

Abstract
Diabetes is an established risk factor for colorectal cancer. However, colorectal cancer is a heterogeneous disease and it is not well understood whether diabetes is more strongly associated with some tumor molecular subtypes than others. A better understanding of the association between diabetes and colorectal cancer according to molecular subtypes could provide important insights into the biology of this association. We used data on lifestyle and clinical characteristics from the Colorectal Cancer Family Registry (CCFR) and the Genetics and Epidemiology of Colorectal Cancer Consortium (GECCO), including 9756 colorectal cancer cases (with tumor marker data) and 9985 controls, to evaluate associations between reported diabetes and risk of colorectal cancer according to molecular subtypes. Tumor markers included BRAF and KRAS mutations, microsatellite instability and CpG island methylator phenotype. In the multinomial logistic regression model, comparing colorectal cancer cases to cancer‐free controls, diabetes was positively associated with colorectal cancer regardless of subtype. The highest OR estimate was found for BRAF‐mutated colorectal cancer, n = 1086 (ORfully adj: 1.67, 95% confidence intervals [CI]: 1.36‐2.05), with an attenuated association observed between diabetes and colorectal cancer without BRAF‐mutations, n = 7959 (ORfully adj: 1.33, 95% CI: 1.19‐1.48). In the case only analysis, BRAF‐mutation was differentially associated with diabetes (P difference = .03). For the other markers, associations with diabetes were similar across tumor subtypes. In conclusion, our study confirms the established association between diabetes and colorectal cancer risk, and suggests that it particularly increases the risk of BRAF‐mutated tumors.
Keywords: colorectal cancer, diabetes, subtype
What's new?
Diabetes is a well‐known risk factor for colorectal cancer, but colorectal cancer varies widely among patients. To better understand the association between diabetes and particular molecular subtypes of colorectal cancer, these authors analyzed data from 9,756 colorectal cancer cases and 9,985 controls. They found that diabetes appears to increase the risk of tumors with BRAF mutations, which generally have poorer outcomes. The large pooled dataset allowed detection of even small variations among subtypes, but the study also was not able to account for some potentially relevant factors, such as metformin use.
Abbreviations
- BMI
body mass index
- CCFR
Colorectal Cancer Family Registry
- CIMP
CpG island methylator phenotype
- CPS‐II
Cancer Prevention Study‐II
- CRC
colorectal cancer
- DACHS
The German Darmkrebs: Chancen der Verhütung durch Screening Study
- EPIC
The European Prospective Investigation into Cancer and Nutrition Study
- GECCO
Genetics and Epidemiology of Colorectal Cancer Consortium
- HPFS
The Health Professionals Follow‐up Study
- MCCS
The Melbourne Collaborative Cohort Study
- MSI
microsatellite instability
- NFCCR
The Newfoundland Colorectal Cancer Registry
- NHS
The Nurses' Health Study
- NSHDS
The Northern Sweden Health and Disease Study
- OR
odds ratio
1. INTRODUCTION
Metabolic health, and excess body fat in particular, are involved in the development of colorectal cancer. 1 , 2 , 3 Individuals with diabetes mellitus, especially those with type 2 diabetes, have an increased risk of developing colorectal cancer. 4 This connection is likely independent of shared risk factors between diabetes and colorectal cancer. 5 Instead, the association between diabetes and colorectal cancer may depend on other mechanisms such as alterations to the gut microbiome, increased inflammation in the gut, hyperinsulinemia in early stage type 2 diabetes and activation of cancer promoting pathways. 6
Colorectal cancer is a heterogeneous disease, displaying considerable differences in molecular markers, which correlate with anatomical tumor location and other clinical and patient characteristics. For example, BRAF‐mutations are more common in proximal colon cancer, older patients and women and often co‐occur with high‐level microsatellite instability (MSI). 7 This raises the question of whether the risk factors for colorectal cancer also vary by tumor molecular markers. Investigations into potentially variable associations of metabolic factors and molecular subtypes of colorectal cancer have been inconsistent, but some reports suggest a possible association between adiponectin and lower risk of KRAS‐mutated colorectal cancer. 8 , 9 Adiponectin is known to have anti‐inflammatory effects and has been suggested as a potential treatment for obesity and type 2 diabetes. 10 Although studies have examined associations between body mass index (BMI) and different colorectal cancer phenotypes, 11 , 12 to our knowledge, no studies have investigated diabetes in relation to the risk of molecular subtypes of colorectal cancer, despite the fact that high BMI is one of the strongest known predictors of type 2 diabetes risk. 13
The aim of the present study was to investigate self‐reported diabetes status in relation to molecular tumor traits in colorectal cancer (BRAF and KRAS mutations, MSI status and CpG island methylator phenotype [CIMP]). To achieve this, we used pooled data and tissue samples from the Colorectal Cancer Family Registry (CCFR) and the Genetics and Epidemiology of Colorectal Cancer Consortium (GECCO), comprising a total of 9756 colorectal cases, with tumor marker data, and 9985 colorectal cancer free controls, all with information about self‐reported diabetes status.
2. METHODS
2.1. Study participants
The study population consisted of individuals diagnosed with colorectal cancer and controls from observational studies within GECCO and the CCFR, with available tumor marker data and self‐reported information about diabetes status (not distinguishing between type 1 and type 2) (Supplementary Materials and Table S1). Specifically, 9756 CRC cases had characterization of MSI status, BRAF and KRAS mutations, and CIMP status.
Studies with MSI, BRAF, KRAS and CIMP data included the Seattle, Ontario, Australia and Mayo Clinic sites from the Colon Cancer Family Registry (CCFR), the Cancer Prevention Study‐II (CPS‐II), the German Darmkrebs: Chancen der Verhütung durch Screening Study (DACHS), the Swedish centers of the European Prospective Investigation into Cancer and Nutrition study (EPIC‐Sweden), the Health Professionals Follow‐up Study (HPFS), the Melbourne Collaborative Cohort Study (MCCS), the Newfoundland Colorectal Cancer Registry (NFCCR), the Nurses' Health Study (NHS) and the Northern Sweden Health and Disease Study (NSHDS, an EPIC partner but with no overlap with EPIC‐Sweden in our study).
2.2. Tumor marker data
2.2.1. Collection and harmonization of MSI status CIMP status and BRAF and KRAS mutations
Data collection and harmonization of GECCO and CCFR tumor marker data have been described elsewhere. 14 , 15 Details on the analytical approach are described in the Supplementary Materials.
Briefly, MSI testing was primarily conducted using polymerase chain reaction (PCR) following the guidelines of the National Cancer Institute Bethesda Consensus Panel (CCFR, CPS‐II, MCCS, NHS) 16 with ≥4 interpretable markers typically required to classify tumors (Table S2). DACHS used a mononucleotide panel of four markers. Tumors were classified as MSI‐high if at least 30% of the markers showed instability and non‐MSI‐high if less than 30% of the makers showed instability. Other studies used immunohistochemistry (NSHDS, EPIC‐Sweden and subsets of CCFR and MCCS).
