

Abstract
Background:
Transcriptional reconfiguration is central to heart failure, the common cause of which is dilated cardiomyopathy (DCM). However, the impact of three-dimensional (3D) chromatin topology on transcriptional dysregulation and pathogenesis in human DCM remains elusive.
Methods:
We generated a compendium of 3D-epigenome and transcriptome maps from 101 biobanked human DCM and non-failing heart tissues through HiChIP (H3K27ac), in situ Hi-C, ChIP-seq, ATAC-seq and RNA-seq profiling. We employed human iPSC-derived cardiomyocytes (hiPSC-CMs) and mouse models to further interrogate the key transcription factor implicated in 3D chromatin organization and transcriptional regulation in DCM pathogenesis.
Results:
We discovered that the active regulatory elements (H3K27ac peaks) and their connectome (H3K27ac loops) were extensively reprogrammed in DCM hearts and contributed to transcriptional dysregulation implicated for DCM development. For example, we identified that non-transcribing NPPA-AS1 promoter functions as an enhancer and physically interacts with the NPPA and NPPB promoters, leading to the co-transcription of NPPA and NPPB in DCM hearts. We uncovered that DCM-enriched H3K27ac loops largely resided in conserved high-order chromatin architectures (Compartments, Topologically Associating Domains) and unexpectedly their anchors had equivalent chromatin accessibility. Intriguingly, we discovered that the DCM-enriched H3K27ac loop anchors exhibited a strong enrichment for Heart and Neural Crest Derivatives Expressed 1 (HAND1), a key transcription factor involved in early cardiogenesis. In line with this, its protein expression was upregulated in human DCM and mouse failing hearts. To further validate whether HAND1 is a causal driver for the reprogramming of enhancer/promoter connectome in DCM hearts, we performed comprehensive 3D epigenome mappings in hiPSC-CMs. We found that forced overexpression of HAND1 in hiPSC-CM induced a distinct gain of enhancer/promoter connectivity and, correspondingly, increased the expression of their connected genes implicated in DCM etiology, thus recapitulating the transcriptional signature in human DCM hearts. Moreover, electrophysiology analysis demonstrated that forced overexpression of HAND1 in hiPSC-CM induced abnormal calcium handling. Furthermore, cardiomyocyte-specific overexpression of Hand1 in the mouse hearts resulted in a dilated cardiac remodeling with impaired contractility/Ca2+ handling in cardiomyocytes, increased ratio of heart weight/body weight and compromised cardiac function, which were ascribed to recapitulation of transcriptional reprogramming in DCM.
Conclusions:
This study provided novel chromatin topology insights into DCM pathogenesis and illustrated a model whereby a single transcription factor (HAND1) reprograms the genome-wide enhancer/promoter connectome to drive DCM pathogenesis.
Keywords: 3D genome, Epigenetics, Transcriptional regulation, HAND1, Dilated cardiomyopathy
Introduction
Heart failure is the leading cause of death worldwide and affects over 37.7 million individuals globally1. In particular, dilated cardiomyopathy (DCM) is the most commonly diagnosed type of systolic heart failure1. Currently, no effective treatment can prevent the progression of DCM to heart failure. Heart transplantation is the last resort for DCM patients, but it is hampered by limited donors and staggering medical and economic burdens2. Thus, studies are urgently needed to demystify its etiology and develop effective management strategies.
DCM is characterized by pathogenic structural remodeling of the left ventricle (i.e., enlargement of the chamber and a thin ventricular wall) and poor contractility. Previous studies indicated that genetic variation in protein-coding genes (e.g., sarcomeric and cytoskeletal genes) contributed to the pathogenesis of DCM in a minority of cases3, suggesting that nongenetic (epigenetic) mechanisms may play a crucial role in DCM development4. As cell identity is primarily determined by coordinated gene transcription, novel insights into the mechanisms by which the DCM transcriptome is controlled will enable us to develop novel strategies to diagnose and treat DCM.
Gene transcription is regulated by a variety of determinants, and cis-regulatory elements (CREs), such as enhancers and promoters, are being recognized as the key determinants that shape gene expression5. Dysregulation of CREs has been implicated in the pathogenesis of human diseases, such as thalassemia, polydactyly and various types of cancers6. During heart failure progression, MYH6/MYH7 ratio is known to shift, as characterized by a decrease in MYH6 expression and an increase in MYH7 expression. Recently, a key enhancer has been identified to regulate the switch of MYH6 expression to MYH7 expression. Deletion of the predicted enhancer reduces MYH7 and increases MYH6 expression, leading to faster contraction in human engineered heart7. To identify the potential CREs in human failing hearts, Cap Analysis of Gene Expression (CAGE)-seq was performed in 3 healthy and 4 failed human left ventricles to map the initiation sites of both capped coding and noncoding RNAs8. This study identified the transcribing promoters and the first intronic enhancer in failed ventricles but most distal enhancers (e.g., intergenic enhancers) in heart failure were not explored yet. By performing ChIP-seq for H3K27ac (active enhancer and promoter mark9) in non-failing and end-stage failing human heart tissues, a recent study identified specific enhancers and promoters implicated for established molecular pathways in heart failure10. Nevertheless, the specific cis-regulome in DCM implicated for its pathogenesis remains unexplored.
Moreover, in many cases, enhancers regulate transcription via long-range interactions with target gene promoters, but not nearby gene promoters11. Thus, it is incorrect to assign the enhancer-promoter pair based on only the distance between the enhancer and promoter on the linear genome. Standard methods that map the enhancer-promoter interactome at high resolution (kilobase) require a large number of cells/tissue samples and an extremely deep sequencing depth (billion reads per sample). New approaches to protein-mediated chromatin interactions (e.g., HiChIP12–14, PLAC-seq15, 16, and in situ ChIA-PET17) have recently been developed to enable the generation of high-resolution chromatin contact maps for CREs with substantially reduced cell numbers and sequencing depth and can thus be applied to clinical samples, such as human DCM tissues, with high resolution.
Furthermore, the enhancer-promoter interactome is embedded in topologically associated domains (TADs), which further aggregate into large-scale nuclear architectures called A/B compartments18. In recent years, reorganization of higher-order chromatin architectures have been implicated in aberrant enhancer-promoter interactions in human diseases (e.g., abnormalities in limb development19 and gastrointestinal stromal tumors20) and transcriptional derepression during cardiac lineage specification21. However, the enhancer-promoter interactome and its link to higher higher-order chromatin architectures in DCM remains unexplored.
In this study, we aimed to develop the 3D epigenomic maps of DCM hearts and determine the main factors driving pathological gene transcription in DCM. We leveraged these datasets to a) map the differential active CREs in DCM and NF hearts; b) identify the differential active CREs interactome and determine how the components are linked to transcriptional changes in DCM; c) investigate whether the formation of DCM-enriched CREs interactome is related to higher-order chromatin structure (TADs and compartments) or chromatin accessibility; and d) uncover the key transcription factor that facilitates DCM-enriched CREs interactions and how it drives the pathogenesis of DCM.
Methods
The data, analytic methods, and study materials will be/have been made available to other researchers for purposes of reproducing the results or replicating the procedure. 3D epigenome mapping, bioinformatic analysis, hiPSC-CMs, mouse models, and all experimental approaches are detailed in the Supplemental Methods.
Animal studies
All research protocols conformed to the Guidelines for the Care and Use of Laboratory Animals published by the National Institutes of Health (National Academies Press, eighth edition, 2011). All animal use protocols and experiments were performed according to the guidelines approved by the Institutional Animal Care and Use Committee at Guangdong Provincial People’s Hospital, South China University of Technology, and University of Cincinnati.
Human studies
Acquisition of 101 human heart samples was approved by the Institutional Review Board (IRB) for the protection of human subjects at Duke University in US. De-identified frozen human heart samples with pathological characterization were provided by the Duke Human Heart Repository (DHHR), Department of Surgery at Duke University School of Medicine. In brief, the left ventricular (LV) heart samples were collected from DCM patients during surgery and the LV tissues were immediately dissected and snap frozen in liquid nitrogen. The LV samples from non-failing (NF) donor hearts were served as controls. The heart sample characteristics are provided in Supplemental Table 1.
Statistical Analysis
Hypergeometric test was used to detect significant chromatin interactions in HiChIP data. Negative binomial generalized log-linear model was used to model read counts in sequencing data and quasi-likelihood (QL) F-test was used to identify differentially abundant features (chromatin interactions, expressed genes, transcription factor binding peaks, etc). Statistical analysis was performed with R version 3.5.1 or Prism 7.0 (GraphPad Inc). The statistical tests performed for each statistical graph were listed in the individual figure legends. For further details, see Supplemental Methods.
Results
Genome-Wide Identification of the Active CREs in DCM and NF Hearts
We used 50 human DCM and 51 non-failing (NF) biobanked heart tissues (Supplemental Table 1) to uncover novel mechanisms that regulate DCM pathogenesis by integrative analysis of multi-layered 3D epigenomic features: transcriptome, epigenome and 3D chromatin connectome. To accomplish this, we performed RNA-seq (n=101), ChIP-seq (H3K27ac) (n=20), HiChIP (H3K27ac) (n=20), in situ Hi-C (n=4) and ATAC-seq (n=20) mapping (Figure 1A). Principal component analysis (PCA) from RNA-seq data depicted a separation between DCM and NF hearts, suggesting that the transcriptome of DCM is globally reconfigured compared to NF (Figure S1A). As CREs have been recognized as essential sequences for cell-specific transcription, we performed H3K27ac ChIP-seq9, 22 on 10 DCM and 10 NF human heart samples (left ventricle) to annotate active CREs. Similarly, PCA analysis showed a separation between the DCM and NF samples (Figure S1B), indicating that the active CREs were differentially used in DCM and NF hearts.
Figure 1. Genome-Wide Identification of Active CREs in DCM and NF Hearts.
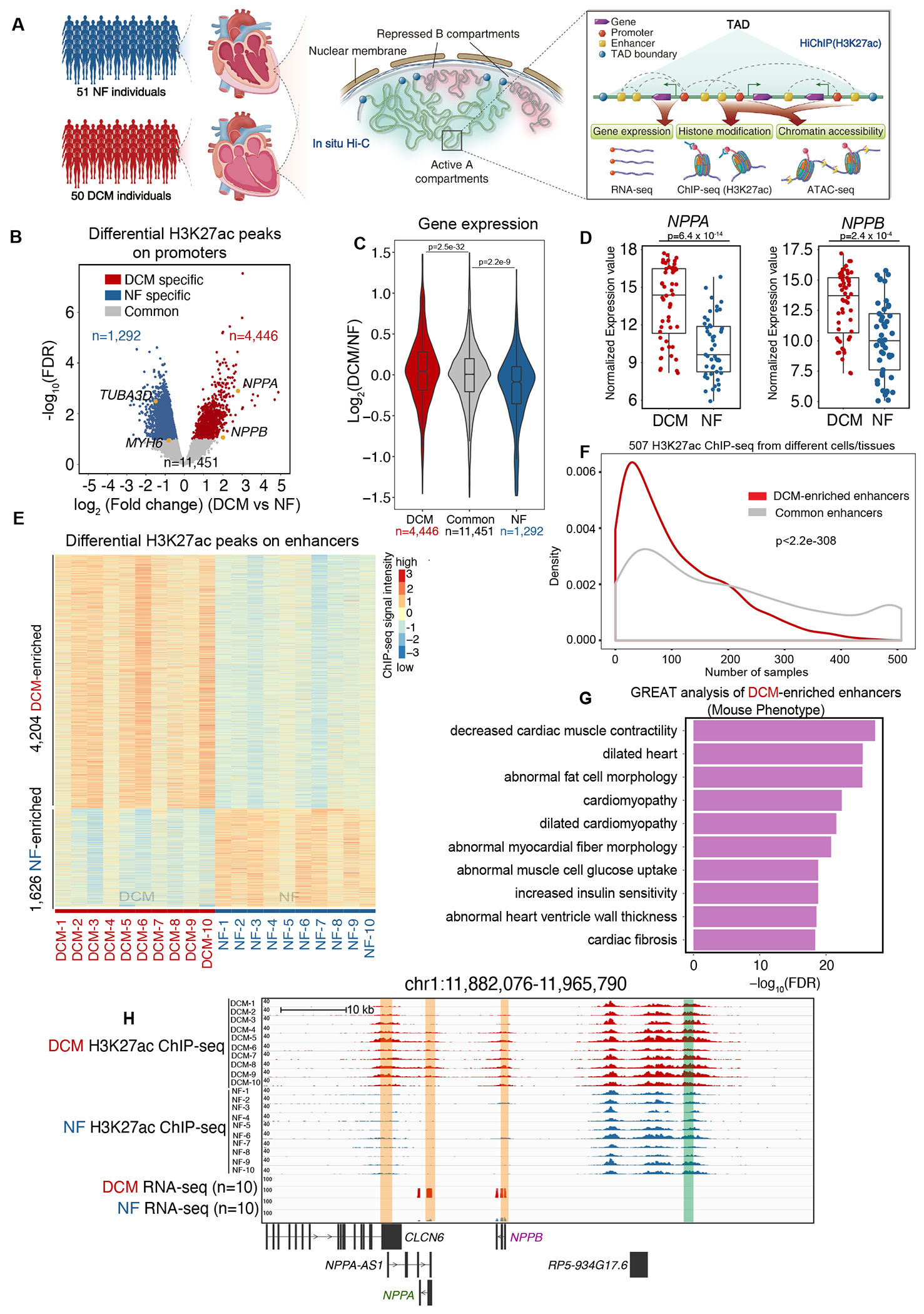
A. Schematic representation of 3D epigenome and transcriptome mapping for 50 DCM and 51 NF left ventricle heart tissue samples (left panel). In situ Hi-C was performed to examine the active A compartment and B compartment and topologically associating domains (TADs) (middle panel). HiChIP (H3K27ac) was performed to map the interactome of the active CREs, which was annotated by H3K27ac ChIP-seq. ATAC-seq was used to identify chromatin accessibility on the CREs. RNA-seq was used to measure transcription output (right panel).
