

Abstract
Germ cells are unique in engendering totipotency, yet the mechanisms underlying this capacity remain elusive. Here, we perform comprehensive and in‐depth nucleome analysis of mouse germ‐cell development in vitro, encompassing pluripotent precursors, primordial germ cells (PGCs) before and after epigenetic reprogramming, and spermatogonia/spermatogonial stem cells (SSCs). Although epigenetic reprogramming, including genome‐wide DNA de‐methylation, creates broadly open chromatin with abundant enhancer‐like signatures, the augmented chromatin insulation safeguards transcriptional fidelity. These insulatory constraints are then erased en masse for spermatogonial development. Notably, despite distinguishing epigenetic programming, including global DNA re‐methylation, the PGCs‐to‐spermatogonia/SSCs development entails further euchromatization. This accompanies substantial erasure of lamina‐associated domains, generating spermatogonia/SSCs with a minimal peripheral attachment of chromatin except for pericentromeres—an architecture conserved in primates. Accordingly, faulty nucleome maturation, including persistent insulation and improper euchromatization, leads to impaired spermatogenic potential. Given that PGCs after epigenetic reprogramming serve as oogenic progenitors as well, our findings elucidate a principle for the nucleome programming that creates gametogenic progenitors in both sexes, defining a basis for nuclear totipotency.
Keywords: 3D genome organization, epigenetic reprogramming, germ cells, lamina‐associated domains, nucleome
Subject Categories: Chromatin, Transcription & Genomics; Development; Stem Cells & Regenerative Medicine
Progressive euchromatization and insulation dynamics safeguard nuclear totipotency during gametogenesis.
Introduction
Germ cells are the origin of totipotency, which in turn is the foundation for individual development. Mechanisms underlying totipotency have been a focus of intensive investigations, ranging from studies involving somatic‐cell nuclear transfer (Gurdon & Wilmut, 2011) to recent efforts exploring the three‐dimensional (3D) chromatin organization in zygotes and early embryos (Zheng & Xie, 2019). The latter works involving chromatin conformation capture have revealed a relaxed chromatin configuration in zygotes in part resulting from unique meiotic intermediates, and the progressive maturation of this configuration in early embryos (Battulin et al, 2015; Du et al, 2017; Flyamer et al, 2017; Ke et al, 2017; Alavattam et al, 2019; Patel et al, 2019; Vara et al, 2019; Wang et al, 2019). On the other hand, the manner by which germ cells elaborate the higher‐order chromatin organization during their mitotic development, and the founding states for gametogenesis and totipotency, remain poorly understood. In‐depth understanding of genome functions requires investigations of the 3D genome organization complemented by thorough epigenome and transcriptome profiling, an approach known as “nucleome” profiling (Dekker et al, 2017). While nucleome profiling has been performed in a few somatic lineages (Bonev et al, 2017; Stadhouders et al, 2018; Zhang et al, 2019), studies applying this approach to germ‐cell development are lacking.
In mammals, germ cells arise as primordial germ cells (PGCs) during early embryonic development (Saitou & Hayashi, 2021). PGCs undergo migration and colonize the embryonic gonads, where they differentiate either into spermatogonia/spermatogonial stem cells (SSCs), the source for spermatogenesis, or oocytes with an immediate entry into the first prophase of meiosis (Griswold, 2016; Spiller et al, 2017; Wen & Tang, 2019). A key event that characterizes PGCs is epigenetic reprogramming, including de‐methylation of genome‐wide DNA to the point that it contains almost no DNA methylation, as well as histone‐modification remodeling, which creates a facultative “naïve” epigenome (Lee et al, 2014; Tang et al, 2016). In males, epigenetic reprogramming is followed by the provision of a distinct spermatogenic epigenome, including global DNA re‐methylation, for spermatogonia/SSC development, whereas in females, the naïve epigenome serves as a direct precursor for the oogenic meiotic entry (Lee et al, 2014). Thus, male germ‐cell development requires at least one additional epigenetic programming step to create spermatogenic progenitors. Here, to explore the principles that create a basis for gametogenic potential, we performed nucleome profiling of an in vitro system that faithfully reconstitutes mouse germ‐cell development from pluripotent precursors to PGCs before and after epigenetic reprogramming and then to spermatogonia/SSCs (Kanatsu‐Shinohara et al, 2003; Hayashi et al, 2011; Ohta et al, 2017, 2021). We show that the in vitro system recapitulates not only gene expression and epigenetic properties but also 3D genome‐organization dynamics during germ‐cell development in vivo, lending credence to our analyses using scalable materials to provide a more complete picture of nucleome dynamics with a high resolution during germ‐cell development. In addition, to delineate the functional significance of appropriate nucleome programming, we analyzed the nucleome of an in vitro counterpart of spermatogonia/SSCs with an impaired spermatogenic potential (Ishikura et al, 2016).
Results
Mouse germ‐cell development in vitro
We analyzed the following male cell types (Fig 1A): mouse embryonic stem cells (mESCs) derived from blastocysts (Ying et al, 2008), epiblast‐like cells (EpiLCs) (Hayashi et al, 2011), mouse PGC‐like cells at day 2 of induction (d2 mPGCLCs) (Hayashi et al, 2011), d4 mPGCLCs expanded in vitro for 7 days for epigenetic reprogramming (d4c7 mPGCLCs; Ohta et al, 2017, 2021), and germline stem cells (GSCs) derived from neonatal spermatogonia (Kanatsu‐Shinohara et al, 2003). These cells show gene expression, epigenetic, and functional properties equivalent to those of their in vivo counterparts, that is, mESCs to epiblast at embryonic day (E) 4.5 with naïve pluripotency (Marks et al, 2012; Boroviak et al, 2014), EpiLCs to epiblast at ~E6.0 with formative pluripotency (Hayashi et al, 2011), d2 mPGCLCs to mPGCs during their specification at ~E7.0 and before epigenetic reprogramming (Hayashi et al, 2011; Kurimoto et al, 2015), d4c7 mPGCLCs to PGCs at E11.5 after epigenetic reprogramming (Ohta et al, 2017, 2021), and GSCs to spermatogonia/SSCs (Ishikura et al, 2016). Note that PGCs before E11.5 do not show overt sexual differences in gene expression and epigenetic properties, except X‐chromosome reactivation in females (Jameson et al, 2012; Ohta et al, 2017). Accordingly, male PGCs bear a capacity to form functional oocytes (Evans et al, 1977), and male mPGCLCs take on the oogenic fate and enter into the meiotic prophase in response to appropriate signals at an efficiency comparable to that of female mPGCLCs (Miyauchi et al, 2017; Nagaoka et al, 2020). Thus, while our present analysis focuses on male germ‐cell development, male d4c7 mPGCLCs can be considered to bear an oogenic potential as well. In addition, to evaluate the functional relevance of proper nucleome programming, we analyzed GSC‐like cells (GSCLCs) that were derived from d4 mPGCLCs in vitro and had an impaired spermatogenic potential (Ishikura et al, 2016; see the “Nucleome programming engenders gametogenic potential” section).
Figure 1. 3D genome programming.
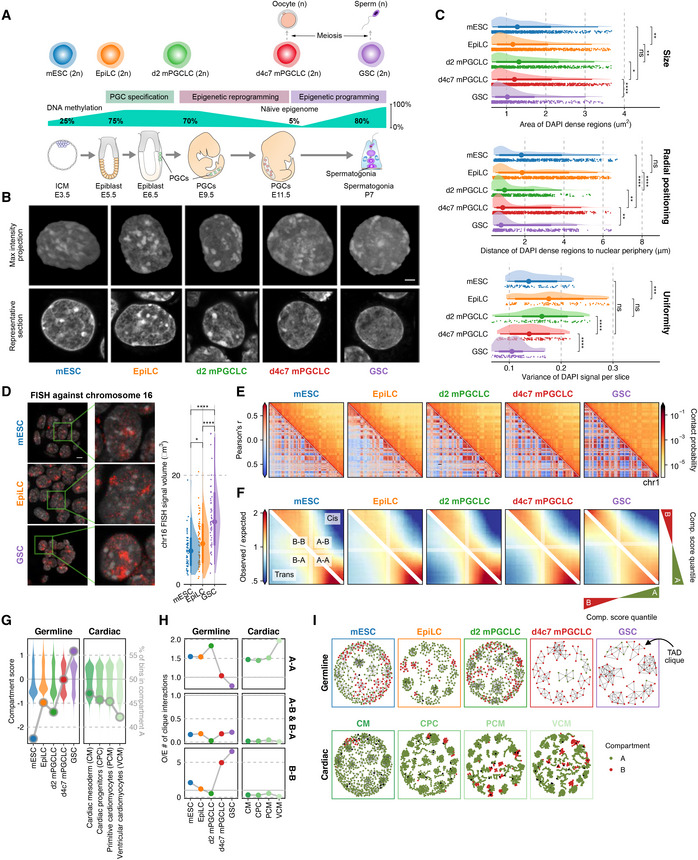
- Scheme for mouse germ‐cell development in vitro (top) and in vivo (bottom), with dynamics of genome‐wide DNA methylation levels (middle).
- Maximum intensity projections (top) and representative sections (bottom) of typical nuclei of the indicated cell types stained with DAPI. Scale bars, 3 μm.
- Areas of DAPI‐dense regions (top), distance of DAPI‐dense regions from the nuclear periphery (middle), and variance of DAPI signals (bottom). The point marks the median while the thick and thin lines correspond to 66% and 95% intervals, respectively. Number of DAPI dense regions = 950/1450/839/1535/736 and number of slices = 90/115/95/135/110 for mESC/EpiLC/d2/d4c7 mPGCLC/GSC. Significances are computed using Wilcoxon rank‐sum tests, p‐values from top to bottom 4.37e−3, 1.62e−3, 2.99e−2, 2.03e−10, 4.03e−1, < 2.2e−16, 1.31e−3, 8.94e−3, 1.06e−4, 5.62e−2, 4.63e−5, 7.65e−13. P‐value symbol brackets: **** = [0, 0.0001); *** = [0.0001, 0.001]; ** = [0.001, 0.01); * = [0.01, 0.05); ns = [0.05, 1].
- (left) Fluorescence in situ hybridization (FISH) against chromosome 16 (red) with DAPI staining (gray). Z‐stacked representative images are paired with magnified views. (right) Distributions of surface volumes for chr16. The point marks the median while the thick and thin lines correspond to 66% and 95% intervals, respectively. Number of cells = 51/68/53 for mESC/EpiLC/GSC. Scale bars, 5 μm. Significances are computed using Wilcoxon rank‐sum tests, P‐values from left to right: 4.16e−2, 4.33e−6, 8.68e−9.
- Hi‐C maps of chromosome 1. (upper right triangle) 250 kb‐resolution balanced contact probability matrices; (lower left triangle) matching Pearson’s correlation matrices.
- Compartmentalization saddle plots for the average interaction frequency between pairs of 50 kb genomic bins belonging to various compartment‐score quantiles in cis (upper right triangle) and trans (lower left triangle).
- Transitions in euchromatin‐vs‐heterochromatin bias during the development of different lineages (cardiomyocyte differentiation (Zhang et al, 2019)) at 100 kb resolution. (left axis: violin plots) Distribution of compartment scores; (right axis: dots) ratio of A:B compartment bins.
- Enrichment of TAD–TAD interactions involved in max cliques (size ≥ 3) during the development of different lineages. A dispersal of active hubs was specifically observed during epigenetic reprogramming. Inter‐compartmental TAD–TAD interactions are under‐represented in all cases.
- Network representation of TAD cliques and their compartment identity during germ cell and cardiomyocyte differentiation.
Higher‐order genome organization: maturation toward a highly euchromatized state
We first examined the nuclear morphology of the five cell types (mESCs, EpiLCs, d2 mPGCLCs, d4c7 mPGCLCs, and GSCs) stained with DAPI (4′,6‐diamidino‐2‐phenylindole) using high‐resolution confocal microscopy. Counterintuitive to GSCs’ acquisition of a distinct spermatogenic epigenome, including global DNA re‐methylation, on the epigenome of naïve PGCs, the areas of high DAPI density (peri‐centromeric heterochromatin; Guenatri et al, 2004), the variances of DAPI density (chromatin condensation heterogeneity), and the distances of the DAPI‐dense areas from the nuclear periphery (chromosome radial positioning), all exhibited a monotonically decreasing transformation toward GSCs (Fig 1B and C). This indicates that chromatin de‐condensation (i.e., euchromatization), as well as peripheral tethering of centromeres, proceeds progressively beyond the canonical epigenetic reprogramming period. Notably, formative EpiLCs showed more discrete chromatin condensation than naïve mESCs, while mESCs and d4c7 mPGCLCs (latent pluripotency; Surani et al, 2007) exhibited significant differences in chromosome radial positioning (Fig 1B and C). Fluorescence in situ hybridization (FISH) confirmed that, in line with chromatin de‐condensation, GSCs bore larger chromosome volumes than mESCs and EpiLCs (Figs 1D and EV1A).
Figure EV1. Investigation of global nuclear architecture dynamics through Hi‐C and FISH.
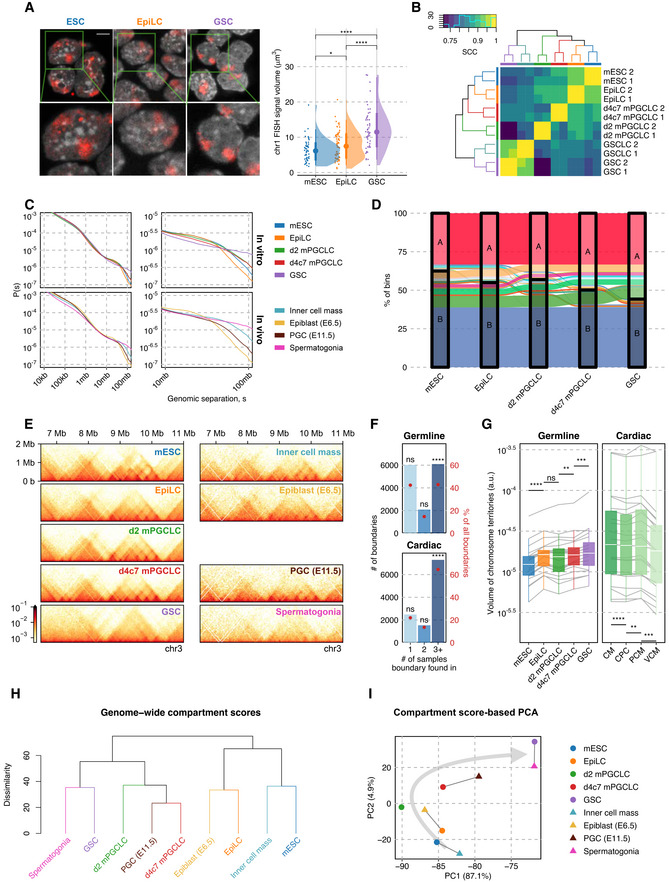
- Fluorescence in situ hybridization (FISH) against chromosome 1 (red) with DAPI counterstaining (gray). (Left) Z‐stacked representative images (top left) are paired with magnified views (bottom left). (Right) The distribution of “surface” volumes for chr1, as seen for chr16, validates chromosomal decondensation in GSCs. Number of cells = 51/68/53 for mESC/EpiLC/GSC. Wilcoxon rank‐sum test P‐values (left to right): 4.16e−2, 4.33e−6, 8.68e−9. P‐value symbol brackets: **** = [0, 0.0001); *** = [0.0001, 0.001]; ** = [0.001, 0.01); * = [0.01, 0.05); ns = [0.05, 1].
- Hierarchical clustering of stratum‐adjusted correlation coefficients (SCC) between samples validating the reproducibility of biological replicates.
- Contact probability decay across different inter‐loci separation distances for various cell types throughout in vivo and in vitro germ cell differentiation, demonstrating a gain of distal interactions along with differentiation, especially at distances > 50 Mb.
- Sankey diagram of compartment identities in 50 kb bins across cell types. Compartment A regions newly acquired by GSCs are formed through a unidirectional switch of B‐A with relatively little reversal.
- 25 kb‐resolution balanced contact maps spanning chr3:5–12.5 mb.
- Degree of TAD boundary conservation in different lineages. Consistent across different lineages, more than 40% TAD boundaries are significantly conserved across differentiation. One‐sided permutation tests were carried out by shuffling sample labels 100,000 times, with P‐values (left to right, top to bottom): 1, 1, 1e−5, 1, 1, 1e−5.
- Convex hull volumes of CSynth‐produced chromosome 3D models during the development of different lineages, after normalization to unit backbone length. n = 22/19 for cardiac/germline. Wilcoxon signed‐rank test P‐values (left to right): 1.91e−6, 1.89e−1, 1.69e−3, 2.61e−4, 4.77e−6, 1.86e−3, 2.93e−4.
- UHC based on Euclidean distance between 100 kb compartment score tracks for cell types from in vitro and in vivo germ cell differentiation, with comparable stages consistently grouped together.
- PCA of compartment scores at 100 kb resolution for various cell types throughout in vivo and in vitro germ cell differentiation, with comparable stages consistently grouped together.
We next analyzed the five cell types by in situ Hi‐C (~5 kb resolution) with reproducible biological replicates (Fig EV1B, Dataset EV1). Consistent with the morphological observations, 3D genome organization was transformed in a unidirectional manner during germ‐cell development: the chromosomal contact profile shifted progressively from the conventional proximal contact‐enriched state to a more uniform profile with heightened distal interactions (Figs 1E and EV1C, Appendix Fig S1A), and the compartment score distributions and euchromatin‐to‐heterochromatin balance exhibited a monotonical increase (Figs 1G and EV1D). Notably, while the vast majority (~33.3% genome‐wide) of the A compartment in mESCs remained an A compartment, more than one third (~38.9% genome‐wide) of the B compartment in mESCs progressively turned into A, with the largest B‐compartment fraction (~7.5% genome‐wide) turning into A upon the d4c7 mPGCLC‐to‐GSC transition. In stark contrast, the compartment scores exhibited a gradual decrease during somatic development, including neuronal, B‐cell, and cardiomyocyte differentiation (Fig 1G, Appendix Fig S1B; Bonev et al, 2017; Stadhouders et al, 2018; Zhang et al, 2019). The brief decrease in the compartment score upon EpiLCs‐to‐d2 mPGCLCs differentiation (Fig 1G) is consistent with the transient activation of a somatic program during mPGCLC specification (Kurimoto et al, 2015). Accordingly, principal component analysis (PCA) of the compartment scores segregated the germline from somatic development (Appendix Fig S1C). Along with the expansion of the A compartment (Figs 1G and EV1D), euchromatic A–A interactions became less intense, while the reduced B compartment exhibited stronger B–B interactions both within (cis) and between (trans) chromosomes, implying the formation of repressive condensates (Fig 1F).
On a smaller scale, topologically associating domain (TAD) boundaries exhibited a substantial overlap during germ‐cell development, with the degree of their conservation being similar to that of somatic lineages (Fig EV1E and F, Appendix Fig S1D). However, inter‐TAD interactions involving the simultaneous aggregations of multiple non‐neighboring TADs, referred to as “TAD‐cliques” (Paulsen et al, 2019), became dramatically less prevalent in the A compartments, while they were over‐represented in the B compartments in both d4c7 mPGCLCs and GSCs, which was in stark contrast to their opposite/relatively stable behaviors in somatic lineages (Fig 1H and I, Appendix Fig S1E). Through polymer simulations, we generated representative 3D structures of whole chromosomes (Todd et al, 2021), which similarly demonstrated the progressive expansion of chromosome volume during germ‐cell development (Fig EV1G, Appendix Fig S1F, Movie EV1).
To examine whether the five cell types recapitulate their in vivo counterparts at the 3D genome organization level, we retrieved published Hi‐C data of the inner cell mass at ~E4.0, epiblast at E6.5, PGCs at E11.5, and spermatogonia in adults, which were generated from small numbers of samples (Du et al, 2017, 2020; Luo et al, 2020). Remarkably, not only at the transcriptomic and epigenomic level that we reported previously (Hayashi et al, 2011; Ishikura et al, 2016; Ohta et al, 2017), the in vitro cell types exhibited a strong concordance with their in vivo counterparts at the 3D genome organization level (Fig EV1C and E) (despite the elevated noise of contact matrices from in vivo samples), with unsupervised hierarchical clustering (UHC) and PCA using compartment scores consistently placing corresponding cell types next to one another (Fig EV1H and I). Thus, the in vitro system faithfully captures the nucleome dynamics of in vivo germ‐cell development, further empowering our strategy for using scalable in vitro materials to delineate a more complete picture of nucleome dynamics during germ‐cell development. We conclude that, beyond the canonical epigenetic reprogramming period, higher‐order genome organization undergoes a continuous maturation and culminates in a largely euchromatic genome and peripherally positioned centromeres in spermatogonia/SSCs (GSCs). Thus, global DNA methylation and euchromatization are separable events. Moreover, our findings revealed that, despite their profound epigenomic differences, PGCs (d4c7 mPGCLCs) with both oogenic and spermatogenic potential and spermatogonia/SSCs (GSCs) show relatively similar higher‐order genome organization.
Epigenome profiling: epigenetic reprogramming for highly open chromatin with enhanced insulation
To explore the mechanism underlying the higher‐order genome organization unique to the germ line, we conducted comprehensive epigenome profiling of the five cell types. We performed mass spectrometry (MS) of histones; chromatin immunoprecipitation followed by deep sequencing (ChIP‐seq) of 13 different targets, including 9 histone modifications; assay for transposase‐accessible chromatin with deep sequencing (ATAC‐seq) for open chromatin; and native elongating transcript–cap analysis of gene expression (NET‐CAGE) for transcribed cis‐regulatory elements (Dataset EV1). For some assays, we analyzed d4 mPGCLCs, which are in the middle of epigenetic reprogramming, as an intermediate between d2 and d4c7 mPGCLCs and mouse embryonic fibroblasts (MEFs) as a somatic control.
Mass spectrometry revealed dynamic changes in histone‐modification levels with high reproducibility (Fig 2A, Dataset EV2). Consistent with previous observations (Kurimoto et al, 2015; Ohta et al, 2017), histone H3 lysine 9 di‐methylation (H3K9me2) was substantially reduced and H3K27 tri‐methylation (H3K27me3) was strongly up‐regulated in d4c7 mPGCLCs (Figs 2A and EV2A and B). With respect to active modifications, H3K27 acetylation (H3K27ac: active cis‐regulatory elements) and H3K18ac were the most abundant in EpiLCs, whereas H3K4 mono‐methylation (H3K4me1: poised enhancers), H3K14ac, and H3K23ac were the most abundant in d4c7 mPGCLCs, and, interestingly, H3K4me3 (promoters) was the least prevalent in d4c7 mPGCLCs (Fig 2A). UHC based on H3‐modification abundance segregated each cell type with their unique sets of associated H3 modifications (Fig 2B), and PCA demonstrated characteristic transitions of epigenetic properties, with the transition from d2 to d4c7 mPGCLCs representing the epigenetic reprogramming to latent pluripotency and the transition from d4c7 mPGCLCs to GSCs signifying the acquisition of a spermatogenetic epigenome (Fig 2C). We proceeded to normalize all histone modification ChIP‐seq signals with MS‐based scaling factors for subsequent analyses (Fig EV2C and D; Farhangdoost et al, 2021).
Figure 2. Epigenome profiles and CTCF insulation.
