

Abstract
Autophagy is a cellular degradative pathway that plays diverse roles in maintaining cellular homeostasis. Cellular stress caused by starvation, organelle damage, or proteotoxic aggregates can increase autophagy, which uses the degradative capacity of lysosomal enzymes to mitigate intracellular stresses. Early studies have shown a role for autophagy in the suppression of tumorigenesis. However, work in genetically engineered mouse models and in vitro cell studies have now shown that autophagy can be either cancer‐promoting or inhibiting. Here, we summarize the effects of autophagy on cancer initiation, progression, immune infiltration, and metabolism. We also discuss the efforts to pharmacologically target autophagy in the clinic and highlight future areas for exploration.
Keywords: ATG, autophagy, cancer, chloroquine, metabolism
Subject Categories: Autophagy & Cell Death, Cancer
This review summarizes the diverse contributions of the cellular degradation pathway autophagy to cancer initiation, progression, immune infiltration, and metabolism, and discusses related potential therapeutic intervention points.

Introduction
Macroautophagy (hereafter referred to as autophagy) is a lysosome‐dependent catabolic process conserved from yeast to human. The basic function of autophagy is to maintain the integrity of proteins and organelles, and balance cellular energy and nutrient needs. The autophagy pathway is activated by a variety of intracellular stressors including damaged mitochondria, pathogen infection, protein aggregates, and nutrient limitation (Parzych & Klionsky, 2014). Short‐term activation of autophagy is homeostatic and supports cell survival under stress by removing or counteracting the intracellular stressor. This activity is achieved through the formation of a double‐membraned vesicle called an autophagosome that captures intracellular components for degradation (Zhao et al, 2021). Autophagic breakdown of encapsulated proteins, lipids, nucleic acids, or carbohydrates occurs after fusion with the lysosome, which provides the acid hydrolases to break down diverse macromolecules so that they can be transported back into the cytoplasm to support essential cellular processes such as translation and metabolism.
Mutational inactivation of the autophagy pathway is not common in cancer, suggesting that a complete loss of autophagy is not a major contributor to cancer development or is harmful to cell viability (Lebovitz et al, 2015). However, dysregulation of selective or bulk autophagy has been observed in several cancers and is linked to disease progression (Klionsky et al, 2021). The role of autophagy in cancer is complicated by several factors including stage of the disease, driver mutation, tissue types, and metabolic sensitivity. However, trends have emerged showing autophagy can play a tumor‐suppressive role in cellular transformation or an oncogenic role in established disease. The tumor‐suppressive role of autophagy has been linked to inhibiting stress and reactive oxygen species, while its oncogenic potential has been described to support cancer cell metabolism and inhibit immune infiltration (Amaravadi et al, 2016).
This review will focus on the role of autophagy in cancer, with particular emphasis on recent advances in the field. We will explore how cancer‐driving pathways regulate autophagy activity and discuss the roles of autophagy in oncogenesis. Further, we will cover animal models of oncogenesis which perturb tumor autophagy or host autophagy. For those interested in general reviews of the autophagy pathway, please see the following reviews (Johansen & Lamark, 2020; King et al, 2021; Zhao et al, 2021).
ATG proteins and autophagy receptors
The formation and maturation of autophagosomes are supported by over 30 autophagy‐related (ATG) proteins. ATG proteins can be categorized into 5 major functional groups (Fig 1). First is the Unc‐51 Like kinase 1 (ULK1) protein kinase complex, which includes FAK family‐interacting protein of 200 kDa (FIP200), ATG13, and ATG101. In cancer models, FIP200 is the most common component disrupted to inhibit autophagy. Second is the phosphatidylinositol‐3‐kinase catalytic subunit type 3 (PIK3C3; hereafter referred to as VPS34) complex, which includes the core subunits of phosphatidylinositide‐3‐kinase regulatory subunit 4 (PIK3R4) and Beclin 1 (BECN1). Additional regulatory binding partners of the VPS34 complex include ATG14, UV radiation resistance associated gene (UVRAG), SH3 domain‐containing GRB2‐like protein B1 (SH3GLB1), activating molecule in BECN1‐regulated autophagy protein 1 (AMBRA 1) and Run domain Beclin 1‐interacting and cysteine‐rich domain‐containing protein (RUBCN). Beclin 1 disruption is the most prevalent method used to inhibit this complex in cancer models. Third is the ubiquitin‐like conjugation system, which contains two subgroupings of proteins: i. ATG3, ATG4, and ATG7 and ii. ATG7, ATG10, ATG5, ATG12, and ATG16L1. Together, these protein cascades are responsible for the conjugation of Atg8 family members to the autophagosomal membrane. ATG5 and ATG7 knockout mouse models are commonly used in autophagy studies, although an inducible dominant negative version of ATG4 has recently been used quite widely. Fourth is the trafficking of the transmembrane ATG9 protein. Fifth is the ATG2/WD repeat domain phosphoinositide‐interacting protein (WIPI) lipid transfer system. In this review, we will pay particular attention to the first 3 groups of autophagy proteins as they are often targeted in transgenic mouse models of cancer.
Figure 1. The autophagy pathway.
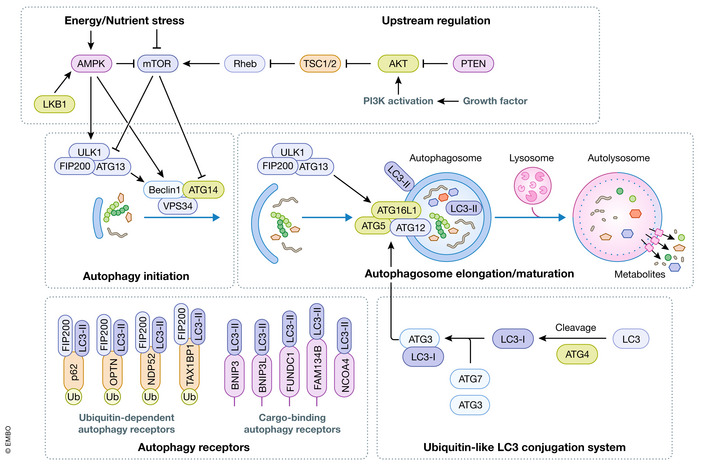
Autophagosome initiation is driven by protein and lipid kinase activity, which is sensitive to nutrient sensitive kinases mTORC1 and AMPK, and the PI3K‐AKT pathway. Expansion of the autophagosome requires the activity of a ubiquitin‐like conjugation system that lipidated LC3 to the autophagosomal membrane. Autophagy receptors bind LC3 to recruit cargo to the autophagosome. Recruitment of FIP200 by autophagy receptors can stimulate autophagosome biogenesis. Lysosomal fusion promotes degradation of autophagic cargo.
ULK1 and VPS34 kinases
One of the earliest events in autophagosome biogenesis is the recruitment of ULK1 to the phagophore, a membrane structure that will expand to form the autophagosome. ULK1 and related ULK2 can both function as mammalian homologs to yeast Atg1. ULK1 and ULK2 are the most upstream components in the autophagy machinery and are the only protein kinases among the Atg proteins (Zhang et al, 2021). Inhibition or deletion of both ULK1 and ULK2 results in a profound defect in stress‐induced autophagy. ULK1 promotes autophagy by phosphorylating several ATG protein targets including multiple subunits of VPS34 lipid kinase complexes (Fig 1; King et al, 2021). VPS34 is the catalytic component of these complexes phosphorylating phosphatidylinositol to form phosphatidylinositol 3‐phosphate (PI3P). VPS34 is not exclusive to the autophagy pathway as it also plays a key role in cellular vesicle trafficking. The specific cellular function of VPS34 is driven by regulatory binding partners within complexes. VPS34 promotes autophagosome formation and maturation when complexed with Beclin 1 binding partners ATG14 and UVRAG, respectively (Kim et al, 2013). PI3P acts as an anchor to recruit downstream autophagy proteins and promotes autophagosome biogenesis and maturation (Zhang et al, 2021).
Ubiquitin‐like conjugation systems
Autophagosome formation is dependent on two ubiquitin‐like conjugation cascades that promote the coupling of Atg8 homologs to phosphatidylethanolamine (PE) of the autophagosomal membrane. Mammals have 7 isoforms of Atg8 including the microtubule‐associated protein light chain 3 (LC3) family (LC3A, LC3B, LC3B2, and LC3C) and the GABA type A receptor‐associated protein (GABARAP) family (GABARAP, GABARAP1, and GABARAPL2/GATE16), which are all conjugated to PE on the autophagosomal membrane (Kumar et al, 2020; Fig 1). The first ubiquitin‐like system involves the activity of ATG7 (acting as E1) and ATG10 (acting as E2), which facilitates an isopeptide bond formation between ATG12 to ATG5 (resembling that of ubiquitin to its substrate). ATG5‐ATG12 then associates with ATG16L1 to form the core of the enzymatic complex (acting as E3) that conjugates LC3 to PE. The processing of LC3 and other Atg8 family members depends on the second ubiquitin‐like pathway required for lipidation (Zhang et al, 2021). LC3 is first processed by C‐terminal cleavage by ATG4 (acting as a protease) and is then activated by ATG7 (E1). Activation by ATG7 promotes transfer of LC3 to ATG3 (E2) to form an ATG3‐LC3 conjugate. ATG3‐LC3 is then recruited to the autophagosomal membrane by the ATG12‐ATG5‐ATG16L1 complex (acting as an E3), where LC3 is finally lipidated with PE (Fig 1; Kabeya et al, 2000). The ATG12‐ATG5‐ATG16L1 enzyme is phosphorylated directly by ULK1 in response to most autophagy‐inducing stressors (Tian et al, 2020). Ablation of lipidation of Atg8 isoforms in mammals inhibits nearly all autophagy. As a result, deletion of proteins required for lipidation are very common targets for inhibiting the autophagy pathway in vitro and in vivo.
