

Abstract
The ClpP serine peptidase is a tetradecameric degradation molecular machine involved in many physiological processes. It becomes a competent ATP‐dependent protease when coupled with Clp‐ATPases. Small chemical compounds, acyldepsipeptides (ADEPs), are known to cause the dysregulation and activation of ClpP without ATPases and have potential as novel antibiotics. Previously, structural studies of ClpP from various species revealed its structural details, conformational changes, and activation mechanism. Although product release through side exit pores has been proposed, the detailed driving force for product release remains elusive. Herein, we report crystal structures of ClpP from Bacillus subtilis (BsClpP) in unforeseen ADEP‐bound states. Cryo‐electron microscopy structures of BsClpP revealed various conformational states under different pH conditions. To understand the conformational change required for product release, we investigated the relationship between substrate hydrolysis and the pH‐lowering process. The production of hydrolyzed peptides from acidic and basic substrates by proteinase K and BsClpP lowered the pH values. Our data, together with those of previous findings, provide insight into the molecular mechanism of product release by the ClpP self‐compartmentalizing protease.
Keywords: acyldepsipeptide, ClpP, cryo‐EM, pH drop, protein degradation
Subject Categories: Post-translational Modifications & Proteolysis, Structural Biology
Conformational changes linked to peptide‐mediated acidification suggest a mechanism for proteolytic product release by a key bacterial and mitochondrial ATP‐dependent Clp protease system.
Introduction
Energy‐dependent proteases are molecular machines conserved in all kingdoms of life (Goldberg, 1990; Groll et al, 2005). They play an essential role in protein quality and quantity control by degrading misfolded, damaged, short‐lived, or regulatory proteins by using energy from ATP hydrolysis (Gottesman, 2003; Sauer & Baker, 2011). Clp proteases are an energy‐dependent protease system in bacteria and the mitochondria of eukaryotic cells (Baker & Sauer, 2012). They contain two distinct functional components: a Clp‐ATPase and the ClpP proteolytic core. The energy‐consuming hexameric AAA+ ATPase (an ATPase associated with a variety of cellular activities) is responsible for substrate selection, unfolding, and translocation into the proteolytic core. Its representative molecules include ClpA, ClpC, ClpE, and ClpX. The central proteolytic machine ClpP is a barrel‐like serine protease complex composed of two stacked heptameric rings that form an enclosed degradation chamber (Yu & Houry, 2007). Both ClpP heptamers have a central axial pore where the unfolded polypeptide chains are translocated.
The structures of ClpP from various species, from E. coli to humans, have been solved, and information regarding the folding, axial entrance pore, and catalytic triad of ClpP is well defined (Wang et al, 1997; Kang et al, 2004; Gribun et al, 2005; Yu & Houry, 2007; Kim & Kim, 2008; El Bakkouri et al, 2010; Lee et al, 2010; Geiger et al, 2011). The activation mechanism of ClpP by its Clp‐ATPase partner was a long‐standing question in the corresponding field and partially revealed by structures of ClpP in complex with acyldepsipeptides (ADEPs) (Lee et al, 2010; Li et al, 2010), which are compounds that have antibiotic activities through the dysregulation of ClpP activity and regarded as Clp‐ATPase mimics (Brotz‐Oesterhelt et al, 2005; Kirstein et al, 2009; Sass et al, 2011). Very recently, more direct evidence was obtained from the complex structures formed between the ClpX ATPase and ClpP protease using cryo‐electron microscopy (cryo‐EM) (Gatsogiannis et al, 2019; Fei et al, 2020; Ripstein et al, 2020a). These studies showed how the symmetry‐mismatched ATPase activates the proteolytic molecular machine by translocating substrate molecules. The critical IGF/L (Ile‐Gly‐Phe in ClpX and Ile‐Gly‐Leu in ClpA) loops of the Clp‐ATPases bind to the hydrophobic pockets between the ClpP monomers, as shown in the ADEP‐bound structures. The asymmetric ClpAP complexes showed one or two empty IGL‐loop binding pockets during the engagement and disengagement cycles (Lopez et al, 2020), which had not been clear in the previously determined structures of ClpP fully occupied with ADEP compounds (Lee et al, 2010; Li et al, 2010; Gersch et al, 2015). The activation mechanism of ClpP by its cognate ATPases has now been proposed. However, the detailed and coherent steps of ClpP motion, from substrate entry to product release, are still elusive, although several conformational equilibria between its active and inactive states have been reported, such as pH‐dependent switching, active site perturbation, and the regulation of the N‐terminal loop (Gersch et al, 2012; Vahidi et al, 2018, 2020; Ripstein et al, 2020b).
Once substrates reach the tetradecameric proteolytic chamber of ClpP, the resulting degraded products need to be eliminated for efficient processing. The equatorial pore for peptide release was structurally revealed by the same observation of the major conformational changes of ClpP within different strains (Geiger et al, 2011; Lee et al, 2011; Zhang et al, 2011; Gersch et al, 2012), which was corroborated by an elegant biophysical study using NMR (Sprangers et al, 2005). Currently, several other factors have been proposed for the regulation of equatorial pore openings (Gersch et al, 2012; Li et al, 2016; Vahidi et al, 2018, 2020; Ripstein et al, 2020b). However, those studies could not fully explain the peptide product release mechanism of ClpP coupled with its activators, AAA+ ATPases or ADEPs. Herein, we present the crystal and cryo‐EM structures of ADEP‐bound ClpP in various states and a plausible link between the decrease in pH and peptide hydrolysis, which reveal conformational changes and the peptide release mechanism of ClpP. These data, in combination with all available structural information, show the complete processing of substrates by ClpP, from recognition to unfolding, translocation, degradation, and release.
Results
Lowering the pH by the accumulation of hydrolyzed peptide products
As described, there are significant conformational changes in the handle region of ClpP from Bacillus subtilis (BsClpP), as shown in our previous studies (Lee et al, 2010, 2011), and those from other species (Zhang et al, 2011; Gersch et al, 2015; Vahidi et al, 2018, 2020; Ripstein et al, 2020b). It has been proposed that His145 interacting with the catalytic aspartate of an adjacent protomer in N. meningitidis ClpP serves as a key determinant for its pH‐dependent conformational switch (Ripstein et al, 2020b). However, the equivalent residue in BsClpP is alanine, and therefore, there must be differences in the conformational changes of ClpP among species (Fig EV1). In M. tuberculosis ClpP1P2, the peptide binding at the active site triggers its transition between active and inactive conformations (Vahidi et al, 2018). Therefore, it is tempting to speculate that there must be a relationship between the pH change and the peptide accumulation in the proteolytic chamber of ClpP. Furthermore, there have been reports that the pH drop can be correlated with peptide bond hydrolysis (Mozersky & Panettieri, 1983; Ge & Zhang, 1993), and naturally, the numerous newly generated amino‐ and carboxy‐terminal groups contribute to the pH drop from neutral pH values. Therefore, we hypothesized that peptide bond hydrolysis decreases the pH value of a protein solution. To test this hypothesis, we measured the pH change during protein hydrolysis. An initial pH of 6.41 was achieved using diluted PBS (phosphate‐buffered saline) with 80 μM α‐casein, which decreased to a terminal pH of 5.94 by the addition of proteinase K. The drop of 0.47 pH units occurred very fast because of the high enzymatic activity of proteinase K for the partially unfolded α‐casein substrate (Figs 1A and EV2A). We observed a similar pH change with BsClpP protease in the presence of ADEP1 (Fig 1A). In contrast to that of monomeric proteinase K, the proteolytic activity of BsClpP is lower, and thus, the pH drop was slower. Nonetheless, a substantial drop of 0.46 pH units occurred from the initial pH 6.58 to pH 6.12 after 300 min (Fig 1A). The same experiment performed with bovine serum albumin (BSA) showed an even larger pH drop of 0.76, from 6.80 to 6.04 (Fig 1B). In the absence of a protease, the pH values were quite stable (Fig 1B). In parallel to the pH measurement, protein degradation was measured with SDS–PAGE (Fig EV2B). The pIs of intact α‐casein and BSA are 4.91 and 5.32, respectively. The average pI values of bacterial proteomes are also slightly acidic (Kozlowski, 2017).
Figure EV1. Sequence alignment among ClpPs.

The UniProt IDs for the ClpP sequences are Bacillus subtilis ClpP (BsClpP) P80244, Neisseria meningitidis ClpP (NmClpP) Q9JZ38, Mycobacterium tuberculosis ClpP1 (MtuClpP1) P9WPC5, Mycobacterium tuberculosis ClpP2 (MtuClpP2) P9WPC3, and Staphylococcus aureus ClpP (SaClpP) Q2G036. Secondary structure elements are indicated (α‐helices ‐ coil; β‐strands ‐ arrow) and labeled above the sequence. Filled blue circles indicate the catalytic triad (S97‐H122‐D171), and the flexible handle region is marked with a black dashed box. His145 in NmClpP and its equivalent residues are highlighted in yellow. Identical residues are shaded in red, and highly conserved residues are marked in blue boxes. The sequence number for B. subtilis ClpP is indicated above the sequence.
Figure 1. pH change during protein hydrolysis.