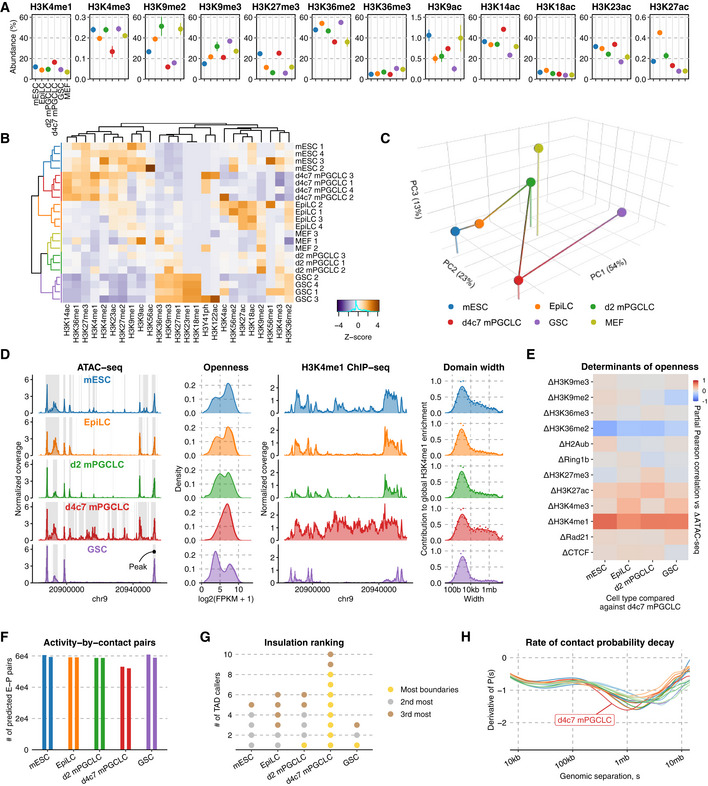
- Relative abundance (%) of key histone modifications as measured by mass spectrometry. The point marks the mean while error bars indicate standard errors. Three biological replicates in each cell type were analyzed.
- UHC of H3 modification abundances. Numeric suffixes indicate biological replicates.
- PCA of average H3 modifications abundances in each cell type.
- Chromatin accessibility landscape throughout germline development. (left) ATAC‐seq coverage tracks at a representative locus, with peaks highlighted; (second left) distribution of read counts per each in the union peak set; (second right) H3K4me1 ChIP‐seq coverage tracks at the same locus; (right) Distribution of domain widths for H3K4me1‐enriched regions based on cross‐correlation, as implemented in MCORE.
- Partial Pearson correlation matrix for inter‐cell type ATAC‐seq differences against d4c7 mPGCLCs versus differences in other epigenetic signals.
- Number of E‐P pairs with ABC score > 0.02 (Fulco et al, 2019). Two biological replicates in each cell type were analyzed.
- Cell type insulation ranking. 10 different TAD‐calling algorithms were used to determine the cell types' rank in terms of insulation (gold: most insulated; silver: 2nd most insulated; bronze: 3rd most insulated).
Figure EV2. Quantitative epigenome analysis by mass spectrometry and chromatin accessibility analysis by ATAC‐seq.
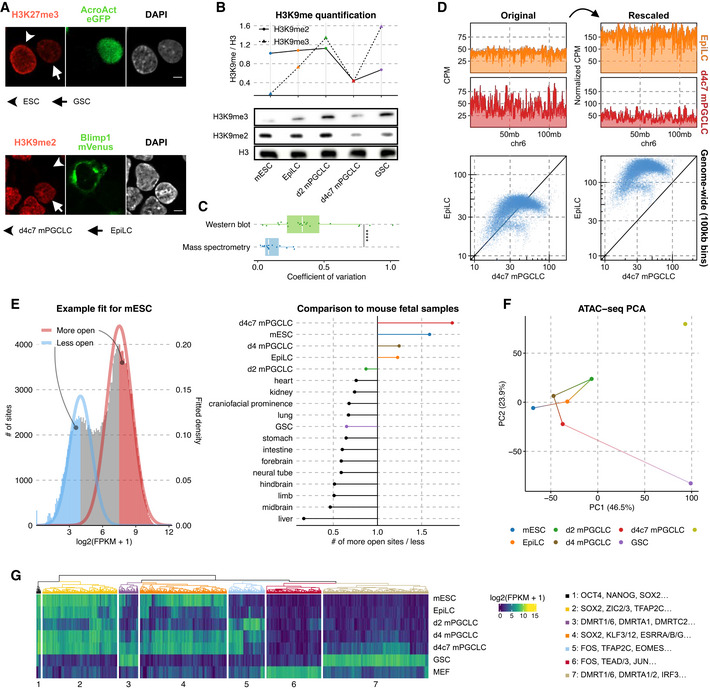
- (Top) Immunofluorescence against H3K27me3 in mESCs and GSCs; the shaftless arrow marks a GFP+ GSCs and the shaftless arrowhead indicates mESCs. (Bottom) Immunofluorescence against H3K9me2 in EpiLCs and d4c7 mPGCLCs; the shaftless arrowhead marks a Blimp1‐mVenus+ d4c7 mPGCLCs and the arrow indicates EpiLCs. Scale bars = 10 μm.
- Western blot against H3K9me3, H3K9me2, and histone H3 in each cell type (bottom) and H3‐normalized quantification (top).
- Coefficients of variation across replicates of histone modification abundance as measured by quantitative histone mass spectrometry versus western blot for H3K9me2, H3K9me3, and H3K27me3. Mass spectrometry measurements consistently exhibit higher reproducibility. Number of biological replicates = 15/21 for mass spectrometry/western blot. Wilcoxon rank‐sum test P‐value: 5.34e−5. P‐value symbol brackets: **** = [0, 0.0001).
- Schematic of normalizing histone modification ChIP‐seq via mass spectrometry‐derived coefficients. With only depth‐normalization (left), EpiLCs and d4c7 mPGCLCs appear to have comparable H3K9me2 profiles both in terms of coverage tracks (top) and in a pairwise scatter plot comparing the two cell types (bottom); after the multiplication of their relative abundances based on mass spectrometry, the comparatively lower levels of H3K9me2 in d4C7GCLCs become apparent (right).
- Comparison of regions with greater (“more open”) and reduced (“less open”) accessibility in the union peak set of germline samples and E14.5 mouse fetal tissues (Gorkin et al, 2020) (left). Through fitting two‐component gaussian mixture models, d4c7 mPGCLCs stand out as possessing the most permissive genome (right).
- PCA of ATAC‐seq signals in the top 10,000 most variable peaks from the union peak set including MEFs (Di Giammartino et al, 2019).
- UHC of the top 2,000 most variable ATAC‐seq peaks in the union peak set including MEFs. (left) Clustered ATAC‐seq enrichment heatmap; (right) overrepresented TF‐binding motifs in each cluster.
We first scrutinized the open‐chromatin landscape. Consistent with d4c7 mPGCLCs being globally DNA demethylated (~5%) (Fig 1A; Ohta et al, 2017, 2021), they exhibited pervasively open chromatin with coincident up‐regulation of H3K4me1, bearing large open domains in a genome‐wide manner (Fig 2D and E). Indeed, among a diverse panel of mouse fetal tissues (Gorkin et al, 2020), d4c7 mPGCLCs showed the highest degree of openness (Fig EV2E). Consistent with the analysis of the abundance of H3 modifications (Fig 2C), PCA with the most variable open sites (Fig EV2F) and UHC revealed that d4c7 mPGCLCs share open sites for pluripotency with mESCs and those for germ‐cell identity with GSCs: the former (clusters 1, 2, 4) being enriched in transcription‐factor (TF)‐binding sites for POU5F1, NANOG, SOX2, ZIC2/3, and KLF3/12, and the latter (clusters 3 and 7) in those for DMRTs (Fig EV2G, Dataset EV3).
Despite their genome‐wide DNA demethylation, PGCs and d4c7 mPGCLCs do not exhibit transcriptional hyperactivity or promiscuousness (Seisenberger et al, 2012; Ohta et al, 2017, 2021). To explore higher‐order regulatory mechanisms, we identified enhancer‐promoter (E‐P) pairs using the activity‐by‐contact model by integrating ATAC‐seq, H3K27ac, and Hi‐C data (Fig EV3A; Fulco et al, 2019). Notably, d4c7 mPGCLCs showed a reduced number and range of active E‐P pairs as compared to the other cell types (Figs 2F and EV3B). Furthermore, NET‐CAGE revealed an under‐representation of E‐P co‐transcription in d4c7 mPGCLCs (Fig EV3C). d4c7 mPGCLCs were also predicted to bear the largest numbers of insulating TAD boundaries (Figs 2G and EV3D), showed the smallest genomic separation (Fig 2H) and exhibited the broadest compartment profile (Fig EV3E), in agreement with the notion that heightened insulation can mask smaller compartments (Schwarzer et al, 2017). While CTCF and RAD21, a key component of cohesin, exhibited comparable enrichment at TAD boundaries across the five cell types (Fig EV3F) (we discuss the CTCF depletion in GSCs below), ATAC‐seq revealed that d4c7 mPGCLCs uniquely exhibited lower chromatin information content around regions with co‐localized CTCF/RAD21 bindings (Fig EV3G), suggesting that d4c7 mPGCLCs bore a shorter CTCF/RAD21 residence time (D'Oliveira Albanus et al, 2021). Taken together, these findings support the idea that, due to a reduced residence time of the loop extrusion machinery with no major changes in global binding sites, d4c7 mPGCLCs bear shorter chromatin loops and enhanced insulation (Fig EV3H and I). Additionally, E13.5 male PGCs in vivo also demonstrate similarly enhanced insulation (Fig EV3J and K). We conclude that PGCs with a naïve epigenome bear highly open chromatin, but undergo enhanced insulation to ensure their transcriptional integrity.
Figure EV3. Exploration of the cis‐regulatory element by NET‐CAGE combined with Hi‐C and comparison against public Hi‐C datasets.

- An example of enhancer‐promoter interactions for Nanog in mESCs as predicted by ABC, all of which correspond to known associations including super‐enhancers.
- (Top) Distribution of distances separating ABC‐predicted enhancer‐promoter pairs in each replicate. The central band of boxplots indicates median values, while the lower and upper hinge corresponds to the first and third quartile, and the upper whiskers extend to the largest value % 1.5 * IQR and vice versa for the lower whiskers. Notches correspond to 1.58 * interquartile range of distances / (# of E‐P pairs)1/2, comparable to 95% confidence intervals around the median. d4c7 mPGCLCs’ E‐P pairs are significantly shorter in range than those of other cell types. (Bottom) Magnified view from 60 kb to 100 kb. Number of ABC E‐P pairs from left to right: 60,535, 59,312, 59,116, 59,075, 58,702, 58,704, 53,092, 52,074, 60,867, 58,858.
- (Top) Co‐transcription of enhancer‐promoter pairs with correlated NET‐CAGE expression. The observed number of correlated E‐P pairs involving tag clusters transcribed (TPM > 1) in a given cell type (points) are compared against a permuted background in which tag clusters are sampled from the union tag cluster set. (Bottom) Observed/expected number of E‐P pairs with correlated NET‐CAGE expression and co‐expressed (> 1 TPM) in a given cell type. Two‐sided permutation tests were carried out by sampling 100,000 times from the set of elements expressed in at least 1 cell type, with p‐values (left to right): 2e−5, 2e−5, 2e−5, 2e−5, 2e−5, 2e−5, 7.64e−2, 6.44e−3. P‐value symbol brackets: **** = [0, 0.0001); ** = [0.001, 0.01); ns = [0.05, 1]. Two biological replicates in each cell type were analyzed.
- Number of TAD boundaries in each cell type across 10 different algorithms. Dots correspond to values produced by a specific algorithm for a given cell type and are grouped into lines by the algorithm.
- Auto‐correlation of compartment scores (25 kb bins), with a slower decay indicative of broader compartments.
- Aggregate plots of S3V2‐normalized ChIP‐seq profiles for CTCF and Rad21 around the union set of TAD boundaries.
- Mean f‐VICE across replicates (error bars indicate standard errors) for CTCF motifs overlapping both Rad21 and CTCF peaks within the union set of TAD boundaries. Two biological replicates per cell type were analyzed.
- Representative locus demonstrating the emergence of smaller insulated domains in d4c7 mPGCLCs within otherwise homogeneous wider TADs observed in earlier stages.
- Proposed mechanism for elevated insulation via the reduction of loop extrusion factor's residence time, leading to shorter loops and domains.
- (Top) Slope of contact decay (P(s)) curves as a function of genomic separation in log–log space for in vivo germline development (Du et al, 2017, 2020); (bottom) genomic separation with the most negative second derivative of P(s) in log‐log space, corresponding to the distance of fastest decline in contact frequency.
- Genomic separation with the fastest decline in contact frequency for cell types across in vivo and in vitro germ cell differentiation.
Insulation erasure for spermatogonia development and oogenesis
We next classified ATAC‐seq peaks (open sites) based on their combinatorial epigenetic states. Building on the Ensembl Regulatory Build and ENCODE’s registry of candidate Cis‐Regulatory Elements (cCREs), we applied uniform manifold approximation and projection (UMAP) in combination with hierarchical density‐based spatial clustering of applications with noise (HDBSCAN) in a semi‐supervised manner through iterative sub‐clustering (Tables EV4 and EV5). This framework classified the open sites into 19 distinct sets (Fig 3A), which we grouped into 6 broader categories (Figs 3B and EV4A). While d4c7 mPGCLCs showed the largest number of enhancer elements (clusters 5, 6, 15, and 18) (Fig 3B and C), GSCs exhibited a relatively large number (~> 10,000) of non‐promoter bivalent open sites (clusters 8, 9, 10, and 13). Additionally, we uncovered a set of open sites with unique trivalency of H3K4me3, H3K27ac, and H3K9me3 that were enriched in EpiLCs (cluster 19; Fig 3B) and overlapped not only with the promoter of long interspersed nuclear elements 1 (LINE1) but also with the binding site of YY1 (Fig EV4B, Dataset EV5), underscoring the capacity of our epigenetic compendium for uncovering biologically distinct regulatory regions. A vast majority of enhancers were cell‐type specific, whereas most CTCF bindings were conserved upon each cell‐fate transition until d4c7 mPGCLCs; strikingly, however, a majority of CTCF‐bound sites in d4c7 mPGCLCs were lost in GSCs (Fig 3C) (see below).
Figure 3. Open‐site characterizations and CTCF release.
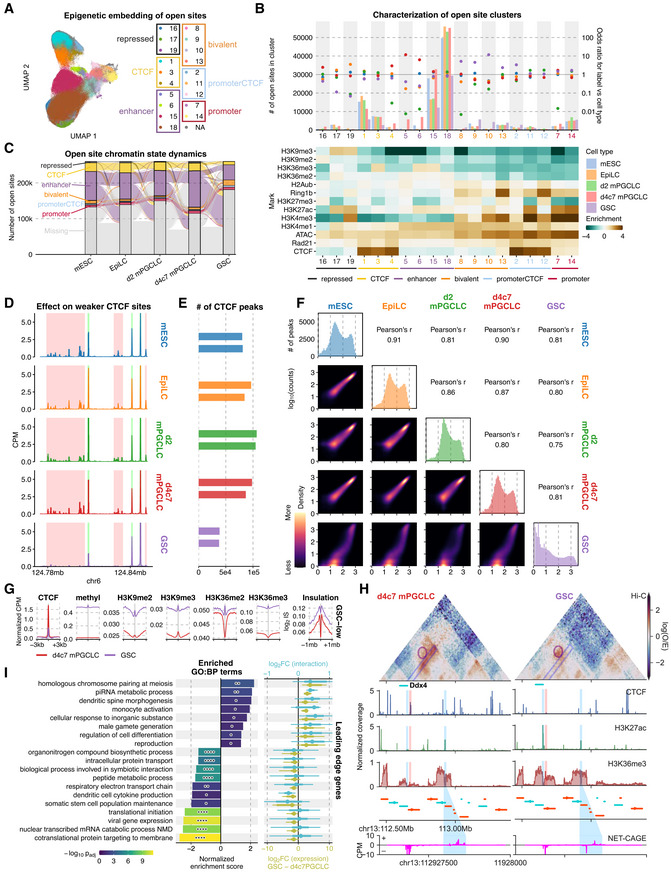
- 2D UMAP embedding based on epigenetic signals in ATAC‐seq peaks for each cell type, with labels derived from semi‐supervised HDBSCAN.
- Association between open‐site clusters and cell types. (top) Number of open sites per cell type in each cluster (left axis: bars) and their enrichment as odds ratios (right axis: dots); (bottom) enrichment of epigenetic signals in each cluster.
- Dynamics of open site classes. Classification of the same open sites peak is compared between adjacent stages and shown as flows. Open sites that could not be reliably clustered or were not called peaks are labeled as “Missing.”
- ChIP‐seq coverage tracks CTCF in each cell type.
- Number of CTCF peaks called in each cell type. GSCs have considerably fewer CTCF peaks. Two biological replicates in each cell type were analyzed.
- Correlograms of CTCF binding in the union peak set. (Upper right panels) Pearson’s correlation coefficients between log2 transformed signals. (Diagonal) Histograms of CTCF signal intensity in the union peak set. (Lower left panels) 2D density plots of CTCF binding in pairs of cell types.
- Aggregate plots of ChIP‐seq enrichment for various targets and insulation score (IS) around CTCF‐binding sites depleted in GSCs as compared to d4c7 mPGCLCs. n = 39,408.
- 3D epigenetic landscape re‐wiring near Ddx4. Observed/expected contact maps at 10 kb resolution for d4c7 mPGCLCs and GSCs are shown alongside select ChIP‐seq and NET‐CAGE coverage tracks. A strong insulating CTCF peak (highlighted in red) upstream of the Ddx4 TSS (upstream blue highlight) is lost in GSCs, facilitating the interaction between the Ddx4 promoter and an active enhancer (downstream blue highlight) demonstrating pronounced bidirectional nascent transcription (bottom).
- GSEA using genes ranked by concomitant differential expression and promoter interaction. (left) ABC‐defined E‐P pairs overlapping GSC‐depleted CTCF peaks are used to rank genes based on coordinated E‐P interaction and expression differences; (right) log2 fold changes for leading‐edge genes of enriched gene sets. Significances computed using pre‐ranked multilevel GSEA, P‐values from top to bottom: 0.00106, 0.00343, 0.0173, 0.0269, 0.0382, 0.0439, 0.0109, 0.0439, 5.19e−6, 6.26e−5, 1.79e−6, 1.91e−6, 0.00931, 0.0454, 0.0125, 6.24e−10, 2.17e−11, 6.24e−10, 6.18e−12. P‐value symbol brackets: **** = [0, 0.0001); ** = [0.001, 0.01); * = [0.01, 0.05); ns = [0.05, 1].
Figure EV4. Open site chromatin state dynamics and differential CTCF binding throughout germ cell differentiation.

- Overlap enrichment analysis of consolidated open site clusters against annotations from the Ensembl Regulatory build. P‐values were computed using Fisher’s exact tests. P‐value symbol brackets: **** = [0, 0.0001); ** = [0.001, 0.01); * = [0.01, 0.05); ns = [0.05, 1].
- Select ChIP‐seq coverage tracks around a representative cluster 2 loci.
- Western blot against CTCF in the chromatin‐bound fraction (top row) and whole‐cell lysate (middle row) as well as α‐tubulin (bottom row) in each cell type. The signals of CTCF from whole‐cell lysates were normalized by α‐Tubulin, while those of the chromatin‐bound fraction was normalized by the mean across all cell types (top panel).
- 2D UMAP embedding based on epigenetic signals in promoters for each cell type, with labels derived from semi‐supervised HDBSCAN.
- Enrichment of epigenetic signals in each promoter cluster and expression of the cognate gene.
- Association between promoter clusters and cell types. Number of open sites per cell type in each cluster (top axis: bars) and their enrichment as odds ratios (bottom axis: dots). Error bars indicate 95% confidence intervals.
- Pile‐up plots of intra‐class promoter‐promoter interactions.
- Contributors of differential CTCF binding. The aggregate plot of various ChIP‐seq enrichment signals (left) as well as the insulation score (right) near CTCF‐binding sites found both in cell types (“constitutive”) or only GSCs but not in d4c7 mPGCLCs (“GSC‐high”) appear largely identical in their chromatin state yet distinct from those lost in GSCs. n = 35,692/13,364 for constitutive/GSC‐high peaks.
- 3D epigenetic landscape rewiring near Ddx4. Observed/expected contact maps at 10 kb resolution for mESCs, EpiLCs, and d2 mPGCLCs are shown alongside select ChIP‐seq coverage tracks. A strongly insulating CTCF peak (highlighted in red) upstream of Ddx4’s TSS is found in all earlier stages and prevents spurious activation.
- Coordinated differential expression and E‐P looping between d4c7 mPGCLCs and GSCs. A strong correlation was observed when applying stratified rank‐rank hypergeometric overlap to genes ranked by differential expression versus differential E‐P interactions straddling sites depleted of CTCF binding in GSCs. While increased E‐P looping is correlated with elevated expression regardless of whether the interaction spans differential CTCF‐bound sites, the degree of coordination is stronger (i.e., more significant/brighter) for those that do straddle GSC‐depleted sites.
We performed the same analyses for promoters (Fig EV4D–F, Dataset EV4). In accord with our previous finding (Kurimoto et al, 2015), EpiLCs bore the largest number of bivalent promoters (Fig EV4F). Evaluation of the promoter–promoter (P–P) interactions revealed that active as well as bivalent promoters exhibited significantly enriched interactions in all cell types, but to lesser extents in d4c7 mPGCLCs bearing elevated insulation (Figs 2G and EV4G).
We next explored the depletion of CTCF binding upon d4c7 mPGCLCs‐to‐GSCs transition (Fig 3C). In GSCs, decreased CTCF protein expression accompanied a dramatic reduction in the number of CTCF peaks (Fig 3D–F). In particular, CTCF was depleted from relatively weak binding sites (Fig 3E and F). These CTCF‐depleted sites exhibited elevated DNA methylation as well as enrichment of H3K9me2/me3 and H3K36me2/me3, whereas CTCF peaks enriched in GSCs showed divergent patterns (Figs 3G and EV4H). Importantly, despite relatively weak bindings, CTCF depletion from such sites resulted in a reduction in insulation (Fig 3G), leading to a rewiring of neighboring cis‐regulatory interactions as exemplified for Ddx4, a key gene up‐regulated upon d4c7 mPGCLCs‐to‐GSCs transition, whose promoter strengthened its long‐range interaction with a distal enhancer (Figs 3H and EV4I). We then systematically identified E‐P pairs straddling CTCF sites depleted in GSCs and ranked the target genes according to coordinated expression up‐regulation and increased E–P interactions (Fig EV4J). Genes with coordinated activation were enriched in gene ontology (GO) functional terms such as “homologous chromosome pairing at meiosis,” and “piRNA metabolic process,” and included Ddx4, Mael, Piwil2, Piwil4, Zbtb16, Sycp1, Syce3, Mei4, and Prdm9 (Fig 3I, Dataset EV6) [these genes are referred to as “germline genes” (Borgel et al, 2010); also, see below], indicating a critical role of the insulation erasure in spermatogonia development and the acquisition of meiotic competence.
To explore whether insulation erasure may also occur upon oogenesis, we re‐analyzed published Hi‐C data for E11.5 PGCs (d4c7 mPGCLC counterparts) and E13.5 germ cells initiating their male or female differentiation (Du et al, 2017, 2020). We found that a majority of E13.5 male germ cells were still in the mitotic phase and bear similar properties to E11.5 PGCs, whereas most E13.5 female germ cells were in the leptotene stage of the meiotic prophase (Western et al, 2008; Nagaoka et al, 2020). Consistent with our comprehensive analyses (Figs 2 and 3), the point of fastest decline in the chromosomal cis‐contact decay rate, an index for TAD width (preprint: Polovnikov et al, 2022), occurred at the smallest genomic separation in E11.5 PGCs and d4c7 mPGCLCs (Figs 2H and EV3J and K), suggesting that, similar to d4c7 mPGCLCs, E11.5 PGCs bear enhanced insulation. Notably, while the fastest point of decline of E13.5 male germ cells was in a range comparable to E11.5 PGCs and d4c7 mPGCLCs, that in E13.5 female germ cells occurred at a much longer distance, suggesting a rapid weakening of insulation upon the initiation of oogenesis. We conclude that insulation erasure occurs both for spermatogonia development and oogenesis, with the latter having an earlier onset.