Autophagy receptors
Autophagosome cargo is often enriched for substrates that have been ubiquitinated. Selective capture of ubiquitinated substrates is facilitated by a class of proteins called autophagy receptors (Fig 1). Autophagy receptors bind ubiquitinated proteins through ubiquitin binding domains and recruit them to the autophagosome by associating with LC3 family members through a LC3‐interacting region (LIR) (Johansen & Lamark, 2020). Some membrane‐embedded autophagy receptors that promote the degradation of membranous organelles lack a ubiquitin binding domain. These transmembrane adaptors can be activated by phosphorylation or increased protein levels to trigger the uptake of damaged organelles into autophagosomes (Palikaras et al, 2018). More recently, autophagy receptors have been shown to bind LC3 outside the LIR domain, adding another layer of selectivity and complexity to selective autophagy (Marshall et al, 2019). Since autophagy receptors are digested by lysosomal proteases along with other autophagy cargos, their levels tend to be inversely correlated with autophagic flux, making them useful markers for studying autophagy.
The best characterized autophagy receptor is p62 (also called sequestosome 1). p62 is implicated in several forms of selective autophagy, including, but not limited to, aggrephagy (degradation of protein aggregates), xenophagy (degradation of pathogens), lipophagy (degradation of lipids), and mitophagy (degradation of mitochondria; Johansen & Lamark, 2020). In addition to binding LC3 and ubiquitin p62 can bind other signaling molecules such as raptor (a subunit of the mTORC1 complex) and Kelch‐like ECH‐associated protein 1 (KEAP1), which is a regulator of nuclear factor erythroid 2‐related factor 2 (NRF2; Katsuragi et al, 2015). Notably, NRF2 and KEAP1 are frequently mutated in cancer (Rojo de la Vega et al, 2018). As a result of these additional interactions, p62 functions as a regulator in amino acid sensing and oxidative stress pathways (Katsuragi et al, 2015). Some functions of p62, such as the positive regulation of mTORC1 through raptor, are autophagy‐independent. Chromosomal amplification of 5q in renal cancer leads to increased p62 levels, which fortifies the cell against redox stress and enhances oncogenesis (Li et al, 2013). Like members of the Atg8 family there is a significant degree of functional overlap possible among the autophagy receptors, which typically contain ubiquitin binding and LIR domains to promote the targeting of autophagy cargo (Johansen & Lamark, 2020).
Autophagy receptors have more recently been shown to promote the formation of autophagosomes by recruiting FIP200 to autophagic cargo. When p62 is recruited to ubiquitinated autophagic cargo, it promotes degradation by binding the claw domain of FIP200 to stimulate the recruitment of autophagy‐initiating enzymes (Turco et al, 2019). The ability to recruit FIP200 is shared by many autophagy receptors, including NBR1, NDP52, TAX1BP1, CCPG1, and OPTN (Fig 1; Ravenhill et al, 2019; Vargas et al, 2019; Turco et al, 2021; Zhou et al, 2021). Importantly, the recruitment of FIP200 by these receptors has been shown to be important, and in some cases sufficient, to promote the degradation of specific cargo including mitochondria, ER, protein aggregates, and pathogens (Ravenhill et al, 2019; Vargas et al, 2019; Zhou et al, 2021). Not all autophagy receptors bind ubiquitin to identify cargo for autophagosomal capture. For example, some autophagy receptors are localized to the lipid bilayer of targeted organelles such as mitochondria or ER (Fig 1). In addition, NCOA4 is stabilized to transfer ferritin to autophagosomes when iron is low, but ferritin does not require ubiquitination for NCOA4 binding (Gubas & Dikic, 2022). The selective degradation of cargo such as mitochondria or antigen presenting receptors without a concomitant activation of bulk autophagy has significant effects on tumor biology, which will be discussed in further detail in subsequent sections.
Upstream signals and autophagy regulators
The formation of autophagosomes and therefore the rate of cellular degradation is tightly controlled by the activity of ULK1, VPS34, and ATG16L1‐containing enzyme complexes described above. Accordingly, several signal transduction pathways converge on these three enzymes to control the initiation and rate of autophagy (King et al, 2021). Autophagy is stress responsive and as such many of the upstream pathways regulating autophagy‐promoting enzymes are controlled by well characterized stress‐sensitive kinases. For example, nutrient deprivation is one of the earliest and best characterized stresses capable of activating autophagy. One of the first nutrients showed to regulate autophagy levels was branched chain amino acids (Mortimore & Schworer, 1977). Withdrawal of amino acids inhibits the activity of mechanistic target of rapamycin (mTOR) complex 1 (mTORC1), which is prominently linked to autophagy regulation (Ravikumar et al, 2003). In addition to amino acids, mTORC1 activity is enhanced by growth factors through the class I phosphatidylinositol 3‐kinase (PI3K)‐Akt pathway. mTORC1 is a potent repressor of the autophagy pathway. Under nutrient and growth factor sufficiency, mTORC1 blocks autophagy through inhibitory phosphorylation of ULK1 and the ATG14 subunit of VPS34 complexes (Fig 1; King et al, 2021). However, upon starvation mTORC1 activity is rapidly inhibited and the repressive phosphorylation on ULK1 or VPS34 complexes is removed, thereby activating these enzymes and autophagy. Dephosphorylation of ULK1 and VPS34 complexes under nutrient withdrawal is facilitated by the serine/threonine‐protein phosphatase 2A catalytic subunit alpha isoform (PP2A)‐B55α phosphatase complex, which is activated upon starvation (Wong et al, 2015; Fujiwara et al, 2016).
In cancer, mTORC1 activity is often elevated, promoting cellular growth through control of translation and other biosynthetic processes. Pathological mTORC1 activation is promoted through activation of PI3K signaling, which is achieved through activation of receptor tyrosine kinase signaling or inactivation of the lipid phosphatase phosphatidylinositol 3,4,5‐trisphosphate 3‐phosphatase and dual‐specificity protein phosphatase (PTEN; Jewell & Guan, 2013). PI3K signaling promotes AKT‐mediated phosphorylation and inactivation of the tuberous sclerosis complex (TSC), thus relieving the inhibitory effect of TSC on mTORC1. Analysis of precancerous lesions from patients harboring TSC mutations revealed a repression of autophagy, which was reversible upon pharmacological mTORC1 inhibition (Reis et al, 2021).
Another important stress responsive kinase upstream of the autophagy pathway is AMP activated protein kinase (AMPK). AMPK is a serine/threonine kinase, whose activity is sensitive to intracellular ratios of AMP/ADP to ATP. In times of energetic stress, cellular AMP and ADP levels rise. The binding of AMP and ADP to a regulatory subunit of the AMPK kinase complex increases phosphorylation on the activation loop of the AMPK catalytic subunit, thereby promoting kinase activity of the complex (Xiao et al, 2007; Dunlop & Tee, 2013). Notably, serine/threonine–protein kinase STK11 (LKB1) is a tumor suppressor kinase that directly phosphorylates and activates AMPK. In addition to the regulatory effects of AMP and ADP on AMPK, intracellular levels of an intermediate metabolite in glycolysis, fructose‐1‐6‐bisphosphate can affect AMPK activity (Zhang et al, 2017). Specifically, lower levels of fructose‐1‐6‐bisphosphate increases the activity of LKB1 on AMPK localized to the lysosome, thereby providing another stimulatory input to AMPK upon glucose scarcity (Zhang et al, 2017). Under energetic stress, AMPK regulates many metabolic processes, including inhibiting lipid synthesis, glycolysis, and protein synthesis. AMPK also promotes autophagy by targeting many of the same autophagy components as mTORC1. However, AMPK‐mediated phosphorylation of autophagy components is activating rather than inhibitory (Kim et al, 2011). Specifically, AMPK can activate ULK1 through direct phosphorylation, or VPS34 lipid kinase through phosphorylation of the Beclin 1 subunit (Kim et al, 2013). AMPK is also capable of indirectly activating autophagy through inhibition of mTORC1. AMPK inhibits mTORC1 through direct phosphorylation of the complex on the raptor subunit, or through activation of the inhibitory TSC complex (Inoki et al, 2003; Gwinn et al, 2008). Taken together, AMPK can act as a powerful autophagy inducer by relieving the repressive effects of mTORC1 signaling and simultaneous activation of key autophagy enzymes.
Together, the regulation of autophagy machinery by the coordinated activity of mTORC1 and AMPK ensures that during acute starvation autophagy is upregulated to generate sufficient nutrients to support essential cellular processes. In solid tumors, the ability of autophagy to compensate for nutrient limitation promotes survival and cancer progression. Autophagy‐deficient mice are born phenotypically normal, but die within 24 h after birth (Kuma et al, 2004). Postnatal lethality poses difficulties in studying the role of autophagy in disease, and as a result, most studies of the role of autophagy in cancer utilize a tissue‐specific knockout of autophagy genes.