CIMP status was determined using methylation analyses as described in the Supplementary Materials and Table S3. Briefly, the CCFR, CPS‐II, HPFS, MCCS, NSHDS, EPIC Sweden and NHS used MethyLight to determine CIMP status. CPS‐II, HPFS, NSHDS, EPIC Sweden and NHS used an 8‐gene panel; CCFR and MCCS used a 5‐gene panel. DACHS determined CIMP status using a different 5‐gene panel and methods described by Warth et al. 17 We created two CIMP categories for this analysis: CIMP‐high and CIMP‐low/negative. In instances in which studies categorized tumors as CIMP‐high, CIMP‐low and CIMP‐negative, we combined CIMP‐low and CIMP‐negative into the CIMP‐low/negative category.
Studies assessed BRAF and KRAS mutations using PCR, sequencing and immunohistochemistry (Supplementary Materials). Most studies evaluated BRAF via c.1799T>A (V600E) mutations in exon 15 and KRAS via mutations in codons 12 and 13, although any mutation identified by one of the studies in BRAF and KRAS genes was included.
We further defined five combined colorectal tumor subtypes consistent with previously suggested classifications 18 , 19 , 20 : Type 1 (MSI‐high, CIMP‐high, BRAF‐mutated, KRAS‐wildtype), Type 2 (non MSI‐high, CIMP‐high, BRAF‐mutated, KRAS‐wildtype), Type 3 (non MSI‐high, CIMP‐low/negative, BRAF‐wildtype, KRAS‐mutated), Type 4 (non MSI‐high, CIMP‐low/negative, BRAF‐wildtype, KRAS‐wildtype) and Type 5 (MSI‐high, CIMP‐low/negative, BRAF‐wildtype, KRAS‐wildtype. Given our large sample size, we grouped additional marker combinations together into 11 additional types including: Type 6 (non MSI‐high, CIMP‐low/negative, BRAF‐mutated, KRAS‐wildtype), Type 8 (non MSI‐high, CIMP‐high, BRAF‐wildtype, KRAS‐mutated), Type 9 (MSI‐high, CIMP‐low/negative, BRAF‐wildtype, KRAS‐mutated), Type 11 (non MSI‐high, CIMP‐low/negative, BRAF‐wild type, KRAS‐mutated) and Type 14 (MSI‐high, CIMP‐high, BRAF‐wild type, KRAS‐wild type). Subtypes with fewer than 50 cases were dropped from analyses or were grouped together with the other marker combinations. These included: Type 7 (non MSI‐high, CIMP‐high, BRAF‐wildtype, KRAS‐wildtype), Type 10 (MSI‐high, CIMP‐high, BRAF‐wildtype, KRAS‐wildtype), Type 12 (MSI‐high, CIMP‐low/negative, BRAF‐mutated, KRAS‐wildtype), Type 13 (MSI‐high, CIMP‐low/negative, BRAF‐mutated, KRAS‐mutated), Type 15 (MSI‐high, CIMP‐high, BRAF‐wildtype, KRAS‐mutated) and Type 16 (MSI‐high, CIMP‐high, BRAF‐mutated, KRAS‐mutated). A summary of the combined marker classifications can be found in Figure 1 and Table S4.
FIGURE 1.
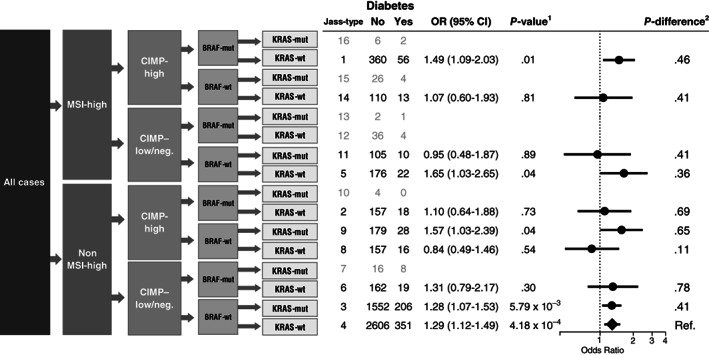
Case‐control associations between individuals reporting diabetes and individuals not reporting diabetes with risk of colorectal cancer subtypes defined by combined marker status. Error bars represent 95% confidence intervals. Adjusted for: study, sex, age at crc diagnosis, energy intake, family history, BMI, red meat, processed meat, vegetables, fruit, alcohol, smoking, exercise and aspirin/NSAID use. 1 P‐values were calculated using multinomial logistic regression, comparing colorectal cancer cases to cancer free controls separately for each defined Jass‐type with more than 50 cases included. 2 P difference was calculated using multinomial logistic regression, comparing cases of each Jass‐type to all additional cases not belonging to that type
2.3. Exposure data
Data collection and harmonization of GECCO and CCFR epidemiologic data have been described elsewhere. 14 , 15 , 21 Briefly, demographic and environmental risk factor data were self‐reported at in‐person interviews or via structured self‐administered questionnaires. Data were collected at study entry, or 1 to 2 years prior to sample ascertainment. A multistep iterative data‐harmonization procedure was applied, reconciling each study's unique protocols and data collection instruments. Multiple quality‐control checks were performed, and outlying values of variables were truncated to the minimum or maximum value of an established range for each variable. Variables were combined into a single dataset with common definition, standardized coding and standardized permissible values.
Diabetes status was obtained through self‐reported answers to questions about diabetes diagnoses (summarized in Table S1) and includes, but does not distinguish between, both type 1 and type 2. We defined age at the time of a colorectal cancer diagnosis for cases and time of enrolment for controls. Missing covariate data were assumed to be missing at random, conditional on observed data and were imputed using mean imputation.
2.4. Statistical analyses
We used multinomial models to estimate odds ratios (OR) and 95% confidence intervals (CIs) for the association between diabetes and the risk of each molecular tumor marker among colorectal cancer cases, defined as MSI‐high vs non‐MSI‐high, CIMP‐high vs low/negative and BRAF or KRAS mutated vs nonmutated. To test for differences related to subtype within the case‐only analysis we used unconditional logistic regression. In the combined marker analysis, Type 4 (non‐MSI‐high, CIMP‐low/negative, BRAF‐wildtype, KRAS‐wildtype) was used as a reference group in the case‐only analysis, whereas in the polytomous analysis, cancer‐free controls were used as the reference group. Both analyses used multinomial logistic regression to compare each molecular pathological subtype to the reference group. We also used multinomial logistic regression to estimate the association between diabetes and risk of colorectal cancer stratified by tumor location (colon, rectum, proximal colon, distal colon) and sex (male and female), and compared case‐combinations of marker and tumor site with controls. For case‐only analyses, we compared marker combinations stratified by tumor site.
Minimally adjusted models (presented in the Supplementary Materials) included study, age and sex as covariates, and fully adjusted models additionally included energy intake, family history of colorectal cancer, BMI, red and processed meat consumption, vegetable consumption, fruit consumption, alcohol use, smoking status, exercise and aspirin/NSAID use. Variables were first selected based on their theoretical relevance as potential confounders of an association between diabetes and colorectal cancer risk. We then performed a forward direction selection of suitable covariates resulting in fiber intake being dropped from the analysis. For polytomous and case‐only analyses of the primary subtypes (MSI, BRAF, KRAS and CIMP‐status), we considered a two sided P‐value of <.05 to be significant (in both minimally and fully adjusted models). However, when testing associations between diabetes and combined marker subtypes, we used the alpha of 0.5% as recommended by Benjamin et al. 22 All analyses were performed using R version 4.0.0 (R Foundation for Statistical Computing, Vienna).