Mechanism for euchromatization: dynamics of LADs, pericentromeric heterochromatin, and H3K9 methylation
We next explored potential mechanisms for progressive euchromatization unique to germ‐cell development (Fig 1G). While the five cell types exhibited relatively conserved correlations between their compartment scores and epigenetic modification profiles, there nevertheless existed cell‐type‐specific variations (Fig 4A). We noted that the binding profiles of lamin B1, which forms the nuclear lamina and tethers chromosomes to create lamina‐associated domains (LADs; Guelen et al, 2008), were the strongest predictor for compartment‐score differences between mESCs and GSCs (Fig 4B), and the LADs changed dramatically with a sweeping reduction across regions that undergo euchromatization in GSCs (Fig 4C). Consequently, among a number of other cell types (Peric‐Hupkes et al, 2010; Robson et al, 2016; Poleshko et al, 2017; Yattah et al, 2020), GSCs bore the smallest genomic coverage of LADs (~10%) (Fig 4D), a vast majority of which were a subset of constitutive LADs found across all other cell types (Fig 4E and F). Indeed, GSCs exhibited low lamin B1 levels and enrichments (Fig 4G and H). Thus, GSCs constitute a cell type with minimal LADs.
Figure 4. Generation of minimal LADs.

- Correlation between compartment score and ChIP‐seq enrichment at 50 kb resolution.
- Correlation between differential compartment score and differential ChIP‐seq enrichment between mESCs and GSCs at 50 kb resolution.
- Representative chromosome‐wide distributions of compartment score and lamin B1 enrichment for mESCs and GSCs.
- IF analysis for lamin B1 in (left) EpiLCs and d4c7 mPGCLCs, as well as (right) EpiLCs and GSCs. Symbols for each cell type are as indicated. Scale bars, 10 μm.
- Western blot for lamin B1 in different cell types (bottom) and quantification normalized by β‐actin (top).
- Average distributions of lamin B1 enrichment across all chromosomes (1–19, X). Ribbons correspond to 95% confidence intervals of fitted GAMs.
- Lamin B1 ChIP‐seq enrichment in the first (left/p‐ter) and the last (right/q‐ter) 300 kb of each chromosome. The point marks the median while the thick and thin lines correspond to 66% and 95% intervals, respectively. Number of chromosomes = 20 (autosomes and chromosome X).
- Representative chromosome‐wide distributions of ChIP‐seq enrichment for lamin B1 and H3K9me3/me2.
- (top) Representative images of FISH against major satellite repeats in EpiLCs and GSCs. Scale bars, 10 μm; (bottom) percentage of the pericentromeres detached from the nuclear lamina in EpiLCs and GSCs. The point marks the median while the thick and thin lines correspond to 66% and 95% intervals, respectively. Number of cells = 18/22 for EpiLC/GSC.
While LADs were prominent toward the distal ends of long arms in mESCs and EpiLCs, they became more uniformly distributed in d2/d4c7 mPGCLCs with a reduction in their coverage in d4c7 mPGCLCs, and they eventually become depleted around the distal ends of long (q) arms in GSCs, where they were only retained toward the opposing (p/short) end, that is, around centromeres of the telocentric mouse chromosomes (Fig 4C, I and J). This is consistent with the progression of nuclear peripheral association of DAPI‐dense areas along with germ‐cell development (Fig 1B and C). Accordingly, DNA FISH for major satellite repeats, a pericentromere marker, revealed that while such regions were localized mainly within the nuclear interior in EpiLCs, they were predominantly positioned around the nuclear periphery in GSCs (Fig 4L).
To explore whether the peripherally positioned centromeres and extensive euchromatization in other chromosomal regions in GSCs are a conserved feature in mouse spermatogonia in vivo and in other mammals such as primates, we re‐analyzed relevant published datasets (Wang et al, 2019; Du et al, 2020). The distributions of chromosome‐wide compartment‐score differences between GSCs and EpiLCs were very similar to those between spermatogonia and fibroblasts in both mice and rhesus monkeys, with spermatogonia showing the lowest compartment score around centromeres and widespread euchromatization across other regions (note that rhesus monkeys bear metacentric chromosomes; Fig EV5A and B). We conclude that higher‐order genome organization in GSCs is conserved in spermatogonia in vivo and, through evolution, in monkeys.
Figure EV5. Inter‐species comparison of germ‐cell specific chromatin structure and characterization of H3K9me3‐enriched repeats.
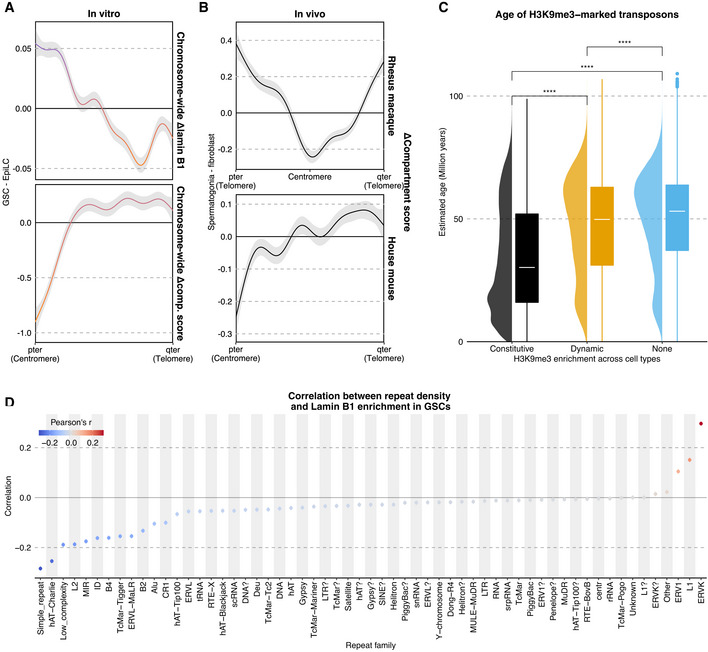
- Average distributions of differential (GSC—EpiLC) lamin B1 enrichment (top) or compartment score (bottom) across all chromosomes (1–19, X). Ribbons correspond to 95% confidence intervals of fitted GAMs.
- Average distributions of compartment score (spermatogonia—fibroblast) across all chromosomes (excluding Y) for Macaca mulatta (top) and Mus musculus (bottom).
- Estimated age of families overlapping H3K9me3 domains based on age = divergence/substitution rate with 4.5 × 10−9 as the rate and milliDiv from RepeatMasker as the divergence (Bourque et al, 2008). Wilcoxon rank‐sum tests p‐values, from left to right: 0, 0, 0. P‐value symbol brackets: **** = [0, 0.0001). Number of TE instances, from left to right: 227,732, 982,369, 2,671,107.
- Correlation between lamin B1 enrichment and density for different repeat families.
As a mechanism that gives rise to the minimal LADs, we noted significant changes in the abundance and distributions of H3K9me2/me3, hallmarks of chromatin anchored to the nuclear lamina (Bian et al, 2013; Chen et al, 2014; Harr et al, 2015). The abundance of both H3K9me2/me3 increased progressively from mESCs to d2 mPGCLCs, and then decreased dramatically in d4c7 mPGCLCs (Fig 2A). While the low abundance of H3K9me2 persisted in GSCs, the abundance of H3K9me3 increased in GSCs to the highest level among the five cell types (Fig 2A). The distributions of H3K9me2 were widespread across the chromosomes and well conserved among the five cell types except in d4c7 mPGCLCs, which, unlike the other cell types, retained H3K9me2 at a relatively high level around the pericentromeres (Fig 4K). On the other hand, in all cell types, H3K9me3 showed a unique and conserved distribution with a characteristic enrichment around the pericentromeres, with GSCs bearing broader/expanded H3K9me3 domains that bridge several peaks present in other cell types (Figs 4K and 5A and B). Notably, consistent with the increased B–B interactions, the broad H3K9me3 domains in GSCs exhibited elevated intra‐ as well as inter‐domain aggregations (Fig 5C).
Figure 5. Heterochromatin re‐organization.
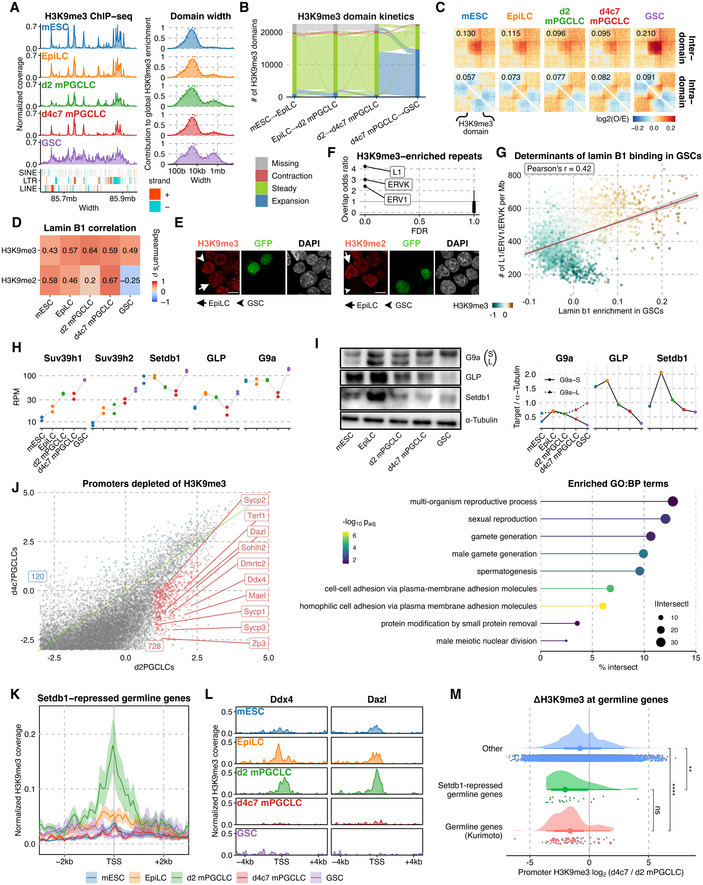
- (left) H3K9me3 ChIP‐seq tracks, with TEs in different classes shown below; (right) Distribution of domain widths for H3K9me3‐enriched regions based on cross‐correlation, as implemented in MCORE.
- Spatial‐temporal dynamics of H3K9me3 domains (> 10 kb) analyzed using ChromTime.
- (Top) Enrichment of interaction between (upper) and within (lower) broad H3K9me3 domains (> 50 kb; identified in GSCs and overlap peaks in all other cell types).
- Correlation between H3K9me2/3 and lamin B1 ChIP‐seq enrichment.
- IF analysis for H3K9me3 (left) and H3K9me2 (right) in EpiLCs and GSCs. Arrowheads: GFP+ GSCs; arrows: EpiLCs. Scale bars, 10 μm.
- Odds ratio and significance of the overlap between H3K9me3 domains conserved across all cell types and different repeat families. Error bars denote 95% confidence intervals.
- Scatter plot of lamin B1 enrichment in GSCs vs the aggregated density of select TEs (L1, ERV1, and ERVK) in 1 mb bins, with points colored by H3K9me3 enrichment in GSCs.
- (left) Western blot for G9a, GLP, Setdb1, and α‐tubulin; (right) quantification normalized by α‐Tubulin.
- (left) Scatter plot of H3K9me3 enrichment across all promoters in d2 mPGCLCs and d4c7 mPGCLCs, with 728 genes (red) showing substantially higher H3K9me3 levels in d2 mPGCLCs than d4c7 mPGCLCs; (right) pathway enrichment of the 728 genes using g:Profiler.
- Aggregate plot of H3K9me3 around the TSSs of Setdb1‐repressed germline genes (Karimi et al, 2011). The thick line marks the mean while the upper and lower limits indicate standard errors.
- Normalized H3K9me3 tracks around the TSSs of Dazl and Ddx4.
- Distribution of differences in promoter H3K9me3 between d2 and d4c7 mPGCLCs for germline genes (Kurimoto et al, 2015), Setdb1‐repressed germline genes (Karimi et al, 2011), and other genes. The point marks the median while the thick and thin lines correspond to 66% and 95% intervals, respectively. From top to bottom, Number of genes = 19,559, 21, 99. Significances are computed using Wilcoxon rank‐sum tests, P‐values from top to bottom: 2.36e−3, 1.14e−9, 4.43e−1. P‐value symbol brackets: **** = [0, 0.0001); ** = [0.001, 0.01); ns = [0.05, 1].
Lamina‐associated domains consistently showed positive correlations with both H3K9me2/me3, except in GSCs, which had minimal LADs showing a positive correlation only with H3K9me3 (Fig 5D). IF analysis verified that GSCs showed a nuclear peripheral enrichment of H3K9me3 but not me2, while EpiLCs bore peripheral H3K9me2 but not me3 enrichment (Fig 5E). Interestingly, regions constitutively enriched with H3K9me3 across all five cell types, that is, putative nucleation sites for H3K9me3 expansion in GSCs, were enriched with evolutionarily young transposable elements (TEs) including ERVK, ERV1, and LINE1 (Figs 5F and EV5C, Dataset EV5). Accordingly, the densities of these TEs were highly predictive of the minimal LADs in GSCs (Figs 5G and EV5D). Thus, minimal LADs in GSCs are the regions that show consistent attachment to the nuclear lamina across all cell types, likely contributing to the continued repression of evolutionarily young TEs and the maintenance of genome fidelity. Collectively, these results indicate that, during germ‐cell development, LADs progressively remodel toward a minimal state, positionally shifting from the distal ends of long arms predominantly associated with H3K9me2 to the opposite ends of the chromosomes, the centromeres. These pericentromeric regions, with newfound peripheral attachment in GSCs, are predominantly associated with H3K9me3 and are populated with evolutionarily young TEs, enabling extensive euchromatization on the opposing chromosome arm (long/q arm).
Next, to gain insights into the mechanisms underlying H3K9 methylome dynamics, we examined the expression of major H3K9 methyltransferases (K9MTases). At the transcriptional level, Suv39h1 and h2, which are responsible for the H3K9 methylation in the peri‐centromeric heterochromatin and other B compartment regions (Fukuda et al, 2021), showed progressive up‐regulation, whereas Setdb1, Ehmt1 (Glp1), and Ehmt2 (G9a), which are involved in the H3K9 methylation in both A and B compartments (Fukuda et al, 2021), were gradually repressed until d4c7 mPGCLCs and then up‐regulated in GSCs (Fig 5H). At the protein level, SETDB1, EHMT1, and EHMT2 were repressed until d4c7 mPGCLCs and remained at a low level in GSCs as well (we were not able to determine the SUV39H1/H2 levels due to the lack of appropriate antibodies; Fig 5I). These findings are consistent with the dynamics of the H3K9me2/me3 levels and distributions, suggesting that the H3K9 methylome is regulated at least in part by the differential expression of K9MTases.
Additionally, we explored the impact of the global remodeling of H3K9me3 on gene expression. In particular, we noted that during the d2‐to‐d4c7 mPGCLC transition, 728 promoters showed H3K9me3 down‐regulation (Fig 5J), and they were enriched with GO terms such as “multi‐organism reproductive process,” “sexual reproduction,” and “gamete generation,” and included Dazl, Ddx4, Sycp1, Sohlh2, and Mael (Fig 5J, Appendix Table S6). These genes, which included many subject to insulation erasure upon spermatogonia development (Fig 3I and J), are referred to as “germline genes” (Borgel et al, 2010), and are known to be repressed by DNA methylation in somatic cells and by H3K27me3 and H3K9me2 in mPGCLCs (Borgel et al, 2010; Kurimoto et al, 2015). Furthermore, a recent report has shown that the germline genes were repressed in EpiLCs with H3K9me3 imposed by Setdb1 (Mochizuki et al, 2021). In good agreement, the transcriptional start sites (TSSs) of germline genes repressed by Setdb1 up‐regulated H3K9me3 in EpiLCs and, more prominently, in d2 mPGCLCs, and lost it in d4c7 mPGCLCs (Fig 5K and L). The TSSs of germline genes defined in another study (Kurimoto et al, 2015) exhibited a comparable reduction of H3K9me3 during d2‐to‐d4c7 mPGCLC transition (Fig 5M). Thus, the germline genes are endowed with multiple layers of mechanisms, including higher‐order genome organization involving the insulation by CTCF and compound repressive epigenetic modifications, to prevent their activation in somatic cells, and such mechanisms are exempted in a stepwise manner— that is, erasure of DNA and H3K9 methylation occurs first and then release from H3K27me3/H2AK119u1 and CTCF insulation ensues—during germ‐cell development.
Heterochromatin compaction excludes H3K36me2 to create PMDs and Y‐chromosome hypomethylation
A unique epigenetic characteristic of male germ cells (pro‐spermatogonia, spermatogonia, and spermatozoa) is the presence of large partially methylated domains (PMDs) in intergenic regions (Kubo et al, 2015). PMDs can be defined as broad genomic domains with a comparatively lower methylation level than the rest of the genome and typically cover a substantial fraction of the genome (Lister et al, 2009). They were first identified in a human cultured cell line (Lister et al, 2009) and subsequently found to be prevalent in cancers, aged cells, and tissues such as the placenta (Hansen et al, 2011; Hon et al, 2012; Schroeder et al, 2013). While evidence suggests that PMDs arise from imperfect maintenance of methylation during mitosis (Salhab et al, 2018), the mechanism that engenders PMDs in mitotically arrested pro‐spermatogonia and their subsequent maintenance in male germ cells remains unclear.
We found that GSCs bore PMDs larger than 140 mb in total, a majority (~86%) of which overlapped with those in spermatogonia (Fig 6A; Kubo et al, 2015). The PMDs in GSCs consisted almost entirely of B compartments and were enriched with heterochromatic modifications such as H3K9me3, while depleted of active modifications including H3K36me2, H3K27ac, and H3K4me1/3 (Appendix Fig S2). The epigenomic profiles revealed that the epigenome of d4c7 mPGCLCs exhibited the greatest predictive power for PMDs in GSCs (greater than that of the epigenome of GSCs themselves; Fig 6B), and among individual epigenetic markers, H3K9me2/me3 and lamin B1 in d4c7 mPGCLCs were the strongest negative predictors (Fig 6C), suggesting that the constitutive heterochromatin in d4c7 mPGCLCs contributes to the subsequent formation of PMDs. Accordingly, we found that H3K36me2, which is catalyzed by NSD1 and serves as a recruiter of the androgenetic DNA methylome (Shirane et al, 2020), showed a specific depletion in the B compartments and the regions retaining H3K9me3, but not H3K27me3, in d4c7 mPGCLCs (Fig 6D–F), resulting in an exquisite concordance of H3K36me2 with the A compartments and a near‐complete exclusion from LADs in d4c7 mPGCLCs (Fig 6G). We found that the TADs involved in larger‐sized TAD cliques in d4c7 mPGCLCs exhibited the greatest H3K9me3 enrichment (Fig 6H). Given that the heterochromatic TAD‐cliques become dominant in d4c7 mPGCLCs and GSCs (Fig 1H and I), these findings suggest an increased aggregation of constitutive heterochromatin in d4c7 mPGCLCs may exclude the recruitment of NSD1 and hence the deposition of H3K36me2, leading to the formation of PMDs in GSCs.
Figure 6. Mechanism of PMD formation via balancing H3K36me2 versus H3K9me‐marked LADs and Y chromosome hypomethylation.

- (top) Overlap of PMDs between spermatogonia (Kubo et al, 2015) and GSCs; (bottom) Representative locus demonstrating colocalization of H3K9me3 and lamin B1 enrichment with DNA hypomethylation.
- The area under the receiver operating characteristic (AUROC) of classifiers predicting 50 kb bins as either PMD or not in GSCs using each cell type’s own epigenome. Error bars denote 95% confidence intervals.
- Correlation of GSCs’ DNA methylation levels in GSC LADs with epigenetic signals in different cell types.
- Aggregate plots of H3K36me2, H3K9me2, H3K9me3, and lamin B1 enrichment as well as DNA methylation around PMDs in GSCs. The thick line marks the mean while the upper and lower limits indicate standard errors.
- Scatter plot of d4c7 mPGCLCs’ H3K9me3 and H3K27me3 enrichment in 50 kb bins colored by differential H3K36me2 (EpiLCs−d4c7 mPGCLCs).
- Representative chromosome‐wide distributions of compartment score, lamin B1 enrichment, and H3K36me2 coverage.
- Correlation between H3K36me2 and compartment scores or lamin B1 enrichment in 50 kb bins.
- Relationship between the max clique size involving a given TAD and the average H3K9me3 enrichment in that TAD in d4c7 mPGCLCs. Number of TADs with specific max clique sizes, from left to right: 798/269/94/35/18/11. The central band of boxplots indicates median values, while the lower and upper hinge corresponds to the first and third quartile, and the upper whiskers extend to the largest value % 1.5 * IQR and vice versa for the lower whiskers.
- IP/input ratio of H3K9me3 and lamin B1 alignments per chromosome.
- Enrichment tracks of H3K9me3 and lamin B1 as well as DNA methylation in EpiLCs and d4c7 mPGCLCs on chromosome Y.
- (top) FISH against the Y chromosome; (bottom) sphericity of the Y chromosome FISH signals; (right) distributions of Y chromosome surface volumes. The point marks the median while the thick and thin lines correspond to 66% and 95% intervals, respectively. Number of cells = 89/76/69 for mESC/EpiLC/GSC. Scale bar, 10 μm. Significances are computed using Wilcoxon rank‐sum tests, P‐values from left to right: 1.7e−5, 0.025, 1.7e−5, 1.3e−11, 7.9e−21, 4.3e−13. P‐value symbol brackets: **** = [0, 0.0001); * = [0.01, 0.05).
- Proportion of the genome occupied by PMDs in GSCs with stratification by chromosome.
- 2D density plots of DNA methylation level (mCG/CG) between EpiLCs and GSCs in 10 kb bins.
In this regard, we noted that, as compared to the autosomes and the X chromosomes, the Y chromosomes, which bear a highly repetitive structure (Soh et al, 2014), were the most enriched with H3K9me3 in all five cell types, and interestingly, exhibited a progressive enrichment of lamin B1 during germ‐cell development, with the Y chromosomes in GSCs showing the highest lamin B1 enrichment level (Fig 6I). In addition, we found that the Y chromosome in GSCs was hypo‐methylated across almost its entire length, with ~75% of it identified as falling within PMDs—a much greater proportion than in autosomes (4%) or the X chromosome (21%) (Fig 6 J, L and M). Indeed, by alternatively mapping directly to the consensus repeat sequences of the Y chromosome, we found that all repetitive units demonstrate reduced methylation levels in GSCs as compared to EpiLCs (Appendix Fig S3A and B). Consistent with the de‐condensation of chromatin in GSCs (Fig 1B–D), the Y chromosomes in GSCs exhibited loose structures and were associated with the nuclear periphery with lower sphericity (Fig 6K), indicating greater surface contact with the nuclear lamina through chromosome elongation. Thus, the Y chromosome in GSCs achieves chromosome‐wide hypomethylation likely via a convergent mechanism with PMDs in autosomes. Together, these results lead us to conclude that the unique 3D epigenomic character of the progenitors (d4c7 mPGCLCs) serves as a blueprint for the formation of PMDs in male germ cells.
Nucleome programming engenders gametogenic potential
To delineate the functional significance of a proper nucleome for gametogenesis, we performed nucleome analyses (morphology; in situ Hi‐C; MS; ChIP‐seq for 13 targets; ATAC‐seq; and NET‐CAGE) of GSC‐like cells (GSCLCs), which were derived from d4 mPGCLCs with their differentiation into spermatogonia‐like cells in reconstituted testes followed by expansion under a GSC derivation condition (Ishikura et al, 2016) (Fig 7A). GSCLCs derived under this condition bore a morphology, transcriptome, and DNA methylome similar to those of GSCs, but showed a severely impaired capacity for spermatogenesis for unclear reasons (Ishikura et al, 2016) (Appendix Fig S4A). We hypothesized that aberrant nucleome programming during the derivation of GSCLCs might underlie their impaired function.
Figure 7. Nucleome differences between GSCs and GSCLCs.