Selective autophagy
Selective autophagy relies on the binding of autophagy receptors to membrane‐associated LC3/GABARAP family members and ubiquitin. Localized activation of autophagy enzymes coupled with the ubiquitination of damaged organelles or proteins drives many types of selective autophagy (Johansen & Lamark, 2020). Unlike starvation‐induced autophagy, activation of selective autophagy is not always accompanied by a reduction in total protein levels for the autophagy receptors involved. For example, degradation of a single invading bacteria or a few mitochondria does not require enough p62 to affect protein levels, which also complicates the analysis of selective autophagy in disease (Losier et al, 2019). However, the lack of alteration in levels of proteins associated with autophagic flux does not indicate that defects in selective autophagy are devoid of biological impact. For example, inhibition of mitophagy can affect cellular metabolism and viability. Similarly, defects in clearance of protein aggregates because of aggrephagy inhibition can stimulate cancer cell survival and exacerbate neurodegeneration. Here, we will limit our discussion of selective autophagy to mitophagy and aggrephagy. However, there are several in‐depth reviews of selective autophagy in normal and disease states (Mancias & Kimmelman, 2016; Lamark et al, 2017; Kirkin & Rogov, 2019; Liu et al, 2020).
Mitophagy
The autophagic degradation of mitochondria (hereafter referred to as mitophagy) is selective for damaged mitochondria. Specifically, depolarization of the outer mitochondrial membrane (OMM) or hypoxia‐induced accumulation of mitophagy receptors can block mitochondrial fusion and trigger isolation from the rest of the mitochondrial compartment, thereby promoting its engulfment into an autophagosome (Liu et al, 2012; Palikaras et al, 2018). Efficient removal of damaged mitochondria is essential to limit the release of reactive oxygen species (ROS), mitochondrial DNA, and activators of intrinsic apoptosis from damaged mitochondria into the cytosol.
Ubiquitin dependent mitophagy
The best characterized mechanism for the clearance of damaged mitochondria involves the PTEN‐induced putative kinase 1 (PINK1) and the E3 ubiquitin ligase Parkin. This pathway is stimulated by the depolarization of the mitochondrial membrane, which results in stabilization and accumulation of PINK1 on the OMM (Jin et al, 2010; Shi & McQuibban, 2017). On the OMM, PINK1 phosphorylates and activates Parkin and ubiquitin (Vives‐Bauza et al, 2010; Kane et al, 2014; Kazlauskaite et al, 2014; Koyano et al, 2014). Activated Parkin targets mitochondrial proteins for ubiquitination, which promotes the recruitment of ubiquitin binding domain‐containing autophagy receptors (Fig 1; Geisler et al, 2010; Chen & Dorn, 2013; Sarraf et al, 2013). Despite being well characterized in vitro, the requirement for PINK1/Parkin in physiological mitophagy remains more elusive. For example, knocking out PINK1 in mice does not result in a profound change in the number of mitochondria (Gautier et al, 2008; McWilliams et al, 2018). This may be a result of PINK1/Parkin‐mediated mitophagy being activated in rare instances or compensation by other mitophagy‐inducing pathways, which are discussed below.
Ubiquitin‐independent mitophagy
Ubiquitin‐independent mitophagy uses mitochondria‐localized adaptors that have transmembrane domains and LIRs. These adaptors are regulated by protein stability and post‐translational modification. BCL2/adenovirus E1B 19 kDa protein‐interacting protein 3 (BNIP3) and BNIP3‐like (BNIP3L; also known as NIX; Mellor & Harris, 2007) are two such mitophagy adaptors. BNIP and BNIP3L are BH3‐only containing members of the Bcl2 family that also contain LIR domains (Bellot et al, 2009). Transcription of BNIP3 and BNIP3L is sensitive to hypoxia via stabilization of the hypoxia‐inducible factor transcriptional coactivator. BNIP3 is also transcriptionally regulated by forkhead box O3a (FOXO3a) and nuclear factor kappa B (NF‐kappaB) signaling (Mellor & Harris, 2007; Dhingra et al, 2013; Chaanine et al, 2016). Phosphorylation BNIP3 and BNIP3L near the LIR motif can increase their affinity to LC3 and upregulate mitophagy (Zhu et al, 2013; Rogov et al, 2017; Poole et al, 2021).
Like BNIP3, FUN14 domain‐containing 11 (FUNDC1) is an OMM protein that binds LC3 to promote mitophagy under hypoxia (Liu et al, 2012; Lampert et al, 2019). FUNDC1 is subject to inhibitory phosphorylation by the Src kinase and activating phosphorylation by ULK1. In addition to the pathways involving adaptor proteins like FUNDC1, many more proteins have been implicated in mitophagy regulation that have been described in depth (Palikaras et al, 2018; Vara‐Perez et al, 2019).
Mitophagy defects in cancer
Alterations in mitophagy may play an important role in supporting the metabolic requirements of a cancer cell. For example, deletion of the mitophagy adaptor BNIP3 is found in triple‐negative breast cancer and was shown to have tumor‐suppressive functions in the MMTV‐PyMT murine breast cancer model (Chourasia et al, 2015). In this model, BNIP3 deletion resulted in a reduction in the rate of mitophagy and an increase in ROS‐induced HIF1α expression (Chourasia et al, 2015). Concordantly, siRNA‐mediated knockdown of BNIP3 in breast cancer cell lines injected into the fat pad of syngeneic mice resulted in a reduction of primary tumor growth and metastases (Manka et al, 2005). Reduction of BNIP3 has been linked to poor mitochondrial health and an increased reliance on glycolysis (Fig 2) (Chourasia et al, 2015; Lyons et al, 2017). While aerobic glycolysis had been linked to metastasis, the release of reactive oxygen species by the accumulation of defective mitochondria has also been shown to promote metastatic potential (Ishikawa et al, 2008; Porporato et al, 2014). In triple‐negative breast cancer, BNIP3 repression is predictive of poor metastasis free survival (Chourasia et al, 2015).
Figure 2. The dynamic role of autophagy in oncogenesis.
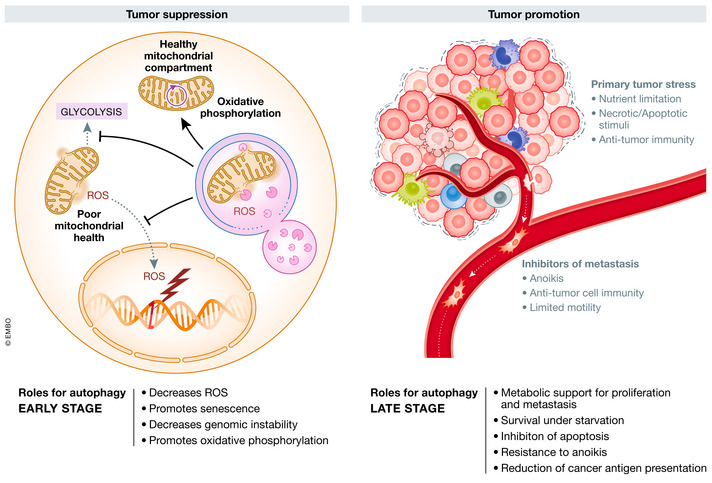
Autophagy can inhibit tumor initiation through efficient removal of damaged mitochondria, which reduces glycolysis, ROS levels, and DNA damage. Autophagy supports later stage tumor growth by promoting metabolic remodeling, inhibiting apoptosis and anoikis, and suppressing anti‐tumor immunity (see text for details).
Conversely, in several cancers expression of BNIP3 and family member BNIP3L are positively correlated with poor prognosis, which may suggest an oncogenic role for these mitophagy receptors (Petrova et al, 2015; Chen et al, 2017; Macher‐Goeppinger et al, 2017). However, this correlation may be in part due to the hypoxic signature of advanced malignancies and the role of the HIFα transcription factor in driving expression of BNIP3 and BNIP3L. The best evidence for an oncogenic role for BNIP3L comes from a study of KRAS proto‐oncogene (KRAS) and tumor suppressor protein p53 (hereafter referred to as p53)‐driven pancreatic cancer. In this model, KRAS activation drives BNIP3L expression and enhances redox capacity (Humpton et al, 2019). Deletion of BNIP3L resulted in an increase in mitochondrial number and improved survival. The expression of BNIP3 and BNIP3L is tissue specific and, in PDAC, BNIP3 expression was not observed except in BNIP3L knockout tumors, indicating a partial compensatory mechanism in these cells. Therefore, it will be important in the future to tease apart the role of these two mitophagy receptors in the promotion or inhibition of malignancy while also mediating the oncogenic effects of tumor hypoxia.
Parkin is encoded by the PARK2 gene, a large gene that is in the genomically unstable region FRAG6E, which is a fragile region and mutational hotspot in cancer (Toma et al, 2013). In kidney cancer driven by HIF stabilization, loss of Parkin results in a reduced overall survival (Toma et al, 2013). Deletion of Park2 in a ras‐driven model of pancreatic cancer accelerated oncogenesis, further underscoring the potential tumor‐suppressive functions of Parkin (Li et al, 2018). Ablation of Parkin in this model resulted in an increase of mitochondrial iron and HIF stabilization, which in turn promoted glycolysis (Li et al, 2018). Due to the location of PARK2 in a genomically fragile site Parkin loss has also been found in several cancers, although in many instances the role of mitophagy disruption has not been explored (Vara‐Perez et al, 2019). It is important to determine the biological consequences of Parkin loss because Parkin has functions that extend beyond promoting the recycling of damaged mitochondria. For example, Parkin forms multiple E3–ligase complexes to ubiquitinate cell cycle proteins including polo‐like kinase (PLK1), Aurora A&B, and cyclins E1, D, B1 (Gong et al, 2014; Lee et al, 2015). The discovery that Parkin can directly ubiquitinate cell cycle proteins explained the observations that Parkin deletion can increase cell division and lead to errors in chromosomal segregation (Gong et al, 2014; Lee et al, 2015). In addition to a role in regulating cell cycle through chromosomal segregation and control of cyclin stability, Parkin may also control damage‐sensitive impediment of cell cycle. This role was identified through a genetic interaction with ataxia telangiectasia mutated (ATM), which is a kinase involved in DNA damage response (Sarraf et al, 2019). The deleterious effects of deleting both ATM and Parkin were described through a mechanism involving a Parkin‐dependent sequestration of activated TANK‐binding kinase 1 (TBK1) to damaged mitochondria, thereby inhibiting TBK1 function at the centrosome (Sarraf et al, 2019). Interestingly, this mechanism serves to blur the lines between the classical mitophagy activity of Parkin and its role in regulating chromosomal segregation, further muddling the role(s) Parkin may play in oncogenesis.