B. Volcano plot showing NF-enriched (n=1,292), DCM-enriched (n=4,446) and common (n=11,451) H3K27ac ChIP-seq peaks in promoter regions (within +/−2.5 kb of known transcription start sites (TSSs)) of DCM and NF hearts. Cardiac hypertrophy marker genes (NPPA, NPPB), normal cardiac marker gene (MYH6) and the cytoskeleton marker gene (TUBA3D) are labeled in yellow.
C. Violin plot showing expression bias for genes exhibiting specific H3K27ac peaks in promoter regions (n=4,446 DCM-enriched H3K27ac peaks, n=1,292 NF heart-enriched H3K27ac peaks and n=11,451 common H3K27ac peaks). Data were analyzed by Wilcoxon rank sum test with continuity correction.
D. Expression values (log2(normalized counts)) for NPPA and NPPB in 50 DCM and 51 NF heart RNA-seq datasets. Data were analyzed by Wald test.
E. Heatmap of DCM-enriched (n=4,204) and NF-enriched (n=1,626) H3K27ac ChIP-Seq peaks on enhancer regions in 10 DCM and 10 NF heart samples (rows represent peaks, and columns represent samples).
F. Enhancer breadth (number of cells/tissues in which an enhancer is activated) of DCM-enriched and common enhancers. DCM-enriched enhancers (red) are significantly more represented as tissue-enriched than common enhancers (grey). Data were analyzed by Kolmogorov-Smirnov test.
G. Bar chart showing the top 10 Ontology terms (Mouse Phenotype, from GREAT analysis) enriched for genes associated with DCM-enriched enhancers (ontology terms were ranked by negative log10(FDR) from smallest to largest).
H. Genome browser view of the genes NPPA and NPPB and their associated promoters and enhancers in DCM and NF hearts. Enhancer is highlighted in the green box, while promoter is highlighted in the orange box.
Since H3K27ac is associated with both promoters and distal enhancers9, we then separated H3K27ac peaks into promoter proximal peaks and enhancer peaks based on their distances (+/−2.5 kb) to known transcription start sites (TSSs). Among the promoter regions, we identified 4,446 DCM-enriched H3K27ac peaks (e.g., promoters of the cardiac hypertrophy marker genes NPPA and NPPB23), 1,292 NF-enriched H3K27ac peaks (e.g., promoters of the normal cardiac marker gene MYH624 and the cytoskeleton marker gene TUBA3D25), and 11,451 H3K27ac peaks common to both DCM and NF hearts (Figure 1B). By integrating the RNA-seq data for those samples, we found that the expression of genes with DCM-enriched H3K27ac peaks on their promoters was significantly upregulated in DCM hearts compared to NF hearts, while the expression of genes with NF-enriched H3K27ac peaks was significantly downregulated, and the expression of genes with common H3K27ac peaks was unchanged (Figure 1C). We further corroborated these results by analyzing the expression of NPPA, NPPB, MYH6 and TUBA3D in human DCM and NF heart tissues. NPPA and NPPB were significantly upregulated (Figure 1D), whereas MYH6 and TUBA3D were significantly downregulated in DCM compared to NF hearts (Figure S1C). These trends are consistent with the roles of H3K27ac in active transcription. Interestingly, the expression levels of NPPA and NPPB were highly correlated (Pearson correlation coefficient=0.751) (Figure S1D), suggesting that they are co-regulated at the transcriptional level and may share common regulatory elements.
Next, we identified 4,204 DCM-enriched enhancers, 1,626 NF-enriched enhancers (Figure 1E) and 38,018 common enhancers present in both DCM and NF hearts (Figure S1E). By integrating 507 H3K27ac ChIP-seq data derived from different cells/tissues from the ENCODE project, we further validated that the 4,204 DCM-enriched enhancers displayed high cell specificity compared to 38,018 common enhancers (Figure 1F, Supplemental Table 2). In addition, we split the H3K27ac ChIP-seq data into cardiac (n=18) and non-cardiac (n=489) cell types and found that H3K27ac peak regions in cardiac-cell types overlapped significantly more DCM-enriched enhancers compared to non-cardiac cell types (Figure S1F). These results suggested that DCM-enriched enhancers could play a role in orchestrating cell-type-specific regulatory activity in DCM hearts. In line with this, GREAT analysis showed that DCM-enriched enhancers are located near genes essential for decreased muscle contractility, dilated cardiomyopathy, abnormal myocardial fiber morphology etc. (Figure 1G), suggesting the regulatory potential of these genes in DCM pathogenesis. DCM-enriched CREs can be exemplified by a genomic region on chromosome 1 (Figure 1H). This region contained a DCM-enriched enhancer near the coding gene NPPB. In addition, the NPPA-AS1, NPPA, and NPPB promoters had specific H3K27ac peaks. Previous study demonstrated a cis-regulatory role for NPPA-AS1 transcript in the repression of NPPA expression in atria26. However, NPPA-AS1 was transcriptionally inactive, as shown from RNA-seq data, suggesting that this promoter may function in trans to regulate the transcription of other genes in DCM ventricle.
Taken together, the above results highlight that gene expression in DCM hearts is regulated by CREs and suggest that their interactions will bring to light the functions and target genes of these regulatory elements.
Characterization of the 3D Chromatin Organization in DCM and NF Hearts
The 3D connectome brings together regulatory chromatin regions and hence can potentially contribute to the regulation of gene expression. To map the high-resolution chromatin contact maps of active CREs, we performed H3K27ac HiChIP on 10 DCM and 10 NF samples. We obtained ~45 million high-quality paired-end reads (i.e., Unique valid interaction pairs) on average from the HiChIP libraries (Supplemental Table 3). For the HiChIP data, we found that the inter-group variation was greater than intra-group variation (Figure S2). HiChIP data contain both 1D (binding sites of the targeted proteins) and 3D (chromatin interactions between binding sites) information12, 13. We found that the 1D signal of the HiChIP data was comparable to that of the H3K27ac ChIP-seq datasets (Figure S3A). Inspection of the combined interaction matrix at different resolutions showed typical compartments (500 kb resolution), TADs (25 kb resolution) and focal loops (5 kb resolution) for both DCM and NF hearts, similar to those reported in previous HiChIP data12, 13 (Figure 2A). We then identified high-confidence chromatin interactions among gene promoters and distal enhancers in DCM and NF hearts (Figure 2B). We found that 13,562 promoters and 11,308 enhancers were involved in chromatin interactions in DCM and that anchor genes had significantly higher expression than that of non-anchor genes (Figure 2C), suggesting that chromatin interactions may play a role in transcriptional activation.
Figure 2. Characterization of the 3D Chromatin Organization of the Active CREs by H3K27ac HiChIP in DCM and NF Hearts.
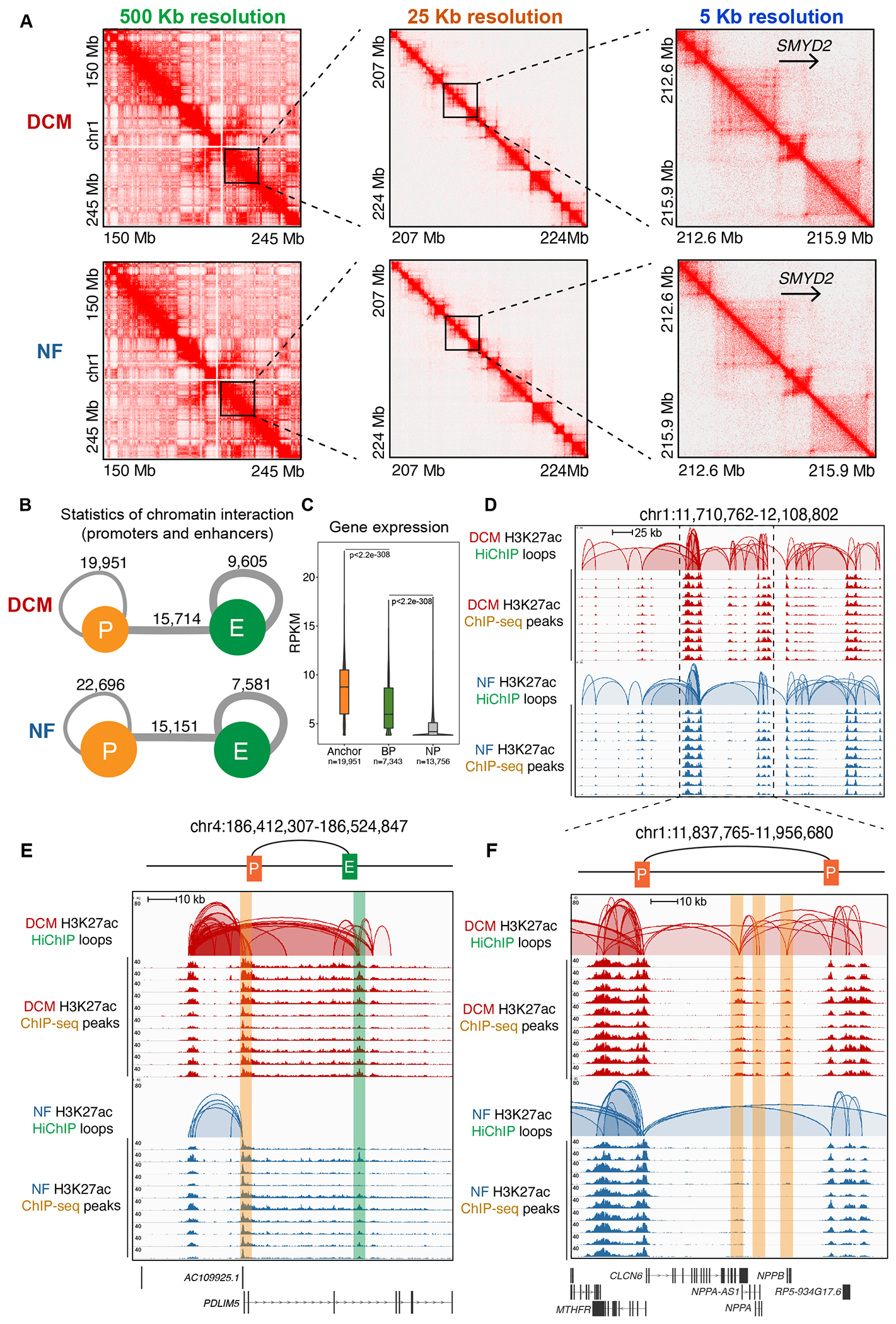
A. Knight–Ruiz (KR) matrix–balanced interaction maps of merged DCM (top) and NF (bottom) HiChIP valid interaction pairs of typical compartments at 500 kb resolution (left), TADs at 25 kb resolution (middle) and focal loops at 5 kb resolution (right).
B. Summary for the number of chromatin interactions between promoters (P) and enhancers (E) in DCM (upper) and NF (lower) hearts.
C. Violin plot of expression levels for anchor genes (n=19,951), basal promoter (BP) genes (n=7,343) and non-promoter (NP) genes (n=13,756) in DCM. Data were analyzed by Wilcoxon rank sum test with continuity correction.
D. Genome browser view of CREs interactions in chr1:11,710,762-12,108,802. The top two rows represent the CREs interactions in DCM hearts, and the bottom two rows represent those in NF hearts. The HiChIP loops and ChIP-seq peaks are shown for each.
E. Genome browser view of enhancer-promoter interactions for PDLIM5 (chr4:186,412,307-186,524,847). The top two rows represent the interactions in DCM hearts, and the bottom two rows represent those in NF hearts. The HiChIP loops and ChIP-seq peaks are shown for each. The enhancer is highlighted in the green box, while the promoter is highlighted in the orange box.
F. Genome browser view of promoter-promoter interaction for NPPA-AS-1, NPPA and NPPB. The top two rows represent the interactions in DCM hearts, and the bottom two rows represent those in NF hearts. The HiChIP loops and ChIP-seq peaks are shown for each. Enhancer is highlighted in the green box, while promoter is highlighted in the orange box.