3. RESULTS
The main characteristics of the study participants are described in Table 1. Individuals reporting a diabetes diagnosis were generally more likely than those not reporting diabetes to be male (P < .01) and to use aspirin regularly (P < .01), and less likely to have a reported family history of colorectal cancer, especially among colorectal cancer cases (P < .01). Participants with diabetes were also more often nondrinkers (P < .01), were more likely to exercise (P < .01), had a history of smoking (P < .01) and were more often obese (P < .01). These relationships were consistent among both colorectal cancer cases and controls.
TABLE 1.
Participant characteristics
| Characteristics | CRC cases | P a | Controls | P a | ||
|---|---|---|---|---|---|---|
| Diabetes | Diabetes | |||||
| No | Yes | No | Yes | |||
| Total | 8639 | 1117 | 9101 | 883 | ||
| Study, n (row %) | <.01 | <.01 | ||||
| CCFR_Australia | 713 (94.1) | 45 (5.9) | 175 (96.2) | 7 (3.8) | ||
| CCFR_Ontario | 1073 (91.3) | 102 (8.7) | 1178 (90.1) | 121 (9.9) | ||
| CCFR_Seattle | 1612 (87.5) | 231 (12.5) | 705 (93.0) | 53 (7.0) | ||
| CPSII | 798 (93.0) | 60 (7.0) | 901 (93.0) | 68 (7.0) | ||
| DACHS | 1879 (81.4) | 430 (18.6) | 2940 (86.0) | 481 (14.0) | ||
| EPIC_Sweden | 142 (96.6) | 5 (3.4) | 384 (99.7) | 1 (0.3) | ||
| HPFS1 | 241 (96.0) | 10 (4.0) | 251 (98.8) | 3 (1.2) | ||
| HPFS2 | 354 (93.7) | 24 (6.3) | 192 (93.7) | 13 (6.3) | ||
| MCCS | 467 (95.3) | 23 (4.7) | 658 (97.6) | 16 (2.4) | ||
| NFCCR | 405 (78.9) | 108 (21.1) | 401 (86.1) | 65 (13.9) | ||
| NHS1 | 204 (95.8) | 9 (4.2) | 749 (97.5) | 19 (2.5) | ||
| NHS2 | 519 (89.5) | 61 (10.5) | 283 (90.1) | 31 (9.9) | ||
| NSHDS | 232 (96.3) | 9 (3.7) | 284 (98.2) | 5 (1.8) | ||
| Age of CRC diagnosis | <.01 | <.01 | ||||
| Mean (range) | 62.57 (20‐96) | 67.27 (27‐90) | 65.75 (20‐102) | 69.74 (27‐97) | ||
| Sex, n (column %) | <.01 | <.01 | ||||
| Female | 4157 (48.1) | 451 (40.4) | 4505 (49.5) | 318 (36) | ||
| Male | 4482 (51.9) | 666 (59.6) | 4596 (50.5) | 565 (64) | ||
| Family history of CRC, n (column%) | <.01 | .38 | ||||
| No | 6594 (76.3) | 902 (80.8) | 7656 (84.1) | 780 (88.3) | ||
| Yes | 1571 (18.2) | 169 (15.1) | 947 (10.4) | 87 (9.9) | ||
| Aspirin, n (column %) | <.01 | <.01 | ||||
| No | 5949 (68.9) | 668 (59.8) | 5404 (59.4) | 446 (50.5) | ||
| Yes | 1976 (22.9) | 412 (36.9) | 2736 (30.1) | 419 (47.5) | ||
| Dietary intake, mean (range) | ||||||
| Energy intake, kcal/day | 2046 (357‐4958) | 2155 (310‐4959) | .01 | 1964 (379‐4958) | 2043 (482‐4684) | .06 |
| Redmeat, servings/day | 0.72 (0‐8) | 0.77 (0‐5) | .01 | 0.67 (0‐8) | 0.75 (0‐5.2) | <.01 |
| Process meat, servings/day | 0.48 (0‐4) | 0.66 (0‐2.9) | <.01 | 0.45 (0‐4) | 0.64 (0‐2.5) | <.01 |
| Vegetable, servings/day | 2.21 (0‐20) | 1.77 (0‐14) | <.01 | 2.24 (0‐18) | 1.76 (0.03‐17) | <.01 |
| Fruit, servings/day | 1.6 (0‐20) | 1.41 (0‐9) | <.01 | 1.69 (0‐20) | 1.36 (0‐11) | <.01 |
| Fiber, g/day | 22.5 (3.2‐80) | 23.4 (5.6‐70.4) | .11 | 22.7 (1.8‐80) | 23.0 (3.8‐80) | .65 |
| Alcohol intake, n (column %) | <.01 | <.01 | ||||
| >28 g/day | 1074 (12.4) | 124 (11.1) | 946 (10.4) | 89 (10.1) | ||
| 1‐28 g/day | 3854 (44.6) | 412 (36.9) | 4624 (50.8) | 393 (44.5) | ||
| Nondrinker | 3135 (36.3) | 539 (48.3) | 3078 (33.8) | 356 (40.3) | ||
| Smoke ever, n (column %) | <.01 | <.01 | ||||
| No | 3597 (41.6) | 419 (37.5) | 4311 (47.4) | 324 (36.7) | ||
| Yes | 4810 (55.7) | 672 (60.2) | 4547 (50) | 533 (60.4) | ||
| Exercise, n (column %) | <.01 | <.01 | ||||
| No | 332 (3.8) | 25 (2.2) | 427 (4.7) | 20 (2.3) | ||
| Yes | 2642 (30.6) | 471 (42.2) | 3892 (42.8) | 532 (60.2) | ||
| BMI, n (%) | <.01 | <.01 | ||||
| Normal | 3096 (35.8) | 157 (14.1) | 3792 (41.7) | 180 (20.4) | ||
| Overweight | 3613 (41.8) | 459 (41.1) | 3778 (41.5) | 396 (44.8) | ||
| Obese | 1632 (18.9) | 469 (42) | 1307 (14.4) | 287 (32.5) | ||
| Stage, n (%) | .19 | |||||
| Stage 1 or local | 1746 (20.2) | 219 (19.6) | — | — | ||
| Stage 2/3 or regional | 4735 (54.8) | 687 (61.5) | — | — | ||
| Stage 4 or distant | 928 (10.7) | 124 (11.1) | — | — | ||
| Site, n (%) | .02 | |||||
| Proximal | 3173 (36.7) | 460 (41.2) | — | — | ||
| Distal | 2525 (29.2) | 298 (26.7) | — | — | ||
| Rectal | 2759 (31.9) | 339 (30.3) | — | — | ||
| BRAF, n (%) | .02 | — | ||||
| Wildtype | 7053 (81.6) | 906 (81.1) | — | — | ||
| Mutated | 937 (10.8) | 149 (13.3) | — | — | ||
| KRAS, n (%) | .74 | — | ||||
| Wildtype | 4675 (54.1) | 603 (54) | — | — | ||
| Mutated | 2315 (26.8) | 306 (27.4) | — | — | ||
| MSI, n (%) | .92 | — | ||||
| Non MSI‐high | 6843 (79.2) | 877 (78.5) | — | — | ||
| MSI‐high | 1152 (13.3) | 149 (13.3) | — | — | ||
| CIMP, n (%) | .49 | — | ||||
| Low/negative | 5741 (66.5) | 737 (66) | — | — | ||
| High | 1162 (13.5) | 159 (14.2) | — | — | ||
P‐values from a χ 2 test for categorical variables and ANOVA for continuous variables.