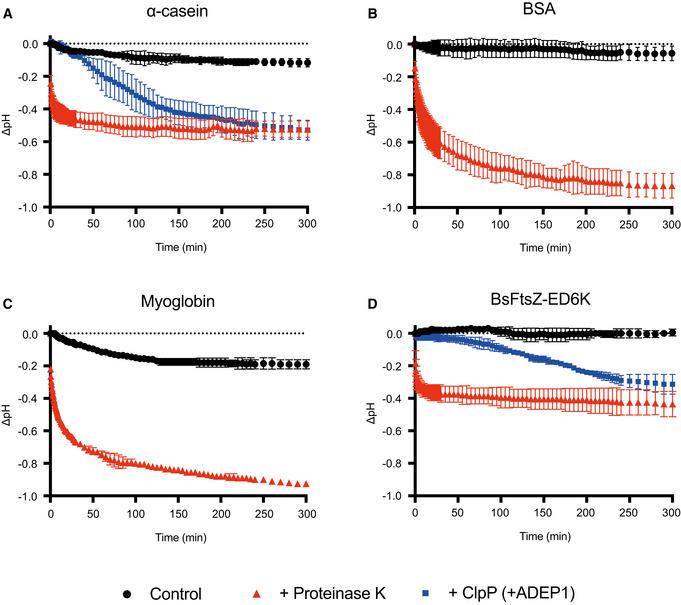
- Monitoring of the pH change during the degradation of α‐casein by proteinase K and BsClpP in the presence of ADEP1.
- Monitoring of the pH change during the degradation of bovine serum albumin (BSA) by proteinase K.
- Monitoring of the pH change during the degradation of myoglobin by proteinase K.
- Monitoring of the pH change during the degradation of the BsFtsZ‐ED6K mutant by proteinase K and BsClpP in the presence of ADEP1.
Data information: In all panels (A–D), black circles represent the control, only substrates without proteases. Red (triangle) and blue (square) symbols represent proteinase K and BsClpP+ADEP1, respectively. Data points represent the mean value of three measurements, and the error bars show the standard deviations.
Figure EV2. SDS–PAGE results showing protein hydrolysis in the course of the pH‐drop experiment presented in Fig 1 .

-
ASubstrates α‐casein (red arrowhead) and bovine serum albumin (BSA: black arrowhead) were incubated with proteinase K (ProK: white arrowhead).
-
BSubstrate α‐casein (red arrowhead) was incubated with BsClpP (green arrowhead) in the presence of ADEP1.
-
C, DSubstrate myoglobin (blue arrowhead) was incubated with (C) and without proteinase K (white arrowhead) (D).
-
E, FBasic BsFtsZ‐ED6K substrate (yellow arrowhead) was incubated with proteinase K (white arrowhead) (E) and BsClpP (green arrowhead) + ADEP1 (F).
Data information: In all panels (A–F), the samples were collected at each time point. The “M” lane indicates the molecular weight marker (240, 140, 100, 70, 50, 35, 25, 20, 15, 7 kDa as shown in panel (A)).
To test whether basic substrates are also degraded by proteases (Fig EV2, EV3, EV4, EV5) and whether their hydrolysis lowers the pH value, we used myoglobin from equine skeletal muscle as a model basic protein (pI of 7.36). The pH values dropped significantly in the presence of proteinase K, although they were slightly lowered in the absence of protease (Fig 1C). To check the same results with BsClpP, we used a well‐established physiological substrate, FtsZ, from Bacillus subtilis (BsFtsZ) (Sass et al, 2011). Unfortunately, the pI value of BsFtsZ is 5.01, and therefore, we generated a basic mutant of BsFtsZ (BsFtsZ‐ED6K) in which six acidic residues (Glu345, Glu349, Glu350, Asp366, Asp367, and Asp370) were replaced with basic lysine residues, which became a basic protein with a pI of 7.90. As shown in Figs 1D and EV2F, BsClpP degraded the BsFtsZ mutant, but not completely, and it is evident that the pH values of the proteolysis reaction mixture of the basic BsFtsZ mutant and BsClpP were decreased. These values are derived from the summation of the pKa values of all side‐chain groups and amino‐ and carboxy‐terminal groups. However, when proteases cleave the numerous peptide bonds in proteins, many new α‐amino and carboxylic groups are generated. Subsequently, the pH value of the environment is mainly governed by the total summation of all the pKa values of the α‐amino and carboxylic groups, which is slightly acidic (see Discussion for more details). The cleaved peptide products must be highly concentrated in the proteolytic chamber of ClpP, and therefore, the local pH of the handle region near the active site becomes acidic. Consequently, we conclude that the conformation of BsClpP under low‐pH conditions mimics its conformation during peptide accumulation inside the proteolytic chamber.
Figure EV3. Structural analyses of 2ADEP and 5ADEP related to Fig 3 .
- Side view of the 2ADEP structure obtained by rotating the top view (Fig 3A) 90° about the horizontal axis (middle). Left and right, approximately 120° (counterclockwise (−) and clockwise (+)) rotation along the vertical axis showing asymmetric side pores. The side exit pores are marked with transparent red ovals. The colors and numbers indicating the subunit environments are the same as those in Fig 3A.
Figure EV4. Cryo‐EM data processing workflow.
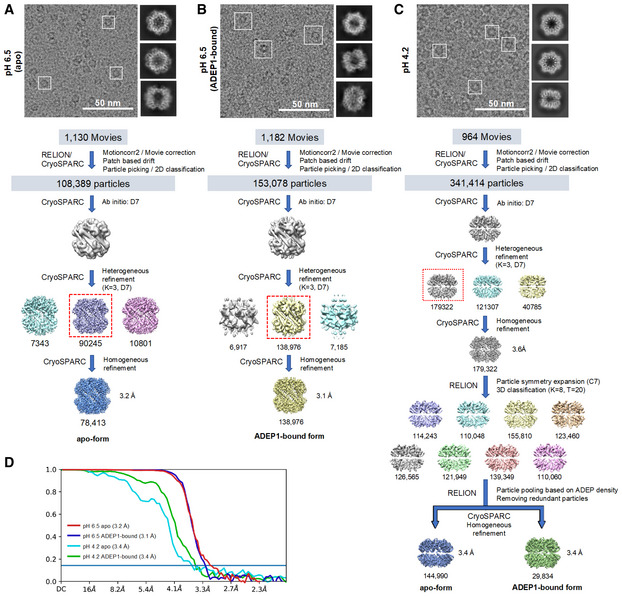
-
A–CRepresentative electron micrograph, 3‐D classification and map refinement workflow of the apo form of BsClpP at pH 6.5 (A), 14 ADEP‐bound form of BsClpP at pH 6.5 (B), and apo and 14 ADEP‐bound forms of BsClpP at pH 4.2 (C).
-
DMap resolution for each BsClpP cryo‐EM structure according to the gold‐standard Fourier shell correlation (FSC) at the 0.143 criterion.
Figure EV5. Comparison of the activator binding sites of ClpP.
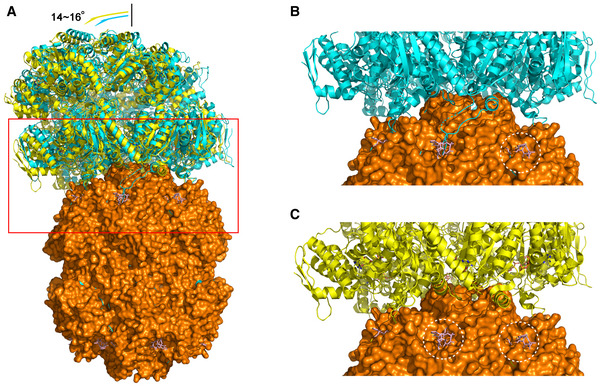
- Overall view of two conformations (6W21 in cyan and 6W20 in yellow) of the E. coli ClpAP complex. The ClpAP complex was superimposed with BsClpP at pH 6.5 (orange) complexed with ADEP (pink). The red boxed region indicates the interaction region between ClpP and the activators ClpA or ADEP.
- Six IGL loops of ClpA overlap with the ADEP molecules, and thus, only one site of ClpP is empty in this conformation of the ClpAP complex (cyan). Enlarged view of the red boxed region in (A).
- Only five IGL loops of ClpA overlap with the ADEP molecules; thus, two sites of ClpP are empty in this conformation of the ClpAP complex (yellow). The binding sites left empty by ClpAP are marked with white dotted circles in ClpAP, whereas ADEP occupies these sites in the ADEP‐BsClpP structure. The conformation of ClpAP in panel (C) shows slightly more tilted angles (14 versus 16°), as shown in panel (A) (Lopez et al, 2020). Enlarged view of the red boxed region in (A).