We then constructed chromatin interaction networks for DCM and NF hearts separately, as genome-wide chromatin interactions are organized into giant, modular interaction networks27. Promoters and enhancers were organized into chromatin interaction domains (Figure 2D) across the genomes of both DCM and NF hearts; in addition, those interaction domains spanned similar genomic distances (Figure S3B) and contained most of the active promoters and enhancers (Figure S3C, D). The numbers of connected enhancers were positively correlated with increase in expression levels of their connected genes (Figure S3E). Chromatin interactions within the domains can first be classified as promoter-enhancer interactions, i.e., gene promoters regulated by one or multiple enhancers. For example, the promoter of PDLIM5, a gene involved in actin cytoskeleton organization in heart development28 and DCM29, was regulated by an enhancer embedded in its intron (Figure 2E). Second, these chromatin interactions may be classified as promoter-promoter interactions. For example, both the NPPA and NPPB promoters have strong interactions with the NPPA-AS1 promoter as shown by paired-end tags (PETs) (Figure 2F), a finding that supports the conclusion that NPPA and NPPB may be co-regulated by NPPA-AS1 promoter. NPPA-AS1 transcript is shown to have an inhibitory effect on NPPA expression in atria26, but our RNA-seq revealed that the NPPA-AS1 gene is transcriptionally repressed in DCM ventricle. Interestingly, recent study showed that non-transcribing promoter can have enhancer activity to regulate other gene transcription via chromatin looping30, in line with previous identification of enhancer-like promoters31–33. To test whether NPPA-AS1 promoter can function as an enhancer to regulate NPPA and NPPB expression, we performed luciferase reporter assays, a commonly used method for promoter and enhancer characterization34 (Figure S3F). NPPA or NPPB promoter fragment was cloned upstream of the luciferase gene as the driving promoter, and the NPPA-AS1 promoter was cloned downstream of the luciferase gene as a presumed enhancer, as enhancers generally function in a manner independent of orientation. The analysis of transfection and luciferase activity was performed in an in vitro cardiac hypertrophy model induced by entothelin-1 (10 nM, 48 hours) in human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs). We found that the NPPA-AS1 promoter alone was not able to activate luciferase gene expression. However, the NPPA-AS1 promoter could act in concert with NPPA/NPPB promoter to significantly enhance the luciferase activity compared to NPPA/NPPB promoter alone (Figure S3G, H). This result indicated that the NPPA-AS1 promoter could function as an enhancer to regulate NPPA/NPPB transcription. Overall, we identified the interactome of active CREs in DCM and NF in high resolution and uncovered that active promoters and enhancers were organized by multiple types of chromatin loops.
Specific Enhancer-Promoter Connectome Regulates DCM-Specific Transcription
To dissect how CRE interactions contribute to DCM-specific transcription, we performed differential analysis of HiChIP loops. Two classes of differential loops were identified based on H3K27ac signal on loop anchors: Class I (change of H3K27ac signal on loop anchors) and Class II (no change of H3K27ac signal on loop anchors). For Class I, we identified 670 DCM-enriched H3K27ac loops (DCM-enriched anchors) and 1,035 NF-enriched H3K27ac loops (NF-enriched anchors) (Figure 3A, Supplemental Table 4). We found that these DCM-enriched H3K27ac loop-connected genes were significantly upregulated in DCM hearts (Figure 3B), while the NF-enriched H3K27ac loop-connected genes were significantly downregulated in DCM hearts (Figure 3C). For Class II, we identified 4,883 DCM-enriched H3K27ac loops (Common anchors) and 3,712 NF-enriched H3K27ac loops (Common anchors) (Figure S4A, Supplemental Table 4). However, these differential loops have less interaction intensity (PET counts) compared to their counterparts in Class I (Figure S4B) and their connected genes are not significantly linked to transcription change (Figure S4C, D). Hence, in downstream analysis, we focus on Class I loops to explore the transcriptional regulation in DCM.
Figure 3. DCM-Enriched Enhancer/Promoter Connectome Contributes to DCM-Specific Transcription.
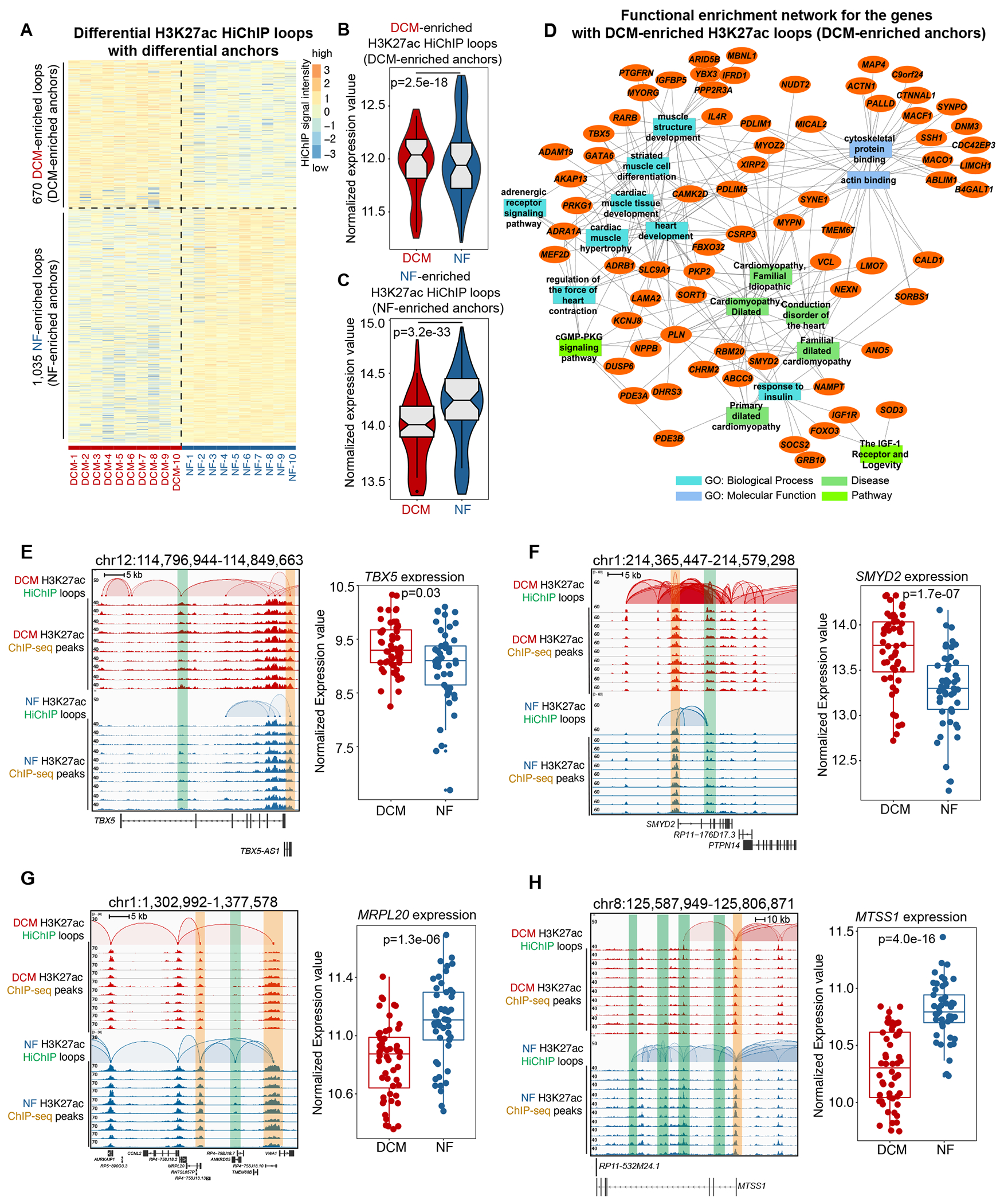
A. Heatmap showing the intensity of differential H3K27ac loops with differential loop anchors (H3K27ac signal) among 10 DCM and 10 NF heart samples.
B. Normalized expression values of the genes with DCM-enriched H3K27ac loops (DCM-enriched anchors). Data were analyzed by Wilcoxon rank sum test with continuity correction.
C. Normalized expression values of the genes with NF-enriched H3K27ac loops (NF-enriched anchors). Data were analyzed by Wilcoxon rank sum test with continuity correction.
D. Network representation of functional enrichment of the genes with DCM-enriched H3K27ac loops (DCM-enriched anchors). Functional enrichment was done using the ToppFun application. Orange nodes represent DCM-enriched genes, while the different colored rectangles represent enriched terms. Only select enriched terms are shown here. Network was generated using the Cytoscape application.
E. Left panel: browser screenshot showing TBX5 with DCM-enriched E-P interactions (chr12:114,796,944-114,849,663). The top two rows represent the interactions in DCM hearts, and the bottom two rows represent those in NF hearts. The HiChIP loops and ChIP-seq peaks are shown for each. Enhancer is highlighted in the green box, while promoter is highlighted in the orange box; Right panel: expression values (log2(normalized counts)) for TBX5 in 50 DCM and 51 NF heart RNA-seq datasets. Data were analyzed by Wald test.
F. Left panel: browser screenshot showing SMYD2 with DCM-enriched E-P interactions (chr1:214,365,447-214,579,298). The top two rows represent the interactions in DCM hearts, and the bottom two rows represent those in NF hearts. The HiChIP loops and ChIP-seq peaks are shown for each. Enhancer is highlighted in the green box, while promoter is highlighted in the orange box; Right panel: expression values (log2(normalized counts)) for SMYD2 in 50 DCM and 51 NF heart RNA-seq datasets. Data were analyzed by Wald test.
G. Left panel: browser screenshot showing MRPL20 with NF-enriched E-P interactions (chr1:1,302,992-1,377,578). The top two rows represent the interactions in DCM hearts, and the bottom two rows represent those in NF hearts. The HiChIP loops and ChIP-seq peaks are shown for each. Enhancer is highlighted in the green box, while promoter is highlighted in the orange box; Right panel: expression values (log2(normalized counts)) for MRPL20 in 50 DCM and 51 NF heart RNA-seq datasets. Data were analyzed by Wald test.
H. Left panel: browser screenshot showing MTSS1 with NF-enriched E-P interactions (chr8:125,587,949-125,806,871). The top two rows represent the interactions in DCM hearts, and the bottom two rows represent those in NF hearts. The HiChIP loops and ChIP-seq peaks are shown for each. Enhancer is highlighted in the green box, while promoter is highlighted in the orange box; Right panel: expression values (log2(normalized counts)) for MTSS1 in 50 DCM and 51 NF heart RNA-seq datasets. Data were analyzed by Wald test.
To understand the functional implications of the genes regulated by DCM/NF-enriched H3K27ac loops, we represent their functional enrichment network35 (Figure 3D, Figure S4E). For DCM-enriched H3K27ac loops, the most highly represented GO terms included (1) molecular functions related to cytoskeletal protein binding and actin binding and (2) biological processes related to heart development, cardiac muscle development, adrenergic receptor signaling pathway, cardiac muscle hypertrophy, regulation of the force of heart contraction, response to insulin, muscle structure development, and striated muscle and differentiation. Pathway terms included cGMP-PKG signaling pathway, the IGF-1 receptor and longevity. Disease terms included, for example, cardiomyopathy (dilated), familial dilated cardiomyopathy, primary dilated cardiomyopathy, cardiomyopathy (familial idiopathic), and conduction disorder of the heart. These results suggested that specific enhancer/promoter connectome regulated specific transcription programs implicated in DCM development.
We uncovered specific examples of DCM-enriched E-P interactions. TBX5 encodes a transcription factor crucial for proper cardiac development36, and we found that its promoter interacted with an activated enhancer embedded in its 7th intron in DCM hearts (Figure 3E). SMYD2 (histone lysine methyltransferase gene) is robustly expressed in neonatal heart and regulates cardiac development and regeneration37, 38. Similar to TBX5, we also found that its promoter interacted with an activated enhancer embedded in its 3rd intron in DCM hearts (Figure 3F). Other DCM-enriched E-P examples (MEF2D, regulator for myogenesis39 and cardiac stress40; MYPN, regulator for maintaining sarcomeric integrity41) are displayed in Figure S4F, G respectively. These specific examples of DCM-enriched E-P interactions were validated by 3C-qPCR (Figure S4H). In addition to DCM-enriched E-P interactions, we also examined NF-enriched E-P interactions. For example, MRPL20 encodes the large 39S subunit of mitochondrial ribosomes for protein synthesis within the mitochondria. We identified NF-enriched enhancer-promoter and promoter-promoter interactions for MRPL20, which are lost in DCM, suggesting its potential impact on mitochondrial dysfunction in DCM42 (Figure 3G). In addition, a recent study employing integrated multiomics approaches like RNA-seq and methylation assays identified MTSS1 as one of the key genes associated with left ventricular systolic function43. We found that enhancer connectome associated with MTSS1 promoter was lost in DCM as compared to NF (Figure 3H), thus indicating its possible association with cardiac dysfunction in DCM. Other NF-enriched E-P examples (AIF1L, regulator for actomyosin contractility44; LRRC10B, whose paralog LRRC10 was ablated to induce DCM in mice45) are displayed in Figure S4I, J respectively. Collectively, these findings indicate that DCM and NF-enriched E-P interactions regulate genes involved in cardiac homeostasis.
DCM-Enriched Enhancer/Promoter Connectome Largely Resides in Conserved Higher-Order Chromatin Architectures and Pre-Accessible Chromatin Sites Bound by Reactivated HAND1
Next, to investigate whether the altered H3K27ac looping in DCM is associated with changes in higher-order chromatin structures, we performed in situ Hi-C on DCM and NF heart tissue samples (left ventricles) (Supplemental Table 3, Figure S5). In situ Hi-C can capture the multilevel chromatin architecture, including the A and B compartments and TADs. We found that both the DCM and NF heart genomes were partitioned into A and B compartments (Figure 4A). We compared the A and B compartments in the genomes of NF and DCM hearts; overall, only a small fraction (A-to-B, 13.3%; B-to-A, 9.4%) of the genome switched compartments from NF to DCM hearts (Figure 4B). Gene expression was reduced for the genes that switched from the A-to-B compartment, while expression was increased for the genes that switched from the B-to-A compartment (Figure 4C); these results agree with previous findings showing that the A compartment corresponds to active transcription and that the B compartment corresponds to repressed transcription18.