Aggrephagy
Cancer cells are routinely under oxidative stress and have activated unfolded protein response pathways. To prevent the deleterious effects of these pathways, cancer cells use autophagy and the ubiquitin proteasome pathway to control the levels of protein aggregates. Protein aggregates form from accumulation of misfolded proteins, which protects the cell from the adverse effects of soluble cytosolic misfolded proteins. Protein aggregates are often ubiquitinated, which links these aggregates to autophagy receptors that promote their clearance (Ravikumar et al, 2002; Bjørkøy et al, 2005). The E3‐ubiquitn ligase TRIM16 has been shown to play an important role in both the biogenesis and clearance of proteotoxic aggregates. TRIM16 promotes both the ubiquitination and autophagic clearance of proteotoxic aggregates in conjunction with p62 (Jena et al, 2018). In a xenograft mouse model, tumors engineered with a deletion of TRIM16 regressed upon induction of oxidative stress when compared to the parental line (Jena et al, 2018).
The complex role of autophagy in cancer
Cancer cells grow despite a constant exposure to intrinsic and extrinsic stressors, which would kill or halt growth in normal cells. The ability of autophagy to counteract stress can have diverse effects. For example, the ability of autophagy to clear‐damaged mitochondria reduces ROS and inhibits glycolysis, contributing to tumor suppression. Conversely, autophagy inhibits apoptosis and promotes survival in the nutrient poor tumor microenvironment, contributing to oncogenesis. These conflicting effects of stress‐reduction by the autophagy pathway have led to the theory that autophagy may initially play a role in tumor suppression by maintaining cellular homeostasis, which switches to a protooncogenic role as the tumor develops (Fig 2). Beyond the intrinsic effects of autophagy in the cancer cell is a more recent focus on the effects of autophagy in the tumor stroma, including its role in supporting the metabolic requirements of the tumor and immune surveillance. Lastly, autophagy may contribute to the metastatic potential of the tumor by either suppressing detachment induced death (referred to as anoikis) or by promoting cancer stem cells (Fig 2). These complexities will be discussed in further detail below.
Tumor‐suppressive functions of autophagy
The mutation of autophagy genes in cancer is rare and as a result much of what we know about autophagy and cancer is gleaned from genetically engineered mouse models (GEMM). The earliest hint of a role for autophagy in cancer came from the characterization of Beclin 1 as a candidate tumor suppressor in breast cancer (Liang et al, 1999). However, the proximity of BECN to BRCA1 on 17q21 meant that its monoallelic loss in cancer may be partly driven by co‐deletion of BRCA1. Supporting this notion, BECN1 deletion is not observed independently of BRCA1 in breast cancer, while deletions of BRCA1 without BECN1 inactivation are observed (Laddha et al, 2014). Conditional deletion of Fip200, which encodes a binding protein required for both ULK1 and ULK2 activity, resulted in a repression of tumor growth in a MMTV‐PyMT model of breast cancer, indicating that the tumor‐suppressive effects of Beclin 1 may be partially autophagy‐independent (Wei et al, 2011). Similarly to Becn1, deletion of Atg5 or Atg12 leads to an increase in metastases in PyMT‐driven mouse model of breast cancer that was driven by the accumulation of the autophagy receptor neighbor of BRCA1 gene 1 (NBR1; Marsh et al, 2020). Notably, this increase in metastasis upon Atg5 or Atg12 deletion underscores the diverse roles that autophagy plays in metastasis. When the effects of autophagy inhibition on primary tumor growth are controlled for, an increase in metastatic outgrowth could be observed upon inducible ATG5 or ATG12 disruption, uncovering a specific role for autophagy in the suppression of metastatic outgrowth (Marsh et al, 2020). Several additional studies have implicated BECN1 in the development of ovarian and breast cancer (Qu et al, 2003; Yue et al, 2003; Cicchini et al, 2014). BECN1 deletion results in genomic instability, thereby contributing to cancer development (Fig 2; Karantza‐Wadsworth et al, 2007; Delaney et al, 2020).
While additional clarity is needed on the mechanisms underlying Beclin 1 tumor suppression, it seems that in some genetically driven mouse models of breast cancer that wnt family member 1 (WNT1) is involved (Cicchini et al, 2014). A recent unbiased screen of the proteome identified the involvement of e‐cadherin and alpha catenin in Beclin 1 tumor‐suppressive function (Wijshake et al, 2021). Study of other organs in Becn1 +/− mice has uncovered an increased susceptibility to tumor development in the lung and liver (Qu et al, 2003; Yue et al, 2003), in addition to some type of breast tumors (Cicchini et al, 2014). Interestingly, the lung and livers of Becn1 +/− mice exhibit reduced levels of p53. The link between the tumor suppressors p53 and Beclin 1 is through regulation of p53 deubiquitination. Mechanistically, this is achieved by Beclin 1‐dependent regulation of ubiquitin‐specific protease USP10, which stabilizes p53 through cleavage of p53 ubiquitin chains (Yuan et al, 2010; Liu et al, 2011). Collectively, work on Beclin 1 has yielded some clues into the tumor‐suppressive functions of the autophagy pathway, while also identifying some autophagy‐independent functions of Beclin 1 that may contribute to its tumor‐suppressive function.
Since Beclin 1 has well‐established autophagy‐independent functions, a concerted effort has been made to explore the tumor‐suppressive functions of the autophagy pathway by knocking out genes that are both essential and autophagy specific. These studies have provided evidence that autophagy can play an oncogenic or tumor‐suppressive function. Additional models that show tumor suppression by autophagy include the conditional deletion of Atg5 and Atg7 in the liver, which resulted in limited tumor development consisting of age‐dependent benign adenomas rather than the well‐developed hepatocellular carcinoma in the Becn1+/− background (Qu et al, 2003; Takamura et al, 2011). Reduction of ATG5 by monoallelic deletion or promoter methylation have both been described to reduce autophagy in melanoma and are associated with poor survival (Liu et al, 2013; García‐Fernández et al, 2016). Stable knockdown of ATG5 in melanoma cells reduced senescence in BRAF‐induced oncogenesis, indicating a role for ATG5 suppression in the tumor initiation (Young et al, 2009; Liu et al, 2013). In BRAF‐driven melanoma, autophagy is required to overcome the senescence triggered by BRAF inhibitors. Deletion of Atg7 in BrafV600E ‐driven melanoma resulted in a reduction in tumor initiation, further strengthening the link between autophagy and tumor initiation in melanoma (Fig 2; Young et al, 2009; Xie et al, 2015). Notably, the tumor suppressive effects of Atg7 disruption are lost if the BrafV600E ‐driven melanoma also has disruption of Pten, underscoring some of the underlying complexities in trying to characterize the role of autophagy in oncogenesis (Xie et al, 2015).
Autophagy‐mediated promotion of oncogenesis
Autophagy is a stress‐responsive pathway and in most cases promotes survival and a return to cellular homeostasis. Perhaps the most intuitive example of this is the recycling of cytosolic components to produce compounds to support metabolism and metabolic adaptation when nutrients become limiting. A solid tumor expanding from a preneoplastic lesion into a malignant or invasive tumor will experience limitations in oxygen, glucose, and amino acids as it grows beyond the ability of nutrients to freely diffuse from the normal vasculature. The ability to recycle nutrients gives tumor cells that upregulate autophagy a competitive advantage under starvation pressure. More information on cell competition in the tumor microenvironment is reviewed here (Parker et al, 2020). Malignant solid tumors often have areas that are anoxic or hypoxic because tumor angiogenesis is haphazardly organized compared to the vasculature of normal tissue. In addition to nutrient stress, DNA damage, activation of the unfolded protein response (UPR), and organelle damage are all capable of activating autophagy in tumors. To compensate for the growth‐suppressing effects of these cancer‐related stresses autophagy is frequently elevated in cancer. Autophagy also plays tumor cell non‐autonomous roles in promoting cancer growth. For example, in Kras‐driven lung cancer a conditional whole‐body deletion of ATG7 was more potent in promoting tumor regression than inhibiting autophagy in the cancer cells (Karsli‐Uzunbas et al, 2014). Tumor regression was linked to metabolic deficiencies in the tumor cells that were reliant on host nutrient availability, which was altered due to autophagy inhibition in the liver and tumor microenvironment (Fig 2; Karsli‐Uzunbas et al, 2014). This trend of autophagy supporting the metabolic requirements of an aggressive tumor, either intrinsically or non‐autonomously, have been reproduced in several models and expose a pattern by which autophagy supports cancer progression (Amaravadi et al, 2016, 2019).