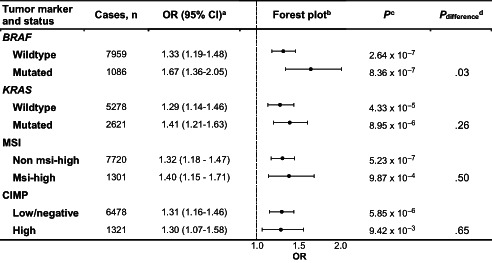
In the case‐control analysis, we observed an association between diabetes and colorectal cancer (ORall crc[fully adj.] = 1.34, 95% CI: 1.21‐1.48). This relationship was consistent across all molecular subtypes as shown in Table 2. However, it was stronger in cases with BRAF‐mutated tumors (ORfully adj. = 1.67, 95% CI: 1.36‐2.05) as compared to tumors without BRAF mutations (ORfully adj. = 1.33, 95% CI: 1.19‐1.48) (P difference = .03) (Tables 2 and S5). For additional comparisons of subtype‐specific associations, differences were not significant.
TABLE 2.
Associations between diabetes and risk of different molecular subtypes of colorectal cancer
|
Adjusted for: study, sex, age at CRC diagnosis, energy intake, family history, BMI, red meat, processed meat, vegetables, fruit, alcohol, smoking, exercise and aspirin/NSAID use.
Error bars represent 95% confidence intervals.
Multinomial logistic regression was used to compare colorectal cancer cases to cancer free controls separately for each molecular pathological subtype (polytomous analysis, P).
Multinomial logistic regression was used to compare cases of each molecular pathological subtype to all additional cases not belonging to that subtype (case only analysis, P difference).
Analyses were then further stratified by tumor subsite and sex (Table S6).
We observed significant differences between the association of diabetes and colorectal cancer by BRAF‐mutation status for colon (P difference = .04) and proximal colon cancers (P difference = .02). The difference between BRAF‐mutated and wild‐type OR point estimates was also retained for rectal tumors (but not for distal tumors) and among both men and women, but without reaching significance (Table S6). However, the number of BRAF‐mutated tumors in the rectum was much lower than in the colon, which may explain why the observed difference in the association between diabetes and colorectal cancer risk by BRAF‐mutation status was not statistically significant. The association between diabetes and colorectal cancer risk did not differ by other tumor markers in any of the stratified analyses.
Results from analyses of combined marker subtypes are presented in Figure 1 and Table S7. Among the 10 subtype combinations tested, diabetes was statistically significantly associated with risk of five different types (1, 3‐5, and 9) in case‐control analyses, but only type 4 remained significant after adjustments for multiple comparisons. No significant differences in subtype‐specific associations were noted in case‐only analyses.
4. DISCUSSION
This large collaborative effort is the first study to investigate the impact of diabetes on subtypes of colorectal cancer, while also considering both distinct molecular markers and tumor subtypes based on marker combinations. Our primary finding was a statistically significant difference in the strength of the association between reported diabetes status and colorectal cancer by BRAF status, with a stronger association for BRAF‐mutated than nonmutated tumors. This was consistent in both our minimally and fully adjusted models. Reporting a diabetes diagnosis was more strongly associated with BRAF‐mutated tumors in both the proximal colon and rectum indicating that tumor location does not explain this result.
BRAF mutations, found in 8% to 12% of colorectal cancers, are generally associated with a poor prognosis and are more common in proximal tumors. 23 , 24 , 25 BRAF is a serine‐threonine kinase activated by KRAS as part of the MAPK signaling cascade. 26 Both BRAF and KRAS are oncogenes that are commonly mutated in colorectal cancer, and mutations in these genes are often considered mutually exclusive. Previous studies have found some evidence that medical drugs can differentially affect the risk of different molecular subtypes. For example, a study from 2013 27 found that aspirin use (which has consistently been shown to decrease colorectal cancer risk 28 ) seemed to specifically lower the risk of BRAF‐nonmutated colorectal cancer but not BRAF‐mutated colorectal cancer. More related to diabetes, a study from 2012 29 found that metformin, an antidiabetic drug previously shown to have antitumor activity in several different cancers, 30 did not affect BRAF‐mutated melanoma cells, but instead seemed to accelerate their growth in a xenograft mice model of melanoma. The authors suggested that the accelerated growth of the BRAF‐mutated tumors could be attributed to improved angiogenesis. Although this is in line with our own results, where individuals reporting diabetes had a higher incidence of BRAF‐mutated tumors compared to other molecular markers, later studies have been unable to replicate the findings. 31 In addition, at least one study 32 specifically examining the association between metformin and colon or colorectal cancer, but not by subtype, did not report any evidence of metformin acting differently depending on tumor site. It should also be noted that although metformin is commonly prescribed to individuals with type 2 diabetes mellitus, we lacked information about metformin use among our participants.
Colorectal cancer is known to often develop through a specific number of events along the so‐called conventional pathway, which is a multistep process initiated by mutations in APC, KRAS or BRAF genes. It is also well known that other important pathways exist, such as the serrated pathway and the alternate pathway, which have other characteristics as well as distinct risk factors. 33 For example, the serrated pathway is associated with older age at onset, female sex and smoking and is also characterized primarily by BRAF mutations and CIMP‐high status. 34 , 35 Previous studies originating from the GECCO consortia have investigated whether other established risk factors for colorectal cancer are associated with specific molecular subtypes, sometimes supporting development through distinct pathways. One recent study focused on dietary factors did show some evidence of heterogeneity related to fruit and especially fiber intake. 15 In polytomous analyses, higher fruit intake was associated with a decreased risk of developing BRAF‐mutated tumors. High fiber intake, on the other hand, was associated with decreased risk of non‐MSI‐high, CIMP‐low/negative, BRAF‐wildtype and KRAS‐wildtype subtypes, although none of these associations retained significance in case‐only analyses. In another study, it was found that smoking was strongly associated with subtype combinations that included CIMP‐high and MSI‐high tumors. 36 As a result of these findings, the authors suggest that smoking might specifically increase the risk of tumors developing through the serrated pathway. Our study however, despite showing an increased risk of developing BRAF‐mutated tumors, did not find any evidence of diabetes resulting in any pathway specific development. Both previous studies and ours underline the importance of using large enough sample sets to be able to adequately assess patterns of associations that differ depending on molecular subtype.