Figure 4. DCM-Enriched Enhancer/Promoter Connectome Largely Resides in Conserved Higher-Order Chromatin Architectures and Pre-accessible Chromatin Sites Bound by Reactivated HAND1.
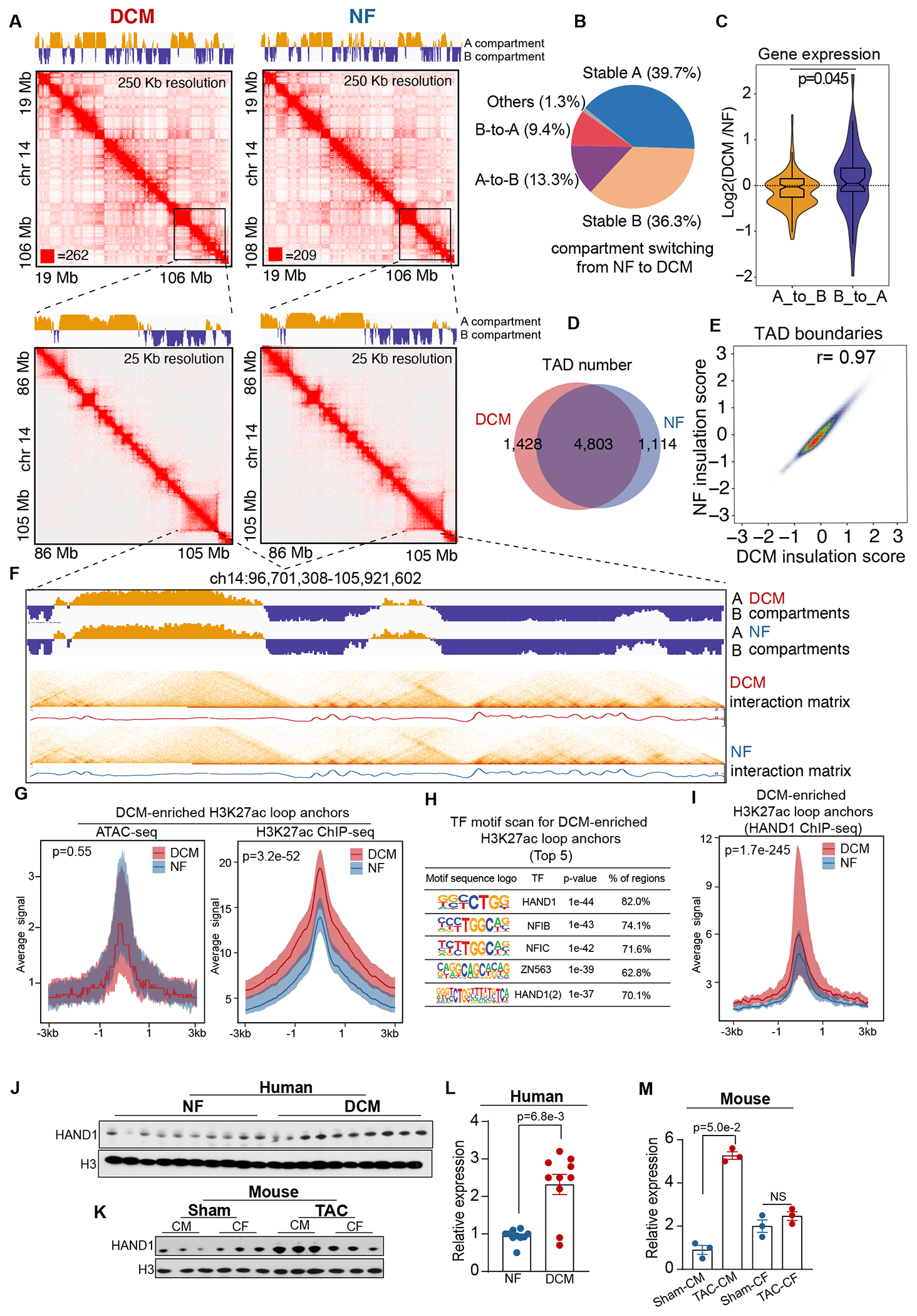
A. Knight–Ruiz (KR) matrix–balanced interaction maps of merged DCM (top) and NF (bottom) in situ Hi-C valid pairs for DCM and NF hearts at 250 kb and 25 kb resolution. The top tracks indicate the A (yellow) and B (blue) compartments.
B. Proportion of genome switching between the A and B compartments (A-to-B (13.3%), B-to-A (9.4%), stable A (39.7%), and stable B (36.3%)) from NF to DCM hearts.
C. Expression bias in A-to-B and B-to-A compartment-switched genomes in DCM hearts normalized to expression in NF hearts. Data were analyzed by Mann-Whitney U test.
D. Venn diagram showing the number of TADs in DCM hearts (n=1,428) and NF hearts (n=1,114) and in both (n=4,803).
E. Correlation of insulation scores for DCM and NF hearts; the Pearson correlation coefficient (R) is indicated (r=0.97).
F. A/B compartment, Hi-C contact map (heatmap) and corresponding insulation profiles for DCM and NF hearts in the genomic region (ch14:96,701,308-105,921,602).
G. Left panel: aggregation plot of DCM (n=20) and NF (n=20) normalized ATAC-seq signals ± 3 kb centered on the DCM-enriched H3K27ac loop anchors; Right panel: Left panel: aggregation plot of DCM (n=20) and NF (n=20) normalized H3K27ac ChIP-seq signals ± 3 kb centered on the DCM-enriched H3K27ac loop anchors. Data were analyzed by Mann-Whitney U test.
H. Transcription factor motif scan for DCM-enriched loop anchors. The top 5 transcription factors are shown along with their motifs, p-values and percentage of regions.
I. Aggregation plot of normalized HAND1 ChIP-seq signals ± 3 kb centered on the DCM-enriched H3K27ac loop anchors in DCM (n=4) and NF (n=4) hearts. Data were analyzed by Mann-Whitney U test.
J. Evaluation of HAND1 expression (normalized to H3 expression) in human non-failing (NF) hearts and failing hearts (DCM) by Western blot analysis.
K. Evaluation of HAND1 expression (normalized to H3 expression) in mouse cardiomyocytes (CMs) and cardiac fibroblasts (CF) obtained from non-failing hearts (Sham) and failing hearts (8 weeks post-transverse aortic constriction (TAC)) by Western blot analysis.
L. Quantification of HAND1 expression in human NF and DCM hearts. Data were analyzed by Mann-Whitney U test.
M. Quantification of HAND1 expression in mouse CMs and CFs isolated from Sham and TAC hearts. Data were analyzed by Mann-Whitney U test.
Then, we examined the TAD distribution and identified 1,428 DCM-specific TADs, 1,114 NF-specific TADs, and 4,803 common TADs (Figure 4D, Supplemental Table 4). Comparison of genome-wide insulation scores suggested that TADs were highly conserved between DCM and NF hearts (correlation coefficient = 0.97) (Figure 4E) and presented similar insulation profiles (Figure 4F). Hence, the overall higher-order chromatin structures remain largely invariant from NF to DCM hearts. Moreover, we found that the A and B compartments occupied similar portions of the genome, with TADs distributed evenly in the A and B compartments in both DCM and NF hearts, while H3K27ac loops located mostly in the A compartment, suggesting that H3K27ac-associated chromatin interactions are involved in active transcription (Figure S6A, B).
Furthermore, we aimed to determine the distributions of enriched H3K27ac loops in altered compartments and TADs in DCM and NF hearts. We found that only 1.3% of DCM-enriched H3K27ac loops (Figure S6C) and 5.8% of NF heart-enriched H3K27ac loops (Figure S6D) were in switched compartments. In addition, 9.4% of DCM-enriched H3K27ac loops (Figure S6E) and 15.5% of NF-enriched H3K27ac loops (Figure S6F) were in changed TADs. The small percentage of specific loops within altered compartments and TADs suggested that factors other than higher-order chromatin structures might underlie the formation of specificH3K27ac loops in DCM.
As chromatin accessibility is essential for the function and interaction of CREs5, we aimed to test whether DCM/NF-enriched H3K27ac loop formation could be explained solely by chromatin accessibility. To this end, we developed an optimized ATAC-seq protocol46 for frozen heart tissues (see Supplemental Methods) and performed the experiments on the same 20 DCM/NF heart tissues described above (Figure S6G). Compared to NF hearts, DCM hearts had decreased chromatin accessibility for the NF-enriched H3K27ac loop anchors (Figure S6H). However, chromatin accessibility was equivalent for the DCM-enriched H3K27ac loop anchors in DCM and NF hearts (Figure 4G). These results suggested that DCM-enriched H3K27ac loops were formed on pre-accessible chromatin regions.
Because transcription factors are key determinants shaping the 3D genome47, we scanned the transcription factor motifs on the DCM-enriched H3K27ac loop anchors that overlapped with their ATAC-seq peaks. The motifs for the transcription factors SMAD348 and MEF49, known to be involved in heart failure or DCM, were among the 20 most significantly enriched motifs (ranked No. 15 and No. 19, respectively) (Figure S6I). The most significantly enriched transcription factor motif on the DCM-enriched H3K27ac loop anchors was HAND1 (Figure 4H), which is not enriched in NF-enriched H3K27ac loop anchors (Figure S6J). HAND1 is a basic helix-loop-helix (bHLH) transcription factor that is highly expressed in the embryonic heart and essential for heart development and homeostasis. In humans and rodents, HAND1 transcription in the adult heart is maintained at a much lower level than that in the embryonic heart50. Recently, a report identified a heterozygous HAND1 mutation in the blood DNA from some Chinese familial DCM patients51. However, the functional roles of HAND1 in 3D epigenome reprogramming and transcription regulation within DCM hearts remains unexplored.
To further uncover the link of HAND1 with DCM-enriched H3K27ac looping, we performed HAND1 ChIP-seq (Figure S6K). We found that HAND1 binding was preferably enriched on DCM-enriched H3K27ac loop anchors (Figure 4I) in DCM hearts compared to randomly permuted regions (Figure S6L), NF-enriched H3K27ac loop anchors (Figure S6M) or NF/DCM-all loop anchors (Figure S6N). Although we demonstrated no significant difference of HAND1 RNA expression between human DCM and NF hearts (Figure S6O), HAND1 protein expression in human DCM hearts was significantly upregulated compared to that in NF hearts (Figure 4J, L), suggesting post-transcriptional/post-translational modifications of HAND1 in DCM. To further analyze HAND1 protein expression in mouse failing hearts, we constructed transverse aortic constriction (TAC) model and isolated cardiomyocytes (CMs) and cardiac fibroblasts (CFs) at 8 weeks after TAC. We found that HAND1 protein expression was significantly upregulated in TAC-CMs but not in TAC-CFs as compared to Sham controls (Figure 4K, M). These data suggested that HAND1 might be the potential transcription factor linked to the DCM-enriched CREs interactions in cardiomyocytes.
HAND1 Rewires Enhancer/Promoter Connectome in Concordance with Transcriptional Changes Associated with DCM Etiology in Human iPSC-Derived Cardiomyocytes
To dissect the functionality of HAND1 in organizing enhancer/promoter connectome of human cardiomyocytes, we transduced Ctrl and HAND1-overexpressing (HAND1OE) adenovirus into human iPSC-derived cardiomyocytes (hiPSC-CM) for 72 hrs, which have emerged as invaluable platforms for disease modeling and therapeutic screening in DCM52. Subsequently, we performed 3D epigenome mapping (H3K27ac HiChIP, H3K27ac ChIP-seq, HAND1 ChIP-seq and RNA-seq) (Figure 5A, Figure S7A–C) in these cells. HAND1-overexpression induced extensive rewiring of enhancer/promoter connectome with more gained H3K27ac HiChIP loops (N=5,456, 73%) compared to lost loops (N=1,968, 23%) (Figure 5B; global view of gained loops are demonstrated in Figure S7D). By integrating HAND1 ChIP-seq data, we found that 85% of the gained loops were bound by HAND1 on one or both anchors in HAND1OE hiPSC-CM (Figure 5C). In addition, HAND1 binding intensity is significantly higher in those gained loop anchors of HAND1OE compared to Ctrl hiPSC-CM (Figure 5D). However, HAND1 binding intensity is minimal on permuted regions (Figure S7E) while equivalent on common loop anchors (Figure S7F), and subtly less on lost loops (Figure S7G) in HAND1OE compared to Ctrl hiPSC-CM, which further supported that gained loops are attributed to specific HAND1 binding on the loop anchors. Moreover, HAND1 binding intensity on the gained loop anchors is moderately correlated with loop intensity (Figure 5E). Taken together, these data indicated that the induced enhancer/promoter rewiring is a direct effect of the upregulation of HAND1.
Figure 5. Overexpression of HAND1 Rewires Long-Range Enhancer/Promoter Connectome and Transcriptome in Human iPSC-Derived Cardiomyocytes.
A. Schematic illustration of the experimental system and strategy to examine the roles of HAND1 in 3D chromatin organization and transcriptional control in human iPSC-derived cardiomyocytes (hiPSC-CM).
B. Venn diagram showing the number of H3K27ac HiChIP loops gained, lost and constant in HAND1-overexpressing (HAND1OE) versus Ctrl hiPSC-CM.