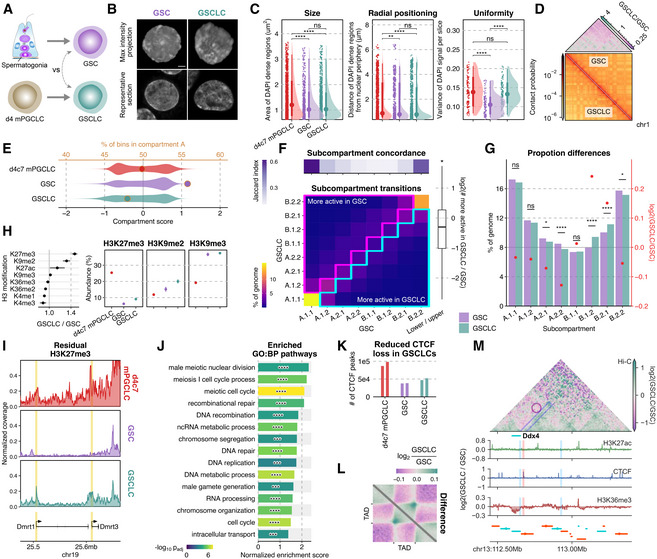
- Scheme for the derivation of GSCs and GSCLCs.
- Maximum intensity projections (top) and representative sections (bottom) of typical nuclei of GSCs and GSCLCs stained with DAPI. Scale bars, 3 μm.
- Areas of DAPI‐dense regions (left), distance of DAPI‐dense regions from the nuclear periphery (middle), and variance of DAPI signals (right). The point marks the median while the thick and thin lines correspond to 66% and 95% intervals, respectively. Number of DAPI dense regions = 1,535/736/1,227 and number of slices = 135/110/120 for d4c7 mPGCLC/GSC/GSCLC. Significances are computed using Wilcoxon rank‐sum tests, P‐values from left to right: 2.03e−10, 1.69e−9, 0.123, 0.00894, 8.02e−8, 0.0707, 7.65e−13, 0.417, 2.07e−12.
- (bottom) 250 kb resolution balanced contact probability matrices of chromosome 1 in GSCs (upper) and GSCLCs (lower); (top) fold change (GSCLCs/GSCs) of contact probability, showing an attenuation of distal interactions in GSCLCs.
- Distribution of compartment scores (bottom axis: violin plots) and the ratio of A:B compartment bins (top axis: dots) at 100 kb resolution.
- Differential subcompartmentalization between GSCs and GSCLCs at 50 kb resolution. (top) Jaccard index between genomic bins belonging to each subcompartment in GSCs vs GSCLCs. (bottom) A comparison of subcompartment labels between cell types reveals a greater proportion of the genome belongs to the upper triangle, in line with GSCLCs being more repressive. (right) Quantification of matched bins in the upper vs lower triangle.
- Comparison of overall subcompartment proportions in GSCs versus GSCLCs. Most significant changes are again observed mostly for the intermediate states and not active euchromatin (A.1) or constitutive heterochromatin (B.2). Significances are computed using two‐proportions z‐tests, p‐values from left to right: 0.0829, 0.107, 0.0169, 3.32e−5, 0.683, 1.09e−16, 7.46e−9, 0.0112.
- (left) Fold change (GSCLCs/GSCs) of different H3 modifications as measured by mass spectrometry, with confidence intervals denoting standard errors; (right) full data for select modifications. Three biological replicates in each cell type were analyzed.
- Normalized H3K27me3 coverage tracks around Dmrt1 and Dmrt3.
- GSEA results for promoters ranked by preferential enrichment in GSCLCs as compared to GSCs. Significances computed using pre‐ranked multilevel GSEA, p‐values from top to bottom: 7.11e−5, 3.31e−6, 5.15e−8, 1.44e−5, 9.71e−5, 3.06e−6, 0.000159, 1.42e−6, 0.000513, 3.66e−7, 0.000185, 8.43e−6, 3.31e−6, 3.7e−7, 0.000194.
- Number of CTCF peaks in each cell type. Two biological replicates in each cell type were analyzed.
- Pile‐up plots of intra‐TAD interactions in GSCs and GSCLCs.
- 3D epigenetic landscape rewiring near Ddx4. Differential (GSCLCs/GSCs) contact maps and ChIP‐seq coverage at the Ddx4 locus are shown. The insulating CTCF peak separating Ddx4 from one of its enhancers is not completely removed in GSCLCs.
Data information: P‐value symbol brackets: **** = [0, 0.0001); ** = [0.001, 0.01); * = [0.01, 0.05); ns = [0.05, 1].
GSC‐like cells were similar to GSCs in terms of the areas of high DAPI density and the distances of the DAPI‐dense areas from the nuclear periphery, but showed greater variances of DAPI density than GSCs (Fig 7B and C), indicating that GSCLCs bear a more heterogeneous chromatin de‐condensation. In situ Hi‐C revealed that, compared to GSCs, GSCLCs exhibited a depletion in long‐range interactions, indicative of incomplete chromatin uniformalization (Fig 7D, Appendix Fig S4B), and notably, failed to acquire the positively skewed compartment score distribution characteristic of GSCs (Fig 7E). A multi‐scale model dividing the genome into the eight subcompartments with distinct epigenetic properties (Liu et al, 2021) revealed that major differences between GSCLCs and GSCs were localized to intermediate compartments, with GSCLCs bearing fewer and more intermediate A and B sub‐compartments, respectively (Fig 7F and G, Appendix Fig S4C).
Accordingly, MS revealed that GSCLCs bore an elevated level of H3K27me3 and H3K9me2, which are associated with a state intermediate between compartments A and B (Johnstone et al, 2020; Fig 7H). The regions with higher H3K27me3 in GSCLCs were enriched in promoters and CpG islands (CGIs) (Appendix Fig S4D, Dataset EV6), which were, importantly, associated with pathways such as “male meiotic nuclear division,” and “recombinatorial repair,” and included Ddx4, Dmrt1, Dmc1, Stag3, and Spo11 (Fig 7I and J, Dataset EV6). These genes bore higher levels of H3K27me3 on their gene bodies as well (Fig 7I, Appendix Fig S4E). In contrast, the regions with higher levels of H3K9me2 in GSCs were enriched in enhancers and distal active regulatory elements (Appendix Fig S4F and G), and were associated with pathways such as “response to ciliary neurotrophic factor,” “rod bipolar cell differentiation,” and “adrenal cortex formation” (Appendix Fig S4H, Dataset EV6).
Moreover, GSCLCs bore a larger number of the CTCF‐binding peaks coinciding with an insufficient accumulation of H3K9me3 (Fig 7K, Appendix Fig S4I and J), and indeed GSCLCs developed higher intra‐TAD interaction strength compared to GSCs (Fig 7L), indicating that the chromatin of GSCLCs is more insulated than that of GSCs. In a megabase‐scale domain encompassing Ddx4, the insulating CTCF peak separating the Ddx4 promoter from one of its potential enhancers was removed only partially in GSCLCs, resulting in a reduced activation as evidenced by the comparatively lower H3K36me3 levels on Ddx4 (Fig 7M). Collectively, these results lead us to conclude that GSCLCs exhibit aberrant nucleome programming, including insulation erasure and epigenome programming, with partial retention of the properties of d4c7 mPGCLCs, resulting in their impaired spermatogenic potential.
Discussion
Germ‐cell development lays the groundwork for nuclear totipotency, creating sexually dimorphic haploid gametes, the oocytes, and the spermatozoa, which unite to form totipotent zygotes. PGCs bear naïve epigenome after epigenetic reprogramming and can serve as a direct precursor for oocyte differentiation; they can also acquire a distinct spermatogenic epigenome, including global DNA re‐methylation, to differentiate into spermatogonia/SSCs, a direct precursor for spermatozoa differentiation (Lee et al, 2014). PGCs and spermatogonia/SSCs therefore exhibit dimorphic epigenomic properties and have been thought to represent highly distinct cellular states. Contrary to this notion, our nucleome analyses have uncovered a smooth and unidirectional maturation of higher‐order genome organization from pluripotent precursors (mESCs/EpiLCs) to PGCs (d2/d4/d4c7 mPGCLCs) and then to spermatogonia/SSCs (GSCs), involving progressive euchromatization and radial chromosomal re‐positioning (Figs 1 and 8). This finding delineates a common nuclear‐architectural foundation toward gamete generation in both sexes, coordination not found in somatic lineages. This widespread euchromatization might underlie the potential of GSCs to de‐differentiate into pluripotent stem cells, albeit at a low frequency (Kanatsu‐Shinohara et al, 2004). Thus, germ‐cell development entails mechanisms that create and preserve a broadly euchromatic genome, while simultaneously accommodating essential epigenetic orchestrations. Our findings also demonstrate that global DNA methylation and euchromatization are dissociable events.
Figure 8. A model for nucleome programming during mouse germ‐cell development.
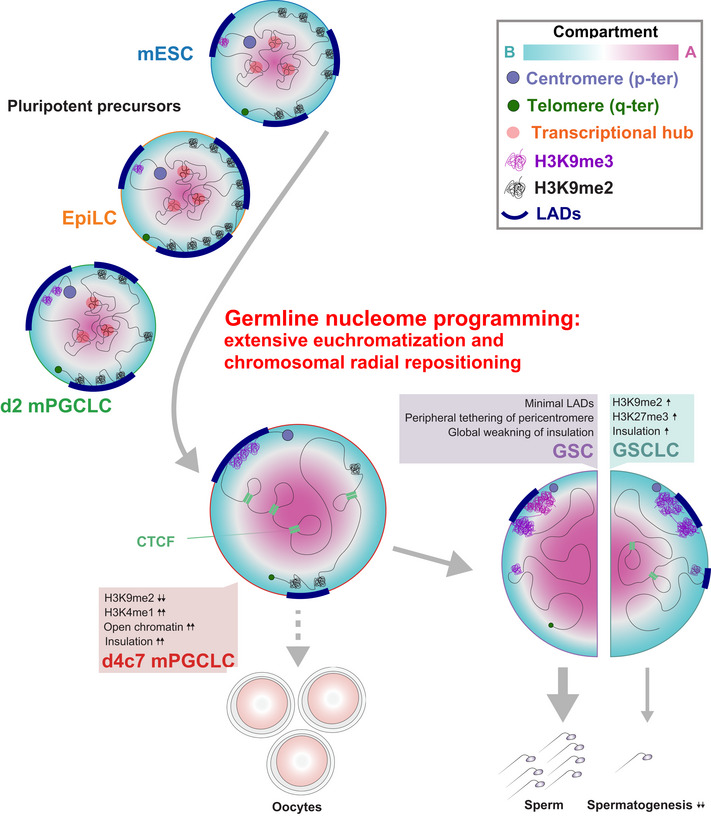
Unlike somatic fates, germline nucleome programming entails extensive euchromatization, which is associated with radial re‐positioning of pericentromeres and peripheral de‐attachment elsewhere. Augmented insulation helps to maintain transcriptional fidelity during global DNA hypomethylation in PGCs (PGCs bear oogenic potential as well; represented by a dashed arrow). Insulators are subsequently erased en masse to activate gametogenic program during the PGCs‐to‐spermatogonia/SSC development. Faulty nucleome maturation involving intermediate compartment states leads to impaired spermatogenic capacity (represented by a thin arrow).
As a key mechanism for global euchromatization, we have shown that germ‐cell development distinctly down‐regulates H3K9me2, an aggregative force for heterochromatin formation (Poleshko et al, 2019), and progressively restricts LADs to around centromeres (Figs 2 and 4). These events would be mediated at least in part through the repression of SETDB1 and EHMT1, K9MTases acting in both the A and B compartments (Fukuda et al, 2021), as well as lamin B1 itself. On the other hand, germ cells up‐regulate Suv39h1 and h2, K9MTases specific to the B compartment and particularly for pericentromeric regions. This results in an expansion of H3K9me3 into broad domains in GSCs with an appreciable increase in both local and distal compaction among such domains (Fig 5), consistent with the notion of a critical threshold of H3K9me3 domain width for phase separation to take place via HP1 (Sanulli et al, 2019). This compaction would also contribute to the formation of PMDs, and most remarkably, those on the Y chromosome, likely by physically excluding spermatogenesis‐associated NSD1 and preventing H3K36me2 depositions (Fig 6). Thus, typical LADs mediated by H3K9me2, which are seen in pluripotent precursors as well as in most somatic lineages, are progressively re‐organized into a minimal state marked by H3K9me3 during germ‐cell development. Importantly, the positional preference of H3K9me3‐associated minimal LADs is in part attributable to the density of evolutionarily young TEs that are enriched near centromeres (Figs 5 and EV5), indicating a critical role of inherent genomic properties in shaping the fundamental nuclear architecture. In good agreement with this concept, cell‐type‐specific LADs have been reported to be enriched in such TEs (preprint: Keough et al, 2021). The involvement of H3K9 demethylases and the interplay among associated machinery for LAD formation warrant further investigation.
Despite adopting a highly permissive epigenome with abundant enhancer‐like open sites, d4c7 mPGCLCs strengthened their chromatin insulation to thwart spurious distal activation, which, combined with a mechanism to ensure low H3K4me3 levels, would prevent the pervasive poised enhancers from realizing their potential (Figs 2 and 3). Thus, epigenetic reprogramming creates PGCs that have almost no DNA methylation and a highly open epigenome, but that are protected by elevated H3K27me3 (Ohta et al, 2017) and CTCF insulation against hyper‐transcription. As to a possible mechanism for the enhanced insulation, we revealed a reduced residence time of the loop extrusion machinery at TAD boundaries in d4c7 mPGCLCs (Fig EV3H–J). Such a reduction in residence time could be achieved through multiple mechanisms, including the use of variant cohesin complexes and modulating the balance between cohesin loading/release factors (Wutz et al, 2017; Cuadrado et al, 2019). Clarification of these potential mechanisms warrants future investigation.
On the other hand, such protective mechanisms must be at least partly disentangled upon male and female germ‐cell specification to eventually achieve full activation of the gametogenic program. Accordingly, a failure of such unraveling and a partial retention/aberrant development of the PGC‐like nucleome together contributed to the limited spermatogenic capacities of GSCLCs (Fig 6, Appendix Fig S4). In the original GSCLC induction strategy, d4 mPGCLCs, which are in the middle of epigenetic reprogramming and bear ~50% genome‐wide DNA methylation, were aggregated with embryonic testicular somatic cells for differentiation into spermatogonia‐like cells (Ishikura et al, 2016). We speculate that precocious testicular sex‐determining signals on mPGCLCs might be a reason for mis‐organized nucleome in the originally reported GSCLCs. In good agreement with this speculation, we have recently succeeded in deriving fully functional GSCLCs using d4c5 mPGCLCs, which have an almost fully complete epigenetic reprogramming, as starting materials for aggregation culture with embryonic testicular somatic cells (Ishikura et al, 2021). The nucleome analysis of these newly established GSCLCs would be important to confirm this hypothesis.
The nucleome programming for germ‐cell development that we have delineated herein, which involves progressive euchromatization with peripheral centromere positioning, is reminiscent of climbing up the Waddington’s landscape of epigenesis (Fig 8), and we propose that it constitutes at least part of the mechanism for creating nuclear totipotency, including meiotic potential. Elucidation of the nucleome programming during germ‐cell development in other mammals, including humans, will be crucial for a more comprehensive understanding of nuclear totipotency and its evolutionary divergence. The rich datasets we have assembled would be invaluable as a benchmark for mammalian in vitro gametogenesis studies (Saitou & Hayashi, 2021) and for future studies aiming to identify unifying principles for the acquisition of unique cellular identities across lineages. Further, they could contribute to the development of powerful computational frameworks, which in turn could help integrate time‐series multi‐omics datasets and unveil hidden insights.
Materials and Methods
Reagents and Tools table
| Reagent/Resource | Reference or Source | Identifier or Catalog Number |
|---|---|---|
| Experimental Models | ||
| AAG 129/B6 GSC2 (Acrosin‐EGFP; beta‐Actin‐EGFP, 129 Sv × C57BL/6, P7 spermatogonia, Germline stem cell line) | Ishikura et al (2016) | https://doi.org/10.1016/j.celrep.2016.11.026 |
| AAG 129/B6 GSCLC16_1 (Acrosin‐EGFP; beta‐Actin‐EGFP, 129SvJ × C57BL6, Germline stem cell‐like line, derived from mESCs) | Ishikura et al (2016) | https://doi.org/10.1016/j.celrep.2016.11.026 |
| BVSC BDF1‐2‐1 mESCs (Blimp1‐mVenus; Stella‐ECFP, DBA/2 × C57BL/6, embryonic stem cell line) | Ohta et al (2021) | https://doi.org/10.1093/biolre/ioaa195 |
| m220‐5 (sub‐cloned from Sl/Sl4‐m220, resistant to mitomycin C, expressing membrane‐bound SCF, stromal cell) | Ohta et al (2017) | https://doi.org/10.15252/embj.201695862 |
| MEF (ICR, mitomycinC‐treated mouse embryonic fibroblasts prepared from E12.5 fetuses) | N/A | N/A |
| Antibodies | ||
| Anti‐CTCF | CST | #3418 |
| Anti‐G9a | R&D Systems | PP‐A8620A‐00 |
| Anti‐GFP(Rat IgG2a), Monoclonal(GF090R), CC | Nacalai Tesque | 04404‐84 |
| Anti‐GLP | R&D Systems | PP‐B0422‐00 |
| Anti‐H2Aub | CST | #8240 |
| Anti‐H3 | CST | #9715 |
| Anti‐H3K27ac | MBL | MABI0309 |
| Anti‐H3K27me3 | MBL | MABI0323 |
| Anti‐H3K27me3 | Merk | 07‐449 |
| Anti‐H3K36me2 | CST | #2901 |
| Anti‐H3K36me3 | Active Motif | 61101 |
| Anti‐H3K4me1 | CST | #5326 |
| Anti‐H3K4me3 | MBL | MABI0304 |
| Anti‐H3K9me2 | MBL | MABI0317 |
| Anti‐H3K9me3 | MBL | MABI0318 |
| Anti‐Laminb1 | Proteintech | 12987‐1‐AP |
| Anti‐Laminb1 | Abcam | ab16048 |
| Anti‐mouse IgG (whole molecule)–peroxidase antibody produced in sheep affinity isolated antibody, buffered aqueous solution | Sigma | A5906‐1ML |
| Anti‐normal mouse IgG | Santa Cruz | sc‐2025 |
| Anti‐normal rabbit IgG | Santa Cruz | sc‐2027 |
| Anti‐rabbit IgG (whole molecule)–peroxidase antibody produced in goat affinity isolated antibody, buffered aqueous solution | Sigma | A6154‐1ML |
| Anti‐Rad21 | Abcam | ab992 |
| Anti‐Ring1b | CST | #5694 |
| Anti‐Setdb1 | Proteintech | 11231‐1‐AP |
| Anti‐α‐tubulin | Sigma | T9026 |
| Anti‐β‐actin | MBL | M177‐3 |
| Goat anti‐mouse IgG (H + L) highly cross‐adsorbed secondary antibody, Alexa Fluor 568 | Invitrogen | A‐11031 |
| Goat anti‐rabbit IgG (H + L) cross‐adsorbed secondary antibody, Alexa Fluor 568 | Invitrogen | A‐11011 |
| Goat anti‐rat IgG (H + L) cross‐adsorbed secondary antibody, Alexa Fluor 488 | Invitrogen | A‐11006 |
| Oligonucleotides and sequence‐based reagents | ||
| XMP 1 orange | MetaSystems | D‐1401‐050‐OR |
| XMP 16 orange | MetaSystems | D‐1416‐050‐OR |
| XMP Y orange | MetaSystems | D‐1421‐050‐OR |
| Chemicals, enzymes and other reagents | ||
| 16% Formaldehyde solution | Thermo Fisher Scientific | 28906 |
| 2‐Mercaptoethanol | Nacalai Tesque | 21438‐82 |
| 20xSCC | Sigma | S6639 |
| 37% Formaldehyde(FA) | Sigma | 252549 |
| 4% Paraformaldehyde | Nacalai Tesque | 26126‐25 |
| 4X Laemmli sample buffer | Bio‐Rad | #1610747 |
| Activin A (human/mouse/rat) | Peprotech | 120‐14 |
| AlbuMaxⅠ | Gibco | 11020062 |
| Amanitin 1 mg | Wako | 1022961 |
| Apo transferrin | Sigma | T1147 |
| Axygen® AxyPrep MAG PCR Clean‐Up Kit | Corning | MAG‐PCR‐CL‐250 |
| B27 | Thermo Fisher Scientific | 12587010 |
| bFGF | Invitrogen | 13256029 |
| Biotin‐14‐dATP | Thermo Fisher Scientific | 19524‐016 |
| Bovine serum albumin cold ethanol fraction, pH 5.2, ≥ 96% | Sigma | A4503‐10G |
| BSA fraction V | Gibco | 15260‐037 |
| CHIR99021 | Bio Vision | 4423 |
| cOmplete™, protease inhibitor cocktail | Roche | 4693116001 |
| cOmplete™, EDTA‐free protease inhibitor cocktail | Roche | 11873580001 |
| cOmplete™, mini, EDTA‐free | Roche | 4693159001 |
| Cyclosporin A | Sigma | 30024 |
| DAPI | Wako | 342‐07431 |
| Difco™ skim milk | BD Biosciences | 232100 |
| Digitonin | Promega | G9441 |
| DMEM/F12 | Gibco | 11330‐057 |
| DMEM/F12 (phenol red free) | Thermo Fisher Scientific | 21041025 |
| DNA polymerase I, large (Klenow) fragment | NEB | M0210S |
| DNaseI 1 unit/μl, RNase‐free | Thermo Fisher Scientific | 89836 |
| DpnII | NEB | R0543L |
| DTT 100 μl | Promega | P1171 |
| Dynabeads M‐280 sheep anti‐mouse IgG | Thermo Fisher Scientific | DB11201 |
| Dynabeads protein A | Thermo Fisher Scientific | DB10001 |
| Dynabeads® MyOne™ Streptavidin C1 | Thermo Fisher Scientific | 65001 |
| EGF, mouse, recombinant, carrier‐free | RSD | 2028EG |
| Fetal bovine serum (FBS) | Hyclone | SH30910.03 |
| Fibronectin (human) | Merck Millipore | FC010 |
| Formamide | Nacalai Tesque | 16228‐05 |
| Forskolin | Sigma | F3917 |
| GDNF, rat, recombinant | RSD | 512GF |
| Glasgow's MEM (GMEM) | Thermo Fisher Scientific | 11710035 |
| GlutaMAX supplement | Life Technologies | 35050061 |
| Immobilon‐P PVDF membrane | Merck Millipore | IPVH00010 |
| Insulin | Sigma | #I‐1882 |
| Insulin‐transferrin‐selenium (ITS)‐G | Gibco | 41400045 |
| KnockOut™ serum replacement | Gibco | 10828028 |
| l‐Glutamine | Thermo Fisher Scientific | 25030149 |
| Laminin | BD Bioscience | 354232 |
| LIF(ESGRO®) | Merck Millipore | ESG1107 |
| MEM non‐essential amino acids solution | Thermo Fisher Scientific | 11140‐050 |
| Minimum Essential Medium (MEM) Vitamin Solution | Thermo Fisher Scientific | 11120052 |
| Mitomycin C | kyowakirin | KWN‐057039107 |
| NEBNext High‐Fidelity 2× PCR Master Mix | NEB | M0541S |
| Neurolbasal™ medium | Invitrogen | 2113‐049 |
| Nuclei EZ Prep | Sigma | NUC101 |
| Orange‐dUTP | Abbott | 02N33‐050 |
| PD325901 | Stemgent | 04‐2006 |
| Penicillin‐Streptomycin (10,000 units/ml, 10,000 µg/ml) | Thermo Fisher Scientific | 15140148 |
| PhosSTOP™ | Roche | 4906837001 |
| Pierce™ Protease Inhibitor Mini Tablets, EDTA‐free | Thermo Fisher Scientific | A32955 |
| Poly‐L‐ornithine | Sigma | P3655 |
| Progesterone | Sigma | P8783 |