Metabolic dysfunction, often defined as the presence of three or more of the criteria for metabolic syndrome (central obesity, hypertension, dysglycemia and dyslipidemia), is associated with an increased risk of developing both diabetes (type 2) and colorectal cancer. Several studies have aimed to assess the relationship between metabolic abnormalities (such as high BMI, hypertension and dysglycemia) and colorectal cancer, focusing on metabolic syndrome, inflammation and specific colorectal cancer subtypes. 37 , 38 , 39 One of several plausible links between diabetes and colorectal cancer could be related to insulin resistance and hyperinsulinemia, a state of heightened insulin levels and a hallmark of untreated type 2 diabetes in its earlier natural history. High insulin levels have been linked to an increased risk of multiple cancers, including colorectal cancer. 40 This may relate to its growth promoting effects as well as its ability to increase circulating IGF1 levels. 41 Several studies have also identified a link between markers of heightened insulin levels (eg, C‐peptide) and colorectal cancer risk, 42 , 43 but without finding any evidence of heterogeneity. 44 However, the most important metabolic factor likely to affect both diabetes and colorectal cancer risk is obesity and the relationship between BMI and colorectal cancer has also shown consistent directions of associations across studies, although just as for hyperglycemia, evidence of heterogeneity between subtypes has been somewhat inconsistent. 11 , 45 , 46 , 47 , 48 , 49 , 50 , 51 In the current study however, we find some evidence of diabetes increasing the risk of specifically BRAF‐mutated tumors, but taking into account previous studies, this subtype‐specific risk difference is probably not related to the shared metabolic risk factors connecting diabetes and colorectal cancer.
An important strength of our study is the ability to pool data from multiple observational studies with readily available information on diabetes status. This pooling of datasets enabled us to detect even modestly differential associations between diabetes status and risk of colorectal cancer by molecular subtypes. However, there are also limitations that have to be taken into account when interpreting the study results. First, we were not able to distinguish between type 1 and type 2 diabetes, or to take into account use of specific medications such as metformin. Time and duration of diabetes diagnosis was also lacking. These are all factors that can have important implications and result in misclassification bias. However, the size of our study, and the fact that type 2 diabetes makes up more than 90% of all diabetes cases, 52 makes it less likely that these have substantially distorted our results. There may also be selection bias related to the cases included in the pooled analysis or to tissue availability potentially depending on tumor stage and size, 53 a limitation that has been previously described. 15 Finally, we did not apply any formal adjustment for multiple comparisons to our primary analyses. Although adding such adjustments would not affect the associations of risk, as they are all highly significant, the reported difference in OR estimates between BRAF‐mutated and nonmutated tumors would fall slightly below the significance threshold (accounting for tests of four different subtypes). This should be considered when interpreting the findings. It is worth noting that individuals reporting diabetes had a higher risk of all subtypes of colorectal cancer and the difference related to BRAF‐mutated tumors only affected the magnitude of the risk, but the direction remained the same.
In summary, our study confirms the established association between diabetes and colorectal cancer risk and suggests that it especially increases the risk of BRAF‐mutated tumors. Additional studies to confirm our finding, and possibly determine if the subtype specific association could be attributed to any particular component (such as metformin treatment), are needed before further conclusions can be drawn.
CONFLICT OF INTEREST
Marios Giannakis reports potential financial conflicts of interest through research funding from Bristol‐Myers Squibb, Merck, Servier and Janssen unrelated to our study. Victor Moreno reports grants from Agency for Management of University and Research Grants (AGAUR) of the Catalan Government grant 2017SGR723; Instituto de Salud Carlos III grant PI17‐00092; Spanish Association Against Cancer (AECC) Scientific Foundation grant GCTRA18022MORE, Consortium for Biomedical Research in Epidemiology and Public Health (CIBERESP), action Genrisk. Jonathan Nowak reports prior research funding (for pilot projects) from NanoString and Illumina and ongoing research funding (for technology development) from Akoya Biosciences, unrelated to our study. Mireia Obón‐Santacana reports a post‐doctoral fellowship from the Spanish Association Against Cancer Scientific Foundation (AECC; POSTD037OBÓN). All other authors declare no conflicts of interest.
DISCLAIMER
Where authors are identified as personnel of the International Agency for Research on Cancer / World Health Organization, the authors alone are responsible for the views expressed in this article and they do not necessarily represent the decisions, policy or views of the International Agency for Research on Cancer / World Health Organization.
AUTHOR CONTRIBUTIONS
The work reported in the article has been performed by the authors, unless clearly specified in the text. Individual author contributions are as follows: Sophia Harlid: Conceptualization, investigation, project administration, visualization, writing‐original draft, writing‐review and editing; Bethany Van Guelpen: Conceptualization, resources, writing‐review and editing; Conghui Qu: Data curation, formal analysis, investigation, methodology, writing‐review and editing; Björn Gylling: Writing‐review and editing; Elom K. Aglago: Writing‐review and editing; Efrat L. Amitay: Writing‐review and editing; Hermann Brenner: Resources, writing ‐ review & editing; Daniel D. Buchanan: Resources, writing‐review and editing; Peter T. Campbell: Resources, writing‐review and editing; Yin Cao: Writing‐review and editing; Andrew T. Chan: Resources, writing‐review and editing; Jenny Chang‐Claude: Writing‐review and editing; David A. Drew: Writing‐review and editing; Jane C. Figueiredo: Writing‐review and editing; Amy J. French: Writing‐review and editing; Steven Gallinger: Resources, writing‐review and editing; Marios Giannakis: Writing‐review and editing; Graham G. Giles: Writing‐review and editing; Marc J. Gunter: Resources, writing‐review and editing; Michael Hoffmeister: Resources, writing‐review and editing; Li Hsu: Methodology, writing‐review and editing; Mark A. Jenkins: Resources, writing‐review and editing; Yi Lin: Data curation, software, writing‐review and editing; Victor Moreno: Resources, writing‐review and editing; Neil Murphy: Writing‐review and editing; Polly A. Newcomb: Resources, writing‐review and editing; Christina C. Newton: Writing‐review and editing; Jonathan A. Nowak: Writing‐review and editing; Mireia Obón‐Santacana: Writing‐review and editing; Shuji Ogino: Resources, writing‐review and editing; John D. Potter: Resources, writing‐review and editing; Mingyang Song: Writing‐review and editing; Robert S. Steinfelder: Data curation, writing‐review and editing; Wei Sun: Writing‐review and editing; Stephen N. Thibodeau: Resources, writing‐review and editing; Amanda E. Toland: Writing‐review and editing; Tomotaka Ugai: Writing‐review and editing; Caroline Y. Um: Writing‐review and editing; Michael O. Woods: Resources, writing‐review and editing; Amanda I. Phipps: Project administration, writing‐review and editing; Tabitha Harrison: Conceptualization, investigation, project administration, writing‐review and editing; Ulrike Peters: Conceptualization, supervision, funding acquisition, methodology, project administration, writing‐review and editing.
ETHICS STATEMENT
All study participants had provided informed consent and included studies had obtained ethical approval from their respective research ethics committee or institutional review board.
Supporting information
Appendix S1Supporting Information.