C. Percentage of gained H3K27ac HiChIP loops, the anchors of which do or do not overlap with HAND1 ChIP-seq peaks in HAND1OE hiPSC-CM.
D. Aggregation plot and heatmap showing the intensity of HAND1 ChIP-seq peaks on the gained H3K27ac HiChIP loop anchors (HAND1OE versus Ctrl hiPSC-CM).
E. Correlation of the intensity of HAND1 ChIP-seq peaks and the intensity of gained H3K27ac HiChIP loops in HAND1OE hiPSC-CM.
F. Expression of genes with H3K27ac HiChIP loops gained, lost and constant in HAND1-overexpressing (HAND1OE) versus Ctrl hiPSC-CM. Data were analyzed by Wilcoxon rank sum test with continuity correction.
G. Normalized expression values for genes with gained H3K27ac HiChIP loops in HAND1OE hiPSC-CM, the promoter anchors of which connect with different numbers of enhancers. Data were analyzed by Wilcoxon rank sum test with continuity correction.
H. Functional annotation (RNA-Seq_Disease_Gene_and_Drug_Signatures_from_GEO) for the genes with gained H3K27ac HiChIP loops.
I. Functional annotation (Mammalian phenotypes) for genes with gained H3K27ac HiChIP loops.
J. Functional annotation (GO-Biological Process) for genes with gained H3K27ac HiChIP loops.
K-N. Examples of H3K27ac gained interactions (identified by H3K27ac HiChIP and ChIP-seq), HAND1 binding (identified by HAND1 ChIP-seq) and transcription (identified by RNA-seq) in HAND1OE versus Ctrl hiPSC-CM.
By integrating RNA-seq data, we found that genes within the gained and lost loops were significantly up- and downregulated, respectively (HAND1OE compared with Ctrl hiPSC-CM) (Figure 5F). In addition, in gained loops, the numbers of enhancers connected to promoters were significantly correlated with expression level (Figure 5G). Functional annotation analysis indicated that gained loops-associated genes overlapped with the transcriptional signatures in public human DCM RNA-seq data (Figure 5H). In line with this, we found that the genes (e.g., NPPA, NPPB) both upregulated in hiPSC-HAND1OE (vs. Ctrl) and DCM hearts (vs. NF) from this study are enriched in the transcriptional signatures of dilated cardiomyopathy and left ventricle hypertrophy (Figure S7H). Moreover, mammalian phenotypes for gained loops-associated genes showed significant enrichment in decreased survival, abnormal myocardial fiber, and abnormal muscle physiology/contractility (Figure 5I). Furthermore, Gene Ontology for gained loops-associated genes showed significant enrichment in biological processes associated with embryonic heart tube development, regulation of ventricular cardiac cell action potential, cardiac muscle hypertrophy, cardiac muscle contractility, and regulation of cardiac muscle contraction by regulating the release of sequestered calcium ion (Figure 5J), further validating that some HAND1 functions through activating chromatin looping and gene transcription involved in these processes during DCM pathogenesis. Examples of gained loops involving the well-established DCM driver gene PDE1C53 and unexplored genes FREM2, SLC16A2, GAS1 are shown in Figure 5K, L, M, N. These results demonstrate that the upregulation of HAND1 in hiPSC-CM results in the genome-wide gain of enhancer/promoter contacts and concordant transcriptional activation of connected genes implicated in DCM development.
HAND1 Drives DCM Pathology in Human iPSC-CM and Mouse Models
To test the pathological effects of HAND1 in DCM, we performed systematic in vitro and in vivo functional assays (Figure 6A). For in vitro study, we investigated the effect of HAND1 overexpression on human cardiac morphology and function using adenoviral vectors to overexpress HAND1 in hiPSC-CMs. At 72 hrs after transduction, we found a significant higher percentage of HAND1 overexpressing hiPSC-CMs (hiPSC-CMHAND1) were larger than null adenovirus transfected hiPSC-CMs (hiPSC-CMNull) (Figure S8A). Furthermore, overexpression of HAND1 (hiPSC-CMHAND1) resulted in significant increases in Ca2+ transient amplitude (Figure 6B), cell shortening, and relaxation velocity respectively, compared to hiPSC-CMNull (Figure S8B–D). These results indicated acute overexpression of HAND1 induced hiPSC-CM hypertrophy development with associated alteration in Ca2+-handling and contractility, which might be a compensation to maintain cardiomyocyte contraction54–56.
Figure 6. HAND1 Induces DCM Phenotypes in Human iPSC-Derived Cardiomyocytes and Mouse Hearts.
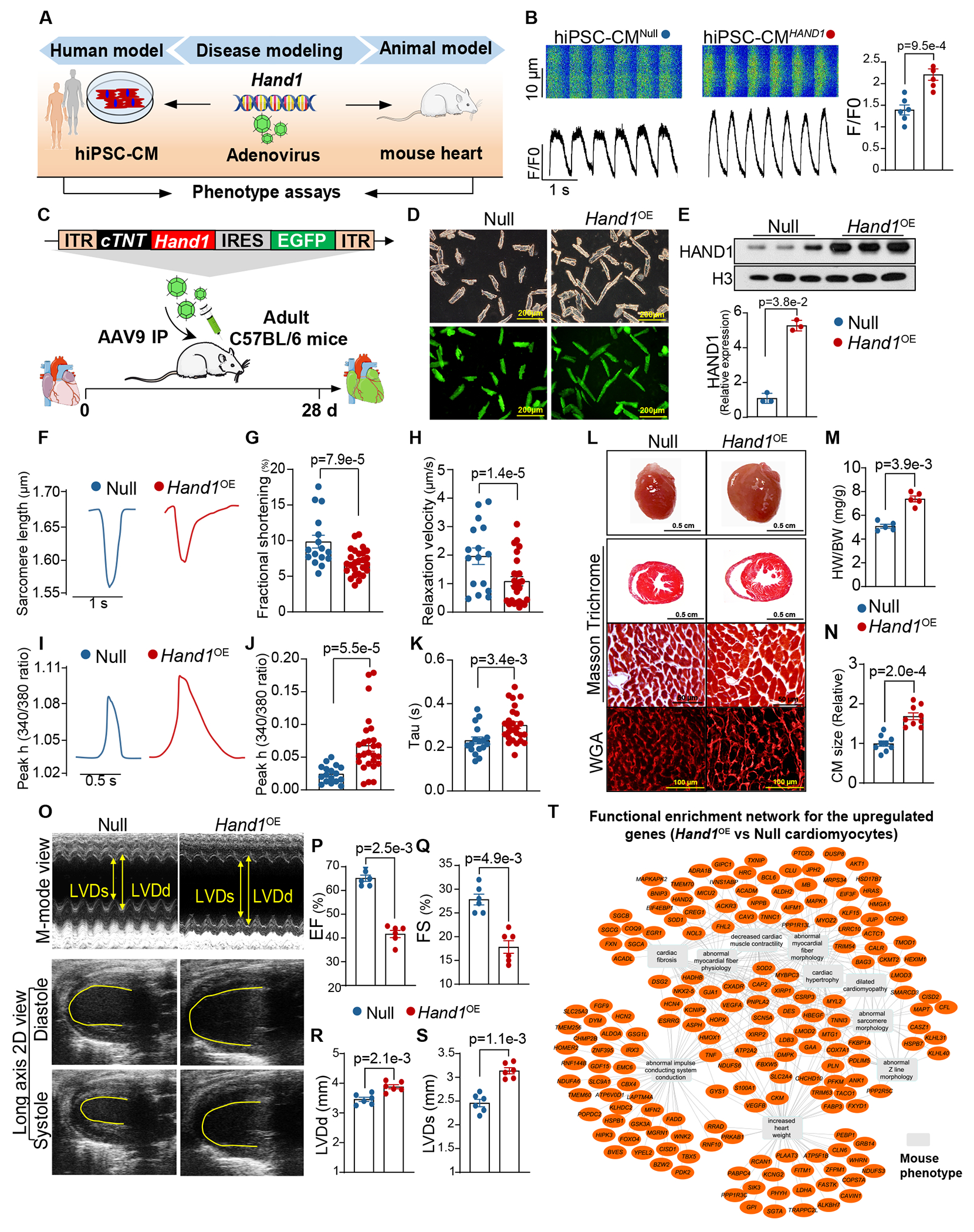
A. Experimental scheme for assessing Hand1 pathogenicity in both human and mouse models with hiPSC-CMs and mouse hearts, respectively.
B. Representative calcium imaging recording traces and calcium transients to electrical pacing at 1 Hz. HiPSC-CMs were transduced with adenoviruses carrying the CMV promoter driving HAND1 (hiPSC-CMHAND1) or adenoviruses carrying the CMV promoter only (hiPSC-CMNull). F/F0, peak amplitude relative to baseline fluorescence. Data were analyzed by Student’s t test.
C. Experimental scheme for evaluating AAV9-driven Hand1 overexpression in cardiac morphology and function. Hand1 under the control of the cTNT promoter was cloned into an AAV9 backbone with an EGFP reporter (AAV9-cTNT-Hand1-EGFP). Animals were injected intraperitoneally and subsequently analyzed 28 days later.
D. Representative images of CMs isolated from AAV treated hearts under light microscopy and fluorescence microscopy. The mice receiving injection of AAV9-cTNT-EGFP were designated as Null and the mice receiving AAV9-cTNT-Hand1-EGFP injection were designated as Hand1OE.
E. Western blot assay for HAND1 protein expression in Null and Hand1OE hearts. Data were analyzed by Mann-Whitney U test.
F-H. Contractility mechanic assay of mouse CMs isolated from Null and Hand1OE hearts. (F) Representative images of sarcomere shortening tracing. (G) Cell shortening (%) at 0.5 Hz. (H) Relaxation velocity of sarcomere at 0.5 Hz. Data were analyzed by Student’s t test.
I-K. Ca2+ kinetics of mouse CMs isolated from Null and Hand1OE hearts. (I) Representative images of Ca2+ transient tracing. (J) Ca2+ transient amplitude (Peak h) as indicated by Fura-2 ratio (340/380 nm) at 0.5 Hz. (K) Ca2+–decay time (Tau) at 0.5 Hz. Data were analyzed by Student’s t test.
L. Gross morphology (first row), Masson’s trichrome staining of gross morphology (second row), Masson trichrome staining of CMs (third row) and wheat germ agglutinin (WGA) staining (bottom panel) of Null and Hand1OE hearts.
M-N. Measurement of heart weight (HW)-to-body weight (BW) ratio (M) and CM size (N) in Null and Hand1OE mice. Data were analyzed by Mann-Whitney U test.
O-S. Heart function was analyzed by echocardiography in Null and Hand1OE mice. Representative images of M-mode and long axis 2D views (O). Qutifications of left ventricular (LV) ejection fraction (EF)(P), fraction shortening (FS)(Q), LV end-diastolic diameter (LVDd)(R), and LV end-systolic diameter (LVDs)(S). Data were analyzed by Mann-Whitney U test.
T. Network representation of functional enrichment of the upregulated genes in Hand1OE cardiomyocytes (vs Null). Functional enrichment was done using the ToppFun application. Orange nodes represent DCM-enriched genes, while the grey colored rectangles represent enriched terms. Top10 enriched terms are shown here. Network was generated using the Cytoscape application.
To validate these results in vivo, we engineered an AAV9 vector with the cTNT promoter to drive cardiac-specific overexpression of Hand1 in mouse CMs (AAV9-cTNT-Hand1-EGFP) (Figure 6C). To assess the specificity and potency of the cTNT promoter, AAV9 virus lacking Hand1 (AAV9-cTNT-EGFP) was delivered to adult wild-type (WT) mice (8 weeks old) (Figure S8E) at 3 different doses (5×1011, 1×1012, and 5×1012 viral genomes (vg) by intraperitoneal injection), and the mice receiving AAV9 were defined as Low, Medium, and High groups, respectively. The mice that received the same volume of saline were defined as the negative control group (designated as the Saline group). Mice injected with the AAV9-cTNT-EGFP viral vector exhibited robust expression of the GFP signal in the CMs (Figure S8F, G). As expected, no GFP signal was observed in saline-treated hearts. Although the AAV dosage was positively correlated with the number of GFP+ CMs, no difference in the GFP protein level was observed between the Medium and High groups (Figure S8F). These data indicate that one systemic dose of 1×1012 vg yields highly efficient transduction in the majority of CMs. We then used the selected dosage to intraperitoneally inject WT adult mice with AAV9-cTNT-Hand1-EGFP (designated as the Hand1OE group) or AAV9-cTNT-EGFP (designated as the Null group). AAV9 transduction in Hand1OE hearts (Figure 6D) resulted in significant upregulation of HAND1, as determined by Western blotting (Figure 6E). To interrogate the functional effect of Hand1 on heart failure progression, contractile mechanics, amplitude and kinetics of intracellular Ca2+ transients were measured in freshly isolated mouse CMs (mCMs) from Hand1OE hearts and Null hearts after AAV9 treatment for 4 weeks in vivo. In contrast to acute overexpression of HAND1 in hiPSC-CM, chronic overexpression of Hand1 resulted in significant decreases in cell shortening and relaxation velocity respectively, compared with the Null CMs (Figure 6F–H). Although assessment of Ca2+ amplitude (peak h) revealed an increase in Hand1OE CMs, the Tau was significantly prolonged as compared to Null CMs (Figure 6I–K), suggesting impaired Ca2+ handling. These results indicated the regulatory effect of Hand1 on CM dysfunction progression, as evidenced by a decline in contractile function and abnormal Ca2+ handling.
