.pone.0127216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang J., Peng X., Peng W., Wu F.-X. Dynamic protein interaction network construction and applications. Proteomics. Mar. 2014;14(4–5):338–352. doi: 10.1002/pmic.201300257. [DOI] [PubMed] [Google Scholar]
- 62.Duan G., Walther D. The Roles of Post-translational Modifications in the Context of Protein Interaction Networks. PLOS Comput. Biol. 2015;11(2) doi: 10.1371/journal.pcbi.1004049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chuffa L.G. de A., et al. A meta-analysis of microRNA networks regulated by melatonin in cancer: Portrait of potential candidates for breast cancer treatment. J. Pineal Res. 2020;69(4) doi: 10.1111/jpi.12693. [DOI] [PubMed] [Google Scholar]
- 64.Bossi A., Lehner B. Tissue specificity and the human protein interaction network. Mol Syst Biol. Jan. 2009;5(1):260. doi: 10.1038/msb.2009.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gates A.J., Gysi D.M., Kellis M., Barabási A.-L. A wealth of discovery built on the human genome project—by the numbers. Nature Publishing Group; 2021. [DOI] [PubMed] [Google Scholar]
- 66.Dunham B., Ganapathiraju M.K. Benchmark Evaluation of Protein-Protein Interaction Prediction Algorithms. Molecules. 2022;27(1) doi: 10.3390/molecules27010041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cheng F., et al. Comprehensive characterization of protein–protein interactions perturbed by disease mutations. Nat Genet. 2021;53(3):342–353. doi: 10.1038/s41588-020-00774-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Guala D., Ogris C., Müller N., Sonnhammer E.L.L. Genome-wide functional association networks: background, data & state-of-the-art resources. Brief Bioinform. Jul. 2020;21(4):1224–1237. doi: 10.1093/bib/bbz064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bajpai A.K., et al. Systematic comparison of the protein-protein interaction databases from a user’s perspective. J Biomed Inform. 2020;103 doi: 10.1016/j.jbi.2020.103380. [DOI] [PubMed] [Google Scholar]
- 70.Cecchini V., Nguyen T.-P., Pfau T., Landtsheer S.D., Sauter T. 2019 11th International Conference on Knowledge and Systems Engineering (KSE) 2019. An Efficient Machine Learning Method to Solve Imbalanced Data in Metabolic Disease Prediction; pp. 1–5. [DOI] [Google Scholar]
- 71.Shannon P., et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. Nov. 2003;13(11):2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Agrawal M., Zitnik M., Leskovec J. “Large-scale analysis of disease pathways in the human interactome”. Biocomputing WORLD SCIENTIFIC. 2018;2017:111–122. [PMC free article] [PubMed] [Google Scholar]
- 73.Cao M., et al. Going the Distance for Protein Function Prediction: A New Distance Metric for Protein Interaction Networks. PLoS One. 2013;8(10) doi: 10.1371/journal.pone.0076339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Guala D., Sonnhammer E.L.L. A large-scale benchmark of gene prioritization methods. Sci Rep. 2017;7(1):46598. doi: 10.1038/srep46598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shi X., et al. Comprehensive evaluation of computational methods for predicting cancer driver genes. Brief. Bioinform. 2022:bbab548. doi: 10.1093/bib/bbab548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fine R.S., Pers T.H., Amariuta T., Raychaudhuri S., Hirschhorn J.N. Benchmarker: An Unbiased, Association-Data-Driven Strategy to Evaluate Gene Prioritization Algorithms. Am J Hum Genet. 2019;104(6):1025–1039. doi: 10.1016/j.ajhg.2019.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nguyen H., Shrestha S., Tran D., Shafi A., Draghici S., Nguyen T. A Comprehensive Survey of Tools and Software for Active Subnetwork Identification. Front Genet. 2019;10 doi: 10.3389/fgene.2019.00155. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