Table S1. Association between diabetes and risk of colorectal cancer stratified by tumor marker
Table S2. Association between diabetes and risk of individual molecular subtypes of colorectal cancer, stratified by tumor location and sex
Table S3. Minimally and fully adjusted case‐control associations between individuals with and without diabetes with risk of colorectal cancer subtypes defined by combined marker status. A two‐sided Wald test was used to calculate the P‐values from the case‐only analysis (P difference). Error bars represent 95% confidence intervals.
ACKNOWLEDGEMENTS
CCFR: The Colon CFR graciously thanks the generous contributions of their study participants, dedication of study staff and the financial support from the US National Cancer Institute, without which this important registry would not exist. The authors would like to thank the study participants and staff of the Seattle Colon Cancer Family Registry and the Hormones and Colon Cancer study (CORE Studies). CPS‐II: The authors thank the CPS‐II participants and Study Management Group for their invaluable contributions to this research. The authors would also like to acknowledge the contribution to our study from central cancer registries supported through the Centers for Disease Control and Prevention National Program of Cancer Registries, and cancer registries supported by the National Cancer Institute Surveillance Epidemiology and End Results program. DACHS: We thank all participants and cooperating clinicians, and everyone who provided excellent technical assistance. Harvard cohorts (HPFS, NHS): The study protocol was approved by the institutional review boards of the Brigham and Women's Hospital and Harvard T.H. Chan School of Public Health, and those of participating registries as required. We would like to thank the participants and staff of the HPFS and NHS for their valuable contributions as well as the following state cancer registries for their help: AL, AZ, AR, CA, CO, CT, DE, FL, GA, ID, IL, IN, IA, KY, LA, ME, MD, MA, MI, NE, NH, NJ, NY, NC, ND, OH, OK, OR, PA, RI, SC, TN, TX, VA, WA, WY. The authors assume full responsibility for analyses and interpretation of these data. NSHDS investigators thank the Västerbotten Intervention Programme, the Northern Sweden MONICA study, the Biobank Research Unit at Umeå Universitet and Biobanken Norr at Region Västerbotten for providing data and samples and acknowledge the contribution from Biobank Sweden, supported by the Swedish Research Council.
Harlid S, Van Guelpen B, Qu C, et al. Diabetes mellitus in relation to colorectal tumor molecular subtypes: A pooled analysis of more than 9000 cases. Int J Cancer. 2022;151(3):348‐360. doi: 10.1002/ijc.34015
Funding information Genetics and Epidemiology of Colorectal Cancer Consortium (GECCO): National Cancer Institute, National Institutes of Health, U.S. Department of Health and Human Services (U01 CA137088, R01 CA059045, R01CA201407). Genotyping/Sequencing services were provided by the Center for Inherited Disease Research (CIDR) contract number HHSN268201700006I and HHSN268201200008I. This research was funded in part through the NIH/NCI Cancer Center Support Grant P30 CA015704. Scientific Computing Infrastructure at Fred Hutch funded by ORIP Grant S10OD028685.
The Colon Cancer Family Registry (CCFR, www.coloncfr.org) is supported in part by funding from the National Cancer Institute (NCI), National Institutes of Health (NIH) (award U01 CA167551). Support for case ascertainment was provided in part from the Surveillance, Epidemiology and End Results (SEER) Program and the following U.S. state cancer registries: AZ, CO, MN, NC, NH; and by the Victoria Cancer Registry (Australia) and Ontario Cancer Registry (Canada). The CCFR Set‐1 (Illumina 1M/1M‐Duo) and Set‐2 (Illumina Omni1‐Quad) scans were supported by NIH awards U01 CA122839 and R01 CA143247 (to GC). The CCFR Set‐3 (Affymetrix Axiom CORECT Set array) was supported by NIH award U19 CA148107 and R01 CA81488 (to SBG). The CCFR Set‐4 (Illumina OncoArray 600K SNP array) was supported by NIH award U19 CA148107 (to SBG) and by the Center for Inherited Disease Research (CIDR), which is funded by the NIH to the Johns Hopkins University, contract number HHSN268201200008I. Additional funding for the OFCCR/ARCTIC was through award GL201‐043 from the Ontario Research Fund (to BWZ), award 112746 from the Canadian Institutes of Health Research (to TJH), through a Cancer Risk Evaluation (CaRE) Program grant from the Canadian Cancer Society (to SG) and through generous support from the Ontario Ministry of Research and Innovation. The SFCCR Illumina HumanCytoSNP array was supported in part through NCI/NIH awards U01/U24 CA074794 and R01 CA076366 (to PAN). The content of this article does not necessarily reflect the views or policies of the NCI, NIH or any of the collaborating centers in the Colon Cancer Family Registry (CCFR), nor does mention of trade names, commercial products or organizations imply endorsement by the US Government, any cancer registry or the CCFR.
CPS‐II: The American Cancer Society funds the creation, maintenance and updating of the Cancer Prevention Study‐II (CPS‐II) cohort. Our study was conducted with Institutional Review Board approval.
DACHS: This work was supported by the German Research Council (BR 1704/6‐1, BR 1704/6‐3, BR 1704/6‐4, CH 117/1‐1, HO 5117/2‐1, HE 5998/2‐1, KL 2354/3‐1, RO 2270/8‐1 and BR 1704/17‐1), the Interdisciplinary Research Program of the National Center for Tumor Diseases (NCT), Germany and the German Federal Ministry of Education and Research (01KH0404, 01ER0814, 01ER0815, 01ER1505A and 01ER1505B).
EPIC: The coordination of EPIC is financially supported by International Agency for Research on Cancer (IARC) and also by the Department of Epidemiology and Biostatistics, School of Public Health, Imperial College London which has additional infrastructure support provided by the NIHR Imperial Biomedical Research Centre (BRC). The national cohorts are supported by: Danish Cancer Society (Denmark); Ligue Contre le Cancer, Institut Gustave Roussy, Mutuelle Générale de l'Education Nationale, Institut National de la Santé et de la Recherche Médicale (INSERM) (France); German Cancer Aid, German Cancer Research Center (DKFZ), German Institute of Human Nutrition Potsdam‐ Rehbruecke (DIfE), Federal Ministry of Education and Research (BMBF) (Germany); Associazione Italiana per la Ricerca sul Cancro‐AIRC‐Italy, Compagnia di SanPaolo and National Research Council (Italy); Dutch Ministry of Public Health, Welfare and Sports (VWS), Dutch ZON (Zorg Onderzoek Nederland), Statistics Netherlands (The Netherlands), LK Research Funds, Dutch Prevention Funds, Netherlands Cancer Registry (NKR), World Cancer Research Fund (WCRF); Health Research Fund (FIS) ‐ Instituto de Salud Carlos III (ISCIII), Regional Governments of Andalucía, Asturias, Basque Country, Murcia and Navarra and the Catalan Institute of Oncology ‐ ICO (Spain); Swedish Cancer Society, Swedish Research Council and County Councils of Skåne and Västerbotten (Sweden); Cancer Research UK (14136 to EPIC‐Norfolk; C8221/A29017 to EPIC‐Oxford), Medical Research Council (1000143 to EPIC‐Norfolk; MR/M012190/1 to EPIC‐Oxford). (United Kingdom).