Accordingly, compared with the Null hearts, the explanted Hand1OE hearts were markedly enlarged (Figure 6L). In addition, the heart-to-body weight ratios (HW/BW) were higher in Hand1OE mice than in Null mice, indicating a significant increase in heart mass (Figure 6M). Using wheat germ agglutinin (WGA) staining, we also found that the Hand1OE CMs were significantly larger than the Null CMs (Figure 6N), implying that cardiac hypertrophy was developed in the Hand1OE CMs. These effects were not the result of AAV9-induced toxicity since no changes in CM size were observed in mice transduced with AAV9 without Hand1 (AAV9-cTNT-GFP) or in Saline control mice (Figure S8H and I). Next, cardiac function analysis was examined in Hand1OE mice and Null mice by echocardiography (Figure 6O); compared to the Null mice, the Hand1OE mice showed significant reductions in left ventricular (LV) ejection fraction (EF) and fractional shortening (FS) (Figure 6P, Q), which are key parameters of cardiac contractile functions. Cardiac remodeling was also significantly increased in Hand1OE mice, with increased LV end-diastolic diameter (LVDd), and LV end-systolic diameter (LVDs) (Figure 6R, S). In order to interrogate underlying mechanisms for HAND1-induced cardiac dysfunction, we performed RNA-seq in the cardiomyocytes isolated from Null and Hand1OE mice (Figure S8J). Differential analysis showed that HAND1 induced drastic transcriptome reprogramming in Hand1OE cardiomyocytes (Figure S8K) and recapitulated the gene signatures in human DCM hearts (Figure S8L). Functional enrichment network analysis for the upregulated genes in Hand1OE cardiomyocytes further showed that the most highly represented GO terms included dilated cardiomyopathy, decreased cardiac muscle contractility, abnormal impulse conducting system conduction, cardiac hypertrophy, increased heart weight etc. (Figure 6T), which are in line with our observations in functional studies.
These data indicated that overexpression of Hand1 in the adult heart provokes impaired contractility, Ca2+ mishandling, CM hypertrophy, cardiac dilation and dysfunction. Taken together, our functional and mechanistic data uncovered HAND1 as a novel factor in the development of DCM that could also serve as a therapeutic target for this heart disease.
Discussion
Although one-dimensional epigenome mapping techniques (e.g., ATAC-seq and H3K27ac ChIP-seq) can identify CREs in the genome, they do not indicate the connectivity of CREs for transcription regulation. In contrast, high-resolution 3D genome techniques (e.g., H3K27ac HiChIP) can identify the connectome of active CREs, which is crucial since enhancers can skip their nearby promoters to regulate distal promoters.
In this study, we dissected, for the first time, the 3D chromatin architecture in human DCM at high-resolution and unraveled how this reorganization dictates transcriptional deregulation and its pathogenesis (Figure 6). We identified the DCM-enriched enhancer/promoter connectome, which is largely dependent on pre-existing chromatin accessibility and higher-order chromatin architecture, possibly priming their connected genes for rapid transcription in response to cardiac stress. We found that fetal cardiac and stress response genes (e.g., PDLIM5 (Figure 2E), TBX5 (Figure 3E), SMYD2 (Figure 3F), MEF2D (Figure S4F), MYPN (Figure S4G)) were aberrantly activated via gain of enhancer-promoter loops, whose anchors are characterized by increased HAND1 binding. In line with this, overexpression of Hand1 in adult mouse hearts also activated the expression of PDLIM5, TBX5, SMYD2, MEF2D, MYPN etc. (Figure S8L). This suggests that HAND1 may reactivate, at least in part, the early cardiac development and stress response genes in adult mice and humans to drive DCM pathogenesis.
HAND1 has previously not been investigated for a role in mediating long-range chromatin interactions. In order to test whether HAND1 could causally reprogram enhancer/promoter connectome and transcription, we performed extensive 3D epigenome mapping in hiPSC-CM. We found that HAND1 induces a genome-wide gain of distinct H3K27ac HiChIP loops. In addition, we showed that most of the gained loop anchors are characterized by increased HAND1 binding, indicating the direct effect of HAND1-induced chromatin looping. Moreover, the gained enhancer/promoter connections increase the expression of their associated genes implicated for DCM, suggesting a direct functional connection with the disease. Furthermore, we showed that reactivation of HAND1 in hiPSC-CM and mouse heart drives DCM phenotype and cardiac dysfunction, indicating that the precise temporal regulation of HAND1 is critical for cardiac homeostasis. Although the 3D epigenome mapping indicates differential chromatin architectures in DCM compared to NF, these datasets have some limitations. These are bulk sequencing data which were obtained from hearts at the end-stage of the disease at transplantation. As such, it is difficult to determine which of the many observed changes are disease casual, disease compensatory, or non-specific changes that accompany any form of heart failure. Such insight is particularly challenging to obtain in humans due to ethical limitations. Nevertheless, the identification of HAND1 as transcription factor regulating the 3D chromatin connectomes, gene expression in DCM as well as in mouse cardiomyocytes highlights the importance of this factor and its effects in playing an important causal role in this disease.
Interestingly, while we identified the essential roles of HAND1 in chromatin looping, we also noticed that HAND1 binding is not always sufficient for loop formation and gene activation in iPSC-CM, as some enhancers/promoters have strong HAND1 binding, but are not involved in chromatin looping, suggesting that additional factors (e.g., architectural proteins /transcription co-factors or chromatin-associated RNA) are required to facilitate HAND1-mediated enhancer/promoter looping. Regarding this hypothesis, in-situ capture of chromatin interactions by biotinylated dCas9 in iPSC-CM, followed by proteomics and RNA-seq57, could identify HAND1-interacting proteins/RNAs involved in chromatin looping. In addition, how HAND1 and its associated factors work together to regulate 3D chromatin contact (e.g., via phase-separated nuclear condensates58) should be addressed in the future. Moreover, future work could be performed to compare the effect of HAND1 with other transcription factors (e.g., NFIB, NFIC and ZN563) which we explored from motif scan, and investigate whether they cooperate with HAND1 in regulating 3D genome organization in DCM.
An intriguing aspect is the possibility of using our discoveries for predicting the risk for developing DCM. A reactivation of HAND1 protein in DCM could indicate the early activation of signaling pathways involved in mesodermal-cardiac differentiation and HAND1 protein induction. Additionally, the unique HAND1-mediated H3K27ac looping of genes is likely to play a central role in DCM. This could open novel therapeutic strategies for DCM. For example, signaling pathways blocking HAND1 protein levels (i.e., the E3 ligase Fbxo25 for HAND159) or chemical inhibitors that prevent HAND1 from reprogramming the enhancer-promoter connectomes leading to DCM could be attractive targets for therapeutic intervention. Our RNA-sequencing analysis of loop-specific genes uncovered several candidate pathways, such as EZH2 and TGFβ, that could be studied further for their therapeutic potential in the hiPSC-CM model.
Limitation
Several limitations are acknowledged in this study. First, based on the availability of ex vivo hearts at the Duke Human Heart Repository (DHHR) from Duke University, only 101 donor hearts were employed in our study which may not be representative of all human hearts. Second, although these non-failing (NF) samples were collected from the donors without a history of heart failure by DHHR, noncardiac comorbidities and lifestyle modifications in the NF group may affect the results. Third, as genetic testing is a non-standard diagnostic workflow in clinical DCM diagnosis, no genetic information of these samples possibly introducing some selection bias for this analysis. Fourth, the heart samples in this study were not age and gender matched due to the limited sample size. The association of these risk factors with 3D chromatin architecture and transcription regulation in DCM requires future study.
Conclusion
In sum, based on comprehensive 3D epigenome mapping and functional analysis in human DCM hearts, iPSC-CM platform and mouse adult cardiomyocytes, complemented by data from in vivo studies of Hand1-induced cardiomyocyte hypertrophy, cardiac dilation and dysfunction, our study represents a significant advance in the understanding of DCM etiology from a new view (chromatin topology) and points to HAND1 as a novel therapeutic target.
Supplementary Material
Figure 7. Model Illustrating 3D Epigenome Organization in Human DCM and How HAND1 Rewires Enhancer/Promoter Connectome to Contribute to Transcriptome Reprogramming and DCM Pathogenesis.
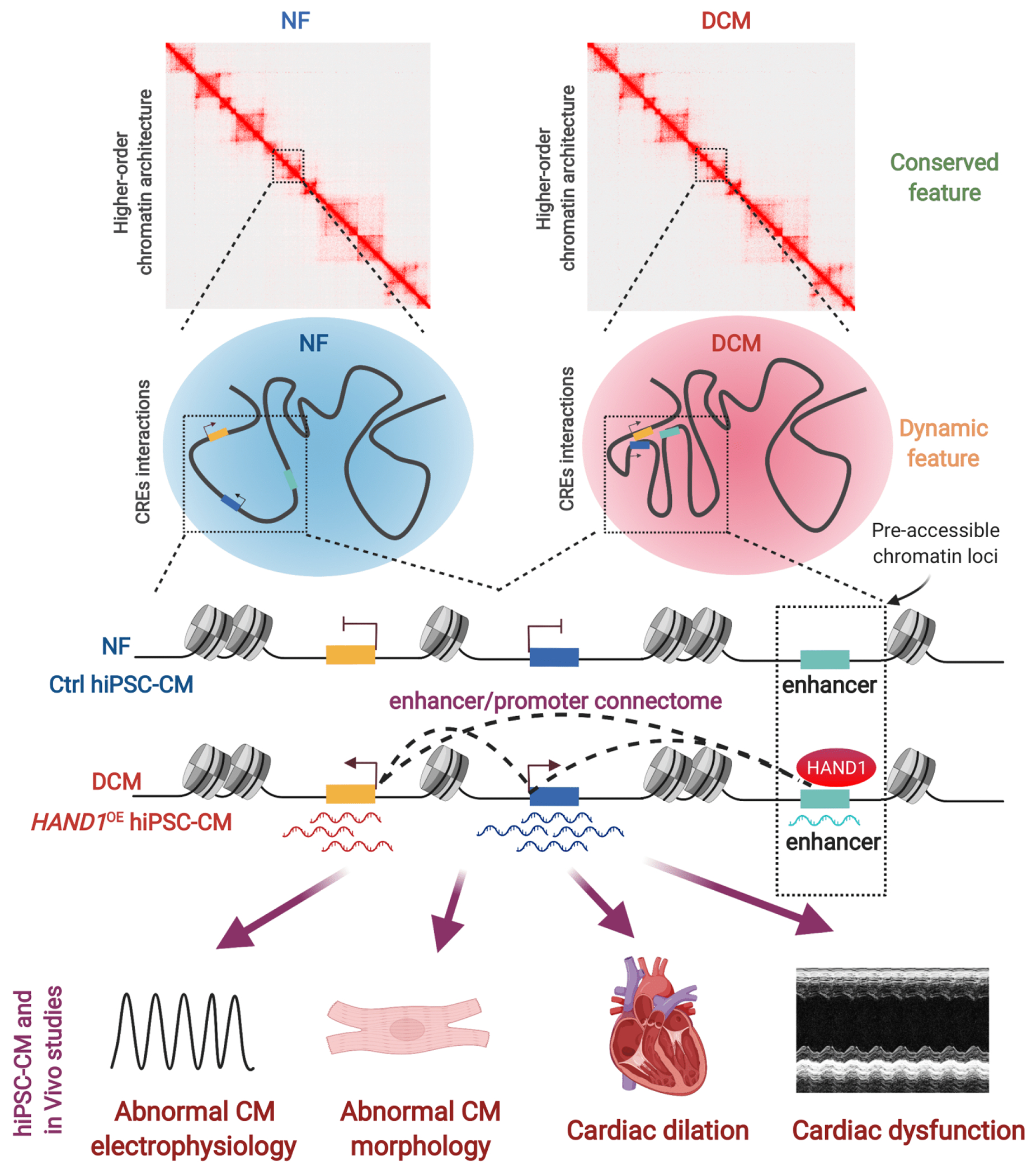
DCM-enriched enhancer/promoter connectome largely resides in pre-established, high-order chromatin architectures and pre-accessible chromatin sites bound by reactivated HAND1. Ectopic expression of HAND1 in human iPSC-derived cardiomyocytes (hiPSC-CM) and DCM hearts induces a distinct gain of enhancer/promoter connectivity and increases the expression of their associated genes implicated for DCM etiology, subsequently leading to abnormal cardiomyocyte electrophysiology/morphology, cardiac dilation and dysfunction. This illustrates a model whereby a single transcription factor rewires genome-wide enhancer/promoter connectome to drive DCM pathogenesis.
Clinical Perspective.
What Is New?
High-resolution 3D-epigenomic mapping and comprehensive computational analyses were performed in human DCM hearts.
Enhancer-promoter connectomes are extensively rewired in human DCM, which reside in pre-accessible chromatin sites.
HAND1 drives the rewiring of enhancer-promoter connectome to induce DCM pathogenesis.
What Are the Clinical Implications?
DCM-enriched enhancer-promoter loops identified in this study could be developed as novel 3D genomic biomarkers for DCM.
Targeting HAND1 might be used as a novel approach for therapeutic intervention of DCM.