| Proteinase K solution | Thermo Fisher Scientific | AM2546 |
| Putrescine | Sigma | P5780 |
| Recombinant Human BMP‐4 | RSD | 314BP01 M |
| Recombinant Mouse SCF | RSD | 455MC |
| RIPA lysis buffer system 50 ml | Santa Cruz | SC‐24948 |
| RNase A | Thermo Fisher Scientific | EN0531 |
| Rolipram | Abcam | AB120029 |
| Sodium pyruvate | Thermo Fisher Scientific | 11360‐070 |
| Sodium selenite | Sigma | S5261 |
| StemPro™‐34 SFM (1×) | Gibco | 10639011 |
| SUPERase | Thermo Fisher Scientific | AM2694 |
| T4 DNA ligase 1 U/µl | Thermo Fisher Scientific | 15224090 |
| T4 DNA polymerase | NEB | M0203L |
| Tks Gflex™ DNA Polymerase | Takara | R060A |
| TryPLE‐Express | Thermo Fisher Scientific | 12604‐021 |
| VECTASHIELD® Antifade Mounting Medium | Vector Laboratories | H‐1000‐10 |
| β‐Mercaptoethanol | Thermo Fisher Scientific | 21985023 |
| Software | ||
| ABC commit 7fd69b0 | Fulco et al (2019) | https://github.com/broadinstitute/ABC‐Enhancer‐Gene‐Prediction |
| BEDTools v2.29.2 | Quinlan and Hall (2010) | https://github.com/arq5x/bedtools2 |
| Bismark v0.22.1 | Krueger and Andrews (2011) | https://www.bioinformatics.babraham.ac.uk/projects/bismark/ |
| Bowtie2 v2.3.4.1 | Langmead and Salzberg (2012) | http://bowtie‐bio.sourceforge.net/bowtie2/index.shtml |
| CAGEfightR v1.7.6 | Thodberg et al (2019) | https://bioconductor.org/packages/release/bioc/html/CAGEfightR.html |
| CAGEr v1.32.0 | Haberle et al (2015) | https://www.bioconductor.org/packages/release/bioc/html/CAGEr.html |
| CALDER commit 32220e8 | Liu et al (2021) | https://github.com/CSOgroup/CALDER |
| Chrom3D | Paulsen et al (2017) | https://github.com/Chrom3D/pipeline |
| ChromA v2.1.1 | Gabitto et al (2020) | https://github.com/marianogabitto/ChromA |
| chromatin_information v1.0 | D'Oliveira Albanus et al (2021) | https://github.com/ParkerLab/chromatin_information |
| Chromosight v1.5.1 | Matthey‐Doret et al (2020) | https://github.com/koszullab/chromosight |
| cooler v0.8.10 | Abdennur and Mirny (2020) | https://github.com/open2c/cooler |
| coolpup.py v0.9.7 | Flyamer et al (2020) | https://github.com/open2c/coolpuppy |
| cooltools v0.4.0 | Venev et al (2021) | https://github.com/open2c/cooltools |
| CSynth commit 26e21fb | Todd et al (2021) | https://github.com/csynth/csynth |
| Cutadapt v1.9.1 | Martin (2011) | https://cutadapt.readthedocs.io/en/stable/ |
| dcHiC commit 7b1727f | preprint: Wang et al (2021) | https://github.com/ay‐lab/dcHiC |
| deepTools v3.5.0 | Ramirez et al (2016) | https://github.com/deeptools/deepTools |
| DESeq2 v1.28.1 | Love et al (2014) | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| DiffBind v3.0.13 | Ross‐Innes et al (2012) | https://bioconductor.org/packages/release/bioc/html/DiffBind.html |
| EDD v1.1.19 | Lund et al (2014) | https://github.com/CollasLab/edd |
| epic2 v0.0.41 | Stovner and Saetrom (2019) | https://github.com/biocore‐ntnu/epic2 |
| EpiProfile v2.0 | Yuan et al (2018) | https://github.com/zfyuan/EpiProfile2.0_Family |
| FACSDiva Software | BD Biosciences | N/A |
| FAN‐C v0.9.13 | Kruse et al (2020) | https://github.com/vaquerizaslab/fanc |
| fastp v0.21.0 | Chen et al (2018) | https://github.com/OpenGene/fastp |
| GimmeMotifs v0.15.3 | preprint: Bruse and Heeringen (2018) | https://github.com/vanheeringen‐lab/gimmemotifs |
| HDBSCAN v0.8.27 | Campello et al (2013) | https://github.com/scikit‐learn‐contrib/hdbscan |
| HiCKey commit 6e282b9 | Xing et al (2021) | https://github.com/YingruWuGit/HiCKey |
| HiCRep.py v0.2.3 | Lin et al (2021) | https://github.com/Noble‐Lab/hicrep |
| HiCRes v1.1 | Marchal et al (2022) | https://github.com/ClaireMarchal/HiCRes |
| HiCSeg v1.1 | Levy‐Leduc et al (2014) | https://cran.r‐project.org/web/packages/HiCseg/index.html |
| HiCUP v0.8.0 | Wingett et al (2015) | https://github.com/StevenWingett/HiCUP |
| IDR2D v1.4.0 | Krismer et al (2020) | https://github.com/kkrismer/idr2d |
| Imaris v9.1.2 | N/A | https://imaris.oxinst.com/ |
| Juicer tools v1.22.01 | Durand et al (2016) | https://github.com/aidenlab/juicer |
| MACS v2.1.1 | Zhang et al (2008) | https://github.com/macs3‐project/MACS |
| OnTAD v1.2 | An et al (2019) | https://github.com/anlin00007/OnTAD |
| Picard Tools v2.18.23 | N/A | https://broadinstitute.github.io/picard/ |
| Python v3.8.8 | N/A | https://www.python.org/ |
| R (v4.0.3) | https://www.r‐project.org/ | https://www.r‐project.org/ |
| RobusTAD | preprint: Dali et al (2018) | https://github.com/rdali/RobusTAD |
| S3V2‐IDEAS commit b7cc2d5 | Xiang et al (2021) | https://github.com/guanjue/S3V2_IDEAS_ESMP |
| Salmon v1.4.0 | Patro et al (2017) | https://github.com/COMBINE‐lab/salmon |
| SAMtools v1.7 | Li et al (2009) | https://github.com/samtools/samtools |
| SpectralTAD v1.4.0 | Cresswell et al (2020) | https://github.com/dozmorovlab/SpectralTAD |
| TADpole 0.0.0.9000 | Soler‐Vila et al (2020) | https://github.com/3DGenomes/TADpole |
| TopDom v0.10.1 | Shin et al (2016) | https://github.com/HenrikBengtsson/TopDom |
| Trim‐Galore! v0.6.3 | Krueger et al (2021) | https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ |
| tximport v1.16.1 | Soneson et al (2015) | https://github.com/mikelove/tximport |
| UMAP v0.5.1 | Mcinnes et al (2018) | https://github.com/lmcinnes/umap |
| Other | ||
| Chemi‐Lumi One Super | Nacalai Tesque | 02230‐14 |
| FastGene Adapter Kit | FastGene | FG‐NGSAD24 |
| Illumina Tagment DNA Enzyme and Buffer, Small Kit | Illumina | 20034197 |
| KAPA Hyper Prep Kit | KAPA | KK8504 |
| KAPA Library Quantification Kit | KAPA | KK4824 |
| MinElute PCR purification Kit (50) | QIAGEN | 28004 |
| miRNeasy Mini Kit 50 | QIAGEN | 217004 |
| NEBNext® Multiplex Oligos for Illumina® (Index Primers Set 1) | NEB | E7335S |
| NEBNext® Ultra™ II DNA Library Prep Kit for Illumina® | NEB | E7645S |
| NextSeq 500/550 High Output Kit v2.5 (150 Cycles) | Illumina | 20024907 |
| NextSeq 500/550 High Output Kit v2.5 (75 Cycles) | Illumina | 20024906 |
| NextSeq 500/550 High‐Output v2 Kit (150 cycles) | Illumina | FC‐404‐2002 |
| NextSeq 500/550 High‐Output v2 Kit (75 cycles) | Illumina | FC‐404‐2005 |
| NextSeq 500/550 Mid Output Kit v2.5 (150 Cycles) | Illumina | 20024904 |
| NextSeq 500/550 Mid‐Output v2 Kit (150 cycles) | Illumina | FC‐404‐2001 |
| NovaSeq 6000 S1 Reagent Kit (200 cycles) | Illumina | 20012864 |
| NovaSeq 6000 SP Reagent Kit (200 cycles) | Illumina | 20040326 |
| QIAquick PCR Purification Kit (50) | QIAGEN | 28104 |
| Qubit RNA HS Assay Kit | Thermo Fisher Scientific | Q32855 |
| RNase‐Free DNase Set | QIAGEN | 79254 |
| Film‐bottom dish | Matsunami Glass | FD10300 |
| MAS‐GP type A | Matsunami Glass | S9901‐9905 |
Methods and Protocols
Culture of mESCs
The BDF1‐2‐1 mouse mESCs bearing Blimp1‐mVenus and Stella‐ECFP (BVSC) transgenes (Ohta et al, 2021) were cultured as described previously (Hayashi et al, 2011). Briefly, mESCs were maintained in N2B27 medium supplemented with PD0325901 (0.4 μM) (Stemgent, 04‐2006), CHIR99021 (3 μM) (Bio Vision, 4423), and leukemia inhibitory factor (LIF) (1,000 U/ml) (Merck Millipore, ESG1107) on a 12‐well plate coated with poly‐l‐ornithine (0.01%) (Sigma, P3655) and laminin (10 ng/ml) (BD Biosciences, 354232). In this study, all cells were cultured at 37°C under an atmosphere of 5% CO2 in the air.
Induction of EpiLCs and mPGCLCs
Induction of EpiLCs and PGCLCs was performed as described previously (Hayashi et al, 2011) with minor modifications. Briefly, the EpiLCs were induced by plating 8 × 104 mESCs on a well of a 12‐well plate coated with human plasma fibronectin (16.7 mg/ml) (Merck Millipore, FC010) in N2B27 medium containing activin A (20 ng/ml) (Peprotech, 120‐14), bFGF (12 ng/ml, 13256029) (Invitrogen), and KSR (1%) (Gibco, 10828028). mPGCLCs were induced from d2 EpiLCs (2 days after induction) under a floating condition in wells of a low‐cell‐binding U‐bottom 96‐well plate in GMEM medium (Thermo Fisher Scientific, 11710035) containing 15% KSR (Gibco, 10828028), 0.1 mM NEAA (Thermo Fisher Scientific, 11140‐050), 1 mM sodium pyruvate (Thermo Fisher Scientific, 11360‐070), 0.1 mM β‐mercaptoethanol (Thermo Fisher Scientific, 21985023), 100 U/ml penicillin, 0.1 mg/ml streptomycin (Thermo Fisher Scientific, 15140148) and 2 mM l‐glutamin (Thermo Fisher Scientific, 25030149) supplemented with BMP4 (500 ng/ml) (RSD, 314BP01 M), LIF (1,000 U/ml) (Merck Millipore, ESG1107), SCF (100 ng/ml) (RSD, 455MC), and EGF (50 ng/ml) (RSD, 2028EG).
Expansion culture of mPGCLCs
The expansion culture of mPGCLCs was performed as previously described (Ohta et al, 2021). Briefly, following incubation in TrypLE™ Express (Gibco, 12604‐021) for 10 min, the aggregates of d4 mPGCLCs (PGCLCs induced for 4 days) were dissociated into single cells by rigorous pipetting. Subsequently, BV‐positive cells were sorted with a FACSAria III cell sorter. Purified d4 mPGCLCs were cultured on m220‐5 cells as the feeder cells in GMEM (Gibco, 11710035) containing 10% KSR (Gibco, 10828028), 0.1 mM NEAA (Thermo Fisher Scientific, 11140‐050), 1 mM sodium pyruvate (Thermo Fisher Scientific, 11360‐070), 0.1 mM β‐mercaptoethanol (Thermo Fisher Scientific, 21985023), 100 U/ml penicillin, 0.1 mg/ml streptomycin (Thermo Fisher Scientific, 15140148), 2 mM l‐glutamin (Thermo Fisher Scientific, 25030149), 2.5% FBS (Hyclone, SH30910.03), SCF (100 ng/ml) (RSD, 455MC), 10 mM forskolin (Sigma, F3917), 10 μM rolipram (Abcam, AB120029), and 5 μM CsA (Sigma, 30024). Half of the culture medium was changed every 2 days.
Culture of GSCs and GSCLCs
Germline stem cells and GSCLCs bearing Acrosin‐EGFP and beta‐Actin‐EGFP (AAG) transgenes (Ohta et al, 2000) were cultured as described previously (Ishikura et al, 2016). Briefly, cells were cultured in Stempro‐34 SFM supplemented with Stempro Supplement (Gibco, 10639011), with 0.1 mM β‐mercaptoethanol (Thermo Fisher Scientific, 21985023), 1% FBS (Hyclone, SH30910.03), 1×MEM vitamin solution (Thermo Fisher Scientific, 11120052), 5.0 mg/ml AlbMAXI (Gibco, 11020062), 0.1 mM NEAA (Thermo Fisher Scientific, 11140‐050), 1 mM sodium pyruvate (Thermo Fisher Scientific, 11360‐070), 0.1 mM β‐mercaptoethanol (Thermo Fisher Scientific, 21985023), 100 U/ml penicillin, 0.1 mg/ml streptomycin (Thermo Fisher Scientific, 15140148), 2 mM l‐glutamin (Thermo Fisher Scientific, 25030149), 1×Insulin‐Transferrin‐Selenium (ITS‐G) (Gibco, 41400045), 10 ng/ml bFGF (Invitrogen, 13256029), 20 ng/ml GDNF rat recombinant (RSD, 512GF), 20 ng/ml EGF (RSD, 2028EG), and 1,000 U/ml LIF (Merck Millipore, ESG1107) in a well of a 6‐well plate on MEFs as feeder cells. Half of the medium was replaced every 2 or 3 days.
Immunofluorescence staining
The following primary antibodies were used at the indicated dilutions: rabbit anti‐Laminb1 (1/1,000; Abcam ab16048); mouse anti‐H3K9me2 (1/500; MBL, MABI0317); mouse anti‐H3K9me3 (1/500; MBL, MABI0318); and mouse anti‐H3K27me3 (1/500; Merk, 07‐449).
The following secondary antibodies from Thermo Fisher Scientific were used at a 1/500 dilution: Alexa Fluor 568 goat anti‐rabbit IgG; Alexa Fluor 488 goat anti‐rabbit IgG; and Alexa Fluor 568 goat anti‐mouse IgG.
Immunofluorescence (IF) staining was performed as previously described (Ohta et al, 2017) with minor modifications. Briefly, cells were fixed in 4% PFA (paraformaldehyde; Nacalai Tesque, 26126‐25) for 30 min at RT. After fixation, cells were washed in PBS three times and then permeabilized in 1% Triton‐X100/PBS for 5 min on ice. Then, they were washed in PBS three times and incubated in 1% BSA (Sigma, A4503‐10G)/PBS for 1 h. The cells were incubated with primary antibodies in 1% BSA/PBS overnight. After incubation with primary antibodies, the cells were washed in PBS three times and then incubated for 2 h with secondary antibodies and DAPI (1 mg/ml) (Wako, 342‐07431) at RT. Then, they were washed three times in PBS and mounted in VECTOR SHIELD (Vector Laboratories, H‐1000‐10). Images were captured with a confocal microscope (LSM780 or LSM980 with Airyscan2; Zeiss).
Probe preparation for DNA‐FISH against major satellite repeats
The probe against major satellite repeats was generated as previously described (Anton et al, 2014) with some modifications. DNA fragments were amplified with forward (5′‐GCGAGAAAACTGAAAATCAC‐3′) and reverse (5′‐TCAAGTCGTCAAGTGGATG‐3′) primers using mouse genomic DNA as a template, and purified using a QIA quick PCR purification kit (QIAGEN, 28104). A 500 ng of the PCR product was labeled with Orange‐dUTP (Abbott, 02N33‐050) using a Nick translation kit (Roche, 10976776001).
DNA‐FISH
DNA‐FISH was performed as described previously (Okamoto et al, 2005). Briefly, cells were cultured in a film‐bottom dish (Matsunami Glass, FD10300) and fixed in 3% PFA/PBS (Nacalai Tesque, 26126‐25) for 10 min at RT. After a brief wash in PBS, cells were permeabilized in 0.5% Triton‐X100 in PBS for 5 min on ice and stored in 70% ethanol at −30°C by the day of use. Then, the DNA was denatured in 50% FA (formamide) (Nacalai Tesque, 16228‐05)/2×SSC pH 7.4 (Sigma, S6639) for 40 min at 80°C and dehydrated through an ice‐cold ethanol series. Hybridization with probes was performed at 37°C overnight. After incubation, the samples were washed in 50% FA/2×SSC followed by 2×SSC. The samples were counterstained with DAPI (1 mg/ml) (Wako, 342‐07431), and mounted and viewed under a confocal microscope (Zeiss LSM980 with Airyscan2). Images were analyzed using Imaris 9.1.2 software (Bitplane).
Western blot analysis
The following primary antibodies were used at the indicated dilutions: rabbit anti‐Lamin b1 (1/1,000; Abcam ab16048); mouse anti‐H3K9me2 (1/500; MBL, MABI0317); mouse anti‐H3K9me3 (1/500; MBL, MABI0318); and mouse anti‐H3K27me3 (1/500; MBL, MABI0323); rabbit anti‐H3 (1/10,000; CST, #9715); rabbit anti‐CTCF (1/500; CST, #3418); mouse anti‐G9a (1/500; R&D, PP‐A8620A‐00); mouse anti‐GLP (1/500; R&D, PP‐B0422‐00); rabbit anti‐Setdb1 (1/1,000; Proteintech, 11231‐1‐AP); mouse anti‐α‐tubulin (1/5,000; Merk, T9026); and mouse anti‐β‐actin (1/5,000; MBL, M177‐3).
The following secondary antibodies from Merk were used at the indicated dilutions: goat anti‐rabbit IgG conjugated with peroxidase (1/8,000) and sheep anti‐mouse IgG conjugated with peroxidase (1/10,000).
Western blot was performed as previously described (Hayashi et al, 2011) with slight modifications. Briefly, cells were lysed by RIPA buffers (Santa Cruz, SC‐24948). After incubation for 30 min at 4°C with rotation, the lysates were sonicated by Bioruptor using 10 cycles of 30 s on/30 s off. Then, the lysates were spun down at 14,000 rpm for 15 min at 4°C and the supernatant was collected. A BCA assay was performed using a Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific, 23227) to measure the protein concentration. For western blot, 4.5 μg of whole cell lysate or 2.25 μg of chromatin fraction was loaded onto each lane. After the addition of 4× Laemmli buffer (Bio‐Rad, #1610747), the sample was run by SDS–PAGE, followed by blotting to PVDF membrane (pore size: 0.45 mm) (Millipore, IPVH00010) in CAPS buffer (10 mM CAPS‐NaOH pH 11, 5% methanol). After blotting, the membrane was incubated for 1 h in 0.1% Tween‐20/PBS (PBST) with 1% skim milk (BD Bioscience, 232100). After blocking, the membrane was incubated overnight with the primary antibodies in 0.1% PBST with 1% skim milk. The membrane was washed in 0.1% PBST, followed by incubation for 2 h with the secondary antibodies in the 0.1% PBST with 1% skim milk. After washing in 0.1% PBST three times, secondary antibodies were detected by Chemilumi One Super (Nacalai Tesque, 02230‐14) using Fusion solo 4S (Vilber). Quantification analysis of the signal intensity was performed in ImageJ v2.1.0 (NIH). Target protein signals were normalized by the loading control.
Chromatin fraction isolation
Chromatin fractionation was performed as previously described (Wutz et al, 2017). In brief, cells were resuspended in extraction buffer (20 mM Tris–HCl pH 7.5, 100 mM NaCl, 5 mM MgCl2, 2 mM NaF, 10% glycerol, 0.2% NP‐40, 20 mM β‐glycerophosphate, 0.5 mM DTT, and protease inhibitor cocktail; Roche, 11873580001). The chromatin pellet was fractionated by centrifugation at 2,000 g for 5 min and washed in the same buffer three times. Then, the chromatin pellet was resuspended in RIPA buffer (Santa Cruz, SC‐24948) and processed along with the whole cell lysate by a downstream BCA assay (Thermo Fisher Scientific, 23227) followed by western blot.
Visualization and analysis of nuclei by DAPI staining
All cells except d2 mPGCLCs were cultured in a film‐bottom dish (Matsunami Glass, FD10300). d2 mPGCLCs were attached on a slide glass (MATSUNAMI, S9901‐9905) using Cyto Spin 4 (Thermo Fisher Scientific) as previously described (Ohta et al, 2017). Cells were fixed in 4% PFA (Nacalai Tesque, 26126‐25) at RT for 30 min and washed in PBS three times. For permeabilization, cells were incubated on ice in 0.5% TritonX‐100/PBS for 5 min. Then, cells were incubated in DAPI solution (1 mg/ml) (Wako, 342‐07431) for 8 min, mounted and viewed under a fluorescence microscope. Confocal z‐series images with an interval of 0.14 µm were captured by Zeiss LSM980 with Airyscan2 using a 405 nm wavelength and a 63×objective oil‐immersion lens. For DAPI‐staining analysis, cells were attached to slides using Cyto Spin 4 (Thermo Fisher Scientific) as previously described (Ohta et al, 2017) to avoid the effect of differences in their colony shapes. DAPI‐staining and image acquisition were performed as described above. Acquired images were processed as follows. The nuclear mask, nuclear rim, and DAPI dense regions were defined in each z‐slice using ImageJ custom script as previously described (Miura, 2020). Then, the slice showing the maximum diameter was decided for each cell as a representative slice, and the representative slice ± 5 slices for each cell (i.e., 11 slices/cell) were used in the downstream analysis. Approximately 20–30 cells were analyzed in each cell type. The parameters presented in the Figures were calculated using the R custom script.
Histone extraction for mass spectrometry
Frozen cell pellets containing 3 million cells were lysed in nuclear isolation buffer (15 mM Tris pH 7.5, 60 mM KCl, 15 mM NaCl, 5 mM MgCl2, 1 mM CaCl2, 250 mM sucrose, 10 mM sodium butyrate, 0.1% v/v b‐mercaptoethanol (Nacalai Tesque, 21438‐82), commercial phosphatase and protease inhibitor cocktail tablets; Roche, 4906837001; Thermo Fisher Scientific, A32955) containing 0.3% NP‐40 alternative on ice for 5 min. Nuclei were washed in the same solution without NP‐40 twice and the pellet was slowly resuspended while vortexing in chilled 0.4 N H2SO4, followed by 3 h of rotation at 4°C. After centrifugation, the supernatants were collected and proteins were precipitated in 20% TCA overnight at 4°C, washed once with 0.1% HCl (v/v) acetone and then twice with acetone only, and resuspended in deionized water. Acid‐extracted histones (20–50 μg) were resuspended in 100 mM ammonium bicarbonate pH 8, derivatized using propionic anhydride and digested with trypsin as previously described (Sidoli et al, 2016). After the second round of propionylation, the resulting histone peptides were desalted using C18 Stage Tips, dried using a centrifugal evaporator and reconstituted using 0.1% formic acid in preparation for liquid chromatography‐mass spectrometry (LC–MS) analysis.