Asymmetric ADEP binding in crystal structures of the BsClpP‐ADEP complex
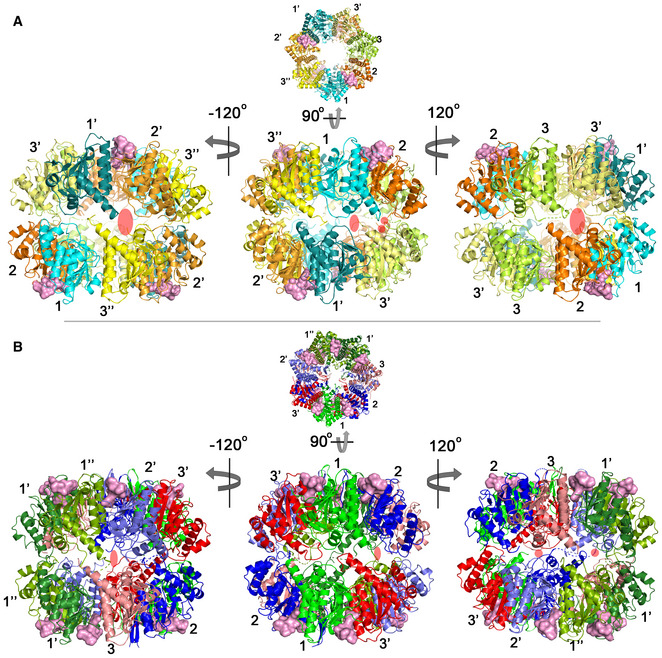
We solved two structures of BsClpP in complex with acyldepsipeptides (specifically ADEP2) at 2.8 and 3.0 Å resolutions (Fig 2 and Appendix Table S1). For molecular replacement, previously compressed BsClpP (PDB ID: 3TT6) was used as a search model (Lee et al, 2011). Very intriguingly, these two structures have two and five ADEP2 compounds, respectively, bound to each heptameric ring (Fig 2 and Appendix Fig S1A and B), which is in stark contrast to the previous 14 ADEP‐bound BsClpP (Lee et al, 2010) as well as all other ADEP‐ClpP complex structures from various organisms published to date (Gersch et al, 2015; Wong et al, 2018; Brotz‐Oesterhelt & Vorbach, 2021; Yang et al, 2021). Our two new BsClpP tetradecamer structures exhibit a large difference in their degrees of compression as well as in their numbers of ADEP molecules bound, four and ten (Fig 2). Hereafter, 2ADEP and 5ADEP indicate 2 and 5 ADEPs for each heptameric ring (4 and 10 ADEPs for a BsClpP tetradecamer). Their axial heights are approximately 86 Å (2ADEPs) and 92 Å (5ADEP), and the state of BsClpP bound to 4 ADEPs (2ADEP) has a similar height to that of the previous compressed apo‐form BsClpP structure (Lee et al, 2011). For consistency, the height was defined as the distance between the outermost two atoms; therefore, it is slightly taller than that in previous reports, which is usually the distance measured between the two most distant Cα atoms. The height of BsClpP in complex with 10 ADEPs (5ADEP) is in the middle of those of the extended and compressed states (Lee et al, 2011), which is similar to that of the state called the “compact state” (Geiger et al, 2011; Liu et al, 2014; Brotz‐Oesterhelt & Vorbach, 2021). Thus, these two different states are referred to as “compressed” for the 2ADEP structure and “compact” for the 5ADEP structure. In both structures, ADEPs bind at the interface of the two subunits in the orders of A‐E‐E‐E‐A‐E‐E (A: ADEP‐bound and E: empty) in the compact state and A‐A‐A‐E‐A‐A‐E in the compressed state (Fig 2).
Figure 2. X‐ray structures of ADEP‐bound BsClpP.
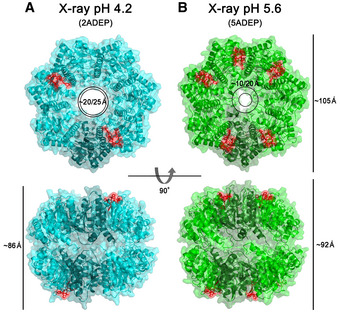
- Ribbon diagram of 4 ADEP‐bound BsClpP (cyan) with a transparent molecular surface and one monomer in each heptameric ring, colored darker for clarity (2ADEP).
- Ribbon diagram of 10 ADEP‐bound BsClpP (green) with a transparent molecular surface and one monomer in each heptameric ring, colored darker for clarity (5ADEP). Tetradecameric BsClpP is viewed along a 7‐fold molecular symmetry axis (upper), and the 2‐fold side view is observed by rotating 90° (lower). The bound ADEP molecules colored red are shown as stick models. The dimensions of the models are indicated. Two diameters are noted due to the asymmetric shape of the entrance pore.
Different compressions of BsClpP control the entrance and exit pores
The aforementioned two states differ in terms of their preservation of N‐terminal residues in addition to their heights. The N‐terminal segments of compact 5ADEP are well structured and resolved, whereas those of 2ADEP are disordered, so the pore sizes in the two states are markedly different (Fig 2). Due to the asymmetric binding of the activator ADEP, the entrance pores showed distorted shapes in both states, and in particular, the compact state possessed even more asymmetry, in that the diameter between the empty subunits was nearly half that of the ADEP‐bound subunits (Fig 2B). More importantly, the pores in the lateral direction, known as the product exit pores, were also different. This correlates with the flexibility or unwinding of the α5‐helix, known as the handle region (Geiger et al, 2011; Lee et al, 2011; Zhang et al, 2011). Since we obtained asymmetric ADEP‐bound structures, each monomer in the different neighboring subunits was compared (Fig 3A–F). In both structures, each subunit has three different environments, and the details of the 2ADEP and 5ADEP structures are different. In the orientation looking down from the substrate entrance pore in the case of 2ADEP (Fig 3A), there are three states: protomers having ADEP bound on the right side (2 subunits), ADEP bound on the left side (2 subunits), and no ADEP binding (3 subunits). The monomeric structures of protomers with ADEP bound on either the right or the left sides are quite similar, whereas those of the protomers with unbound monomers are slightly different, especially regarding the loop immediately before the handle region (Fig 3B). The N‐terminal regions of all subunits are quite flexible and have almost undetectable electron densities; thus, the asymmetry of the entrance pore is only marginal (Fig 2A).
Figure 3. Structural analyses of compressed 2ADEP and compact 5ADEP.
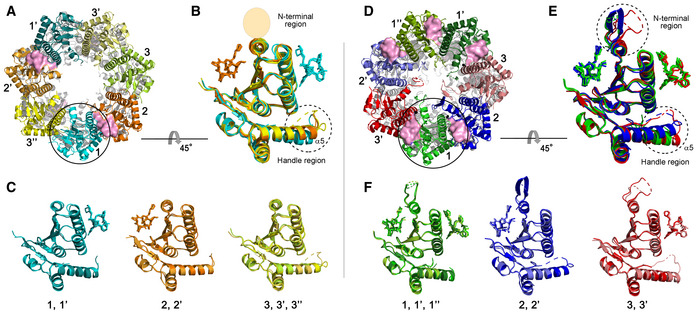
- Three different subunit environments of 2ADEP viewed along a 7‐fold axis: two cyanish subunits (1 and 1′) with the bound ADEP (pink molecular surface) on the right side, two orangish subunits (2 and 2′) with the bound ADEP on the left side, and three yellowish subunits (3, 3′ and 3′′) with no ADEP molecule.
- Superposition of all 7 subunits in the heptameric ring of 2ADEP viewed by rotating panel (A) 45° about the horizontal axis. The invisible N‐terminal region, due to flexibility, is marked with a transparent oval, and the structurally dynamic handle region is marked with a dashed circle.
- Superposition of cyanish subunits (1, 1′), orangish subunits (2, 2′), and yellowish subunits (3, 3′, 3′′). The view is the same as that of panel (B).
- Three different subunit environments of 5ADEP viewed along a 7‐fold axis: three greenish subunits (1, 1′ and 1′′) with the bound ADEP (pink molecular surface) on both the left and right sides, two bluish subunits (2 and 2′) with the bound ADEP on the left side, and two reddish subunits (3 and 3′) with the bound ADEP on the right side.
- Superposition of all 7 subunits in the heptameric ring of 5ADEP viewed by rotating panel (D) 45° about the horizontal axis. The very flexible N‐terminal region and handle region are marked with dashed circles.
- Superposition of greenish subunits (1, 1′, 1′′), bluish subunits (2, 2′), and reddish subunits (3, 3′). The view is the same as that of panel (E).
In the same orientation of 5ADEP (Fig 3D), there are also three different states: ADEP bound on both sides (3 subunits), ADEP bound on the right side (2 subunits), and ADEP bound on the left side (2 subunits). Therefore, the main difference between 2ADEP and 5ADEP is that 2ADEP has 3 monomeric subunits with ADEP bound on neither side and 5ADEP has 3 monomeric subunits with ADEP bound on both sides. In the case of 5ADEP, the monomeric structures with ADEP bound on both sides are quite similar (Fig 3F), whereas the monomeric structures with a single ADEP bound on either the right or the left side are different in regard to the loop immediately before the handle region and the N‐terminal regions (Fig 3E). The superposition of all subunits in 5ADEP shows that the N‐terminal regions and the α5 handle regions of all subunits are very dynamic, and thus, the asymmetry of the entrance pore, as well as the side pores, is augmented (Figs 2B, 3E and EV3A and B).