PPIs considered here are physical interactions between proteins that lead to downstream biological changes in an organism [18]. (Fig. 2 (a)) Such interactions are responsible, for example, for signalling cascades involved in all biological processes. There are several generic and tissue-specific PPI databases available which offer experimentally verified and computationally predicted interactions. These databases document interactions available in the literature, such as those detected experimentally using methods such as yeast two-hybrid screening, affinity purification, etc., as well as those predicted by computational algorithms. Some of the most widely used databases include HPRD, APID, BioGRID, HINT, HIPPIE, STRING, and IntAct [19], [20], [21], [22], [23], [24], [25] Some databases provide confidence scores, which can be used to construct weighted networks, in which an interaction’s weight corresponds to the degree of certainty that the interaction is, indeed, a part of the real network. These generic PPIs contain interactions collected across multiple cell/tissue types, occurring at different times, in multiple biological contexts. However, not all interactions occur in all entities simultaneously, as each cell/tissue type has a characteristic protein expression profile. Contextualizing generic PPINs into tissue-specific networks is based on tissue-specific expression data. For example, in their study, Magger et al., identified 60 tissue-specific PPINs based on gene expression data and a generic PPIN [26]. Such tissue-specific networks are used to study localized conditions, or to identify specific drug targets. A database of cell-line specific networks, BioPLEX, is also under construction [27]. However, gene expression might not always be a good predictor of protein expression, and several studies have reported conflicting results in this regard [28], [29], [30]. Several mechanisms exist, such as transcriptional regulation, ribosomal competition, post-translational modifications, ubiquitin-dependent degradation etc., which regulate the protein levels, especially after the mRNA has been transcribed [31]. Hence, caution must be exercised when using gene expression as a proxy for protein expression. Background on various experimental techniques, an overview of how PPI databases are built, and a recent list of PPI databases can be found here [3], [32]. Some examples of PPI databases are shown in Table 2.
Fig. 2.
Different components of contextualized PPIN construction.
Table 2.
Some PPIN databases.
| Database | Size (Human)* | Type | Organisms | Comments | Website | Ref. |
|---|---|---|---|---|---|---|
| HPRD | 41,327 | Primary | 1 (h. sapiens) | Manually curated from literature. Last updated in 2010 | https://www.hprd.org/ | [19] |
| APID | 667,805 | Secondary | >400 | Experimentally validated interactions. Last updated in 2021. Collection of interactions from IntAct, HPRD, BioGRID, DIP and BioPlex | https://cicblade.dep.usal.es:8080/APID/init.action | [20] |
| BioGRID | 841,206+ 15,642 ++ | Primary | 81 | Lists physical and genetic interactions for various organisms. Contains a ‘muti-validated’ dataset with high confidence interactions, based on presence of multiple evidences of a given interaction. Updated monthly | https://thebiogrid.org/ | [21] |
| IntAct | 3,62,712** | Primary | 16 | Experimentally obtained data, curated data from literature | https://www.ebi.ac.uk/intact/home | [25] |
| BioPlex | ∼120,000 (HEK293T) ∼ 71,000 (HCT116) | Primary | 2 human Cell lines | Experimentally obtained Affinity-Purification Mass Spectrometry (AP-MS) data | https://bioplex.hms.harvard.edu/ | [27] |
| STRING | 1,19,38,498# | Secondary/Predictive | 14,094 | Physical and functional interactions obtained from experiments, computational predictions, text-mining and other databases. Provides confidence scores associated with each interaction. | https://string-db.org/ | [24] |
| HIPPIE | 7,83,182 | Secondary | 1 (h. sapiens) | Provides confidence scores and functional annotation for experimentally verified interactions. Last updated April 2022 | https://cbdm-01.zdv.uni-mainz.de/~mschaefer/hippie/index.php | [23] |
| HINT | 119,526 | Secondary | 12 | Manually curated high-throughput experimental data, curated from 8 different databases | https://hint.yulab.org/ | [22] |
| GeneMANIA | 1,17,49,785^ | Secondary | 9 | Physical and functional interactions. Can be used as a curation tool, e.g for adding missing members in a network; as a tool for functional annotation and interpretation | https://genemania.org/ | [33] |
*As obtained from the database website, May 2022.
** For Human species, only intra-species physical interactions between proteins considered here.
^All human interactions.
Non-Redundant-Physical, ++Non-Redundant – Genetic.
Physical and functional interactions.