Autophagy can also promote certain aspects of tumor cell biology that enhances metastasis, including metabolic remodeling, inhibiting anoikis, evading immune cells, and improving motility (Fig 2; Peng et al, 2013; Chourasia et al, 2015; Sharifi et al, 2016). However, it is important to note that the effects of autophagy on metastasis are also dependent on tumor stage and genetic drivers (Chen et al, 2013; Lock et al, 2014; Marsh et al, 2020). Below we discuss in more detail the tumor‐promoting activities of autophagy, which are broken down by target organ.
Breast cancer
Ablation of the tumor suppressor Palb2 in the mouse mammary gland impedes repair of DNA damage and redox regulation, leading to tumor development (Bowman‐Colin et al, 2013). In this model, impairment of autophagy by monoallelic deletion of Becn1 reduced Palb2‐assocaited tumorigenesis (Huo et al, 2013). Notably, this oncogenic effect of Becn1 deletion was not observed in p53 null mice, which indicates context‐dependent function of Beclin 1 in tumorigenesis (Huo et al, 2013). Becn1 deletion results in only a partial inhibition of autophagy and likely has impacts outside the autophagy pathway, which weakens the link between autophagy and oncogenicity in this model. Further evidence for a role of autophagy in breast cancer came from crossing the conditional knockout of Fip200 in a PyMT‐driven mouse model of breast cancer. Mammary tumors in the Fip200 null mice were significantly smaller and occurred with less frequency than in autophagy proficient mice (Wei et al, 2011). Mechanistically, Fip200 ablation is proposed to suppress oncogenesis through accumulation of the autophagy receptor p62 and regulation of NF‐kappaB signaling (Wei et al, 2014). Various syngeneic breast cancer xenograft models that have disrupted autophagy through deletion of Becn1, Atg5, or Atg7 have also highlighted a role for autophagy in supporting tumor growth (Baginska et al, 2013; DeVorkin et al, 2019; Li et al, 2020). Xenograft studies with breast cancer cells have also uncovered a role for autophagy in the regulation of metastasis. In non‐metastatic cells, the DNA‐binding protein (DEDD) promotes VPS34 stability and autophagic degradation of twist family bHLH transcription factor 1 (TWIST1) and snail family transcriptional repressor 1 (Snail), which are two important regulators of epithelial‐mesenchymal transition (EMT) and metastasis (Lv et al, 2012). The autophagic degradation of EMT‐related transcription factors has also been described in other cancer types (Qiang et al, 2014; Catalano et al, 2015; Qin et al, 2015).
Lung cancer
Non‐small‐cell lung cancer driven by the activation of the oncogenic allele KrasG12D gives rise to the development of adenocarcinomas in the context of a functional immune system (Jackson et al, 2001; Kwon & Berns, 2013). Elevated autophagy in response to KRAS activation is required for mitochondrial homeostasis, metabolic adaptation, and stress adaptation (Guo et al, 2011, 2013; Lock et al, 2011; Yang et al, 2011; Karsli‐Uzunbas et al, 2014). Deletion of Atg7 or Atg5 in Kras‐driven lung tumors reduces tumor size and results in the development of predominantly benign oncocytomas, which exhibited accumulation of defective mitochondria (Guo et al, 2013; Rao et al, 2014). The effects of autophagy have also been explored in the KrasG12D ; p53 −/− model, which shows more aggressive adenocarcinoma than those in the p53 wild‐type mice. Dual ablation of autophagy and p53 resulted in an increase in tumor volume, which indicates that, similarly to work in breast cancer, p53 plays a role in oncogenesis following inhibition of autophagy (Guo et al, 2013; Rao et al, 2014). In addition to KRAS, mutations in the kinase domain of Braf are found in ~3% of lung cancer (Davies et al, 2002). Combinational mutation of ATG7 and BrafV600E resulted in an increase on early‐stage tumor formation, but ultimately resulted in a reduced tumor burden and metabolic dysfunction (Strohecker et al, 2013), highlighting the tumor‐suppressive and oncogenic roles of autophagy (Fig 2). Fasting in conditional whole body Atg7 knockout mice was lethal and lead to muscle wasting and hypoglycemia. However, anti‐tumor activity of autophagy inhibition was achieved prior to systemic metabolic dysfunction in lung cancer models indicating the existence of a therapeutic window (Karsli‐Uzunbas et al, 2014).
Pancreas
Autophagy is elevated in pancreatic cancer and inhibition of autophagy by shRNA knockdown of ATG5 was first used to link autophagy to tumor development (Yang et al, 2011). Similarly to breast and lung cancer, the mutational landscape seems to be a critical factor in determining if autophagy plays a role in pancreatic cancer. Oncogenic activation of KRAS and inactivation of p53 are common in pancreatic cancer (Ying et al, 2016). In the humanized genetically modified mouse model of pancreatic cancer driven by KrasG12D , autophagy inhibition by deletion of Atg5 or Atg7 results in an inhibition of high‐grade pancreatic lesions and pancreatic ductal carcinoma (PDAC) (Rosenfeldt et al, 2013). Importantly, tumors in mice with ablation of both copies of p53 and Kras activation are no longer sensitive to autophagy (Rosenfeldt et al, 2013). However, a subsequent study using a pancreas specific p53 +/−, which is subject to loss of heterozygosity during disease progression, showed that autophagy deficiency was still able to block cancer progression in this model that more closely mimics the tumor development in humans (Yang et al, 2014). Collectively, these studies show that autophagy plays an oncogenic role in the progression of pancreatic cancer, while also underscoring the importance of considering the timing and makeup of accompanying genetic lesions in recapitulating human disease. Additionally, a study in the KrasG12D model showed that reductions in autophagy, as opposed to complete inhibition, exhibit very different results than complete loss of autophagy (Görgülü et al, 2019). To modulate autophagy in the Kras‐driven cancer, Atg5 was either reduced to one allele or completely deleted. While Atg5 −/− co‐deletion reduced tumor formation when compared to Kras controls, a single allele deletion of Atg5 resulted in an increase in tumor formation and metastases (Görgülü et al, 2019). The increase in metastasis and tumor burden with partial autophagy inhibition may raise concerns about unintended results from drug‐induced autophagy inhibition. However, in a genetic model using an inducible and reversible dominant negative point mutant of ATG4BC74A meant to mimic the reversible inhibition of an autophagy‐targeting drug, autophagy inhibition resulted in tumor regression without significant side effects (Yang et al, 2018). While additional details still need to be resolved regarding the role of autophagy in pancreatic cancer, including the effects of autophagy in heterogeneous cell types found in pancreatic cancer (Milan et al, 2021), the preponderance of data suggests that inhibition of autophagy may have a therapeutic role. The potential of inhibiting autophagy has spurred on multiple clinical trials using combination therapy with hydroxychloroquine (NCT04145297, NCT03825289, NCT04132505).
Melanoma
Early work in melanoma showed an elevation of autophagy, which correlated with poor therapeutic response (Ma et al, 2011). But the exact contribution of autophagy in the development of melanoma remains unclear. As described in the tumor‐suppressive section above, Atg7 deletion with wildtype Pten worsens overall survival (Rosenfeldt et al, 2021). BRAF and NRAS mutations are common in cutaneous melanoma (Schadendorf et al, 2015). However, using a model that inhibits autophagy by Atg7 deletion in melanoma driven by Braf activation and allelic loss of Pten, an inhibition of tumor growth was observed as well as significantly extended overall survival (Xie et al, 2015). One possible explanation for this discrepancy is that downregulation of autophagy and Pten deletion have both been implicated in avoidance of oncogene‐induced senescence and the timing or exclusivity of these mechanisms to escape senescence may warrant further study to explain this genetic interaction. While the role of autophagy in breast, lung, pancreatic, and melanoma cancers has been studied in some detail, autophagy has also been examined in additional genetic mouse models and allografts, which were summarized in a recent review (Klionsky et al, 2021).
Chaperone‐mediated autophagy in cancer
Chaperone‐mediated autophagy (CMA) is a type of selective autophagy that uses different proteins to promote cellular degradation and does not involve formation of an autophagosome. Unlike conventional autophagy which targets ubiquitinated and damaged organelles, CMA is known to target fully functional proteins to exert its effects on important cellular processes such as cell cycle (Kon et al, 2011; Hubbi et al, 2014; Zhou et al, 2016) and metabolism (Kon et al, 2011; Lv et al, 2011; Xia et al, 2015; Arias & Cuervo, 2020). In CMA, individual cargo are delivered to the lysosome via a heat shock‐cognate chaperone of 70 kDa (hsc70), which recognizes a pentapeptide motif on the cargo protein (Kirchner et al, 2019; Arias & Cuervo, 2020). The translocation of substrate into the lysosome requires lysosome‐associated membrane protein type 2A (LAMP2A), which acts in concert with hsc70. In this pathway, protein levels of LAMP2A are limiting and are used as a proxy for measurement of CMA capacity (Cuervo & Dice, 2000). Chaperone‐mediated autophagy is upregulated in several cancers and contributes to tumor growth (Kon et al, 2011). Inhibition of CMA by LAMP2A knock‐down suppresses tumor growth (Ding et al, 2016; Han et al, 2017). However, not all roles of CMA support tumor growth and, like autophagy, CMA has been reported to have tumor‐suppressive attributes in early stages of tumor formation (Arias & Cuervo, 2020). Additionally, CMA in cancer cells can promote tumor growth through regulation of immune cells in tumor microenvironment. In glioblastoma, the inactivation of CMA in the tumor suppresses the proinflammatory activity of pericytes, resulting in an increase of anti‐tumor T cells and reduction of tumor growth (Valdor et al, 2019).