LC/LC–MS
Nanoflow liquid chromatography was performed using a Thermo Fisher Scientific Dionex UltiMate 3000 LC system equipped with a 300 mm ID × 0.5‐cm trap column (Thermo) and a 75 mm ID × 20‐cm analytical column packed in‐house using Reprosil‐Pur C18‐AQ (3 mm; Dr. Maisch). Buffer A was 0.1% formic acid and Buffer B was 0.1% formic acid in 80% acetonitrile. Peptides were resolved using a two‐step linear gradient from 5% B to 33% B over 45 min, then from 33% B to 90% B over 10 min at a flow rate of 300 nl/min. The HPLC was coupled online to an Orbitrap QE‐HF mass spectrometer operating in the positive mode using a Nanospray Flex Ion Source (Thermo Fisher Scientific) at 2.3 kV. Two full mass spectrometry scans (m/z 300–1,100) were acquired in the Orbitrap Fusion mass analyzer with a resolution of 120,000 (at 200 m/z) every 8 data‐independent acquisition tandem mass spectrometry (MS/MS) events, using isolation windows of 50 m/z each (e.g., 300–350, 350–400, and 650–700). MS/MS spectra were acquired in the ion trap operating in normal mode. Fragmentation was performed using collision‐induced dissociation in the ion trap mass analyzer with a normalized collision energy of 35. The automatic gain control target and maximum injection time were 5 × 105 and 50 ms for the full mass spectrometry scan, and 3 × 104 and 50 ms for the MS/MS scan, respectively. Raw files were analyzed using EpiProfile 2.0 (Yuan et al, 2018). The area for each modification state of a peptide was normalized against the total signal for that peptide to give the relative abundance of the histone modification.
ChIP‐seq library preparation and sequencing
The ChIP‐seq library preparation was performed as previously described (Lee et al, 2006) with minor modifications. We used harvested mESCs and EpiLCs, and FACS‐sorted BV‐positive cells for d2 mPGCLCs and d4c7 mPGCLCs samples, and FACS‐sorted AAG‐positive cells for GSCs and GSCLCs samples. Briefly, the harvested cells were crosslinked with 1% formaldehyde (Thermo Fisher Scientific, 28906)/PBS for 10 min at RT and quenched with 125 mM glycine. Crosslinked cells were lysed consecutively using LB1 (50 mM HEPES‐KOH pH 7.5, 1 mM EDTA, 140 mM NaCl, 10% glycerol, 0.5% NP‐40, 0.25% Triton‐100, and protease inhibitors (Roche, 11873580001)), LB2 (10 mM Tris–HCl pH 8.0, 1 mM EDTA, 0.5 mM EGTA, 200 mM NaCl, and protease inhibitors), and LB3 (50 mM Tris–HCl pH 8.0, 1 mM EDTA, 0.5 mM EGTA, 100 mM NaCl, 0.1% Na‐deoxycholate, 0.5% N‐lauroylsarcosine, and protease inhibitors) and then sonicated by a picoruptor to achieve a mean DNA fragment size of around 200–400 bp. Sonicated chromatin was incubated with Dynabeads M‐280 Sheep anti‐Mouse IgG beads (Thermo Fisher Scientific, DB11201) or Dynabeads ProteinA beads (Thermo Fisher Scientific, DB10001) for 35 min at 4°C for preclear. Precleared chromatin was then incubated with antibodies that were preincubated with the appropriate Dynabeads in 0.5% BSA (Gibco, 15260‐037) in PBS as follows: a chromatin equivalent of 5 × 105 cells with anti‐H3K4me1 (rabbit monoclonal, CST, #5326, 5 μl), anti‐H3K9me2 (mouse monoclonal, MBL, MABI0317, 5 μl), anti‐H3K27me3 (mouse monoclonal, MBL, MABI0323, 5 μl); 1 × 106 cells with anti‐H3K4me3 (mouse monoclonal, MBL, MABI0304, 5 μl), anti‐H3K9me3 (mouse monoclonal, MBL, MABI0318, 5 μl), anti‐H3K36me2 (rabbit monoclonal, CST, #2901, 5 μl), anti‐H2AK119ub1 (rabbit monoclonal, #8240, 10 μl), anti‐H3K36me3 (rabbit polyclonal, Active Motif, 61101, 2 μl); 1.5 × 106 cells with anti‐H3K27ac (mouse monoclonal, MBL, MABI0309, 5 μl); 2 × 106 cells with anti‐CTCF (rabbit monoclonal, CST, #3418, 5 μl), anti‐Laminb1 (rabbit polyclonal, Proteintech, 12987‐1‐AP, 10 μl); 4 × 106 cells with anti‐Ring1b (rabbit monoclonal, CST, #5694, 10 μl); and 4.5 × 106 cells with anti‐Rad21 (rabbit polyclonal, ab992, 5 μl).
After incubation for 6 h at 4°C, the beads were washed 4 times in wash buffer 1 (20 mM Tris–HCl pH 8.0, 2 mM EDTA, 150 mM NaCl, 1% TritonX‐100, and 0.1% SDS), 2 times in wash buffer 2 (20 mM Tris–HCl pH 8.0, 2 mM EDTA, 500 mM NaCl, 1% TritonX‐100, and 0.1% SDS), and 2 times in wash buffer 3 (10 mM Tris–HCl pH 8.0, 1 mM EDTA, 250 mM LiCl, 1% Na‐Deoxycolate, and 1% NP‐40). Then, the washed beads were eluted in 10 mM Tris–HCl pH 8.0, 5 mM EDTA, 300 mM NaCl, and 1% SDS, and crosslinks were reversed overnight at 65°C. Input samples were treated in a similar manner. The following day, the IP and Input samples were incubated with RNaseA (Thermo Fisher Scientific, EN0531) and proteinase K (Thermo Fisher Scientific, AM2546). IP or Input DNA was purified using a QIA quick PCR purification kit (QIAGEN, 28104).
ChIP‐seq libraries were prepared using a KAPA Hyper Prep Kit (KAPA, KK8504) following the manufacturer’s guidelines. An adaptor kit (Fastgene, FG‐NGSAD24) was used for the sample indexes. The average size and concentration of libraries were analyzed using LabChIP GX (PerkinElmer) and a KAPA library quantification Kit (KAPA, KK4824), respectively. Libraries were sequenced as 75 bp single‐end reads on an Illumina NextSeq 500/550 platform with a NextSeq 500/550 High Output kit (75 cycles) (Illumina, 20024906).
ATAC‐seq library preparation and sequencing
The ATAC‐seq experiment was performed as described previously (Buenrostro et al, 2013; Corces et al, 2017) with minor modifications. We used FACs‐sorted viable cells for mESCs and EpiLCs; FACS‐sorted BV‐positive cells for d2 mPGCLCs, d4 mPGCLCs, and d4c7 mPGCLCs; and FACS‐sorted AAG‐positive cells for GSCs and GSCLCs. 50,000 cells were permeabilized in cold lysis buffer 1 (10 mM Tris–HCl pH8.0, 10 mM NaCl, 3 mM MgCl2, 0.1% NP‐40, 0.1% Tween20, and 0.1% Digitonin; Promega, G9441) for 3 min followed by addition of 1 ml of cold lysis buffer 2 (10 mM Tris–HCl pH8.0, 10 mM NaCl, 3 mM MgCl2, and 0.1% Tween20). Nuclei were centrifuged and resuspended with 50 ml of transposase reaction mixture (25 μl of 2×TD buffer (Illumina, 20034197), 2.5 ml of Transposase (Illumina, 20034197), 16.5 ml of PBS, 0.5 ml of Digitonin, and 0.5 ml of Tween‐20, 5 μl of DDW). After incubation at 37°C for 30 min, the tagged DNA was purified using a Minelute PCR purification kit (QIAGEN, 28004). The purified DNA was amplified for 8 cycles by a PCR reaction (NEB, M0541S) followed by size selection using AMPure XP beads (Corning, MAG‐PCR‐CL‐250) to remove primer dimers. Libraries were sequenced as 2 × 75 bp paired‐end reads on an Illumina NextSeq 500/550 platform with a NextSeq 500/550 Mid Output Kit (150 cycles) (Illumina, 20024904) or NextSeq 500/550 High Output Kit (150 cycles, 20024907) (Illumina).
In situ Hi‐C library preparation and sequencing
In situ Hi‐C library preparation was performed as described previously (Rao et al, 2014; Belaghzal et al, 2017) with minor modifications. We used the whole harvested cells for mESCs and EpiLCs; FACS‐sorted BV‐positive cells for d2 mPGCLCs and d4c7 mPGCLCs; and FACS‐sorted AAG‐positive cells for GSCs and GSCLCs. 2.5 × 106 cells were used for one replicate. The cells were fixed by 1% formaldehyde (Sigma, 252549)/HBSS and lysed in lysis buffer (10 mM Tris–HCl pH 8.0, 10 mM NaCl, 0.2% NP‐40) for 30 min on ice with frequent inversion. The cells were digested by 500 U of DpnII (NEB, R0543L) overnight at 37°C. Following biotin filling (Thermo Fisher Scientific, 19524‐016; NEB, M0210S), proximity ligation (Thermo Fisher Scientific, 15224090) and reverse crosslinking, DNA was purified by ethanol precipitation and sheared to 200–400 bp fragments using a Covaris E220 sonicator (Covaris) at 4°C (10% Duty Factor, 200 cycles/burst, 175 W Peak Incident Power, 110 s). Ligation fragments containing biotin were immobilized on MyOne Streptavidin T1 beads (Thermo Fisher Scientific, 65001) followed by library preparation using an NEB library preparation kit (NEB, E7645S; NEB, E7335S) according to the manufacturer’s guidelines. The libraries were amplified in 8 cycles and DNA fragments of 300–800 bp were selected using AMPure XP beads (Corning, MAG‐PCR‐CL‐250). Libraries were sequenced as 2 × 100 bp paired‐end reads on an Illumina NovaSeq 6000 platform with a NovaSeq 6000 S1 Reagent Kit (200 cycles) (Illumina, 20012864).
NET‐CAGE library preparation and sequencing
Native elongating transcript–cap analysis of gene expression library preparation was performed as described previously (Hirabayashi et al, 2019) with minor modifications. For extraction of nascent RNA, cells were first lysed with 1,400 μl of Buffer A, which is Nuclei EZ Lysis Buffer (Sigma, NUC101‐1KT) supplemented with 25 μM α‐amanitin (Wako, 1022961), 1×cOmplete Protease Inhibitor Cocktail (Roche, 4693116001) and SUPERase•IN RNase Inhibitor (20 units; Thermo Fisher Scientific, AM2694), and then incubated on ice for 10 min and centrifuged at 800 g for 5 min at 4°C followed by washing once with the same buffer. Washed pellets were resuspended in 200 μl of Buffer B, containing 1% NP‐40, 20 mM HEPES pH 7.5, 300 mM NaCl, 2 M urea, 0.2 mM EDTA, 1 mM dithiothreitol (DTT) (Promega, P1171), 25 μM α‐amanitin, 1×cOmplete Protease Inhibitor Cocktail and SUPERase•IN RNase Inhibitor (20 units), and incubated for 10 min on ice. The suspension was centrifuged at 3,000 g for 2 min at 4°C. After removing the supernatant, the nuclear insoluble fraction was washed once with 100 μl of Buffer B. DNase I solution (50 μl) containing DNase I (10 units; Thermo Fisher Scientific, 89836), 1×DNase I Buffer (Thermo Fisher Scientific) and SUPERase•IN RNase Inhibitor (20 units) was added to the pellets. The samples were incubated for 30 min at 37°C while being pipetted up and down several times at 10‐min intervals. QIAzol (700 μl) was then added and the solution was thoroughly mixed. RNA was extracted with a miRNeasy Mini kit (QIAGEN, 217004) according to the manufacturer’s instructions. On‐column DNase I digestion was carried out with an RNase‐free DNase set (QIAGEN, 79254). RNA was eluted in 30 μl RNase‐free water, and its quality and quantity were measured with a Qubit RNA HS assay kit (Thermo Fisher Scientific, Q32855) and 2100 BioAnalyzer (Agilent). cDNA was synthesized from 200 ng of nascent RNA. CAGE libraries were generated according to the no amplification non‐tagging CAGE libraries for Illumina next‐generation sequencers (nAnT‐iCAGE) protocol (Murata et al, 2014) with PCR amplifications (Takara, R060A). All CAGE libraries were sequenced in 75 bp single‐end reads on an Illumina NextSeq 500 platform.
ChIP‐seq data processing
Single‐end reads were processed using Trim‐Galore! v0.4.1/cutadapt v1.9.1 (Martin, 2011; Krueger et al, 2021) to remove adaptor sequences. The truncated reads were then aligned to (GRCm38p3) using Bowtie2 v2.3.4.1 (Langmead & Salzberg, 2012) with the “‐very‐sensitive” option. Reads aligned to chromosomes 1–19, X, and Y were converted to the BAM format by SAMtools v1.7 (Li et al, 2009). BED files were obtained from the BAM files using the bamtobed command of BEDTools v2.29.2 (Quinlan & Hall, 2010). BigWig files were generated from the BAM files using bamcoverage for raw count with the “‐‐normalizeUsing CPM ‐bs 25” or bamcompare for IP/Input command with the “‐‐pseudocount 1 ‐bs 1000” option of deepTools v3.5.0 (Ramirez et al, 2016) In both cases, the blacklist regions (Amemiya et al, 2019) were excluded.
The regions enriched by epigenetic marks were identified using peak calling tools. For CTCF peaks, MACS v2.1.1 (Zhang et al, 2008) was used with the “‐q 0.01 ‐‐nomodel ‐‐keep‐dup all ‐‐extsize 200” option. For H3K9me3 domains, epic2 v0.0.41 (Stovner & Saetrom, 2019) was used with “‐kd ‐fdr 0.01” option. The number of IP or Input reads in 10/25/50/100 kb genomic windows were counted by the intersect command of BEDTools v2.29.2, and normalized by the total million mapped reads (FPM) and transformed to Log2(IP/Input) for the downstream analysis. The bins in which no reads were detected in the Input samples were excluded.
ATAC‐seq data processing
Assay for transposase‐accessible chromatin with deep sequencing data processing including public data was performed as previously described (Buenrostro et al, 2013) with minor modifications. First, adaptor sequences were trimmed from the reads using TrimGalore! v0.4.1/cutadapt v1.9.1. These reads were aligned using Bowtie2 v2.3.4.1 to GRCm38p3 with the “‐‐very‐sensitive ‐X 2000” option. The properly mapped reads with the flag (99, 147, 83 or 163) were extracted by awk, and mitochondrial reads were excluded. Duplicated reads were removed using the MarkDuplicates command of Picard Tools v2.18.23 (https://broadinstitute.github.io/picard/). These de‐duplicated reads were then filtered for high quality (MAPQ ≧ 30). The reads with an insert size of less than 100 bp were extracted as nucleosome‐free region (NFR) reads. Bed files for downstream analysis were generated by the bamtobed command of BEDTools v2.29.2 with the “‐bedpe” option. BigWig files were generated from the BAM files using bamcoverage for raw count with the “‐‐normalizeUsing CPM ‐bs 25” option of deepTools v3.5.0. The blacklist regions (https://www.encodeproject.org/files/ENCFF999QPV/) were excluded.
Peak calling was performed using MACS v2.1.1 with the “‐‐nomodel ‐‐shift −100 ‐‐extsize 200 ‐‐keep‐dup all” option after shifting NFR reads with the offset by +4 bp in the + strand and by −5 bp in the − strand. Then, confident peak sets in each cell type were obtained by the IDR method (https://www.encodeproject.org/software/idr/) using two replicates.
PBAT data processing
Public read data processing of the methylation levels was performed as described previously (Shirane et al, 2016). In brief, all reads were processed with Trim‐Galore! v0.4.1/cutadapt v1.9.1 with the “‐‐clip_R1 4,” “‐‐trim1” and “‐a AGATCGGAAGAGC” options. Output reads were mapped onto the mouse genome, GRCm38.p6, using Bismark v0.22.1 (Krueger & Andrews, 2011)/Bowtie2 v2.3.4.1 with the "‐‐pbat" option. All public WGBS data were obtained from DDBJ or NCBI SRA ftp sites and processed as described above. Conversion rates were calculated as follows: output reads after Trim‐Galore were mapped onto the lambda phage DNA sequence using Bismark v0.22.1/Bowtie2 v2.3.4.1 with the "‐‐pbat" option. From the Bismark's statistics, conversion rates were determined as 1 − ([total mC counts] / [total C and mC counts]). All CpG sites with a read depth of between 4 and 200 were used for the %mC calculations.
3‐prime RNA sequencing data processing
Raw 3′ RNA‐seq data were directly used with Salmon v1.4.0 (Patro et al, 2017) with default parameters and ‐‐noLengthCorrection to quantify the expression of GENCODE vM25 features on GRCm38.p6. Gene‐level expression estimates were aggregated from transcript‐level abundance using tximport v1.16.1 (Soneson et al, 2015).
In situ Hi‐C data processing
Sequences were first trimmed using fastp v0.21.0 (Chen et al, 2018) with default options and the ‐‐detect_adapter_for_pe flag. Trimmed sequences were then processed using HiCUP v0.8.0 (Wingett et al, 2015) with default options and the di‐tag length range set to 0–800, with bowtie v2.4.2 as the aligner. hicup2juicer was then used to produce pairs files, which were subsequently ingested with Juicer tools v1.22.01 (Durand et al, 2016) for the creation of .hic files. The same set of pairs files was also used to create multi‐resolution cooler files using cooler v0.8.10 (Abdennur & Mirny, 2020) with default options. Additionally, HiCSR commit b13ac41 (preprint: Dimmick et al, 2020) was used to de‐noise 10 kb‐resolution contact maps for visualization. In particular, pooled mESC data from (Bonev et al, 2017) after 10× down‐sampling were used for training with default parameters; the inference was then performed using default parameters. FAN‐C v0.9.13 (Kruse et al, 2020) was finally used for the normalization (with default parameters) and subsequent visualization of the enhanced 10 kb matrices, including virtual 4C profiles.
NET‐CAGE data processing
Sequences were first trimmed using fastp v0.21.0 and then aligned with STAR 2.7.6a (Dobin et al, 2013) using default options. Uniquely mapped reads were converted to coverage bigWig tracks with G‐bias correction using CAGEr v1.32.0 (Haberle et al, 2015) with default options. Tag clusters were identified using CAGEfightR v1.7.6 (Thodberg et al, 2019) with pooledCutoff = 0.1 and mergeDist = 20 for unidirectional clusters as well as balanceThreshold = 0.8 for bidirectional clusters. These clusters were subsequently filtered to require at least 1 sample demonstrating an expression level exceeding 1 TPM. Unidirectional clusters (putative promoters) were removed if they overlapped bidirectional clusters (putative enhancers), and the two region sets were subsequently combined to identify coordinately regulation enhancer‐promoter co‐transcription across stages. In particular, Kendall correlation was used to find putative enhancers within 1 mb of putative promoters that exhibited correlated expression patterns, with TPM as the expression unit.
Global Hi‐C metrics
HiCRes v1.1 (Marchal et al, 2022) with default parameters was used to calculate the resolution of contact maps following the definition in (Rao et al, 2014). Matrix similarity scores were computed using HiCRep.py v0.2.3 (Lin et al, 2021) with ‐‐binSize = 50000 ‐‐dBPMax = 5000000 ‐‐h = 3. Contact probability decay (i.e., the average contact frequency across different genomic separation distances) was assessed using the compute‐expected and logbin‐expected modules from cooltools v0.4.0 (Venev et al, 2021) at all resolutions, in both cis and trans. 3D models of individual chromosomes were produced using CSynth commit 26e21fb (Todd et al, 2021) with balanced 50 kb cis matrices, whose coordinates are normalized to achieve unit backbone length (i.e., the sum of Euclidean distance between adjacent beads being 1); and the size of these predicted structures are taken to be the volume of their 3D convex hulls.
Compartment‐related analysis
For analyses involving data across multiple studies, eigendecomposition was performed at 100 kb resolution using the call‐compartments module from cooltools v0.4.0 with GC content for orientating the track sign to achieve a positive correlation. For analyses strictly focusing on data generated within this study, dcHiC commit 7b1727f (preprint: Wang et al, 2021) was used with default parameters to perform simultaneous compartment score calculation across all samples at 50 kb resolution to facilitate statistical comparison across cell types while integrating replicate data. Though the values produced by dcHiC showed high correlation with those generated by cooltools, dcHiC was not applied to public datasets due to a lack of replication in certain datasets. Quantile‐binned saddle plots were produced using dcHiC‐generated compartment scores and the outputs of compute‐expected described above at 50 kb resolution. Binarization of compartment score tracks was carried out using A := score > 0 and B := score < 0. PCA of compartment scores to contrast lineages was done using 100 kb resolution data and bins non‐masked in all samples. The average size of compartments was assessed using an auto‐correlation function, where the signal profile is shifted and correlated against the original, using the acf function from R library stats 4.0.3 with na.action = na.pass.
Subcompartment‐related analysis
8‐state subcompartment labels were assigned to 50 kb bins with balanced contact frequencies using CALDER commit 32220e8 (Liu et al, 2021). The strength of epigenetic signals in each subcompartment was subsequently examined by converting enrichment values to Z‐scores genome‐wide, after which the average across all bins with the same label was computed. Significant differences in subcompartment proportions were evaluated using the prop.test function from R library stats 4.0.3.
TAD‐related analysis
Insulation scores were computed at 10 kb resolution with a window size of 100 kb using the diamond‐insulation module of cooltools v0.4.0. Consensus TADs in each dataset were derived by taking the set of bins with boundary prominence scores > 0.2 in at least half the cell types present and subsequently pairing neighboring boundaries, with those exceedingly 2 mb filtered out, consistently yielding ~4,000–5,000 domains for each dataset. The significance and strength of TAD–TAD interactions were evaluated using a non‐central hypergeometric (NCHG) test implemented as a part of the Chrom3D pipeline (Paulsen et al, 2017). Biological replicates (the two deepest ones in case there were more than two) were then used to identify highly reproducible TAD–TAD interactions using IDR2D v1.4.0 (Krismer et al, 2020) with default parameters. In particular, TAD–TAD interactions with NCHG P > 0.01 were first filtered out, and then the odds ratio was used as the ranking statistic for IDR analysis, with the final filter criteria being IDR P < 0.01. Treating significant TAD–TAD interactions as edges of a graph, cliques were identified using the max_cliques function from R library igraph v1.2.6 (Csardi & Nepusz, 2006). The over‐representation of A–A versus B–B clique interactions was compared against an expected value based on the proportion of A versus B TADs across all TADs, with the identity of compartment assignment of TADs based on having more 25 kb bins labeled as one compartment versus the other. Confidence intervals were derived from bootstrapping the set of clique interactions. The degree of TAD boundary conservation was evaluated using a permutation test, where the number of boundaries being shared across cell types was compared against a background derived from merging the list of boundaries and shuffling cell type labels. Additionally, 9 other TAD identification algorithms (Levy‐Leduc et al, 2014; Rao et al, 2014; Shin et al, 2016; preprint: Dali et al, 2018; An et al, 2019; Cresswell et al, 2020; Matthey‐Doret et al, 2020; Soler‐Vila et al, 2020; Xing et al, 2021) were used with default parameters to validate trends observed with insulation scores, all at 50 kb resolution.
Histone mass spectrometry analysis
Single histone modification abundances are summed from their individual occurrences as well as co‐occurrences (e.g., H3K27me3 = H3K27me3 + H3K27me3&H3K36me1 + H3K27me3&H3K36me2 + H3K27me3&H3K36me3). PCA of these relative abundance measures for all quantifiable H3 modifications (at least one sample exhibiting abundance > 0.1%) was used as input for PCA using the prcomp function from the R library stats v4.0.3 with default parameters to assess epigenome‐wide tendencies. Abundance measures were further Z‐score transformed for hierarchical clustering using the hclust function from R library stats v4.0.3 with default parameters.
Normalization of epigenetic signals
Histone mass spectrometry‐derived abundances were used to scale corresponding ChIP‐seq tracks by directly multiplying the library‐size normalized (counts/million mapped reads) values with the relative abundance. For targets lacking mass spectrometry data (e.g., transcription factors), we applied S3V2‐IDEAS commit b7cc2d5 (Xiang et al, 2021) to derive scaling factors using default parameters at a bin size of 200 bp.