Diverse ADEP‐bound states found in cryo‐EM structures
Although the 2ADEP and 5ADEP states in crystals must be intermediate structures in the reaction cycle of ClpP, we performed cryo‐EM experiments at the pH of the crystallization buffer to rule out the possibility of crystal packing artifacts. We obtained 2ADEP and 5ADEP structures at pH values of 4.2 and 5.6, respectively. Unfortunately, at pH 5.6, BsClpP formed an aggregate, probably due to its pI of 5.2; therefore, we collected cryo‐EM data at pH 6.5 and 4.2. From datasets of the BsClpP particles, we were able to reconstruct 4 different structures of BsClpP at 3.1, 3.2, 3.4, and 3.4 Å maps (Fig EV4A–D and Appendix Table S2). The initial electron micrographs showed heterogeneous ADEP‐bound states. The 1:1 mixture between BsClpP and ADEP1 at a pH of 6.5 showed that the majority of the particles were in apo forms (Figs 4A and EV4A), whereas in the 1:3 mixture (i.e., three‐fold surplus of ADEP1) at the same pH, the majority of particles were in 14 ADEP‐bound forms (Figs 4B and EV4B). The apo structure at pH 6.5 is similar to that of the previous extended structure, and the ADEP‐bound structure showed a very clear N‐terminal region (Appendix Fig S2A and B). In the electron micrographs of the 1:3 mixture of BsClpP and ADEP at pH 4.2, both the apo and all ADEP‐bound BsClpPs were shown as significant populations (Fig EV4C); thus, we determined the two structures with one dataset (Fig 4C and D, and Appendix Table S2). However, as shown in Fig EV4C, other complex states also existed, and we believe that the 2ADEP and 5ADEP found in crystals are intermediates that are energetically stable in the conformational transition from extended to compressed states.
Figure 4. Cryo‐EM structures of the BsClpP‐ADEP1 complex.
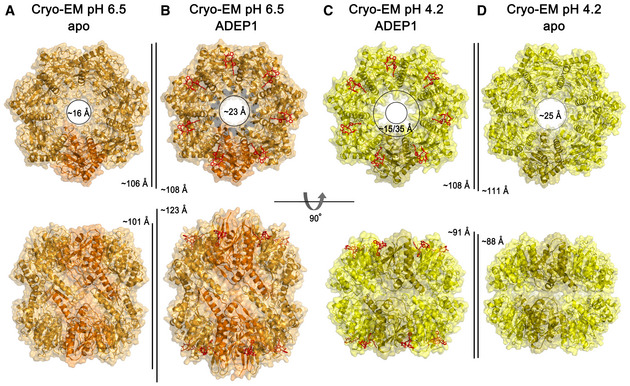
- Ribbon diagram, with the transparent molecular surface of the extended apo‐BsClpP structure at pH 6.5 viewed along a 7‐fold molecular symmetry axis (upper row) and viewed with 90° rotation to display the side view of the 2‐fold symmetry axis (lower row). One monomer is shown in darker orange color for clarity.
- Same representation as that of panel (A) showing the 14 ADEP1‐bound BsClpP at pH 6.5. The bound ADEP1 molecules are shown as stick models colored red.
- Same representation as that of the compressed BsClpP with 14 ADEP1 molecules at pH 4.2. One monomer is shown in darker yellow color for clarity.
- Same representation as that of the compressed apo‐BsClpP at pH 4.2. The dimensions of the models are indicated.
The 14 ADEP‐bound BsClpP cryo‐EM structures at the two different pH values, 6.5 and 4.2, are markedly different in dimension (Fig 4B and C). The structure at pH 6.5 shows an extended conformation, with a well‐ordered N‐terminal region slightly tilted to the outward position, whereas the 14 ADEP‐bound structure at pH 4.2 is similar to the compact structure, with a relatively ordered N‐terminal region tilted to the inward position, suggesting that the N‐terminal region might be involved in the substrate feeding process. The N‐terminal region of ClpP acts as a gate for controlling substrate access with Clp‐ATPases as well as ADEP activators (Jennings et al, 2008; Malik et al, 2020; Vahidi et al, 2020; Brotz‐Oesterhelt & Vorbach, 2021). This 14 ADEP‐bound structure at pH 4.2 is classified as a compact conformation based on its dimension, and it is very similar to 5ADEP (Fig 2B). However, the entrance pore is symmetric because all subunits are virtually identical (Fig 4C). The apo‐BsClpP structure at pH 4.2 is structurally similar to that of 2ADEP (Fig 2A), although some structural details, such as the entrance pore and handle regions, are subtly different. A summary of structural features is shown in Appendix Table S3.
Structural changes in the proteolytic chamber
To understand more about the various structural states at different pH conditions, we focused on the interior of the proteolytic chamber. Although the key determinants for the maintenance of the active extended conformation of ClpP, including the β‐sheets in the handle regions of two subunits from each heptameric ring and the critical salt‐bridge pairs (Asp169–Arg170′:Asp169′–Arg170) at the tip of the handle region, have been extensively studied, the charge property of the chamber has not been examined thoroughly. We analyzed the electrostatic potential surface of all the different structural states from the perspective of hydrolyzed substrates (Fig 5). In general, the surface shows negatively charged features in all states, and it can provide an environment in which the negatively charged products are energetically unfavorable, although they are just slightly acidic due to the summation of the pK1 (−COOH), pK2 (−NH3 +), and pKR (−R: side chain) of all the peptide fragments, as we described in an earlier section (i.e., the lowering of the pH by the accumulation of hydrolyzed peptide products). To release this strained state derived from product generation, a conformational change must be triggered. Usually, the side chain of the histidine residue (pKR = 6.0) is thought to be a sensor for the pH‐dependent transition. Previously, His145 in N. meningitidis ClpP was proposed to be a switch regulating the pH‐dependent conformational change, and it forms an intersubunit H‐bond with the catalytic Asp178′ residue of a neighboring ClpP protomer (Ripstein et al, 2020b). However, the equivalent residues in ClpPs from B. subtilis, M. tuberculosis, and S. aureus are alanine and glutamine, which are not conserved (Fig EV1). Assuming the conservation of the molecular motion of ClpP among all species, the key switch residues must be strictly conserved. Therefore, we thought that another histidine residue, particularly the strictly conserved catalytic His122, might be a candidate (Fig EV1). The catalytic triad Ser97‐His122‐Asp171 is an active configuration in the extended conformation (Fig 5A and B), whereas this triad is an inactive configuration in the compact and compressed conformations (Fig 5C–F). Naturally, the imidazole ring of His122 is protonated at low pH, and subsequently, the catalytic triad is severely distorted. In the compact and compressed structures, the catalytic triads in the upper and lower heptameric rings close, and the previously identified key residues do not participate in the maintenance of the extended structure. The generation of exit side pores is coupled with the transition from the extended state to the compact state (Fig 5C and D), and the pores for peptide release are gradually enlarged during this transition (Fig 5E and F).
Figure 5. Electrostatic potential of the proteolytic chamber of various structural states.
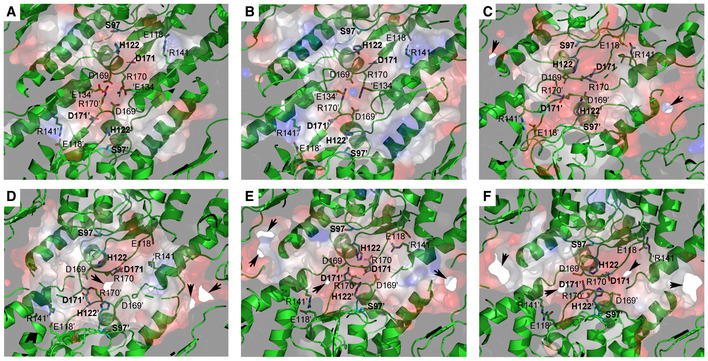
- Ribbon diagram, with the transparent electrostatic potential surface of the extended apo‐BsClpP viewed from the center of the proteolytic chamber. The catalytic triad (S97‐H122‐D171) is shown in the fully active configuration.
- Same representation for the 14 ADEP‐bound BsClpP cryo‐EM structure at pH 6.5. The residues maintaining the extended conformation are now slightly loosened, and the catalytic triad is slightly distorted.
- Same representation for the 14 ADEP‐bound BsClpP at pH 4.2. The conformation is now compact, and side exit pores (black arrows) start to appear.
- Same representation for 5ADEP at pH 5.6. The conformation of BsClpP is compact, and the catalytic triads in the upper and lower heptameric rings become closer and exhibit an inactive configuration.
- Same representation for 2ADEP at pH 4.2. The conformation of BsClpP is compressed, and the catalytic triads in the upper and lower heptameric rings are very close.
- Same representation for the compressed apo‐BsClpP. The exit pores are wide open for product release.
Data information: In all panels (A–F), the residues maintaining the extended conformation of ClpP are shown as stick models and labeled. For clarity, the residues in the neighboring subunits of the different heptameric rings are marked with prime (′) after the residue number, and those in the neighboring subunits of the same heptameric ring are marked with prime (′) before a single letter code used to represent the amino acid. Positively and negatively charged surfaces are colored blue and red, respectively. The inner surface of the proteolytic chamber is virtually negatively charged (red color), and the acidic proteolytic products must be energetically very unfavorable.