In the future, it will be interesting to determine if the next generation of autophagy inhibitors have an effect on CMA, or whether the development of specific inhibitors of CMA are feasible and beneficial in cancer treatment.
Autophagy in cancer stem cells
Cancer stem cells (CSCs, also known as tumor‐initiating cells) are found in many types of cancer and contribute to tumor heterogeneity, metastases, and chemotherapeutic resistance (Kaur et al, 2014; Smith & Macleod, 2019). The first CSC was identified in acute myeloid leukemia (Lapidot et al, 1994), but have since been identified in several cancer types including solid tumors. In normal stem cells, autophagy has been reported to promote stemness through regulation of metabolic stress, hypoxia, mitochondrial turnover, and oxidative stress (Liu et al, 2010; Tra et al, 2011; Oliver et al, 2012; Vázquez et al, 2012; Warr et al, 2013; García‐Prat et al, 2016; Naik et al, 2019; He et al, 2021). It is through some of these same mechanisms that autophagy supports CSCs survival. However, it was the ability of autophagy to preserve the CSC population in chronic myelogenous leukemia (CML) that raised a significant amount of interest in studying the therapeutic potential of targeting autophagy to selectively kill CSCs. The ability of CSCs to upregulate autophagy to survive chemotherapeutics was first clearly illustrated in CML treated with imatinib, a tyrosine kinase inhibitor of the BCR‐ABL fusion protein. Treatment with imatinib in combination with either pharmacological or genetic inhibition of the autophagy pathway resulted in much better killing of CSCs than imatinib alone (Bellodi et al, 2009). This study spurred on several clinical trials targeting autophagy in combination with front‐line therapeutics. However, most of these studies used hydroxychloroquine, a lysosomotropic agent that only indirectly block autophagy, and have met with limited success (Horne et al, 2020). The use of more targeted drugs against autophagy is underway and their functionality in combinational therapy is of intense interest to the field. It has since been shown that upregulation of autophagy is a common mechanism by which CSCs survive pharmacological stressors and the nutrient limitation of the stem cell niche (Nazio et al, 2019).
From genetic models, we know that autophagy plays a role in supporting the CSC population in non‐myelogenous cancers. For example, in breast cancer inhibition of autophagy by deletion of Becn1 or Fip200 results in reduced CSC number and inhibition of tumor growth (Gong et al, 2013; Yeo et al, 2016). While these studies indicate that inhibition of autophagy may be a viable approach to inhibit CSCs, it may take the use of new and more specific autophagy inhibitors in order to successfully target CSC population in susceptible tumors.
Autophagy in the regulation of anti‐tumor immunity
Therapeutic promotion of anti‐tumor immunity has had a profound impact on the treatment of a variety of cancers in numerous organs, including kidney, skin, lung, head and neck, and bladder cancers (Egen et al, 2020). An analysis of genetic circuits used by diverse cancer cells to evade detection and killing by immune cells has identified a conserved role for autophagy genes (Lawson et al, 2020). The strength of an anti‐tumor immune response is governed by a variety of factors, including presentation of cancer antigens to prime T cells, secretion of pro‐tumor and anti‐tumor chemokines, T cell infiltration, T cell activation and killing, and clearance of tumor cell debris. Existing therapies focus heavily on inhibitory checkpoint proteins. Immune checkpoints are ligand–receptor pairs, which elicit a suppression of the immune cell activity (Egen et al, 2020). A well‐studied example is programmed cell death protein 1 (PD‐1) that is expressed on T cells and its ligand PD‐L1 that is expressed on cancer cells. Activation of this immune checkpoint greatly dampens the tumor‐killing activity of T cells. Drugs that exploit this pathway block the checkpoint, often with monoclonal antibodies, and activate the anti‐tumor immune response (Braun et al, 2021). Another checkpoint protein, Cytotoxic T lymphocyte‐associated protein 4 (CTLA‐4) has also been targeted in immunotherapy with good success. However, even in tumors that have seen markedly improved outcomes using checkpoint inhibitors, such as in kidney cancer, a large proportion of patients remain resistant leading to the search for new inhibitory receptors (Andrews et al, 2019; Braun et al, 2021). While the use of checkpoint inhibitors has been a game changer for some diseases, there remains a large fraction of patients who do not respond. Some factors that contribute to checkpoint inhibitor resistance are changes in immune effectors or antigen presentation (Sucker et al, 2017). Inhibition of autophagy by deletion of Fip200 or Becn1 in PyMT‐driven breast cancer and B16‐F10 melanoma leads to an increase in cytokines that recruit anti‐tumor immune cells, indicating that inhibition of autophagy may be a viable approach to sensitizing some cancers to immunotherapy (Wei et al, 2011; Mgrditchian et al, 2017). Additionally, autophagy‐dependent control of extracellular ATP has been shown to play an important role in the control of immune surveillance (Kepp et al, 2021). Recent work in the autophagy field has uncovered links between the autophagy pathway and key steps in the anti‐tumor immune response.
One common cause of immune evasion is mutations that can lead to the inhibition of major histocompatibility complex class 1 (MHC‐I; Rooney et al, 2015). However, in absence of genetic alterations to MHC‐I related proteins, cancer cells can also downregulate MHC‐I through an autophagy‐dependent process. In pancreatic cancer, MHC‐I was specifically targeted for autophagic degradation through its interaction with the autophagy receptor NBR1 (Fig 3; Yamamoto et al, 2020). In this model inhibition of autophagy restores MHC‐I and increases antigen presentation, thus enhancing T‐cell function and reducing tumor growth in a syngeneic model (Yamamoto et al, 2020). Autophagy inhibition by knockdown of ATG3 or ATG7, or pharmacologically using chloroquine (CQ) or ULK1 inhibitors also resulted in a sensitization of pancreatic and lung cancer to checkpoint inhibitors (Yamamoto et al, 2020; Deng et al, 2021). Autophagy has also been linked to the increased production of CD8+ memory T cells and reduction in tumor growth in autophagy‐deficient syngeneic mouse cancer models (Fig 3; DeVorkin et al, 2019). Mechanistically, it was shown that ablation of autophagy by deletion of ATG5 resulted in elevated production of interferon gamma (IFNγ) and tumor necrosis factor alpha (TNF‐α; DeVorkin et al, 2019). Interestingly, a high‐throughput CRISPR screen in multiple cancers identified autophagy as a key pathway contributing to evasion of T‐cell killing and resistance to TNF‐induced cytotoxicity (Lawson et al, 2020). Pharmacological inhibition of autophagy using VPS34 inhibitors has been shown to increase inflammation and T‐cell infiltration (Noman et al, 2020).
Figure 3. Roles for autophagy in anti‐tumor immunity.
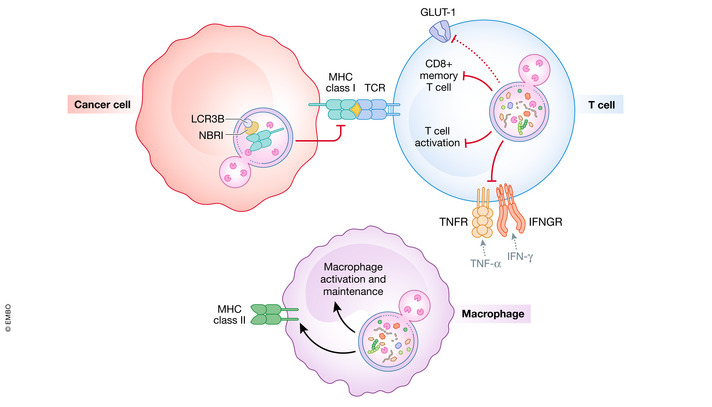
Autophagy inhibition promotes anti‐tumor immunity through a variety of factors. Tumor cells can degrade MHC Class I through the autophagy receptor NBRI reducing T‐cell activation. Autophagy inhibition in T cells also promotes activation of T cells. Autophagy in macrophages creates substrates for MHC Class II and promotes differentiation and maintenance. Systemic inhibition of autophagy in macrophages inhibits tumor‐associated macrophage differentiation and increases macrophage infiltration in the tumor.
Autophagy also plays a role in macrophage‐dependent anti‐tumor immunity. Autophagy has been described to promote M2 macrophage polarization, which is generally considered to be anti‐inflammatory and promote tumorigenesis (Fig 3; Sanjurjo et al, 2018; Wu et al, 2020). Inhibition of autophagy in pancreatic cancer using transient induction of a dominant‐negative ATG4B mutant resulted in an increase in macrophage tumor infiltration. Depletion of macrophages resulted in an impairment of tumor regression upon autophagy induction in this model (Yang et al, 2018). While acute inhibition of autophagy does not seem to damage adaptive immunity, autophagy plays an important role in the differentiation and survival of stimulated monocytes (Zhang et al, 2012; Starobinets et al, 2016). Autophagy has been implicated in the antigen delivery to MHC Class II on macrophages, thereby stimulating T cells (Dengjel et al, 2005). Autophagy and LC3‐associated phagocytosis (LAP) have been linked to the polarization of M2 macrophages, which are generally considered to promote oncogenesis (Liu et al, 2015; Cunha et al, 2018). The contribution of autophagy to the immune response to cancer can also be cell non‐autonomous (Yang et al, 2018; Poillet‐Perez et al, 2020). In a model of hepatic cancer with high mutational burden, the conditional deletion of Atg7 in the host resulted in an increase in inflammatory cytokine production and greater CD4+ T‐cell tumor infiltration (Poillet‐Perez et al, 2020). This indicates that host autophagy plays a role in enhancing the growth of hepatic tumors with high mutational burden by inhibiting T cell‐mediated anti‐tumor immunity (Poillet‐Perez et al, 2020).