Harvard cohorts (HPFS, NHS): HPFS is supported by the National Institutes of Health (P01 CA055075, UM1 CA167552, U01 CA167552, R01 CA151993, R35 CA197735 and R35 CA253185), and NHS by the National Institutes of Health (R01 CA137178, P01 CA087969, UM1 CA186107, R01 CA151993, R35 CA197735 and R35 CA253185).
MCCS cohort recruitment was funded by VicHealth and Cancer Council Victoria. The MCCS was further supported by Australian NHMRC grants 209057, 396414 and 1074383 and by infrastructure provided by Cancer Council Victoria. Cases and their vital status were ascertained through the Victorian Cancer Registry and the Australian Institute of Health and Welfare, including the National Death Index and the Australian Cancer Database.
NFCCR: This work was supported by an Interdisciplinary Health Research Team award from the Canadian Institutes of Health Research (CRT 43821); the National Institutes of Health, U.S. Department of Health and Human Services (U01 CA74783); and National Cancer Institute of Canada grants (18223 and 18226). The authors wish to acknowledge the contribution of Alexandre Belisle and the genotyping team of the McGill University and Génome Québec Innovation Centre, Montréal, Canada, for genotyping the Sequenom panel in the NFCCR samples. Funding was provided to Michael O. Woods by the Canadian Cancer Society Research Institute.
NSHDS: The research was supported by Biobank Sweden through funding from the Swedish Research Council (VR 2017‐00650, VR 2017‐01737), the Swedish Cancer Society (CAN 2017/581), Region Västerbotten (VLL‐841671, VLL‐833291), Knut and Alice Wallenberg Foundation (VLL‐765961) and the Lion's Cancer Research Foundation (several grants) and Insamlingsstiftelsen, both at Umeå University.
DATA AVAILABILITY STATEMENT
Tumor marker and epidemiologic data is available upon request and permission. Please contact gro.hctuhderf@occeg to request the standardized proposal form. The principal investigators of each contributing study will evaluate and approve the proposal, and data access will be managed centrally. Further information is available from the corresponding authors upon request.
REFERENCES
- 1. Choi YJ, Lee DH, Han K‐D, Shin CM, Kim N. Abdominal obesity, glucose intolerance and decreased high‐density lipoprotein cholesterol as components of the metabolic syndrome are associated with the development of colorectal cancer. Eur J Epidemiol. 2018;33:1077‐1085. [DOI] [PubMed] [Google Scholar]
- 2. Jinjuvadia R, Lohia P, Jinjuvadia C, Montoya S, Liangpunsakul S. The association between metabolic syndrome and colorectal neoplasm: systemic review and meta‐analysis. J Clin Gastroenterol. 2013;47:33‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ma Y, Yang Y, Wang F, et al. Obesity and risk of colorectal cancer: a systematic review of prospective studies. PLoS ONE. 2013;8:e53916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. González N, Prieto I, Del Puerto‐Nevado L, et al. 2017 update on the relationship between diabetes and colorectal cancer: epidemiology, potential molecular mechanisms and therapeutic implications. Oncotarget. 2017;8:18456‐18485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rey‐Reñones C, Baena‐Díez JM, Aguilar‐Palacio I, Miquel C, Grau M. Type 2 diabetes mellitus and cancer: epidemiology, physiopathology and prevention. Biomedicine. 2021;9:1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vigneri P, Frasca F, Sciacca L, Pandini G, Vigneri R. Diabetes and cancer. Endocr Relat Cancer. 2009;16:1103‐1123. [DOI] [PubMed] [Google Scholar]
- 7. Salem ME, Weinberg BA, Xiu J, et al. Comparative molecular analyses of left‐sided colon, right‐sided colon, and rectal cancers. Oncotarget. 2017;8:86356‐86368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Myte R, Gylling B, Häggström J, et al. One‐carbon metabolism biomarkers and genetic variants in relation to colorectal cancer risk by KRAS and BRAF mutation status. PLoS One. 2018;13:e0196233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Inamura K, Song M, Jung S, et al. Prediagnosis plasma adiponectin in relation to colorectal cancer risk according to KRAS mutation status. J Natl Cancer Inst. 2016;108:djv363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Achari AE, Jain SK. Adiponectin, a therapeutic target for obesity, diabetes, and endothelial dysfunction. Int J Mol Sci. 2017;18:1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hanyuda A, Cao Y, Hamada T, et al. Body mass index and risk of colorectal carcinoma subtypes classified by tumor differentiation status. Eur J Epidemiol. 2017;32:393‐407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hanyuda A, Ogino S, Qian ZR, et al. Body mass index and risk of colorectal cancer according to tumor lymphocytic infiltrate. Int J Cancer. 2016;139:854‐868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ganz ML, Wintfeld N, Li Q, Alas V, Langer J, Hammer M. The association of body mass index with the risk of type 2 diabetes: a case‐control study nested in an electronic health records system in the United States. Diabetol Metab Syndr. 2014;6:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Labadie JD, Harrison TA, Banbury B, et al. Postmenopausal hormone therapy and colorectal cancer risk by molecularly defined subtypes and tumor location. JNCI Cancer Spectr. 2020;4:pkaa042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hidaka A, Harrison TA, Cao Y, et al. Intake of dietary fruit, vegetables, and fiber and risk of colorectal cancer according to molecular subtypes: a pooled analysis of 9 studies. Cancer Res. 2020;80:4578‐4590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Boland CR, Thibodeau SN, Hamilton SR, et al. A National Cancer Institute workshop on microsatellite instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998;58:5248‐5257. [PubMed] [Google Scholar]
- 17. Warth A, Kloor M, Schirmacher P, Bläker H. Genetics and epigenetics of small bowel adenocarcinoma: the interactions of CIN, MSI, and CIMP. Mod Pathol. 2011;24:564‐570. [DOI] [PubMed] [Google Scholar]
- 18. Jass JR. Classification of colorectal cancer based on correlation of clinical, morphological and molecular features. Histopathology. 2007;50:113‐130. [DOI] [PubMed] [Google Scholar]
- 19. Phipps AI, Limburg PJ, Baron JA, et al. Association between molecular subtypes of colorectal cancer and patient survival. Gastroenterology. 2015;148:77‐87.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. McCarthy AJ, Serra S, Chetty R. Traditional serrated adenoma: an overview of pathology and emphasis on molecular pathogenesis. BMJ Open Gastroenterol. 2019;6:e000317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zaidi SH, Harrison TA, Phipps AI, et al. Landscape of somatic single nucleotide variants and indels in colorectal cancer and impact on survival. Nat Commun. 2020;11:3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Benjamin DJ, Berger JO, Johannesson M, et al. Redefine statistical significance. Nat Hum Behav. 2018;2:6‐10. [DOI] [PubMed] [Google Scholar]
- 23. Sanz‐Garcia E, Argiles G, Elez E, Tabernero J. BRAF mutant colorectal cancer: prognosis, treatment, and new perspectives. Ann Oncol. 2017;28:2648‐2657. [DOI] [PubMed] [Google Scholar]