Acknowledgements
Y.F., S.P., W.H., L.J. conceptualized, supervised this project and wrote the manuscript. L.C. analyzed all the human NGS data and wrote the manuscript. C.W. analyzed the mouse RNA-Seq data. W.H., and W.H. performed the experiments, analyzed data, and wrote the manuscript. X.W. isolated adult mouse cardiomyocytes. N.T., Y.W., H.C., S.W., F.L., F.H., R.A.P. C.G., G-C.F., S.S., and C.Z. provided expertise in cardiovascular biology and chromatin biology. M.W. performed statistical analysis and assisted with interpretation of results. J.M., M.K., and J.F. performed contractility assay, calcium transient assay and interpreted data. Y.M., Y.Y., and C.L. cultured the cells and performed the in vivo study. A.J. performed functional enrichment network analysis and provided valuable suggestions for the overall bioinformatics analysis pipeline. M.W. provided patient samples and clinical data.
Sources of Funding
This work was supported by the National Natural Science Foundation of China (No.81800262 to L.J.; 81670334 to N.T; 81560046 and 81760057 to F.H.; 81670398 and 91639102 to C.X.Z.; 31622031, 31671254, and 91749110 to H.C, Natural Science Foundation of Guangdong Province (No.2018A030313029 to L.J.), Science and Technology Planning Project of Guangzhou (No.201903010005 to L.J.), High-level Hospital Construction Project of Guangdong Province (DFJH201909 to N.T.). National Institutes of Health (NIH) grants R01HL160811 and GM-132149 (to G-C.F.), NIH grants R01HL143490 and R01HL136025, R01 HL157456 (to Y.W.), NIH grants R01HL139006 (to J.F.). American Heart Association (AHA) Career Development Award (20CDA35310176 to W. H.), AHA Career Development Award (19CDA34630009 to C. G.), AHA Postdoctoral Fellowship (834316 to M. K.). Clarendon Fund and St. Edmund Hall Scholarship SFF1920_CB_MSD_759707, to Y.F.) and Cancer Research UK Career Development Fellowship (C59392/A25064 to S.P.). Dr. Sadayappan has received support from National Institutes of Health grants R01 AR078001, R01 HL130356, R01 HL105826, R38 HL155775 and R01 HL143490. This work was further supported by the AHA 2019 Institutional Undergraduate Student (19UFEL34380251) and Transformation (19TPA34830084) awards, the PLN Foundation (PLN crazy idea) and the Leducq Foundation (Transatlantic Network 18CVD01, PLN-CURE).
Nonstandard Abbreviations and Acronyms
- AAV
adeno-associated virus
- ATAC
assay for transposase-accessible chromatin using sequencing
- CAGE
cap analysis of gene expression
- CFs
cardiac fibroblasts
- ChIA-PET
chromatin interaction analysis by paired-end tag sequencing
- ChIP-seq
chromatin immunoprecipitation sequencing
- CM
cardiomyocyte
- CREs
cis-regulatory elements
- DCM
dilated cardiomyopathy
- EF
ejection fraction
- E-P interaction
enhancer-promoter interaction
- EZH2
enhancer of zeste 2 polycomb repressive complex 2 subunit
- FREM2
FRAS1 related extracellular matrix 2
- FS
fraction shortening
- Fbxo25
F-box only protein 25
- GAS1
growth arrest-specific 1
- GWAS
genome-wide association study
- HAND1
heart and neural crest derivatives expressed 1
- H3K27ac
acetylation of lysine 27 on histone H3
- hiPSC-CMs
human induced pluripotent stem cell-derived cardiomyocytes
- Hi-C
high-throughput chromosome conformation capture
- IGF-1
insulin like growth factor 1
- LVDd
left ventricular end-diastolic diameter
- LVDs
left ventricular end-systolic diameter
- MEF2D
myocyte enhancer factor 2d
- MYH7
myosin heavy chain
- MYH6
myosin heavy chain
- MTSS1
MTSS I-BAR domain containing 1
- MYPN
myopalladin
- NF
non-failing
- NPPA
natriuretic peptide A
- NPPA-AS1
NPPA antisense RNA 1
- NPPB
natriuretic peptide B
- NOTCH1
notch receptor 1
- PCA
principal component analysis
- PDE1C
phosphodiesterase 1c
- PDLIM5
PDZ and LIM domain 5
- PLAC-seq
proximity ligation-assisted ChIP-Seq
- SMYD2
SET and MYND domain containing 2
- TAC
transverse aortic constriction
- TAD
topologically associating domains
- TBX5
t-box transcription factor 5
- TGFβ
transforming growth factor beta
- TTSs
transcription start sites
- WGA
wheat germ agglutinin
- WT
wild-type
- TUBA3D
tubulin alpha 3d
Footnotes
Disclosures
Dr. Sadayappan provides consulting and collaborative research studies to the Leducq Foundation (CURE-PLAN), Red Saree Inc., Greater Cincinnati Tamil Sangam, Pfizer, Novo Nordisk, AstraZeneca, MyoKardia, Merck and Amgen.
References
- 1.Ziaeian B and Fonarow GC. Epidemiology and aetiology of heart failure. Nature reviews Cardiology. 2016;13:368–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Merlo M, Cannata A, Gobbo M, Stolfo D, Elliott PM and Sinagra G. Evolving concepts in dilated cardiomyopathy. European journal of heart failure. 2018;20:228–239. [DOI] [PubMed] [Google Scholar]
- 3.Rosenbaum AN, Agre KE and Pereira NL. Genetics of dilated cardiomyopathy: practical implications for heart failure management. Nature reviews Cardiology. 2019;17:286–297. [DOI] [PubMed] [Google Scholar]
- 4.Yu J, Zeng C and Wang Y. Epigenetics in dilated cardiomyopathy. Current opinion in cardiology. 2019;34:260–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Klemm SL, Shipony Z and Greenleaf WJ. Chromatin accessibility and the regulatory epigenome. Nature reviews Genetics. 2019;20:207–220. [DOI] [PubMed] [Google Scholar]
- 6.Chatterjee S and Ahituv N. Gene Regulatory Elements, Major Drivers of Human Disease. Annual review of genomics and human genetics. 2017;18:45–63. [DOI] [PubMed] [Google Scholar]
- 7.Gacita AM, Fullenkamp DE, Ohiri J, Pottinger T, Puckelwartz MJ, Nobrega MA and McNally EM. Genetic Variation in Enhancers Modifies Cardiomyopathy Gene Expression and Progression. Circulation. 2021;143:1302–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gacita AM, Dellefave-Castillo L, Page PGT, Barefield DY, Wasserstrom JA, Puckelwartz MJ, Nobrega MA and McNally EM. Altered Enhancer and Promoter Usage Leads to Differential Gene Expression in the Normal and Failed Human Heart. Circ Heart Fail. 2020;13:e006926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, Boyer LA, Young RA and Jaenisch R. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:21931–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tan WLW, Anene-Nzelu CG, Wong E, Lee CJM, Tan HS, Tang SJ, Perrin A, Wu KX, Zheng W, Ashburn RJ, Pan B, Lee MY, Autio MI, Morley MP, Tam WL, Cheung C, Margulies KB, Chen L, Cappola TP, Loh M, Chambers J, Prabhakar S, Foo RSY and Charge-Heart Failure Working Group C-EC. Epigenomes of Human Hearts Reveal New Genetic Variants Relevant for Cardiac Disease and Phenotype. Circ Res. 2020;127:761–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schoenfelder S and Fraser P. Long-range enhancer-promoter contacts in gene expression control. Nature reviews Genetics. 2019;20:437–455. [DOI] [PubMed] [Google Scholar]
- 12.Mumbach MR, Rubin AJ, Flynn RA, Dai C, Khavari PA, Greenleaf WJ and Chang HY. HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nature methods. 2016;13:919–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mumbach MR, Satpathy AT, Boyle EA, Dai C, Gowen BG, Cho SW, Nguyen ML, Rubin AJ, Granja JM, Kazane KR, Wei Y, Nguyen T, Greenside PG, Corces MR, Tycko J, Simeonov DR, Suliman N, Li R, Xu J, Flynn RA, Kundaje A, Khavari PA, Marson A, Corn JE, Quertermous T, Greenleaf WJ and Chang HY. Enhancer connectome in primary human cells identifies target genes of disease-associated DNA elements. Nature genetics. 2017;49:1602–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cho SW, Xu J, Sun R, Mumbach MR, Carter AC, Chen YG, Yost KE, Kim J, He J, Nevins SA, Chin SF, Caldas C, Liu SJ, Horlbeck MA, Lim DA, Weissman JS, Curtis C and Chang HY. Promoter of lncRNA Gene PVT1 Is a Tumor-Suppressor DNA Boundary Element. Cell. 2018;173:1398–1412 e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu S, Turner KM, Nguyen N, Raviram R, Erb M, Santini J, Luebeck J, Rajkumar U, Diao Y, Li B, Zhang W, Jameson N, Corces MR, Granja JM, Chen X, Coruh C, Abnousi A, Houston J, Ye Z, Hu R, Yu M, Kim H, Law JA, Verhaak RGW, Hu M, Furnari FB, Chang HY, Ren B, Bafna V and Mischel PS. Circular ecDNA promotes accessible chromatin and high oncogene expression. Nature. 2019;575:699–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fang R, Yu M, Li G, Chee S, Liu T, Schmitt AD and Ren B. Mapping of long-range chromatin interactions by proximity ligation-assisted ChIP-seq. Cell Res. 2016;26:1345–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bertolini JA, Favaro R, Zhu Y, Pagin M, Ngan CY, Wong CH, Tjong H, Vermunt MW, Martynoga B, Barone C, Mariani J, Cardozo MJ, Tabanera N, Zambelli F, Mercurio S, Ottolenghi S, Robson P, Creyghton MP, Bovolenta P, Pavesi G, Guillemot F, Nicolis SK and Wei CL. Mapping the Global Chromatin Connectivity Network for Sox2 Function in Neural Stem Cell Maintenance. Cell stem cell. 2019;24:462–476 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zheng H and Xie W. The role of 3D genome organization in development and cell differentiation. Nature reviews Molecular cell biology. 2019;20:535–550. [DOI] [PubMed] [Google Scholar]
- 19.Lupianez DG, Kraft K, Heinrich V, Krawitz P, Brancati F, Klopocki E, Horn D, Kayserili H, Opitz JM, Laxova R, Santos-Simarro F, Gilbert-Dussardier B, Wittler L, Borschiwer M, Haas SA, Osterwalder M, Franke M, Timmermann B, Hecht J, Spielmann M, Visel A and Mundlos S. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell. 2015;161:1012–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Flavahan WA, Drier Y, Johnstone SE, Hemming ML, Tarjan DR, Hegazi E, Shareef SJ, Javed NM, Raut CP, Eschle BK, Gokhale PC, Hornick JL, Sicinska ET, Demetri GD and Bernstein BE. Altered chromosomal topology drives oncogenic programs in SDH-deficient GISTs. Nature. 2019;575:229–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Poleshko A, Shah PP, Gupta M, Babu A, Morley MP, Manderfield LJ, Ifkovits JL, Calderon D, Aghajanian H, Sierra-Pagan JE, Sun Z, Wang Q, Li L, Dubois NC, Morrisey EE, Lazar MA, Smith CL, Epstein JA and Jain R. Genome-Nuclear Lamina Interactions Regulate Cardiac Stem Cell Lineage Restriction. Cell. 2017;171:573–587 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M, Ku M, Durham T, Kellis M and Bernstein BE. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dirkx E, da Costa Martins PA and De Windt LJ. Regulation of fetal gene expression in heart failure. Biochimica et biophysica acta. 2013;1832:2414–24. [DOI] [PubMed] [Google Scholar]
- 24.Chien KR, Knowlton KU, Zhu H and Chien S. Regulation of cardiac gene expression during myocardial growth and hypertrophy: molecular studies of an adaptive physiologic response. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 1991;5:3037–46. [DOI] [PubMed] [Google Scholar]
- 25.Heinig M, Adriaens ME, Schafer S, van Deutekom HWM, Lodder EM, Ware JS, Schneider V, Felkin LE, Creemers EE, Meder B, Katus HA, Ruhle F, Stoll M, Cambien F, Villard E, Charron P, Varro A, Bishopric NH, George AL Jr., Dos Remedios C, Moreno-Moral A, Pesce F, Bauerfeind A, Ruschendorf F, Rintisch C, Petretto E, Barton PJ, Cook SA, Pinto YM, Bezzina CR and Hubner N. Natural genetic variation of the cardiac transcriptome in non-diseased donors and patients with dilated cardiomyopathy. Genome biology. 2017;18:170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Celik S, Sadegh MK, Morley M, Roselli C, Ellinor PT, Cappola T, Smith JG and Gidlof O. Antisense regulation of atrial natriuretic peptide expression. JCI Insight. 2019;4:e130978–130995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sandhu KS, Li G, Poh HM, Quek YL, Sia YY, Peh SQ, Mulawadi FH, Lim J, Sikic M, Menghi F, Thalamuthu A, Sung WK, Ruan X, Fullwood MJ, Liu E, Csermely P and Ruan Y. Large-scale functional organization of long-range chromatin interaction networks. Cell reports. 2012;2:1207–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang X, Qu R, Ouyang J, Zhong S and Dai J. An Overview of the Cytoskeleton-Associated Role of PDLIM5. Front Physiol. 2020;11:975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang D, Fang J, Lv J, Pan Z, Yin X, Cheng H and Guo X. Novel polymorphisms in PDLIM3 and PDLIM5 gene encoding Z-line proteins increase risk of idiopathic dilated cardiomyopathy. Journal of cellular and molecular medicine. 2019;23:7054–7062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chandra V, Bhattacharyya S, Schmiedel BJ, Madrigal A, Gonzalez-Colin C, Fotsing S, Crinklaw A, Seumois G, Mohammadi P, Kronenberg M, Peters B, Ay F and Vijayanand P. Promoter-interacting expression quantitative trait loci are enriched for functional genetic variants. Nat Genet. 2021;53:110–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li G, Ruan X, Auerbach RK, Sandhu KS, Zheng M, Wang P, Poh HM, Goh Y, Lim J, Zhang J, Sim HS, Peh SQ, Mulawadi FH, Ong CT, Orlov YL, Hong S, Zhang Z, Landt S, Raha D, Euskirchen G, Wei CL, Ge W, Wang H, Davis C, Fisher-Aylor KI, Mortazavi A, Gerstein M, Gingeras T, Wold B, Sun Y, Fullwood MJ, Cheung E, Liu E, Sung WK, Snyder M and Ruan Y. Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell. 2012;148:84–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dao LTM, Galindo-Albarran AO, Castro-Mondragon JA, Andrieu-Soler C, Medina-Rivera A, Souaid C, Charbonnier G, Griffon A, Vanhille L, Stephen T, Alomairi J, Martin D, Torres M, Fernandez N, Soler E, van Helden J, Puthier D and Spicuglia S. Genome-wide characterization of mammalian promoters with distal enhancer functions. Nature genetics. 2017;49:1073–1081. [DOI] [PubMed] [Google Scholar]