Comparison with ClpXP structures
Recently, a long‐awaited structure of the symmetry‐mismatched ATP‐dependent Clp protease was revealed by several independent groups (Gatsogiannis et al, 2019; Fei et al, 2020; Lopez et al, 2020; Ripstein et al, 2020a; Vahidi et al, 2020). The complexes comprised a hexameric Clp‐ATPase, ClpX or ClpA, and heptameric ClpP, and most of the structures show that the six IGF/L‐loops of Clp‐ATPase bind to six out of the seven binding pockets of ClpP with a 10–16° tilt angle of each molecular axis. When we compared the ClpP structures of the ClpXP complex to those of our BsClpP structures, the cryo‐EM structures of the extended ClpP at pH 6.5 were similar. More specifically, the monomeric subunit was very similar, while in the oligomeric state of ClpP, the size of the entrance pore was larger in the ADEP complex than in the ClpX complex (Appendix Fig S3A and B), showing that ClpX does not induce pore‐widening in ClpP (Gatsogiannis et al, 2019). The 7 ADEP‐binding sites overlap with the 6 IGF/L‐loops binding sites and an empty site, as shown in most ClpXP and ClpAP complexes (Gatsogiannis et al, 2019; Fei et al, 2020; Lopez et al, 2020; Ripstein et al, 2020a); however, it has been reported that when only 5 IGF/L loops are involved in the interaction with ClpP, the Clp‐ATPase conformation has a higher tilting angle (Fig EV5A–C) (Lopez et al, 2020), and this so‐called “disengaged conformation” is important for the reaction cycle of the Clp‐ATPase. Therefore, we compared our 5ADEP structure with that of the disengaged conformation. As shown in Fig EV5C, the two empty sites in the disengaged conformation of ClpAP are consecutive, whereas the two empty sites in 5ADEP are not (Fig 2B). Considering the differences in activator binding, the movements and diameters of the entrance pores, and the fact that ADEP cannot actively unfold a protein, the substrate feeding processes realized by two different activators must be different.
Discussion
The major species of the ATP‐dependent complex formed between the Clp‐ATPases and ClpP has been reported to be a 2:1 complex (Ortega et al, 2004), and thus, how the proteolytic products are released from the proteolytic chamber remains unknown. A pioneering, elegant NMR study coined the concept of the lateral exit pore (Sprangers et al, 2005), and subsequent reports established that the compressed structure of ClpP is the state for product release (Geiger et al, 2011; Lee et al, 2011; Zhang et al, 2011) and proposed several factors for the structural transitions from the extended state to the compressed state via the compact state (Gersch et al, 2012; Vahidi et al, 2018, 2020; Ripstein et al, 2020b). Since the first extended structure of ClpP was reported (Wang et al, 1997), numerous crystal structures of ClpP from different species and even in different conformational states have been reported (Kang et al, 2004; Gribun et al, 2005; Yu & Houry, 2007; Kim & Kim, 2008; El Bakkouri et al, 2010; Lee et al, 2010, 2011; Geiger et al, 2011). When we analyzed the crystallization conditions of the reported structures of ClpP, compressed or compact structures were mainly reported for crystals grown under low‐pH conditions (Geiger et al, 2011; Lee et al, 2011), but not always. Therefore, we speculated that there must be a relationship between the physiological conditions and the low pH crystallization that yields the compressed structures. ClpP shows a cylindrical shape with doubly capped Clp‐ATPases. Thus, its proteolytic chamber is almost a closed compartment, and the proteolytic products must accumulate in it. Therefore, the physicochemical properties of this compartmental environment can be maintained locally. As we described earlier, the peptide products possess millions of new amino‐ and carboxy‐termini that govern the overall pH of this chamber. The average pKa value of all amino acid residues is below pH 6.0, and thus, although it depends on the pI values of the substrates and the composition of amino acids, the local pH value must be approximately 6.0. Due to the technical difficulty of directly measuring the pH value of the proteolytic chamber of ClpP during protein hydrolysis, we set up a test experiment to determine whether substrate hydrolysis indeed drops the pH of the system (Fig 1). Instead of the compartmentalizing chamber, we used minimal buffering conditions and a high concentration of the substrate to obtain a large amount of peptide products. Three model proteins, α‐casein, BSA, and myoglobin, were degraded by proteinase K, and the pH of the system dropped gradually (Fig 1A–C). Likewise, when the partially unfolded α‐casein and a well‐established physiological substrate, FtsZ, were degraded by BsClpP in the presence of ADEP1 (Fig EV2B and F), the pH of the system dropped slowly as the hydrolyzed products were released (Fig 1A and D). We speculate that the pH drop of the proteolytic chamber must occur faster because the volume of the self‐compartmentalizing chamber is limited. Therefore, we propose that the main driving force for the opening of the side exit pore of the Clp‐ATPase/ClpP complex is the concentration of protons that regulates the conformational change. The pH change controls countless biological processes, including ATP synthesis, virus maturation, oligomerization, autophagy, and lysosomal activity (Carr et al, 1997; Stock et al, 2000; Burkard et al, 2014; Kim et al, 2015; Kwon et al, 2018, 2019; Banerjee & Kane, 2020; Roh et al, 2020). Herein, we add another example, in which the pH‐dependent conformational change controls the proteolytic activity of the self‐compartmentalizing protease.
There are unique features of ClpP activation caused by the small ADEP molecules, which are distinguished from those of Clp‐ATPases. The ADEP‐binding sites share the IGF/L binding region; however, the numbers and positions are different. Previously, only structural information about all 14 ADEP‐binding or no binding structures has been reported (Lee et al, 2010, 2011; Li et al, 2010; Gersch et al, 2015), and now, we present two other states, 5ADEP (10 ADEPs for a BsClpP tetradecamer) and 2ADEP (4 ADEPs for a BsClpP tetradecamer), which might be energetically stable intermediates among the heterogeneous binding states. However, as shown in recently reported ClpAP cryo‐EM structures (Lopez et al, 2020), depending on the conformational states of ClpA, 6 ADEP‐binding sites in the heptameric ClpP ring are occupied in the engaged conformation, and 5 ADEP‐binding sites are occupied in the disengaged conformation. The other difference is the entrance pore formed by the N‐terminal region of ClpP. Pore‐widening in the ClpXP complex is not necessary because the ATPase fully unfolds the substrate and translocates it through the narrow pore, whereas ADEP triggers pore opening for substrate translocation (Gatsogiannis et al, 2019). It is known that stably folded model proteins are not degraded by the ADEP‐ClpP complex, but partially unfolded substrates, such as casein and peptide substrates, are well degraded (Kirstein et al, 2009; Lee et al, 2010). The antibiotic mechanism of ADEP has been established, and the nascent polypeptide chains emerging from the ribosome and, due to intrinsic instability, also the cell division protein FtsZ are the target substrates of the ADEP‐ClpP complex (Silber et al, 2020). Intriguingly, the intermediate structures 5ADEP and 2ADEP show asymmetric pore shapes due to the molecular asymmetry of ClpP (Fig 2). In addition, ClpXP and ClpAP are symmetry‐mismatched complexes, and thus, ClpP becomes asymmetric during the reaction cycle. Although the substrate feeding steps realized by ADEP and Clp‐ATPases are different, the remaining steps must be similar. Once the substrate reaches the proteolytic chamber, hydrolyzed products are produced, and subsequently, the local pH of the chamber must be lower in both cases. Except for in the extended state, the strictly conserved catalytic triad showed a distorted configuration, most likely owing to the protonation state of His122. The gradual widening of side pores was observed in the transition from the extended, fully occupied 14 ADEP‐ClpP structure through 5ADEP and 2ADEP to the compressed apo‐ClpP structure. In combination with current results and previous reports, we proposed a model for product release (Fig 6). The product release mechanism of ClpP, initiated by substrate hydrolysis followed by a decrease in pH, must be similar to both activators, small chemical compounds and Clp‐ATPases. However, the enlarged entrance pores of ClpP induced by ADEP‐binding might be additional exit sites, whereas ClpP engaged with processive Clp‐ATPase utilizes only side exit pores. Notwithstanding these differences, our current study provides insight into the driving force for the opening of the site exit pore of ClpP.
Figure 6. Model proposed for product release of ClpP.