- 24. Kalady MF, Dejulius KL, Sanchez JA, et al. BRAF mutations in colorectal cancer are associated with distinct clinical characteristics and worse prognosis. Dis Colon Rectum. 2012;55:128‐133. [DOI] [PubMed] [Google Scholar]
- 25. Clarke CN, Kopetz ES. BRAF mutant colorectal cancer as a distinct subset of colorectal cancer: clinical characteristics, clinical behavior, and response to targeted therapies. J Gastrointest Oncol. 2015;6:660‐667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Röring M, Brummer T. Aberrant B‐Raf signaling in human cancer: 10 years from bench to bedside. Crit Rev Oncog. 2012;17:97‐121. [DOI] [PubMed] [Google Scholar]
- 27. Nishihara R, Lochhead P, Kuchiba A, et al. Aspirin use and risk of colorectal cancer according to BRAF mutation status. Jama. 2013;309:2563‐2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Drew DA, Cao Y, Chan AT. Aspirin and colorectal cancer: the promise of precision chemoprevention. Nat Rev Cancer. 2016;16:173‐186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Martin MJ, Hayward R, Viros A, Marais R. Metformin accelerates the growth of BRAF V600E‐driven melanoma by upregulating VEGF‐A. Cancer Discov. 2012;2:344‐355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Evans JMM, Donnelly LA, Emslie‐Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ. 2005;330:1304‐1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cerezo M, Tomic T, Ballotti R, Rocchi S. Is it time to test biguanide metformin in the treatment of melanoma? Pigment Cell Melanoma Res. 2015;28:8‐20. [DOI] [PubMed] [Google Scholar]
- 32. Kamarudin MNA, Sarker MMR, Zhou J‐R, Parhar I. Metformin in colorectal cancer: molecular mechanism, preclinical and clinical aspects. J Exp Clin Cancer Res. 2019;38:491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang L, He X, Ugai T, et al. Risk factors and incidence of colorectal cancer according to major molecular subtypes. JNCI Cancer Spectr. 2021;5:pkaa089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. De Palma FDE, D'Argenio V, Pol J, Kroemer G, Maiuri MC, Salvatore F. The molecular hallmarks of the serrated pathway in colorectal cancer. Cancers (Basel). 2019;11:1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kim SY, Kim TI. Serrated neoplasia pathway as an alternative route of colorectal cancer carcinogenesis. Intest Res. 2018;16:358‐365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang X, Amitay E, Harrison TA, et al. Association between smoking and molecular subtypes of colorectal cancer. JNCI Cancer Spectr. 2021;5:pkab056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Harlid S, Myte R, Van Guelpen B. The metabolic syndrome, inflammation, and colorectal cancer risk: an evaluation of large panels of plasma protein markers using repeated, prediagnostic samples. Mediators Inflamm. 2017;2017:4803156‐4803159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lee J, Lee KS, Kim H, et al. The relationship between metabolic syndrome and the incidence of colorectal cancer. Environ Health Prev Med. 2020;25:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Myte R, Gylling B, Häggström J, et al. Metabolic factors and the risk of colorectal cancer by KRAS and BRAF mutation status. Int J Cancer. 2019;145:327‐337. [DOI] [PubMed] [Google Scholar]
- 40. Gallagher EJ, LeRoith D. Hyperinsulinaemia in cancer. Nat Rev Cancer. 2020;20:629‐644. [DOI] [PubMed] [Google Scholar]
- 41. Godsland IF. Insulin resistance and hyperinsulinaemia in the development and progression of cancer. Clin Sci. 2009;118:315‐332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jenab M, Riboli E, Cleveland RJ, et al. Serum C‐peptide, IGFBP‐1 and IGFBP‐2 and risk of colon and rectal cancers in the European prospective investigation into cancer and nutrition. Int J Cancer. 2007;121:368‐376. [DOI] [PubMed] [Google Scholar]
- 43. Ma J, Giovannucci E, Pollak M, et al. A prospective study of plasma C‐peptide and colorectal cancer risk in men. J Natl Cancer Inst. 2004;96:546‐553. [DOI] [PubMed] [Google Scholar]
- 44. Myte R, Harlid S, Sundkvist A, et al. A longitudinal study of prediagnostic metabolic biomarkers and the risk of molecular subtypes of colorectal cancer. Sci Rep. 2020;10:5336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hughes LAE, Williamson EJ, van Engeland M, et al. Body size and risk for colorectal cancers showing BRAF mutations or microsatellite instability: a pooled analysis. Int J Epidemiol. 2012;41:1060‐1072. [DOI] [PubMed] [Google Scholar]
- 46. Brändstedt J, Wangefjord S, Nodin B, et al. Associations of anthropometric factors with KRAS and BRAF mutation status of primary colorectal cancer in men and women: a cohort study. PLoS One. 2014;9:e98964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Carr PR, Amitay EL, Jansen L, et al. Association of BMI and major molecular pathological markers of colorectal cancer in men and women. Am J Clin Nutr. 2020;111:562‐569. [DOI] [PubMed] [Google Scholar]
- 48. Campbell PT, Jacobs ET, Ulrich CM, et al. Case‐control study of overweight, obesity, and colorectal cancer risk, overall and by tumor microsatellite instability status. J Natl Cancer Inst. 2010;102:391‐400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hoffmeister M, Bläker H, Kloor M, et al. Body mass index and microsatellite instability in colorectal cancer: a population‐based study. Cancer Epidemiol Biomarkers Prev. 2013;22:2303‐2311. [DOI] [PubMed] [Google Scholar]
- 50. Yang J, Nishihara R, Zhang X, Ogino S, Qian ZR. Energy sensing pathways: bridging type 2 diabetes and colorectal cancer? J Diabetes Complications. 2017;31:1228‐1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Morikawa T, Kuchiba A, Lochhead P, et al. Prospective analysis of body mass index, physical activity, and colorectal cancer risk associated with β‐catenin (CTNNB1) status. Cancer Res. 2013;73:1600‐1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mobasseri M, Shirmohammadi M, Amiri T, Vahed N, Hosseini Fard H, Ghojazadeh M. Prevalence and incidence of type 1 diabetes in the world: a systematic review and meta‐analysis. Health Promot Perspect. 2020;10:98‐115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hoppin JA, Tolbert PE, Taylor JA, Schroeder JC, Holly EA. Potential for selection bias with tumor tissue retrieval in molecular epidemiology studies. Ann Epidemiol. 2002;12:1‐6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1Supporting Information.
Table S1. Association between diabetes and risk of colorectal cancer stratified by tumor marker
Table S2. Association between diabetes and risk of individual molecular subtypes of colorectal cancer, stratified by tumor location and sex
Table S3. Minimally and fully adjusted case‐control associations between individuals with and without diabetes with risk of colorectal cancer subtypes defined by combined marker status. A two‐sided Wald test was used to calculate the P‐values from the case‐only analysis (P difference). Error bars represent 95% confidence intervals.
Data Availability Statement
Tumor marker and epidemiologic data is available upon request and permission. Please contact gro.hctuhderf@occeg to request the standardized proposal form. The principal investigators of each contributing study will evaluate and approve the proposal, and data access will be managed centrally. Further information is available from the corresponding authors upon request.