Similarly to how autophagy can play dual roles in tumor suppression and oncogenesis, the role of autophagy in immune surveillance is nuanced. As described above, autophagy can play a role in silencing T‐cell activation (Fig 3). However, it can also increase immune surveillance induced by chemotherapeutics. Fasting, a physiological way to induce autophagy in many organs of the body, can have a beneficial effect when combined with chemotherapeutics that target DNA repair and replication (Mizushima et al, 2004; Lee et al, 2012). Notably, the beneficial effects of starvation were lost in mice that lacked T cells, indicating that starvation can enhance chemotherapy‐induced immunosurveillance (Pietrocola et al, 2016). Like T cell‐deficient mice, knockdown of ATG5 also resulted in an attenuation of starvation‐induced immunosurveillance (Pietrocola et al, 2016). The studies above demonstrate some of the potential effects of autophagy inhibition in the promotion of anti‐tumor immunity. However, autophagy has also been shown to play a role in T‐cell priming by dendritic cells and development of anti‐viral CD8+ T cells. Therefore, careful analysis of the immune response needs to be characterized when testing autophagy inhibition in future pre‐clinical models.
Autophagy and tumor cell metabolism
The recycling of nutrients by autophagy is necessary to meet the altered metabolic demands of some cancers (Nyfeler & Eng, 2016). There are several properties that can contribute to a cancers autophagy dependency including intrinsic factors like switching to anaerobic metabolism or an increased need for the biosynthetic building blocks required for continuous cell division (Strohecker et al, 2013). Extrinsically, autophagy‐dependent cancer cells have to contend with the stressors of hypoxia, immune response, and chemotherapeutics (Kimmelman & White, 2017). In RAS‐driven tumors autophagy was shown to support glycolysis and maintain nucleotide levels by supplying amino acids under starvation (Fig 4; Guo et al, 2011). In KRAS‐driven lung cancer, autophagy supports AMPK signaling (Eichner et al, 2019), which was recently tied to lung cancer growth through promotion of lysosomal gene expression (Eichner et al, 2019). In pancreatic cancer, melanocyte inducing transcription factor (MITF), transcription factor binding to IGHM enhancer 3 (TFE3), and transcription factor EB (TFEB) drive autophagy and are necessary for maintaining intracellular amino acid pools (Perera et al, 2015). As discussed in the section on mitophagy, mutations or deletions of mitophagy adaptors are also linked to metabolic reprogramming of cancer cells. Collectively, we have learned from these studies that autophagy plays an important role in promoting the metabolic alterations in multiple cancer models.
Figure 4. Autophagy supports tumor metabolism.
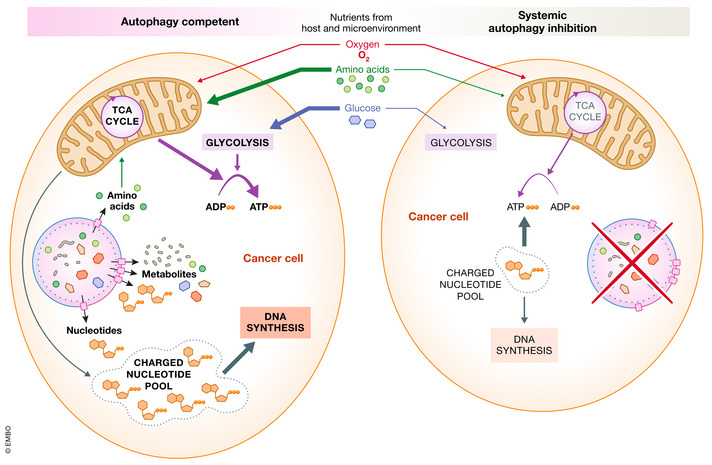
Left: Autophagy supports maintenance of charged nucleotide and adenosine pools within the cancer cell. Host autophagy maintains circulating amino acids and glucose that can be used as metabolic inputs for oxidative phosphorylation and glycolysis for the cancer cell. Right: Systemic autophagy inhibition reduces mitochondrial amino acid oxidative phosphorylation, resulting in a decrease in ATP. Energy stored in charged nucleotide pools is used to support ATP production leading to a depletion in nucleotides that can be used to support cancer cell DNA synthesis.
Host autophagy promotes tumor cell metabolic deficiencies
Cancer cells have altered metabolic requirements that can be supported by metabolites produced by autophagy in distant organs (Fig 4). For example, several cancer cells increase their production of nucleotides by inhibiting expression of argininosuccinate synthase 1 or argininosuccinate lyase, which are necessary for arginine biosynthesis (Fig 5; Soria et al, 2021). As a result, these cells are reliant on circulating arginine that is provided by autophagy in the liver (Dillon et al, 2004; Rabinovich et al, 2015). If Atg7 is conditionally knocked out in the liver or whole body, tumor growth is reduced (Poillet‐Perez et al, 2018). The exact mechanisms by which arginine promotes cancer cell survival remains unclear, but might involve activation of mTOR, polyamine synthesis, or production of nitric oxide (Morris, 2016).
Figure 5. Tumor growth is supported by host autophagy.
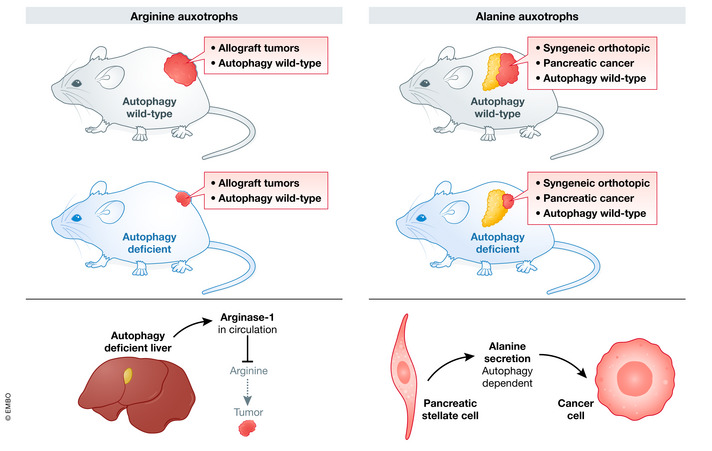
Left: Autophagy inhibition in the liver increases plasma arginase‐1, depleting circulating arginine. Ablation of host autophagy decreases the growth of autophagy competent cancer cells. Right: Autophagy inhibition in pancreatic stellate cells reduces alanine delivery to autophagy competent pancreatic cancer cells.
In addition to distally produced nutrients, the tumor can also stimulate nutrient production from the tumor microenvironment. In pancreatic cancer, the involvement of the stroma in supporting cancer cell metabolism is well established (Feig et al, 2012; Sousa & Kimmelman, 2014). Pancreatic cancer is nutrient poor leading the tumor cells to scavenge protein from its surroundings. The amino acid alanine, rather than glucose or arginine, is the major carbon source for pancreatic cancer (Fig 5; Kamphorst et al, 2015). Pancreatic stromal cell autophagy is required for the release of alanine into the tumor microenvironment, where it is scavenged by the tumor cells (Sousa et al, 2016). Consequently, stable knockdown of ATG5 or ATG7 in pancreatic stellate cells resulted in a reduction in alanine in the tumor microenvironment, inhibition of tumor growth, and an increase in disease‐free survival (Sousa et al, 2016). The discoveries linking systemic autophagy to impairments in tumor metabolism and growth are an exciting advance in the field. Future work to discover the link between an acquired addiction to circulating nutrients and cancer sensitivity to autophagy inhibitors will help in the identification of tumors that should be prioritized inclusion in future clinical trials targeting autophagy.
The therapeutic potential of targeting autophagy
The importance of autophagy in maintaining cellular and organismal homeostasis is well known. For example, conditional knockout of Atg7 in adult mice leads to increased rates of infection, neurodegeneration, and metabolic deficiencies (Karsli‐Uzunbas et al, 2014). These disorders limited mouse survival to 2–3 months due to neurodegeneration and starvation‐induced hypoglycemia and cachexia (Karsli‐Uzunbas et al, 2014). However, this study as well as others identified a therapeutic window where the anti‐cancer effects precede systemic dysfunction (Yang et al, 2011). Moreover, the preponderance of neurological disorders also indicated that an autophagy inhibitor that could not cross the blood brain barrier would be better tolerated (Amaravadi et al, 2019). Another model system, which used a whole‐body inducible dominant negative mutant of the ATG4B enzyme, also found that anti‐cancer effects could be achieved by inhibiting host autophagy, without lethality or increased oncogenesis arising from transient systemic inhibition of autophagy (Yang et al, 2018).
While there is a possible therapeutic window for autophagy inhibitors, evidence from pre‐clinical models indicates that only some tumors will be autophagy sensitive. For example, factors that have been linked “autophagy addiction” in cancer include RAS and BRAF proto‐oncogene activation, MHC I downregulation, and arginine auxotrophy (Yang et al, 2011; Levy et al, 2014; Mulcahy Levy et al, 2014, 2017; Yang & Kimmelman, 2014; Xie et al, 2015; Poillet‐Perez et al, 2018). Because of the involvement of host metabolism and immunity in the sensitivity of cancer to autophagy inhibition, it is important wherever possible that future studies use immune competent animal models to address these variables.