ATAC‐seq analysis
The union set of peaks across all cell types was taken as features against which reads were counted, and the resulting count matrix was further normalized via FPKM to account for variations in peak widths and sequence depth. PCA was then performed on the 10,000 most variable peaks to assess global accessome trends. The 2,000 most variable peaks were additionally clustered using the hclust function from R library stats v4.0.3 with default parameters; visual inspection of the resulting dendrogram suggested 7 as a reasonable number of clusters for cutting. Global openness was assessed by first fitting a two‐component Gaussian mixture model to the log2(FPKM + 1) distribution across the union peak set and then assessing the number of sites exceeding the higher component’s mean versus those below the lower component’s mean.
Motif enrichment analysis
Over‐representation of known transcription factor motifs was assessed in an ensemble manner by combining multiple frameworks (e.g., HOMER and MEME) as implemented in GimmeMotifs v0.15.3 (preprint: Bruse & Heeringen, 2018) using default options. Differential enrichment of motifs between different region sets (e.g., open sites with distinct chromatin states) was examined using the maelstrom module of GimmeMotifs with default options.
Enhancer‐promoter pairing
Cis‐regulatory elements were associated with putative target genes using “activity‐by‐contact” (ABC) commit 7fd69b0 (Fulco et al, 2019). KR‐normalized matrices at 5 kb resolution were combined with H3K27ac and ATAC‐seq data to calculate ABC scores quantile‐normalized to K562 data, after which a stringent cut‐off of 0.02 was applied—corresponding to 70% recall and 60% precision based on previous CRISPRi‐FlowFISH validation (Fulco et al, 2019). Alternatively, enhancer‐promoter pairs identified based on co‐regulated NET‐CAGE tag clusters, as described above, were assessed for their degree of coordination. Specifically, a permutation test was used to compare the number of co‐expressed (> 1 TPM in a specific cell type) enhancer‐promoter pairs versus that of background sets generated by sampling from all tag clusters. Differential interactions between enhancer‐promoter pairs identified by ABC scores were investigated using R library HiCDCPlus v0.99.12 (Sahin et al, 2021) using default parameters at 10 kb resolution. The degree of coordinated differential promoter interaction and differential expression was quantified through the application of RRHO2 v1.0 (Cahill et al, 2018) to gene lists ranked by DESeq2 test statistics; for promoters involved in multiple ABC E‐P pairs, the mean test statistic was used for ranking.
ChIP‐seq analysis
The domain size distributions of histone modifications were determined using MCORE (Molitor et al, 2017) with the maximum shift size set to the chromosome lengths and other parameters kept at their defaults. The resulting cross‐correlation values between replicates were averaged using a cubic spline via the function smooth.spline from R library stats v4.0.3 with default parameters, after which Gardner transformations were applied to decompose the decay spectrum into component exponential functions corresponding to different domain sizes and quantify their contribution. Differential ChIP‐seq analysis was performed using DiffBind (Ross‐Innes et al, 2012) for targets with narrow signals and csaw for broad ones. DiffBind v3.0.13 was applied with union peak sets resized to 500 bp around the summits of MACS peak calls and other options kept at their defaults using both edgeR (Robinson et al, 2010) and DESeq2 (Love et al, 2014) for the underlying statistical framework, after which only concordant results were retained (e.g., up‐regulated with both methods). Unless otherwise stated, “constitutive”/ “conserved” peaks refer to the intersection of MACS peak calls between cell types. csaw v1.24.3 (Lun & Smyth, 2016) was applied with default settings with edgeR as the underlying statistical framework at both a coarse (2 kbp windows with a 500 bp step size for H3K27me3 and 10 kbp windows with a 2 kbp step size for H3K9me2) and a fine resolution (500 bp windows with a 100 bp step size for H3K27me3 and 1 kb windows with a 200 bp step size for H3K9me2), after which the results were consolidated, allowing for a gap size of 100 bp. The domain expansion/contraction kinetics were characterized using ChromTime commit a332dbb (Fiziev & Ernst, 2018) with default settings in broad mode, with a post‐hoc filter applied to exclude regions < 10 kb. Aggregate plots were generated using the module computeMatrix from deepTools v3.5.0 with default options, in the scale‐regions mode for domains and reference‐point mode for focal features such as peaks. Differential H3K9me3 promoters (± 1 kb from TSS) were defined using the mass spectrometry‐derived coefficient‐normalized log2‐transformed FPKM signal with the threshold (log2(FPKM) > 1 in either cell type and log2(FPKM) difference > 1).
Epigenome‐based clustering of cis‐regulatory elements
The log2(enrichment over input) values of ChIP‐seq signals and log2(FPKM + 1) for ATAC‐seq signals in promoters (± 2.5 kb from TSS) or reproducible accessible sites identified using ChromA v2.1.1 (Gabitto et al, 2020; resized to ± 500 bp surrounding the summit) were used as input for dimension reduction through UMAP v0.5.1 (Mcinnes et al, 2018) and subsequently clustered through HDBSCAN v0.8.27 (Campello et al, 2013). For UMAP, manhattan distances were used for promoters and correlation distances for open sites; a grid search over min_dist of [0.0, 0.01, 0.1], n_neighbors of [15, 30, 50] and n_components of 2–10 were all subjected to HDBSCAN clustering to identify epigenetically distinct clusters via visual inspection. For HDBSCAN, a grid search over min_cluster_size and min_samples over [50, 100, 200, 500, 1,000, 2,000, 5,000, and 10,000] were tested. In a semi‐supervised fashion, individual clusters were isolated and subjected to further sub‐clustering until the embedding no longer exhibited distinct segregation of data points for any individual epigenetic signal.
Pathway enrichment analysis
Associations of specific gene lists with particular biological pathways were evaluated using the gost function from R library gprofiler2 v0.2.0 (Kolberg et al, 2020) with default options. The enrichment of pathways towards the extremes of ranked gene lists, on the other hand, was assessed using the fgseaMultilevel function from R library fgsea 1.17.1 (preprint: Korotkevich et al, 2021) with the boundary parameter eps set to 0 and others kept at their default values; redundant terms were collapsed using collapsePathways with an adjusted p‐value threshold of 0.05. To obtain gene lists ranked by multiple metrics (e.g., differential expression and promoter interaction), the mean test statistic was used to rank genes independently for each metric, and an aggregated ranking was then obtained using p‐values produced by the aggregateRanks function from the R library RobustRankAggreg v1.1 (Kolde et al, 2012).
Overlap enrichment analysis
The overlap between genomic regions and annotated intervals was examined using Fisher’s exact tests as implemented in the R library LOLA v1.19.1 (Sheffield & Bock, 2016). Ensembl Regulatory build annotations v20180516 were sourced directly from Ensembl; RepeatMasker annotations were obtained from the rmsk table hosted on the UCSC Genome Browser. ENCODE cCRE annotations were downloaded from SCREEN v13 (http://screen.encodeproject.org/).
Pile‐up analysis
Interaction between specific regions (e.g., promoters of a similar chromatin state) was quantified using the ObsExpSnipper function from cooltools v0.4.0 with default parameters and using the aforementioned diagonal‐wise expected values. For pile‐up of domains (e.g., TADs or broad H3K9me3 domains) rescaled to the same size, coolpup.py v0.9.7 (Flyamer et al, 2020) was used with the option ‐‐rescale and optionally ‐‐local when assessing on‐diagonal patterns, and with all other options kept at their defaults.
Lamin B1‐related analysis
EDD v1.1.19 (Lund et al, 2014) was used to identify lamina‐associated domains from lamin B1 ChIP‐seq with a bin size of 10 kb, gap penalty set to 20, and all other options kept at their defaults. LADetector v8122016 (Harr et al, 2015) was used instead for lamin B1 DamID, with a bin size of 10 kb and max dip size of 25 kb. Generalized linear models with 50 basis functions were used to visualize chromosome‐scale patterns using REML for smoothness selection as implemented in the gam function of R library mgcv v1.8‐31 (Wood, 2011).
Partially methylated domains‐related analysis
Partially methylated domains were identified by calculating median mCG/CG values using a 100 kb sliding window and identifying those falling below 85%; after merging adjacent regions, those wider than 500 kb were called as PMDs. The binary status of whether a bin falls within a GSC PMD or not was modeled using three methods: (i) gradient boosted tree (gbm), (ii) neural network (nnet), and (iii) elastic net (glmnet), each with 10 × 10 cross‐validation using a 70/30 train/test split as implemented in the R library caret v6.0‐86 (Kuhn, 2008). Model performance for predicting PMDs was then assessed on the held‐out test set using the roc function from R library pROC v1.16.2 (Robin et al, 2011).
Mapping to the Y chromosome
Ampliconic sequences on the murine Y chromosome were retrieved from an earlier report describing its assembly (Soh et al, 2014), and were directly used as the reference for alignment. Otherwise, data was processed as described in “PBAT data processing.”
Statistical considerations
P‐values were mapped to symbols as follows: **** = [0, 0.0001); *** = [0.0001, 0.001]; ** = [0.001, 0.01); * = [0.01, 0.05); ns = [0.05, 1]. Wilcoxon rank‐sum tests and T‐tests were carried out using the functions wilcox.test and t.test, respectively, from the R library stats v4.0.3. Bootstrap confidence intervals were computed using the function boot with 100,000 replicates followed by boot.ci from the R library boot 1.3‐28 (Davison & Hinkley, 1997) using default options. For all box plots (i.e., box‐and‐whiskers plots), the lower and upper hinge correspond to the first and third quartile, and the upper whiskers extend to the largest value % 1.5 * IQR and vice versa for the lower whiskers.
Author contributions
Mitinori Saitou: Conceptualization; Resources; Supervision; Funding acquisition; Validation; Investigation; Methodology; Writing—original draft; Project administration; Writing—review and editing. Masahiro Nagano: Conceptualization; Data curation; Formal analysis; Funding acquisition; Validation; Investigation; Visualization; Methodology; Writing—original draft; Writing—review and editing. Bo Hu: Conceptualization; Formal analysis; Validation; Investigation; Visualization; Methodology; Writing—original draft; Writing—review and editing. Shihori Yokobayashi: Conceptualization; Data curation; Supervision; Funding acquisition. Akitoshi Yamamura: Data curation. Fumiya Umemura: Data curation. Mariel Coradin: Data curation; Formal analysis. Hiroshi Ohta: Data curation; Supervision. Yukihiro Yabuta: Formal analysis; Supervision. Yukiko Ishikura: Data curation; Supervision. Ikuhiro Okamoto: Data curation; Supervision. Hiroki Ikeda: Data curation; Supervision. Naofumi Kawahira: Formal analysis; Supervision. Yoshiaki Nosaka: Data curation. Sakura Shimizu: Data curation. Yoji Kojima: Data curation; Supervision. Ken Mizuta: Data curation. Tomoko Kasahara: Data curation. Yusuke Imoto: Formal analysis. Killian Meehan: Formal analysis. Roman Stocsits: Formal analysis; Supervision. Gordana Wutz: Formal analysis; Supervision. Yasuaki Hiraoka: Formal analysis; Supervision. Yasuhiro Murakawa: Data curation; Supervision. Takuya Yamamoto: Data curation; Formal analysis; Supervision. Kikue Tachibana: Supervision; Funding acquisition. Jan‐Michel Peters: Supervision; Funding acquisition. Leonid A Mirny: Supervision; Funding acquisition. Benjamin A Garcia: Data curation; Formal analysis; Supervision; Funding acquisition. Jacek Majewski: Supervision.
In addition to the CRediT author contributions listed above, the contributions in detail are:
MN, BH, SY, and MS conceived the project and designed experiments. MN performed all cell cultures and inductions with assistance from HO, YIs. and YN. MN performed immunofluorescence and its analysis with assistance from HO, NK, and KM. MN and FU performed western blot and its analysis with assistance from YN, SS, and YK. MN performed FISH with assistance from IO. MN performed histone extraction and MC performed mass spectrometry under the supervision of BAG. MN and AY performed ChIP‐seq with assistance from SY and TY. MN performed ATAC‐seq with assistance from SY, HI, and TY. MN performed in situ Hi‐C with assistance from SY, RS, GW, KT, J‐MP, and LAM. MN performed NET‐CAGE with assistance from TK under the supervision of YM. MN and BH performed all data analysis with assistance from YY and JM. BH performed polymer simulation and analysis with assistance from Y. Imoto., KM, and YH. MN, BH, and MS wrote the manuscript with input from all co‐authors. SY, JM, and MS supervised the project.
Disclosure and competing interests statement
M.S. is an EMBO Associate Member.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Dataset EV6
Movie EV1
Acknowledgements
We thank the members of our laboratory for their helpful input on this study. We are grateful to Y. Nagai, N. Konishi, E. Tsutsumi, and M. Kawasaki of the Saitou Laboratory, to the DNAFORM genetic analysis department for NET‐CAGE library preparation and sequencing, to the Single‐Cell Genome Information Analysis Core (SignAC) in ASHBi for their technical assistance and help with all the other sequencing experiments, to R. Maeda of the Tachibana Laboratory for helpful suggestions for ChIP‐seq, to S. Nagaoka and K. Kurimoto of the Kurimoto Laboratory and G. Bourque of the Bourque Laboratory for thoughtful discussions on the data analysis, and to C. Horth from the Majewski Laboratory for her assistance with histone extraction. This work was supported in part by a Grant‐in‐Aid for Specially Promoted Research from JSPS (17H06098, 22H04920), a JST‐ERATO Grant (JPMJER1104), a Grant from HFSP (RGP0057/2018), Grants from the Pythias Fund and Open Philanthropy Project (2018‐193685) to M.S., JSPS KAKENHI Grants (JP18H02613, JP20H05387) to S.Y., and NIH grants (CA196539, NS111997) to B.A.G. M.N. is a fellow of the Takeda Science Foundation. B.H. is supported by studentship awards from the Canadian Institutes of Health Research and the Fonds de recherche du Québec – Santé.
The EMBO Journal (2022) 41: e110600.
Data availability
The accession number for all the sequencing data generated in this study is GSE183828 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE183828) (the GEO database). Scripts used to generate the presented results and additional raw data underlying figures are available at: https://github.com/bhu/germ_nucleome.
References
- Abdennur N, Mirny LA (2020) Cooler: scalable storage for Hi‐C data and other genomically labeled arrays. Bioinformatics 36: 311–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alavattam KG, Maezawa S, Sakashita A, Khoury H, Barski A, Kaplan N, Namekawa SH (2019) Attenuated chromatin compartmentalization in meiosis and its maturation in sperm development. Nat Struct Mol Biol 26: 175–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amemiya HM, Kundaje A, Boyle AP (2019) The ENCODE blacklist: identification of problematic regions of the genome. Sci Rep 9: 9354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- An L, Yang T, Yang J, Nuebler J, Xiang G, Hardison RC, Li Q, Zhang Y (2019) OnTAD: hierarchical domain structure reveals the divergence of activity among TADs and boundaries. Genome Biol 20: 282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anton T, Bultmann S, Leonhardt H, Markaki Y (2014) Visualization of specific DNA sequences in living mouse embryonic stem cells with a programmable fluorescent CRISPR/Cas system. Nucleus 5: 163–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battulin N, Fishman VS, Mazur AM, Pomaznoy M, Khabarova AA, Afonnikov DA, Prokhortchouk EB, Serov OL (2015) Comparison of the three‐dimensional organization of sperm and fibroblast genomes using the Hi‐C approach. Genome Biol 16: 77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belaghzal H, Dekker J, Gibcus JH (2017) Hi‐C 2.0: An optimized Hi‐C procedure for high‐resolution genome‐wide mapping of chromosome conformation. Methods 123: 56–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian Q, Khanna N, Alvikas J, Belmont AS (2013) beta‐Globin cis‐elements determine differential nuclear targeting through epigenetic modifications. J Cell Biol 203: 767–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonev B, Mendelson Cohen N, Szabo Q, Fritsch L, Papadopoulos GL, Lubling Y, Xu X, Lv X, Hugnot J‐P, Tanay A et al (2017) Multiscale 3D genome rewiring during mouse neural development. Cell 171: 557–572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgel J, Guibert S, Li Y, Chiba H, Schubeler D, Sasaki H, Forne T, Weber M (2010) Targets and dynamics of promoter DNA methylation during early mouse development. Nat Genet 42: 1093–1100 [DOI] [PubMed] [Google Scholar]
- Boroviak T, Loos R, Bertone P, Smith A, Nichols J (2014) The ability of inner‐cell‐mass cells to self‐renew as embryonic stem cells is acquired following epiblast specification. Nat Cell Biol 16: 516–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourque G, Leong B, Vega VB, Chen XI, Lee YL, Srinivasan KG, Chew J‐L, Ruan Y, Wei C‐L, Ng HH et al (2008) Evolution of the mammalian transcription factor binding repertoire via transposable elements. Genome Res 18: 1752–1762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruse N, Heeringen SJV (2018) GimmeMotifs: an analysis framework for transcription factor motif analysis. bioRxiv 10.1101/474403 [PREPRINT] [DOI] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ (2013) Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA‐binding proteins and nucleosome position. Nat Methods 10: 1213–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill KM, Huo Z, Tseng GC, Logan RW, Seney ML (2018) Improved identification of concordant and discordant gene expression signatures using an updated rank‐rank hypergeometric overlap approach. Sci Rep 8: 9588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campello R, Moulavi D, Sander J (2013) Density‐based clustering based on hierarchical density estimates. In Advances in Knowledge Discovery and Data Mining, Pei J, Tseng VS, Cao L, Motoda H, Xu G (eds), pp 160–172. Berlin: Springer; [Google Scholar]
- Chen S, Zhou Y, Chen Y, Gu J (2018) fastp: an ultra‐fast all‐in‐one FASTQ preprocessor. Bioinformatics 34: i884–i890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Yammine S, Shi C, Tark‐Dame M, Gondor A, Ohlsson R (2014) The visualization of large organized chromatin domains enriched in the H3K9me2 mark within a single chromosome in a single cell. Epigenetics 9: 1439–1445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott‐Armstrong NA, Vesuna S, Satpathy AT, Rubin AJ, Montine KS, Wu B et al (2017) An improved ATAC‐seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods 14: 959–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cresswell KG, Stansfield JC, Dozmorov MG (2020) SpectralTAD: an R package for defining a hierarchy of topologically associated domains using spectral clustering. BMC Bioinformatics 21: 319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csardi G, Nepusz T (2006) The igraph software package for complex network research. InterJournal, Complex Systems, 1695. [Online]. http://igraph.org
- Cuadrado A, Gimenez‐Llorente D, Kojic A, Rodriguez‐Corsino M, Cuartero Y, Martin‐Serrano G, Gomez‐Lopez G, Marti‐Renom MA, Losada A (2019) Specific contributions of cohesin‐SA1 and cohesin‐SA2 to TADs and polycomb domains in embryonic stem cells. Cell Rep 27: 3500–3510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dali R, Bourque G, Blanchette M (2018) RobusTAD: a tool for robust annotation of topologically associating domain boundaries. bioRxiv 10.1101/293175 [PREPRINT] [DOI] [Google Scholar]
- Davison AC, Hinkley DV (1997) Boostrap methods and their application. Cambridge University Press; [Google Scholar]
- Dekker J, Belmont AS, Guttman M, Leshyk VO, Lis JT, Lomvardas S, Mirny LA, O’Shea CC, Park PJ, Ren B et al (2017) The 4D nucleome project. Nature 549: 219–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giammartino DC, Kloetgen A, Polyzos A, Liu Y, Kim D, Murphy D, Abuhashem A, Cavaliere P, Aronson B, Shah V et al (2019) KLF4 is involved in the organization and regulation of pluripotency‐associated three‐dimensional enhancer networks. Nat Cell Biol 21: 1179–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimmick MC, Lee LJ, Frey BJ (2020) HiCSR: a Hi‐C super‐resolution framework for producing highly realistic contact maps. bioRxiv 10.1101/2020.02.24.961714 [PREPRINT] [DOI] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR (2013) STAR: ultrafast universal RNA‐seq aligner. Bioinformatics 29: 15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Oliveira Albanus R, Kyono Y, Hensley J, Varshney A, Orchard P, Kitzman JO, Parker SCJ (2021) Chromatin information content landscapes inform transcription factor and DNA interactions. Nat Commun 12: 1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Z, Zheng H, Huang BO, Ma R, Wu J, Zhang X, He J, Xiang Y, Wang Q, Li Y et al (2017) Allelic reprogramming of 3D chromatin architecture during early mammalian development. Nature 547: 232–235 [DOI] [PubMed] [Google Scholar]
- Du Z, Zheng H, Kawamura YK, Zhang KE, Gassler J, Powell S, Xu Q, Lin Z, Xu K, Zhou Q et al (2020) Polycomb group proteins regulate chromatin architecture in mouse oocytes and early embryos. Mol Cell 77: 825–839 [DOI] [PubMed] [Google Scholar]
- Durand NC, Shamim MS, Machol I, Rao SS, Huntley MH, Lander ES, Aiden EL (2016) Juicer provides a one‐click system for analyzing loop‐resolution Hi‐C experiments. Cell Syst 3: 95–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans EP, Ford CE, Lyon MF (1977) Direct evidence of the capacity of the XY germ cell in the mouse to become an oocyte. Nature 267: 430–431 [DOI] [PubMed] [Google Scholar]
- Farhangdoost N, Horth C, Hu B, Bareke E, Chen X, Li Y, Coradin M, Garcia BA, Lu C, Majewski J (2021) Chromatin dysregulation associated with NSD1 mutation in head and neck squamous cell carcinoma. Cell Rep 34: 108769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiziev P, Ernst J (2018) ChromTime: modeling spatio‐temporal dynamics of chromatin marks. Genome Biol 19: 109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flyamer IM, Gassler J, Imakaev M, Brandao HB, Ulianov SV, Abdennur N, Razin SV, Mirny LA, Tachibana‐Konwalski K (2017) Single‐nucleus Hi‐C reveals unique chromatin reorganization at oocyte‐to‐zygote transition. Nature 544: 110–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flyamer IM, Illingworth RS, Bickmore WA (2020) Coolpup.py: versatile pile‐up analysis of Hi‐C data. Bioinformatics 36: 2980–2985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda K, Shimura C, Miura H, Tanigawa A, Suzuki T, Dohmae N, Hiratani I, Shinkai Y (2021) Regulation of mammalian 3D genome organization and histone H3K9 dimethylation by H3K9 methyltransferases. Commun Biol 4: 571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulco CP, Nasser J, Jones TR, Munson G, Bergman DT, Subramanian V, Grossman SR, Anyoha R, Doughty BR, Patwardhan TA et al (2019) Activity‐by‐contact model of enhancer‐promoter regulation from thousands of CRISPR perturbations. Nat Genet 51: 1664–1669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabitto MI, Rasmussen A, Wapinski O, Allaway K, Carriero N, Fishell GJ, Bonneau R (2020) Characterizing chromatin landscape from aggregate and single‐cell genomic assays using flexible duration modeling. Nat Commun 11: 747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorkin DU, Barozzi I, Zhao Y, Zhang Y, Huang H, Lee AY, Li B, Chiou J, Wildberg A, Ding BO et al (2020) An atlas of dynamic chromatin landscapes in mouse fetal development. Nature 583: 744–751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griswold MD (2016) Spermatogenesis: the commitment to meiosis. Physiol Rev 96: 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guelen L, Pagie L, Brasset E, Meuleman W, Faza MB, Talhout W, Eussen BH, de Klein A, Wessels L, de Laat W et al (2008) Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature 453: 948–951 [DOI] [PubMed] [Google Scholar]
- Guenatri M, Bailly D, Maison C, Almouzni G (2004) Mouse centric and pericentric satellite repeats form distinct functional heterochromatin. J Cell Biol 166: 493–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurdon JB, Wilmut I (2011) Nuclear transfer to eggs and oocytes. Cold Spring Harb Perspect Biol 3: a002659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberle V, Forrest AR, Hayashizaki Y, Carninci P, Lenhard B (2015) CAGEr: precise TSS data retrieval and high‐resolution promoterome mining for integrative analyses. Nucleic Acids Res 43: e51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KD, Timp W, Bravo HC, Sabunciyan S, Langmead B, McDonald OG, Wen BO, Wu H, Liu Y, Diep D et al (2011) Increased methylation variation in epigenetic domains across cancer types. Nat Genet 43: 768–775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harr JC, Luperchio TR, Wong X, Cohen E, Wheelan SJ, Reddy KL (2015) Directed targeting of chromatin to the nuclear lamina is mediated by chromatin state and A‐type lamins. J Cell Biol 208: 33–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi K, Ohta H, Kurimoto K, Aramaki S, Saitou M (2011) Reconstitution of the mouse germ cell specification pathway in culture by pluripotent stem cells. Cell 146: 519–532 [DOI] [PubMed] [Google Scholar]
- Hirabayashi S, Bhagat S, Matsuki YU, Takegami Y, Uehata T, Kanemaru AI, Itoh M, Shirakawa K, Takaori‐Kondo A, Takeuchi O et al (2019) NET‐CAGE characterizes the dynamics and topology of human transcribed cis‐regulatory elements. Nat Genet 51: 1369–1379 [DOI] [PubMed] [Google Scholar]
- Hon GC, Hawkins RD, Caballero OL, Lo C, Lister R, Pelizzola M, Valsesia A, Ye Z, Kuan S, Edsall LE et al (2012) Global DNA hypomethylation coupled to repressive chromatin domain formation and gene silencing in breast cancer. Genome Res 22: 246–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikura Y, Ohta H, Sato T, Murase Y, Yabuta Y, Kojima Y, Yamashiro C, Nakamura T, Yamamoto T, Ogawa T et al (2021) In vitro reconstitution of the whole male germ‐cell development from mouse pluripotent stem cells. Cell Stem Cell 28: 2167–2179 [DOI] [PubMed] [Google Scholar]
- Ishikura Y, Yabuta Y, Ohta H, Hayashi K, Nakamura T, Okamoto I, Yamamoto T, Kurimoto K, Shirane K, Sasaki H et al (2016) In vitro derivation and propagation of spermatogonial stem cell activity from mouse pluripotent stem cells. Cell Rep 17: 2789–2804 [DOI] [PubMed] [Google Scholar]
- Jameson SA, Natarajan A, Cool J, DeFalco T, Maatouk DM, Mork L, Munger SC, Capel B (2012) Temporal transcriptional profiling of somatic and germ cells reveals biased lineage priming of sexual fate in the fetal mouse gonad. PLoS Genet 8: e1002575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone SE, Reyes A, Qi Y, Adriaens C, Hegazi E, Pelka K, Chen JH, Zou LS, Drier Y, Hecht V et al (2020) Large‐scale topological changes restrain malignant progression in colorectal cancer. Cell 182: 1474–1489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanatsu‐Shinohara M, Inoue K, Lee J, Yoshimoto M, Ogonuki N, Miki H, Baba S, Kato T, Kazuki Y, Toyokuni S et al (2004) Generation of pluripotent stem cells from neonatal mouse testis. Cell 119: 1001–1012 [DOI] [PubMed] [Google Scholar]
- Kanatsu‐Shinohara M, Ogonuki N, Inoue K, Miki H, Ogura A, Toyokuni S, Shinohara T (2003) Long‐term proliferation in culture and germline transmission of mouse male germline stem cells. Biol Reprod 69: 612–616 [DOI] [PubMed] [Google Scholar]
- Karimi M, Goyal P, Maksakova I, Bilenky M, Leung D, Tang J, Shinkai Y, Mager D, Jones S, Hirst M et al (2011) DNA methylation and SETDB1/H3K9me3 regulate predominantly distinct sets of genes, retroelements, and chimeric transcripts in mESCs. Cell Stem Cell 8: 676–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke Y, Xu Y, Chen X, Feng S, Liu Z, Sun Y, Yao X, Li F, Zhu W, Gao L et al (2017) 3D chromatin structures of mature gametes and structural reprogramming during mammalian embryogenesis. Cell 170: 367–381 [DOI] [PubMed] [Google Scholar]
- Keough KC, Shah PP, Gjoni K, Santini GT, Wickramasinghe NM, Dundes CE, Karnay A, Chen A, Salomon REA, Walsh PJ et al (2021) An atlas of lamina‐associated chromatin across twelve human cell types reveals an intermediate chromatin subtype. bioRxiv 10.1101/2020.07.23.218768 [PREPRINT] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolberg L, Raudvere U, Kuzmin I, Vilo J, Peterson H (2020) gprofiler2 – an R package for gene list functional enrichment analysis and namespace conversion toolset g:Profiler. F1000Research 9: 709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolde R, Laur S, Adler P, Vilo J (2012) Robust rank aggregation for gene list integration and meta‐analysis. Bioinformatics 28: 573–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korotkevich G, Sukhov V, Budin N, Shpak B, Artyomov MN, Sergushichev A (2021) Fast gene set enrichment analysis. bioRxiv 10.1101/060012 [PREPRINT] [DOI] [Google Scholar]
- Krismer K, Guo Y, Gifford DK (2020) IDR2D identifies reproducible genomic interactions. Nucleic Acids Res 48: e31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger F, Andrews SR (2011) Bismark: a flexible aligner and methylation caller for Bisulfite‐Seq applications. Bioinformatics 27: 1571–1572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger F, James F, Ewels P, Afyounian E, Schuster‐Boeckler B (2021) FelixKrueger/TrimGalore: v0.6.7. Zenodo.