- 33.Diao Y, Fang R, Li B, Meng Z, Yu J, Qiu Y, Lin KC, Huang H, Liu T, Marina RJ, Jung I, Shen Y, Guan KL and Ren B. A tiling-deletion-based genetic screen for cis-regulatory element identification in mammalian cells. Nature methods. 2017;14:629–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pan YF, Wansa KD, Liu MH, Zhao B, Hong SZ, Tan PY, Lim KS, Bourque G, Liu ET and Cheung E. Regulation of estrogen receptor-mediated long range transcription via evolutionarily conserved distal response elements. The Journal of biological chemistry. 2008;283:32977–88. [DOI] [PubMed] [Google Scholar]
- 35.Chen J, Bardes EE, Aronow BJ and Jegga AG. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic acids research. 2009;37:W305–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Steimle JD and Moskowitz IP. TBX5: A Key Regulator of Heart Development. Current topics in developmental biology. 2017;122:195–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yi X, Jiang XJ and Fang ZM. Histone methyltransferase SMYD2: ubiquitous regulator of disease. Clin Epigenetics. 2019;11:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.She P, Zhang H, Peng X, Sun J, Gao B, Zhou Y, Zhu X, Hu X, Lai KS, Wong J, Zhou B, Wang L and Zhong TP. The Gridlock transcriptional repressor impedes vertebrate heart regeneration by restricting expression of lysine methyltransferase. Development. 2020;147: dev190678–190732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lu J, McKinsey TA, Zhang CL and Olson EN. Regulation of skeletal myogenesis by association of the MEF2 transcription factor with class II histone deacetylases. Mol Cell. 2000;6:233–44. [DOI] [PubMed] [Google Scholar]
- 40.Kim Y, Phan D, van Rooij E, Wang DZ, McAnally J, Qi X, Richardson JA, Hill JA, Bassel-Duby R and Olson EN. The MEF2D transcription factor mediates stress-dependent cardiac remodeling in mice. J Clin Invest. 2008;118:124–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bang ML, Mudry RE, McElhinny AS, Trombitas K, Geach AJ, Yamasaki R, Sorimachi H, Granzier H, Gregorio CC and Labeit S. Myopalladin, a novel 145-kilodalton sarcomeric protein with multiple roles in Z-disc and I-band protein assemblies. J Cell Biol. 2001;153:413–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ramaccini D, Montoya-Uribe V, Aan FJ, Modesti L, Potes Y, Wieckowski MR, Krga I, Glibetic M, Pinton P, Giorgi C and Matter ML. Mitochondrial Function and Dysfunction in Dilated Cardiomyopathy. Front Cell Dev Biol. 2020;8:624216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Andersson C, Lin H, Liu C, Levy D, Mitchell GF, Larson MG and Vasan RS. Integrated Multiomics Approach to Identify Genetic Underpinnings of Heart Failure and Its Echocardiographic Precursors: Framingham Heart Study. Circulation Genomic and precision medicine. 2019;12:e002489. [DOI] [PubMed] [Google Scholar]
- 44.Yasuda-Yamahara M, Rogg M, Yamahara K, Maier JI, Huber TB and Schell C. AIF1L regulates actomyosin contractility and filopodial extensions in human podocytes. PLoS One. 2018;13:e0200487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brody MJ, Hacker TA, Patel JR, Feng L, Sadoshima J, Tevosian SG, Balijepalli RC, Moss RL and Lee Y. Ablation of the cardiac-specific gene leucine-rich repeat containing 10 (Lrrc10) results in dilated cardiomyopathy. PLoS One. 2012;7:e51621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Buenrostro JD, Giresi PG, Zaba LC, Chang HY and Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nature methods. 2013;10:1213–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dixon JR, Gorkin DU and Ren B. Chromatin Domains: The Unit of Chromosome Organization. Molecular cell. 2016;62:668–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, Piolot T, van Berkum NL, Meisig J, Sedat J, Gribnau J, Barillot E, Bluthgen N, Dekker J and Heard E. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature. 2012;485:381–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xu J, Gong NL, Bodi I, Aronow BJ, Backx PH and Molkentin JD. Myocyte enhancer factors 2A and 2C induce dilated cardiomyopathy in transgenic mice. The Journal of biological chemistry. 2006;281:9152–62. [DOI] [PubMed] [Google Scholar]
- 50.Riley P, Anson-Cartwright L and Cross JC. The Hand1 bHLH transcription factor is essential for placentation and cardiac morphogenesis. Nature genetics. 1998;18:271–5. [DOI] [PubMed] [Google Scholar]
- 51.Zhou YM, Dai XY, Qiu XB, Yuan F, Li RG, Xu YJ, Qu XK, Huang RT, Xue S and Yang YQ. HAND1 loss-of-function mutation associated with familial dilated cardiomyopathy. Clin Chem Lab Med. 2016;54:1161–7. [DOI] [PubMed] [Google Scholar]
- 52.Sun N, Yazawa M, Liu J, Han L, Sanchez-Freire V, Abilez OJ, Navarrete EG, Hu S, Wang L, Lee A, Pavlovic A, Lin S, Chen R, Hajjar RJ, Snyder MP, Dolmetsch RE, Butte MJ, Ashley EA, Longaker MT, Robbins RC and Wu JC. Patient-specific induced pluripotent stem cells as a model for familial dilated cardiomyopathy. Science translational medicine. 2012;4:130ra47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Knight WE, Chen S, Zhang Y, Oikawa M, Wu M, Zhou Q, Miller CL, Cai Y, Mickelsen DM, Moravec C, Small EM, Abe J and Yan C. PDE1C deficiency antagonizes pathological cardiac remodeling and dysfunction. Proc Natl Acad Sci U S A. 2016;113:E7116–E7125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mork HK, Sjaastad I, Sande JB, Periasamy M, Sejersted OM and Louch WE. Increased cardiomyocyte function and Ca2+ transients in mice during early congestive heart failure. J Mol Cell Cardiol. 2007;43:177–86. [DOI] [PubMed] [Google Scholar]
- 55.Mork HK, Sjaastad I, Sejersted OM and Louch WE. Slowing of cardiomyocyte Ca2+ release and contraction during heart failure progression in postinfarction mice. Am J Physiol Heart Circ Physiol. 2009;296:H1069–79. [DOI] [PubMed] [Google Scholar]
- 56.Zhao SM, Wang YL, Guo CY, Chen JL and Wu YQ. Progressive decay of Ca2+ homeostasis in the development of diabetic cardiomyopathy. Cardiovasc Diabetol. 2014;13:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu X, Zhang Y, Chen Y, Li M, Zhou F, Li K, Cao H, Ni M, Liu Y, Gu Z, Dickerson KE, Xie S, Hon GC, Xuan Z, Zhang MQ, Shao Z and Xu J. In Situ Capture of Chromatin Interactions by Biotinylated dCas9. Cell. 2017;170:1028–1043 e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Misteli T The Self-Organizing Genome: Principles of Genome Architecture and Function. Cell. 2020;183:28–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jang JW, Lee WY, Lee JH, Moon SH, Kim CH and Chung HM. A novel Fbxo25 acts as an E3 ligase for destructing cardiac specific transcription factors. Biochem Biophys Res Commun. 2011;410:183–8. [DOI] [PubMed] [Google Scholar]
- 60.Ewels P, Magnusson M, Lundin S and Kaller M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 2016;32:3047–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li H and Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Feng J, Liu T, Qin B, Zhang Y and Liu XS. Identifying ChIP-seq enrichment using MACS. Nature protocols. 2012;7:1728–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Robinson MD, McCarthy DJ and Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M and Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R and Genome Project Data Processing S. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liao Y, Smyth GK and Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–30. [DOI] [PubMed] [Google Scholar]
- 67.Love MI, Huber W and Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology. 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Servant N, Varoquaux N, Lajoie BR, Viara E, Chen CJ, Vert JP, Heard E, Dekker J and Barillot E. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome biology. 2015;16:259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Durand NC, Shamim MS, Machol I, Rao SS, Huntley MH, Lander ES and Aiden EL. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell systems. 2016;3:95–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ramirez F, Dundar F, Diehl S, Gruning BA and Manke T. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic acids research. 2014;42:W187–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Phanstiel DH, Boyle AP, Heidari N and Snyder MP. Mango: a bias-correcting ChIA-PET analysis pipeline. Bioinformatics. 2015;31:3092–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li G, Sun T, Chang H, Cai L, Hong P and Zhou Q. Chromatin Interaction Analysis with Updated ChIA-PET Tool (V3). Genes. 2019;10:554–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Naumova N, Smith EM, Zhan Y and Dekker J. Analysis of long-range chromatin interactions using Chromosome Conformation Capture. Methods. 2012;58:192–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hagege H, Klous P, Braem C, Splinter E, Dekker J, Cathala G, de Laat W and Forne T. Quantitative analysis of chromosome conformation capture assays (3C-qPCR). Nature protocols. 2007;2:1722–33. [DOI] [PubMed] [Google Scholar]
- 75.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome research. 2003;13:2498–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Abdennur N and Mirny LA. Cooler: scalable storage for Hi-C data and other genomically labeled arrays. Bioinformatics. 2020;36:311–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Crane E, Bian Q, McCord RP, Lajoie BR, Wheeler BS, Ralston EJ, Uzawa S, Dekker J and Meyer BJ. Condensin-driven remodelling of X chromosome topology during dosage compensation. Nature. 2015;523:240–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wolff J, Rabbani L, Gilsbach R, Richard G, Manke T, Backofen R and Gruning BA. Galaxy HiCExplorer 3: a web server for reproducible Hi-C, capture Hi-C and single-cell Hi-C data analysis, quality control and visualization. Nucleic acids research. 2020;48:W177–W184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gong Y, Lazaris C, Sakellaropoulos T, Lozano A, Kambadur P, Ntziachristos P, Aifantis I and Tsirigos A. Stratification of TAD boundaries reveals preferential insulation of super-enhancers by strong boundaries. Nature communications. 2018;9:542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Quinlan AR and Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yang T, Zhang F, Yardimci GG, Song F, Hardison RC, Noble WS, Yue F and Li Q. HiCRep: assessing the reproducibility of Hi-C data using a stratum-adjusted correlation coefficient. Genome research. 2017;27:1939–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Thorvaldsdottir H, Robinson JT and Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Briefings in bioinformatics. 2013;14:178–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Carroll KJ, Makarewich CA, McAnally J, Anderson DM, Zentilin L, Liu N, Giacca M, Bassel-Duby R and Olson EN. A mouse model for adult cardiac-specific gene deletion with CRISPR/Cas9. Proceedings of the National Academy of Sciences of the United States of America. 2016;113:338–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Huang W, Feng Y, Liang J, Yu H, Wang C, Wang B, Wang M, Jiang L, Meng W, Cai W, Medvedovic M, Chen J, Paul C, Davidson WS, Sadayappan S, Stambrook PJ, Yu XY and Wang Y. Loss of microRNA-128 promotes cardiomyocyte proliferation and heart regeneration. Nature communications. 2018;9:700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kumar M, Haghighi K, Kranias EG and Sadayappan S. Phosphorylation of cardiac myosin-binding protein-C contributes to calcium homeostasis. The Journal of biological chemistry. 2020;295:11275–11291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dai B, Huang W, Xu M, Millard RW, Gao MH, Hammond HK, Menick DR, Ashraf M and Wang Y. Reduced collagen deposition in infarcted myocardium facilitates induced pluripotent stem cell engraftment and angiomyogenesis for improvement of left ventricular function. Journal of the American College of Cardiology. 2011;58:2118–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ma J, Hong K and Wang HS. Progesterone Protects Against Bisphenol A-Induced Arrhythmias in Female Rat Cardiac Myocytes via Rapid Signaling. Endocrinology. 2017;158:778–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.