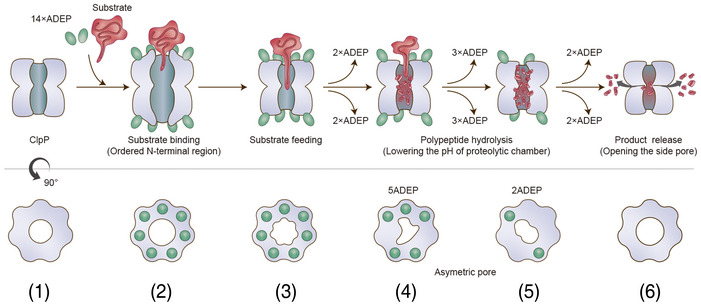
Before (state 1) and immediately after substrate binding (state 2), ClpP is in an active extended conformation, and ADEP widens the entrance pore for better substrate feeding. Our previous crystal structure of the 14 ADEP complex showed a disordered N‐terminal region (Lee et al, 2010), and our current cryo‐EM structure of the same complex demonstrates an ordered N‐terminal region. In both cases, the entrance pore is enlarged. From the extended structure (states 1 and 2) to the compressed structure (states 5 and 6), intermediate conformations were observed. Based on the solution cryo‐EM analysis, these intermediate conformations represented heterogeneous complex species of BsClpP with different numbers of ADEP molecules bound, including a 14 ADEP‐bound compact structure (state 3). Two major species among the intermediate states, 5ADE P (state 4) and 2ADEP (state 5), were crystallized and showed dimensions similar to those of previously known “compact” and “compressed” structures (Geiger et al, 2011; Lee et al, 2011; Zhang et al, 2011; Gersch et al, 2012). Both possess asymmetric entrance pores, especially 5ADEP, which is more distorted. 5ADEP is less compressed, and thus, the number and size of the side pores are fewer and smaller than those of a fully compressed structure, respectively. Apo‐BsClpP at pH 4.2 exhibits compression and larger side exit pores (state 6). The extended states 1 and 2 possess the active catalytic triad configuration (Fig 5A and B). The catalytic triad of the other states (3–6) shows an inactive and distorted configuration (Fig 5C–F). In the model, the red color of the proteolytic chamber represents the lower pH condition derived from substrate hydrolysis.
To establish a more definitive conclusion, the direct pH measurement of the proteolytic chamber, or even the monitoring of its pH in real‐time during protein hydrolysis, must inevitably occur. The small water molecules and ions present under physiological buffer conditions can freely diffuse in and out of the self‐compartmentalized chamber, and therefore, the experiment is not straightforward; however, it is the current rationale that a pH‐dependent conformational switch in a local area can trigger a global change in the molecular shape. We await the elegantly designed experiments, such as the usage of fine‐tuned pH‐sensing probes, that might be helpful in uncovering the detailed molecular motion inside the proteolytic chamber, which can then be a general concept for self‐compartmentalizing proteases.
Materials and Methods
Sample preparation
BsClpP protein was purified as previously described (Lee et al, 2010, 2011). For cryo‐EM experiments, we further purified the proteins using a SuperoseTM 6 Increase 10/300 GL (GE Healthcare, 29‐0915‐96) size‐exclusion column pre‐equilibrated with 50 mM sodium acetate with a pH of 4.2, 500 mM KCl, and 5% (w/v) glycerol [or 50 mM N‐(2‐acetamido)iminodiacetic acid with a pH of 6.5, 200 mM NaCl, and 5% (w/v) glycerol]. A basic BsFtsZ‐ED6K mutant (E345/349/350K and D366/367/370K) with a C‐terminal octa‐histidine tag was generated by using the QuikChange site‐directed mutagenesis method (Strategene). Protein expression of BsFtsZ‐ED6K was induced by the addition of 0.5 mM isopropyl β‐D‐1‐thiogalactopyranoside at 18°C for 24 h. Cells were harvested by centrifugation and resuspended in 50 mM Tris–HCl pH 8.0 buffer containing 200 mM NaCl and 1 mM TCEP [tris(2‐carboxyethyl)phosphine hydrochloride]. The resuspended cells were disrupted by sonication, and the cell lysate was loaded onto a HisTrapTM column (GE Healthcare, 17‐5255‐01). Proteins were eluted using a linear gradient of imidazole concentrations (0‐500 mM). Finally, BsFtsZ‐ED6K was loaded onto a SuperoseTM 6 Increase 10/300 GL (GE Healthcare, 29‐0915‐96) size‐exclusion column pre‐equilibrated with diluted PBS (1.37 mM NaCl, 0.27 mM KCl, 0.1 mM Na2HPO4, and 0.18 mM KH2PO4) for the pH measurement assay. SDS–PAGE and negative stain electron microscopy were used to assess protein purity and quality. All biological materials are available upon request.
pH measurement during protein hydrolysis
For in vitro pH‐change measurements, bovine milk α‐casein (Sigma, C6780), bovine serum albumin (Sigma, A7030), myoglobin from equine skeletal muscle (Sigma, M0630), and BsFtsZ‐ED6K were used as substrates. Reactions were carried out with either 100 μg/ml proteinase K (Sigma, P2308) in diluted PBS (1.37 mM NaCl, 0.27 mM KCl, 0.1 mM Na2HPO4, and 0.18 mM KH2PO4) containing 1 mM CaCl2 or with 5.6 μM BsClpP (monomer) in the presence of 11 μM ADEP1 (Cayman Chemical, A‐54556A, ~2‐fold molar ratio) (Lee et al, 2010) in the same PBS at 40°C. The final concentrations of the substrate were 80 μM (α‐casein), 100 μM (BSA), 110 μM (myoglobin), and 12 μM (BsFtsZ‐ED6K), and the final assay volume was 20 ml. For the degradation of BsFtsZ‐ED6K, a higher concentration of BsClpP was used such as 40 μM monomeric BsClpP. The pH was measured every 30 s for 10 min, every 1 min for 20 min, every 5 min for 210 min, and every 10 min for 60 min, for a total of 300 min, using a pH meter (Thermo Scientific, 13‐644‐928).
Crystallography
A cocrystallization method was used to generate crystals of BsClpP‐ADEP complexes by mixing BsClpP and ADEP2 at a 1:1 molar ratio. Crystals of the compressed and compact forms of the BsClpP‐ADEP complex were grown in hanging drops at 22°C using 100 mM sodium acetate with a pH of 4.6, 500 mM potassium thiocyanate (for the compressed state), 100 mM sodium citrate with a pH of 5.6, 100 mM Li2SO4, and 9–11% (w/v) polyethylene glycol 6,000 (for the compact state). Diffraction data were processed with the program HKL2000 (Otwinowski & Minor, 1997). The phases of the 2ADEP and 5ADEP structures were obtained by molecular replacement using a previously solved compressed BsClpP structure (Lee et al, 2011) as a search model with the program MOLREP in the CCP4 suite (Winn et al, 2011). Model building and refinement were performed using the programs COOT (Casañal et al, 2020) and PHENIX (Afonine et al, 2018), respectively.
Cryo‐EM data collection and processing
The fraction eluted from size‐exclusion chromatography was isolated and incubated with 100 µM ADEP1 (Cayman Chemical, A‐54556A). In particular, the BsClpP pH 6.5 sample was also treated with 4 mg/ml amphipol. After 10 min of incubation, a 5 µl drop was applied to a graphene‐treated and glow‐discharged holey carbon grid (R 1.2/1.3 Quantifoil) for the BsClpP pH 4.2 sample and negatively glow‐discharged holey carbon grid (R 1.2/1.3 Quantifoil) for the BsClpP pH 6.5 sample. They were then blotted for 3 s at 4°C and 100% humidity with Whatman no. 595 filter paper before being plunge‐frozen in liquid ethane using a Vitrobot Mark IV system (Thermo Fisher Scientific Inc., USA). Cryo‐EM images of frozen‐hydrated BsClpP particles were collected at Korea Basic Science Institute with a Titan Krios TEM system (Thermo Fisher Scientific Inc., USA) operated at 300 keV using electron counting mode and automatic data acquisition software (EPU, Thermo Fisher Scientific Inc., USA). Detailed data acquisition conditions and parameters are given in Appendix Table S2.
Cryo‐EM images for BsClpP pH 4.2 were processed using RELION 3.0 (Zivanov et al, 2018) and CryoSPARC v2 (Punjani et al, 2017). Beam‐induced motion correction and dose weighting were performed using MotionCor2 v1.2.1 (Zheng et al, 2017), and CTF estimation was performed using Gctf v1.06 (Zhang, 2016). Then, 341,414 particle images were selected from 964 micrographs after reference‐free 2D class averages. The 3D initial model was generated by CryoSPARC‐implemented ab initio reconstruction. Multiple rounds of successive 3D classifications were performed, and 179,322 homogeneous particles were selected for further processing. Homogeneous refinement with D7 symmetry then yielded a consensus map at 3.6 Å. Of note, particles displayed a significant preferred orientation, showing a dominant top view. Selected particles with angular information were transferred to RELION for further 3D classification without an angular orientation search. To evaluate the possible heterogeneity of ADEP binding, we performed focused 3D classification around ADEP binding sites with expanded particles with C7 symmetry. We only observed the all‐ADEP and apo states from the analysis, and thus, we clustered particles into apo or all‐ADEP states. Finally, each selected particle set was imported into CryoSPARC and refined at 3.4 Å.
The images of BsClpP pH 6.5 were processed using CryoSPARC v2 (Punjani et al, 2017). We originally analyzed ADEP‐binding heterogeneity in the dataset of a 1:3 mixture of BsClpP and ADEP at pH 6.5 using the same procedure as described above for BsClpP pH 4.2. However, the particles were homogeneous, as all ADEP binding sites were occupied in the dataset. Therefore, we obtained two separate imaging sessions for apo (1,130 movies) and ADEP‐bound structures (1,182 movies) at pH 6.5. The recorded movies were subjected to motion correction and CTF estimation using patch‐motion correction and Gctf v1.06 in CryoSPARC. For the apo map at pH 6.5, a total of 108,389 particle images were selected after 2D class averages. After subsequent heterogeneous refinement for suitable particles, 78,413 particles were finally used for 3D reconstruction at 3.2 Å resolution. For the ADEP‐bound map at pH 6.5, a total of 153,078 particle images were selected after 2D class averages. After subsequent heterogeneous refinement, 138,976 particles yielded a map at 3.1 Å resolution. All cryo‐EM images were processed using the computing resources at the Center for Macromolecular and Cell Imaging, Seoul National University.