- Kruse K, Hug CB, Vaquerizas JM (2020) FAN‐C: a feature‐rich framework for the analysis and visualisation of chromosome conformation capture data. Genome Biol 21: 303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo N, Toh H, Shirane K, Shirakawa T, Kobayashi H, Sato T, Sone H, Sato Y, Tomizawa S‐I, Tsurusaki Y et al (2015) DNA methylation and gene expression dynamics during spermatogonial stem cell differentiation in the early postnatal mouse testis. BMC Genom 16: 624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn M (2008) Building predictive models in R using the caret package. J Stat Soft 28: 1–26 [Google Scholar]
- Kurimoto K, Yabuta Y, Hayashi K, Ohta H, Kiyonari H, Mitani T, Moritoki Y, Kohri K, Kimura H, Yamamoto T et al (2015) Quantitative dynamics of chromatin remodeling during germ cell specification from mouse embryonic stem cells. Cell Stem Cell 16: 517–532 [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL (2012) Fast gapped‐read alignment with Bowtie 2. Nat Methods 9: 357–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Hore TA, Reik W (2014) Reprogramming the methylome: erasing memory and creating diversity. Cell Stem Cell 14: 710–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TI, Johnstone SE, Young RA (2006) Chromatin immunoprecipitation and microarray‐based analysis of protein location. Nat Protoc 1: 729–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy‐Leduc C, Delattre M, Mary‐Huard T, Robin S (2014) Two‐dimensional segmentation for analyzing Hi‐C data. Bioinformatics 30: i386–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R (2009) The sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin D, Sanders J, Noble WS (2021) HiCRep.py : Fast comparison of Hi‐C contact matrices in Python. Bioinformatics 37: 2996–2997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti‐Filippini J, Nery JR, Lee L, Ye Z, Ngo Q‐M et al (2009) Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462: 315–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Nanni L, Sungalee S, Zufferey M, Tavernari D, Mina M, Ceri S, Oricchio E, Ciriello G (2021) Systematic inference and comparison of multi‐scale chromatin sub‐compartments connects spatial organization to cell phenotypes. Nat Commun 12: 2439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 15: 550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun AT, Smyth GK (2016) csaw: a Bioconductor package for differential binding analysis of ChIP‐seq data using sliding windows. Nucleic Acids Res 44: e45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund E, Oldenburg AR, Collas P (2014) Enriched domain detector: a program for detection of wide genomic enrichment domains robust against local variations. Nucleic Acids Res 42: e92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Z, Wang X, Jiang H, Wang R, Chen J, Chen Y, Xu Q, Cao J, Gong X, Wu JI et al (2020) Reorganized 3D genome structures support transcriptional regulation in mouse spermatogenesis. iScience 23: 101034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchal C, Singh N, Corso‐Díaz X, Swaroop A (2022) HiCRes: a computational method to estimate and predict the resolution of HiC libraries. Nucleic Acids Res 50: e35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks H, Kalkan T, Menafra R, Denissov S, Jones K, Hofemeister H, Nichols J, Kranz A, Francis Stewart A, Smith A et al (2012) The transcriptional and epigenomic foundations of ground state pluripotency. Cell 149: 590–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011) Cutadapt removes adapter sequences from high‐throughput sequencing reads. Embnetjournal 17: 10 [Google Scholar]
- Matthey‐Doret C, Baudry L, Breuer A, Montagne R, Guiglielmoni N, Scolari V, Jean E, Campeas A, Chanut PH, Oriol E et al (2020) Computer vision for pattern detection in chromosome contact maps. Nat Commun 11: 5795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mcinnes L, Healy J, Saul N, Großberger L (2018) UMAP: uniform manifold approximation and projection. J Open Source Softw 3: 861 [Google Scholar]
- Miura K (2020) Measurements of intensity dynamics at the periphery of the nucleus. In Bioimage Data Analysis Workflows, Miura K, Sladoje N (eds), pp 9–32. Springer International Publishing. 10.1007/978-3-030-22386-1_2 [DOI] [Google Scholar]
- Miyauchi H, Ohta H, Nagaoka S, Nakaki F, Sasaki K, Hayashi K, Yabuta Y, Nakamura T, Yamamoto T, Saitou M (2017) Bone morphogenetic protein and retinoic acid synergistically specify female germ‐cell fate in mice. EMBO J 36: 3100–3119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki K, Sharif J, Shirane K, Uranishi K, Bogutz AB, Janssen SM, Suzuki A, Okuda A, Koseki H, Lorincz MC (2021) Repression of germline genes by PRC1.6 and SETDB1 in the early embryo precedes DNA methylation‐mediated silencing. Nat Commun 12: 7020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molitor J, Mallm JP, Rippe K, Erdel F (2017) Retrieving chromatin patterns from deep sequencing data using correlation functions. Biophys J 112: 473–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata M, Nishiyori‐Sueki H, Kojima‐Ishiyama M, Carninci P, Hayashizaki Y, Itoh M (2014) Detecting expressed genes using CAGE. Methods Mol Biol 1164: 67–85 [DOI] [PubMed] [Google Scholar]
- Nagaoka SI, Nakaki F, Miyauchi H, Nosaka Y, Ohta H, Yabuta Y, Kurimoto K, Hayashi K, Nakamura T, Yamamoto T et al (2020) ZGLP1 is a determinant for the oogenic fate in mice. Science 367: eaaw4115 [DOI] [PubMed] [Google Scholar]
- Ohta H, Kurimoto K, Okamoto I, Nakamura T, Yabuta Y, Miyauchi H, Yamamoto T, Okuno Y, Hagiwara M, Shirane K et al (2017) In vitro expansion of mouse primordial germ cell‐like cells recapitulates an epigenetic blank slate. EMBO J 36: 1888–1907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta H, Yabuta Y, Kurimoto K, Nakamura T, Murase Y, Yamamoto T, Saitou M (2021) Cyclosporin A and FGF signaling support the proliferation/survival of mouse primordial germ cell‐like cells in vitrodagger. Biol Reprod 104: 344–360 [DOI] [PubMed] [Google Scholar]
- Ohta H, Yomogida K, Yamada S, Okabe M, Nishimune Y (2000) Real‐time observation of transplanted 'green germ cells': proliferation and differentiation of stem cells. Dev Growth Differ 42: 105–112 [DOI] [PubMed] [Google Scholar]
- Okamoto I, Arnaud D, Le Baccon P, Otte AP, Disteche CM, Avner P, Heard E (2005) Evidence for de novo imprinted X‐chromosome inactivation independent of meiotic inactivation in mice. Nature 438: 369–373 [DOI] [PubMed] [Google Scholar]
- Patel L, Kang R, Rosenberg SC, Qiu Y, Raviram R, Chee S, Hu R, Ren B, Cole F, Corbett KD (2019) Dynamic reorganization of the genome shapes the recombination landscape in meiotic prophase. Nat Struct Mol Biol 26: 164–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C (2017) Salmon provides fast and bias‐aware quantification of transcript expression. Nat Methods 14: 417–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen J, Liyakat Ali TM, Nekrasov M, Delbarre E, Baudement MO, Kurscheid S, Tremethick D, Collas P (2019) Long‐range interactions between topologically associating domains shape the four‐dimensional genome during differentiation. Nat Genet 51: 835–843 [DOI] [PubMed] [Google Scholar]
- Paulsen J, Sekelja M, Oldenburg AR, Barateau A, Briand N, Delbarre E, Shah A, Sørensen AL, Vigouroux C, Buendia B et al (2017) Chrom3D: three‐dimensional genome modeling from Hi‐C and nuclear lamin‐genome contacts. Genome Biol 18: 21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peric‐Hupkes D, Meuleman W, Pagie L, Bruggeman SWM, Solovei I, Brugman W, Gräf S, Flicek P, Kerkhoven RM, van Lohuizen M et al (2010) Molecular maps of the reorganization of genome‐nuclear lamina interactions during differentiation. Mol Cell 38: 603–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poleshko A, Shah PP, Gupta M, Babu A, Morley MP, Manderfield LJ, Ifkovits JL, Calderon D, Aghajanian H, Sierra‐Pagán JE et al (2017) Genome‐nuclear lamina interactions regulate cardiac stem cell lineage restriction. Cell 171: 573–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poleshko A, Smith CL, Nguyen SC, Sivaramakrishnan P, Wong KG, Murray JI, Lakadamyali M, Joyce EF, Jain R, Epstein JA (2019) H3K9me2 orchestrates inheritance of spatial positioning of peripheral heterochromatin through mitosis. Elife 8: e49278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polovnikov K, Belan S, Imakaev M, Brand HB, Mirny LA (2022) Fractal polymer with loops recapitulates key features of chromosome organization. bioRxiv 10.1101/2022.02.01.478588 [PREPRINT] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dundar F, Manke T (2016) deepTools2: a next generation web server for deep‐sequencing data analysis. Nucleic Acids Res 44: W160–W165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao S, Huntley M, Durand N, Stamenova E, Bochkov I, Robinson J, Sanborn A, Machol I, Omer A, Lander E et al (2014) A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159: 1665–1680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robin X, Turck N, Hainard A, Tiberti N, Lisacek F, Sanchez JC, Muller M (2011) pROC: an open‐source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics 12: 77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robson MI, de Las Heras JI, Czapiewski R, Le Thanh P, Booth DG, Kelly DA, Webb S, Kerr ARW, Schirmer EC (2016) Tissue‐specific gene repositioning by muscle nuclear membrane proteins enhances repression of critical developmental genes during myogenesis. Mol Cell 62: 834–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross‐Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, Brown GD, Gojis O, Ellis IO, Green AR et al (2012) Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 481: 389–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahin M, Wong W, Zhan Y, Van Deynze K, Koche R, Leslie CS (2021) HiC‐DC+ enables systematic 3D interaction calls and differential analysis for Hi‐C and HiChIP. Nat Commun 12: 3366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitou M, Hayashi K (2021) Mammalian in vitro gametogenesis. Science 374: eaaz6830 [DOI] [PubMed] [Google Scholar]
- Salhab A, Nordström K, Gasparoni G, Kattler K, Ebert P, Ramirez F, Arrigoni L, Müller F, Polansky JK, Cadenas C et al (2018) A comprehensive analysis of 195 DNA methylomes reveals shared and cell‐specific features of partially methylated domains. Genome Biol 19: 150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanulli S, Trnka MJ, Dharmarajan V, Tibble RW, Pascal BD, Burlingame AL, Griffin PR, Gross JD, Narlikar GJ (2019) HP1 reshapes nucleosome core to promote phase separation of heterochromatin. Nature 575: 390–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki K, Yokobayashi S, Nakamura T, Okamoto I, Yabuta Y, Kurimoto K, Ohta H, Moritoki Y, Iwatani C, Tsuchiya H et al (2015) Robust in vitro induction of human germ cell fate from pluripotent stem cells. Cell Stem Cell 17: 178–194 [DOI] [PubMed] [Google Scholar]
- Schroeder DI, Blair JD, Lott P, Yu HOK, Hong D, Crary F, Ashwood P, Walker C, Korf I, Robinson WP et al (2013) The human placenta methylome. Proc Natl Acad Sci USA 110: 6037–6042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzer W, Abdennur N, Goloborodko A, Pekowska A, Fudenberg G, Loe‐Mie Y, Fonseca NA, Huber W, Haering CH, Mirny L et al (2017) Two independent modes of chromatin organization revealed by cohesin removal. Nature 551: 51–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seisenberger S, Andrews S, Krueger F, Arand J, Walter J, Santos F, Popp C, Thienpont B, Dean W, Reik W (2012) The dynamics of genome‐wide DNA methylation reprogramming in mouse primordial germ cells. Mol Cell 48: 849–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheffield NC, Bock C (2016) LOLA: enrichment analysis for genomic region sets and regulatory elements in R and Bioconductor. Bioinformatics 32: 587–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin H, Shi Y, Dai C, Tjong H, Gong K, Alber F, Zhou XJ (2016) TopDom: an efficient and deterministic method for identifying topological domains in genomes. Nucleic Acids Res 44: e70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirane K, Kurimoto K, Yabuta Y, Yamaji M, Satoh J, Ito S, Watanabe A, Hayashi K, Saitou M, Sasaki H (2016) Global landscape and regulatory principles of DNA methylation reprogramming for germ cell specification by mouse pluripotent stem cells. Dev Cell 39: 87–103 [DOI] [PubMed] [Google Scholar]
- Shirane K, Miura F, Ito T, Lorincz MC (2020) NSD1‐deposited H3K36me2 directs de novo methylation in the mouse male germline and counteracts Polycomb‐associated silencing. Nat Genet 52: 1088–1098 [DOI] [PubMed] [Google Scholar]
- Sidoli S, Bhanu NV, Karch KR, Wang X, Garcia BA (2016) Complete workflow for analysis of histone post‐translational modifications using bottom‐up mass spectrometry: from histone extraction to data analysis. J Vis Exp 54112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soh Y, Alföldi J, Pyntikova T, Brown L, Graves T, Minx P, Fulton R, Kremitzki C, Koutseva N, Mueller J et al (2014) Sequencing the mouse Y chromosome reveals convergent gene acquisition and amplification on both sex chromosomes. Cell 159: 800–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soler‐Vila P, Cusco P, Farabella I, Di Stefano M, Marti‐Renom MA (2020) Hierarchical chromatin organization detected by TADpole. Nucleic Acids Res 48: e39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soneson C, Love MI, Robinson MD (2015) Differential analyses for RNA‐seq: transcript‐level estimates improve gene‐level inferences. F1000Research 4: 1521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiller C, Koopman P, Bowles J (2017) Sex Determination in the mammalian germline. Annu Rev Genet 51: 265–285 [DOI] [PubMed] [Google Scholar]
- Stadhouders R, Vidal E, Serra F, Di Stefano B, Le Dily F, Quilez J, Gomez A, Collombet S, Berenguer C, Cuartero Y et al (2018) Transcription factors orchestrate dynamic interplay between genome topology and gene regulation during cell reprogramming. Nat Genet 50: 238–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stovner EB, Saetrom P (2019) epic2 efficiently finds diffuse domains in ChIP‐seq data. Bioinformatics 35: 4392–4393 [DOI] [PubMed] [Google Scholar]
- Surani MA, Hayashi K, Hajkova P (2007) Genetic and epigenetic regulators of pluripotency. Cell 128: 747–762 [DOI] [PubMed] [Google Scholar]
- Tang WW, Kobayashi T, Irie N, Dietmann S, Surani MA (2016) Specification and epigenetic programming of the human germ line. Nat Rev Genet 17: 585–600 [DOI] [PubMed] [Google Scholar]
- Thodberg M, Thieffry A, Vitting‐Seerup K, Andersson R, Sandelin A (2019) CAGEfightR: analysis of 5'‐end data using R/Bioconductor. BMC Bioinformatics 20: 487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd S, Todd P, McGowan SJ, Hughes JR, Kakui Y, Leymarie FF, Latham W, Taylor S (2021) CSynth: an interactive modelling and visualization tool for 3D chromatin structure. Bioinformatics 37: 951–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vara C, Paytuví‐Gallart A, Cuartero Y, Le Dily F, Garcia F, Salvà‐Castro J, Gómez‐H L, Julià E, Moutinho C, Aiese Cigliano R et al (2019) Three‐dimensional genomic structure and cohesin occupancy correlate with transcriptional activity during spermatogenesis. Cell Rep 28: 352–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venev S, Abdennur N, Goloborodko A, Flyamer I, Fudenberg G, Nuebler J, Galitsyna A, Akgol B, Abraham S, Kerpedjiev P et al (2021) open2c/cooltools: v0.4.1. Zenodo.
- Wang J, Chakraborty A, Ay F (2021) dcHiC: differential compartment analysis of Hi‐C datasets. bioRxiv 10.1101/2021.1102.1102.429297 [PREPRINT] [DOI] [Google Scholar]
- Wang Y, Wang H, Zhang YU, Du Z, Si W, Fan S, Qin D, Wang M, Duan Y, Li L et al (2019) Reprogramming of meiotic chromatin architecture during spermatogenesis. Mol Cell 73: 547–561 [DOI] [PubMed] [Google Scholar]
- Wen L, Tang F (2019) Human germline cell development: from the perspective of single‐cell sequencing. Mol Cell 76: 320–328 [DOI] [PubMed] [Google Scholar]
- Western PS, Miles DC, van den Bergen JA, Burton M, Sinclair AH (2008) Dynamic regulation of mitotic arrest in fetal male germ cells. Stem Cells 26: 339–347 [DOI] [PubMed] [Google Scholar]
- Wingett S, Ewels P, Furlan‐Magaril M, Nagano T, Schoenfelder S, Fraser P, Andrews S (2015) HiCUP: pipeline for mapping and processing Hi‐C data. F1000Research 4: 1310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood SN (2011) Fast stable restricted maximum likelihood and marginal likelihood estimation of semiparametric generalized linear models. J R Statist Soc B 73: 3–36 [Google Scholar]
- Wutz G, Várnai C, Nagasaka K, Cisneros DA, Stocsits RR, Tang W, Schoenfelder S, Jessberger G, Muhar M, Hossain MJ et al (2017) Topologically associating domains and chromatin loops depend on cohesin and are regulated by CTCF, WAPL, and PDS5 proteins. EMBO J 36: 3573–3599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang G, Giardine BM, Mahony S, Zhang Y, Hardison RC (2021) S3V2‐IDEAS: a package for normalizing, denoising and integrating epigenomic datasets across different cell types. Bioinformatics 37: 3011–3013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing H, Wu Y, Zhang MQ, Chen Y (2021) Deciphering hierarchical organization of topologically associated domains through change‐point testing. BMC Bioinformatics 22: 183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yattah C, Hernandez M, Huang D, Park H, Liao W, Casaccia P (2020) Dynamic lamin B1‐gene association during oligodendrocyte progenitor differentiation. Neurochem Res 45: 606–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying QL, Wray J, Nichols J, Batlle‐Morera L, Doble B, Woodgett J, Cohen P, Smith A (2008) The ground state of embryonic stem cell self‐renewal. Nature 453: 519–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan ZF, Sidoli S, Marchione DM, Simithy J, Janssen KA, Szurgot MR, Garcia BA (2018) EpiProfile 2.0: a computational platform for processing Epi‐proteomics mass spectrometry data. J Proteome Res 17: 2533–2541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Li T, Preissl S, Amaral ML, Grinstein JD, Farah EN, Destici E, Qiu Y, Hu R, Lee AY et al (2019) Transcriptionally active HERV‐H retrotransposons demarcate topologically associating domains in human pluripotent stem cells. Nat Genet 51: 1380–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W et al (2008) Model‐based analysis of ChIP‐Seq (MACS). Genome Biol 9: R137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Xie W (2019) The role of 3D genome organization in development and cell differentiation. Nat Rev Mol Cell Biol 20: 535–550 [DOI] [PubMed] [Google Scholar]