Molecular modeling
Both BsClpP pH 4.2 and pH 6.5 structural models were built manually in COOT (Casañal et al, 2020) by referring to the BsClpP crystallography structure (PDB ID: 3TT6 for the pH 4.2 model and 3KTJ for the pH 6.5 model) and refined using phenix.real_space_refine command in the PHENIX software suite (Liebschner et al, 2019). Additionally, the qualities of the final models were evaluated using the comprehensive model validation section and MolProbity in PHENIX.
Author contributions
Leehyeon Kim: Conceptualization; Data curation; Software; Validation; Investigation; Visualization; Methodology. Byung‐Gil Lee: Conceptualization; Data curation; Software; Validation; Methodology. Minki Kim: Data curation; Formal analysis; Validation. Min Kyung Kim: Data curation; Methodology. Do Hoon Kwon: Validation; Investigation; Visualization; Methodology. Hyunmin Kim: Software; Visualization; Methodology. Heike Brötz‐Oesterhelt: Formal analysis; Supervision; Writing—review & editing. Soung‐Hun Roh: Resources; Software; Supervision; Funding acquisition; Validation; Visualization. Hyun Kyu Song: Conceptualization; Resources; Supervision; Funding acquisition; Validation; Investigation; Visualization; Methodology; Project administration; Writing—review & editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Acknowledgements
We thank the staff at beamlines 5C and 11C at the Pohang Accelerator Laboratory in South Korea and beamline NW12 at the Photon Factory in Japan for their help with X‐ray data collection. We also thank the staff of the cryo‐TEM facility at the Korea Basic Science Institute and the Center for Macromolecular and Cell Imaging, Seoul National University. This study was supported by National Research Foundation of Korea (NRF) grants from the Korean government (grant Nos. 2020R1A2C3008285, 2020R1A5A1019023, 2021M3A9G8024747, and 2021M3A9I4030068 for HKS; 2021M3A9I4021220, 2019M3E5D6063871, and 2020R1A5A1018081 for SHR; 2021R1A6A1A10045235 for LK) and the Deutsche Forschungsgemeinschaft (German Research Foundation, DFG) TRR 261, project‐ID 398967434 and cluster of excellence CMFI, project‐ID 390838134.
The EMBO Journal (2022) 41: e109755.
Contributor Information
Soung‐Hun Roh, Email: shroh@snu.ac.kr.
Hyun Kyu Song, Email: hksong@korea.ac.kr.
Data availability
The cryo‐EM maps have been deposited in the EMDB: ADEP1‐BsClpP complex at pH 4.2 (EMD‐31561, http://www.ebi.ac.uk/pdbe/entry/EMD‐31561), apo‐BsClpP at pH 4.2 (EMD‐31562, http://www.ebi.ac.uk/pdbe/entry/EMD‐31562), ADEP1‐BsClpP complex at pH 6.5 (EMD‐31559, http://www.ebi.ac.uk/pdbe/entry/EMD‐31559), and apo‐BsClpP at pH 6.5 (EMD‐31560, http://www.ebi.ac.uk/pdbe/entry/EMD‐31560). The coordinates have been deposited in the PDB: crystal structures – 2ADEP (7P80, http://www.rcsb.org/pdb/explore/explore.do?structureId=7P80) and 5ADEP (7P81, http://www.rcsb.org/pdb/explore/explore.do?structureId=7P81); cryo‐EM structures – ADEP1‐BsClpP complex at pH 4.2 (7FER, http://www.rcsb.org/pdb/explore/explore.do?structureId=7FER), apo‐BsClpP at pH 4.2 (7FES, http://www.rcsb.org/pdb/explore/explore.do?structureId=7FES), ADEP1‐BsClpP complex at pH 6.5 (7FEP, http://www.rcsb.org/pdb/explore/explore.do?structureId=7FEP), and apo‐BsClpP at pH 6.5 (7FEQ, http://www.rcsb.org/pdb/explore/explore.do?structureId=7FEQ).
References
- Afonine PV, Poon BK, Read RJ, Sobolev OV, Terwilliger TC, Urzhumtsev A, Adams PD (2018) Real‐space refinement in PHENIX for cryo‐EM and crystallography. Acta Crystallogr D Struct Biol 74: 531–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker TA, Sauer RT (2012) ClpXP, an ATP‐powered unfolding and protein‐degradation machine. Biochim Biophys Acta 1823: 15–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee S, Kane PM (2020) Regulation of V‐ATPase activity and organelle pH by phosphatidylinositol phosphate lipids. Front Cell Dev Biol 8: 510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brötz‐Oesterhelt H, Beyer D, Kroll H‐P, Endermann R, Ladel C, Schroeder W, Hinzen B, Raddatz S, Paulsen H, Henninger K et al (2005) Dysregulation of bacterial proteolytic machinery by a new class of antibiotics. Nat Med 11: 1082–1087 [DOI] [PubMed] [Google Scholar]
- Brotz‐Oesterhelt H, Vorbach A (2021) Reprogramming of the caseinolytic protease by ADEP antibiotics: molecular mechanism, cellular consequences. therapeutic potential. Front Mol Biosci 8: 690902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkard C, Verheije MH, Wicht O, van Kasteren SI, van Kuppeveld FJ, Haagmans BL, Pelkmans L, Rottier PJ, Bosch BJ, de Haan CA (2014) Coronavirus cell entry occurs through the endo‐/lysosomal pathway in a proteolysis‐dependent manner. PLoS Pathog 10: e1004502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr CM, Chaudhry C, Kim PS (1997) Influenza hemagglutinin is spring‐loaded by a metastable native conformation. Proc Natl Acad Sci USA 94: 14306–14313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casañal A, Lohkamp B, Emsley P (2020) Current developments in Coot for macromolecular model building of electron cryo‐microscopy and crystallographic data. Protein Sci 29: 1055–1064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Bakkouri M, Pow A, Mulichak A, Cheung KLY, Artz JD, Amani M, Fell S, de Koning‐Ward TF, Goodman CD, McFadden GI et al (2010) The Clp chaperones and proteases of the human malaria parasite Plasmodium falciparum . J Mol Biol 404: 456–477 [DOI] [PubMed] [Google Scholar]
- Fei X, Bell TA, Jenni S, Stinson BM, Baker TA, Harrison SC, Sauer RT (2020) Structures of the ATP‐fueled ClpXP proteolytic machine bound to protein substrate. Elife 9: e52774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatsogiannis C, Balogh D, Merino F, Sieber SA, Raunser S (2019) Cryo‐EM structure of the ClpXP protein degradation machinery. Nat Struct Mol Biol 26: 946–954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge SJ, Zhang LX (1993) Control of the degree of hydrolysis of a protein modification with immobilized protease by the pH‐drop method. Acta Biotechnologica 13: 151–160 [Google Scholar]
- Geiger SR, Bottcher T, Sieber SA, Cramer P (2011) A conformational switch underlies ClpP protease function. Angew Chem Int Ed Engl 50: 5749–5752 [DOI] [PubMed] [Google Scholar]
- Gersch M, Famulla K, Dahmen M, Göbl C, Malik I, Richter K, Korotkov VS, Sass P, Rübsamen‐Schaeff H, Madl T et al (2015) AAA+ chaperones and acyldepsipeptides activate the ClpP protease via conformational control. Nat Commun 6: 6320 [DOI] [PubMed] [Google Scholar]
- Gersch M, List A, Groll M, Sieber SA (2012) Insights into structural network responsible for oligomerization and activity of bacterial virulence regulator caseinolytic protease P (ClpP) protein. J Biol Chem 287: 9484–9494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg AL (1990) ATP‐dependent proteases in prokaryotic and eukaryotic cells. Semin Cell Biol 1: 423–432 [PubMed] [Google Scholar]
- Gottesman S (2003) Proteolysis in bacterial regulatory circuits. Annu Rev Cell Dev Biol 19: 565–587 [DOI] [PubMed] [Google Scholar]
- Gribun A, Kimber MS, Ching R, Sprangers R, Fiebig KM, Houry WA (2005) The ClpP double ring tetradecameric protease exhibits plastic ring‐ring interactions, and the N termini of its subunits form flexible loops that are essential for ClpXP and ClpAP complex formation. J Biol Chem 280: 16185–16196 [DOI] [PubMed] [Google Scholar]
- Groll M, Bochtler M, Brandstetter H, Clausen T, Huber R (2005) Molecular machines for protein degradation. ChemBioChem 6: 222–256 [DOI] [PubMed] [Google Scholar]
- Jennings LD, Bohon J, Chance MR, Licht S (2008) The ClpP N‐terminus coordinates substrate access with protease active site reactivity. Biochemistry 47: 11031–11040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SG, Maurizi MR, Thompson M, Mueser T, Ahvazi B (2004) Crystallography and mutagenesis point to an essential role for the N‐terminus of human mitochondrial ClpP. J Struct Biol 148: 338–352 [DOI] [PubMed] [Google Scholar]
- Kim DY, Kim KK (2008) The structural basis for the activation and peptide recognition of bacterial ClpP. J Mol Biol 379: 760–771 [DOI] [PubMed] [Google Scholar]
- Kim JH, Hong SB, Lee JK, Han S, Roh KH, Lee KE, Kim YK, Choi EJ, Song HK (2015) Insights into autophagosome maturation revealed by the structures of ATG5 with its interacting partners. Autophagy 11: 75–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirstein J, Hoffmann A, Lilie H, Schmidt R, Rubsamen‐Waigmann H, Brotz‐Oesterhelt H, Mogk A, Turgay K (2009) The antibiotic ADEP reprogrammes ClpP, switching it from a regulated to an uncontrolled protease. EMBO Mol Med 1: 37–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlowski LP (2017) Proteome‐pI: proteome isoelectric point database. Nucleic Acids Res 45: D1112–D1116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon DH, Kim L, Song HK (2019) pH‐dependent regulation of SQSTM1/p62 during autophagy. Autophagy 15: 180–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon DH, Park OH, Kim L, Jung YO, Park Y, Jeong H, Hyun J, Kim YK, Song HK (2018) Insights into degradation mechanism of N‐end rule substrates by p62/SQSTM1 autophagy adapter. Nat Commun 9: 3291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BG, Kim MK, Song HK (2011) Structural insights into the conformational diversity of ClpP from Bacillus subtilis . Mol Cells 32: 589–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BG, Park EY, Lee KE, Jeon H, Sung KH, Paulsen H, Rubsamen‐Schaeff H, Brotz‐Oesterhelt H, Song HK (2010) Structures of ClpP in complex with acyldepsipeptide antibiotics reveal its activation mechanism. Nat Struct Mol Biol 17: 471–478 [DOI] [PubMed] [Google Scholar]
- Li DH, Chung YS, Gloyd M, Joseph E, Ghirlando R, Wright GD, Cheng YQ, Maurizi MR, Guarne A, Ortega J (2010) Acyldepsipeptide antibiotics induce the formation of a structured axial channel in ClpP: a model for the ClpX/ClpA‐bound state of ClpP. Chem Biol 17: 959–969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Kandror O, Akopian T, Dharkar P, Wlodawer A, Maurizi MR, Goldberg AL (2016) Structure and functional properties of the active form of the proteolytic complex, ClpP1P2, from Mycobacterium tuberculosis . J Biol Chem 291: 7465–7476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebschner D, Afonine PV, Baker ML, Bunkoczi G, Chen VB, Croll TI, Hintze B, Hung LW, Jain S, McCoy AJ et al (2019) Macromolecular structure determination using X‐rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr D Struct Biol 75: 861–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Ologbenla A, Houry WA (2014) Dynamics of the ClpP serine protease: a model for self‐compartmentalized proteases. Crit Rev Biochem Mol Biol 49: 400–412 [DOI] [PubMed] [Google Scholar]
- Lopez KE, Rizo AN, Tse E, Lin J, Scull NW, Thwin AC, Lucius AL, Shorter J, Southworth DR (2020) Conformational plasticity of the ClpAP AAA+ protease couples protein unfolding and proteolysis. Nat Struct Mol Biol 27: 406–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik IT, Pereira R, Vielberg M‐T, Mayer C, Straetener J, Thomy D, Famulla K, Castro H, Sass P, Groll M et al (2020) Functional characterisation of ClpP mutations conferring resistance to acyldepsipeptide antibiotics in firmicutes. ChemBioChem 21: 1997–2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozersky SM, Panettieri RA (1983) Is pH drop a valid measure of extent of protein hydrolysis? J Agr Food Chem 31: 1313–1316 [Google Scholar]
- Ortega J, Lee HS, Maurizi MR, Steven AC (2004) ClpA and ClpX ATPases bind simultaneously to opposite ends of ClpP peptidase to form active hybrid complexes. J Struct Biol 146: 217–226 [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W (1997) Processing of X‐ray diffraction data collected in oscillation mode. Methods Enzymol 276: 307–326 [DOI] [PubMed] [Google Scholar]
- Punjani A, Rubinstein JL, Fleet DJ, Brubaker MA (2017) cryoSPARC: algorithms for rapid unsupervised cryo‐EM structure determination. Nat Methods 14: 290–296 [DOI] [PubMed] [Google Scholar]
- Ripstein ZA, Vahidi S, Houry WA, Rubinstein JL, Kay LE (2020a) A processive rotary mechanism couples substrate unfolding and proteolysis in the ClpXP degradation machinery. eLife 9: e52158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripstein ZA, Vahidi S, Rubinstein JL, Kay LE (2020b) A pH‐dependent conformational switch controls N. meningitidis ClpP protease function. J Am Chem Soc 142: 20519–20523 [DOI] [PubMed] [Google Scholar]
- Roh SH, Shekhar M, Pintilie G, Chipot C, Wilkens S, Singharoy A, Chiu W (2020) Cryo‐EM and MD infer water‐mediated proton transport and autoinhibition mechanisms of Vo complex. Sci Adv 6: eabb9605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sass P, Josten M, Famulla K, Schiffer G, Sahl HG, Hamoen L, Brotz‐Oesterhelt H (2011) Antibiotic acyldepsipeptides activate ClpP peptidase to degrade the cell division protein FtsZ. Proc Natl Acad Sci USA 108: 17474–17479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer RT, Baker TA (2011) AAA+ proteases: ATP‐fueled machines of protein destruction. Annu Rev Biochem 80: 587–612 [DOI] [PubMed] [Google Scholar]
- Silber N, Pan S, Schakermann S, Mayer C, Brotz‐Oesterhelt H, Sass P (2020) Cell division protein FtsZ is unfolded for N‐terminal degradation by antibiotic‐activated ClpP. MBio 11: e01006‐20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprangers R, Gribun A, Hwang PM, Houry WA, Kay LE (2005) Quantitative NMR spectroscopy of supramolecular complexes: dynamic side pores in ClpP are important for product release. Proc Natl Acad Sci USA 102: 16678–16683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stock D, Gibbons C, Arechaga I, Leslie AG, Walker JE (2000) The rotary mechanism of ATP synthase. Curr Opin Struct Biol 10: 672–679 [DOI] [PubMed] [Google Scholar]
- Vahidi S, Ripstein ZA, Bonomi M, Yuwen T, Mabanglo MF, Juravsky JB, Rizzolo K, Velyvis A, Houry WA, Vendruscolo M et al (2018) Reversible inhibition of the ClpP protease via an N‐terminal conformational switch. Proc Natl Acad Sci USA 115: E6447–E6456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vahidi S, Ripstein ZA, Juravsky JB, Rennella E, Goldberg AL, Mittermaier AK, Rubinstein JL, Kay LE (2020) An allosteric switch regulates Mycobacterium tuberculosis ClpP1P2 protease function as established by cryo‐EM and methyl‐TROSY NMR. Proc Natl Acad Sci USA 117: 5895–5906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Hartling JA, Flanagan JM (1997) The structure of ClpP at 2.3 A resolution suggests a model for ATP‐dependent proteolysis. Cell 91: 447–456 [DOI] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A et al (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr 67: 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong KS, Mabanglo MF, Seraphim TV, Mollica A, Mao Y‐Q, Rizzolo K, Leung E, Moutaoufik MT, Hoell L, Phanse S et al (2018) Acyldepsipeptide analogs dysregulate human mitochondrial ClpP protease activity and cause apoptotic cell death. Cell Chem Biol 25: 1017–1030.e9 [DOI] [PubMed] [Google Scholar]
- Yang T, Zhang T, Zhou X, Wang P, Gan J, Song B, Yang S, Yang CG (2021) Dysregulation of ClpP by small‐molecule activators used against Xanthomonas oryzae pv. oryzae infections. J Agric Food Chem 69: 7545–7553 [DOI] [PubMed] [Google Scholar]
- Yu AY, Houry WA (2007) ClpP: a distinctive family of cylindrical energy‐dependent serine proteases. FEBS Lett 581: 3749–3757 [DOI] [PubMed] [Google Scholar]
- Zhang J, Ye F, Lan L, Jiang H, Luo C, Yang CG (2011) Structural switching of Staphylococcus aureus Clp protease: a key to understanding protease dynamics. J Biol Chem 286: 37590–37601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K (2016) Gctf: real‐time CTF determination and correction. J Struct Biol 193: 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SQ, Palovcak E, Armache JP, Verba KA, Cheng Y, Agard DA (2017) MotionCor2: anisotropic correction of beam‐induced motion for improved cryo‐electron microscopy. Nat Methods 14: 331–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivanov J, Nakane T, Forsberg BO, Kimanius D, Hagen WJ, Lindahl E, Scheres SH (2018) New tools for automated high‐resolution cryo‐EM structure determination in RELION‐3. Elife 7: e42166 [DOI] [PMC free article] [PubMed] [Google Scholar]