To date, clinical trials in cancer that target autophagy, alone or in combination, have relied on chloroquine derivatives. The best studied of these derivatives is hydroxychloroquine (HCQ), which has been used to treat malaria and rheumatoid arthritis for several decades. These drugs block autophagy by deacidifying the lysosome and prevent autophagosome fusion (Mauthe et al, 2018). However, HCQ is not an autophagy‐specific inhibitor and its effect on cancer cells can be autophagy‐independent (Maycotte et al, 2012; Maes et al, 2014; Eng et al, 2016). One of the first trials was a small trial in glioblastoma multiforme, which added chloroquine to surgery, radiotherapy, and chemotherapy (Briceño et al, 2003). Although autophagy was not considered in the execution of this study, they observed that chloroquine improved the response rate to the antineoplastic treatments. Subsequent studies identified the potential anti‐cancer effects of autophagy inhibition, and several clinical trials were initiated with HCQ. The outcomes of these clinical trials are summarized in Table 1 if results were reported (see Table 1).
Table 1.
Clinical trials targeting autophagy.
| Condition | Intervention | Results | References |
|---|---|---|---|
| Refractory metastatic pancreatic Cancer | HCQ | No response to HCQ treatment | NCT01273805 (Wolpin et al, 2014) |
| Previously untreated Pancreatic Cancer | HCQ, Gemcitabine | No overall survival improvement or progression‐free survival with HCQ. Statistically significant response rate was increased in HCQ arm | NCT01506973 (Karasic et al, 2019) |
| Pancreatic Cancer | HCQ, gemcitabine, abraxane | Autophagy inhibition correlates with increased disease‐free survival. 61% of patients exhibited decrease in biomarker CA 19‐9. p53 mutational status did not affect response | NCT01978184 (Boone et al, 2015) |
| Solid Tumor | HCQ prior to surgical resection | Increase in apoptosis in resected tumors, no clinical outcomes reported |
NCT02232243 (Wang et al, 2018) |
| Renal Cell Carcinoma | HCQ, everolimus | Increase in progression free survival with HCQ was observed. | NCT01510119 (Haas et al, 2019) |
| Brain and Central Nervous System Tumors | HCQ, temozolomide, Radiation | Results posted, no associated publications | NCT00486603 |
| Metastatic Renal Cell Carcinoma | HCQ, IL‐2 | Results posted, no associated publications | NCT01550367 |
| Glioblastoma | HCQ, temozolomide, radiation | No improvement in overall survival. Dose limiting toxicity prevented escalation of HCQ. Autophagy inhibition was not consistently achieved | NCT00486603 (Rosenfeld et al, 2014) |
While some of the HCQ trials have shown promise, the efficiency of autophagy inhibition and autophagy‐independent effects have led to a search for more targeted and potent inhibitors. ULK1 inhibitors SBI‐0206965, ULK101 and MRT403, which should also inhibit ULK2, should be much more specific than HCQ in targeting the autophagy pathway. Inactivation of ULK1&2 will abrogate most bulk autophagy. However, it will spare LAP, which may be more beneficial for preserving immune function. In terms of specificity, SBI‐0206965 may also target FAK and aurora kinases, while ULK101 inhibits members of the CAMK family (Egan et al, 2015; Martin et al, 2018). MRT403 was recently shown to sensitize CSCs from patient‐derived CML to imatinib (Ianniciello et al, 2021). This is an exciting development and the sensitivity of CSCs to autophagy inhibition was one of the early drivers for the application of autophagy inhibitors in the clinic (Bellodi et al, 2009).
VPS34 is the catalytic component of multiple lipid kinase complexes, which are necessary for autophagy and endocytosis. Specific inhibitors of VPS34, like SAR405, therefore effects both autophagosome and late endosome compartments (Ronan et al, 2014). Inhibition of VPS34 with SAR405 inhibited the growth of multiple cancer types in a xenograft model and promoted immune infiltration in the B16‐F10 melanoma model (Noman et al, 2020). As a result, VPS34 inhibitors sensitized tumors to anti‐PD‐L1 checkpoint inhibitors. Despite their recent success in pre‐clinical models, VSP34 inhibitors have not yet made it into clinical trials.
ATG4B is a protease that can activate LC3 for conjugation of LC3 to phosphatidylethanolamine and also plays a role in its deconjugation in the final steps of autophagosome closure. Overexpression of mutant ATG4B results in an inhibition of autophagy and accumulation of defective autophagosomes. Inhibitors of ATG4B have been developed and tested in vitro and mouse models (Akin et al, 2014; Chu et al, 2018; Fu et al, 2019). ULK1, VPS34, and ATG4B inhibitors are of interest, but have not yet made it to clinical trials.
As mentioned above, the use of general autophagy inhibitors that cannot cross the blood‐brain barrier is a strategy to limit neurotoxicity. However, this would not address the more uncommon, but potentially serious damage to the liver, retina, and other tissues that can occur with high dose and prolonged use (Geamănu (Pancă) et al, 2014; Madhavan et al, 2016). Another approach is to inhibit selective forms of autophagy that are relevant to the disease in order to retain the majority of autophagy‐dependent activity in normal cells. Autophagy receptors are attractive targets as they recruit specific ubiquitinated cargo to the autophagosome and their individual deletion does not eliminate all forms of autophagy. For example, the p62‐NRF2‐KEAP1 signaling pathway in liver cancer regulates metabolic reprogramming in response to oxidative stress (Saito et al, 2016). In pancreatic cancer, which has high basal autophagy, targeting of specific autophagy receptors has uncovered distinct in tumor‐suppressive effects. Knockdown of NBR1 inhibited autophagic MHC‐I turnover and immune evasion in pancreatic cancer (Yamamoto et al, 2020), while knockdown of optineurin caused an inhibition in colony formation and activation of apoptosis (Ali et al, 2019).
Targeting of mitophagy receptors in cancer has also been the focus of significant study (see preceding section “Mitophagy in cancer” for additional details). These studies have yielded contrasting results with BNIP3 knockdown in triple‐negative breast cancer reducing tumor growth (Chourasia et al, 2015), while Park2 or Bnip3L deletion caused an increase in pancreatic cancer growth (Li et al, 2018; Humpton et al, 2019). In spite of the promising results from inhibition of autophagy receptors by knockout or knockdown, the therapeutic potential of targeting selective autophagy remains to be realized due to a lack of drugs to target these receptors selectively.
Synergy of dual inhibition of autophagy and Ras pathways in cancer
In response to chemotherapeutics, cancer cells can upregulate autophagy to promote survival (Sui et al, 2013). The ability of autophagy inhibition to sensitize cancer cells to targeted therapeutics has been exemplified by, but is not limited to, recent work in RAS‐driven pancreatic cancer. Activation of KRAS signaling is common in pancreatic cancer and drives autophagy induction. In KRAS‐driven cancers pharmacologic or genetic inhibition of RAS‐ERK signaling activates autophagy (Bryant et al, 2019; Kinsey et al, 2019; Lee et al, 2019). While inhibition of KRAS‐signaling alone is not effective in pancreatic cancer, the inhibition of autophagy in conjunction with targeting MAPK or ERK1/2 downstream of RAS results in a decrease in tumor formation (Infante et al, 2014; Bryant et al, 2019; Kinsey et al, 2019; Lee et al, 2019). RAS also activates BRAF, which is frequently mutated in melanoma, central nervous system tumors, and colorectal cancers. Cancers treated with the BRAF inhibitor vemurafenib often develop resistance to the drug and display a concomitant increase in autophagy levels (Ma et al, 2014). Inhibition of autophagy with CQ has been described to re‐sensitize tumors to vemurafenib or other inhibitors targeting kinases downstream of BRAF (Mulcahy Levy et al, 2017). Collectively, these studies show that in “autophagy addicted” cancers the combination of autophagy inhibitors with frontline treatments holds significant therapeutic promise.
Challenges and future directions
Despite intense interest in targeting autophagy in cancer clinical trials, no definitive therapeutic edge has been provided by combination of HCQ to approved therapeutics. It is obvious that autophagy inhibitors are unlikely to be effective as a single agent therapy for cancer. Encouragingly, there seems to be a therapeutic window for autophagy inhibition and use of these antimalarials have largely been devoid of serious metabolic dysfunction, neurological problems, or liver injury. The use of newer and more specific autophagy inhibitors will hopefully be able to exploit autophagy addicted cancers in a more efficacious manner. Work in GEMM has also shown that certain mutations result in sensitivity to autophagy inhibition, while others do not (Karsli‐Uzunbas et al, 2014). Therefore, expanded experimentation in GEMMs may uncover which cancer types may benefit from autophagy inhibitors. Additionally, in the last decade we have seen a rapid advance in our understanding of the regulatory mechanisms controlling selective autophagy. However, the inhibition of selective autophagy in GEMM or with drugs is significantly more challenging than the study of blocking all autophagy. Despite these challenges, targeting selective autophagy, in particular mitophagy, represents an attractive target for future study and therapeutic development.
Disclosure and competing interests statement
K.L.G. is a cofounder of and has equity interest in Vivace Therapeutics. R.C.R. has equity interest in Benchmark Bioscience.
Acknowledgments
The Russell laboratory acknowledges support from the CIHR grants #462274 and #153034; and NIH grant GM51586 (K.L.G.). The authors thank Damian Gatica and Karyn King for their input. Figures created with Biorender.com.
The EMBO Journal (2022) 41: e110031.
This article is part of the Cancer Reviews series.
References
- Akin D, Wang SK, Habibzadegah‐Tari P, Law B, Ostrov D, Li M, Yin X‐M, Kim J‐S, Horenstein N, Dunn WA (2014) A novel ATG4B antagonist inhibits autophagy and has a negative impact on osteosarcoma tumors. Autophagy 10: 2021–2035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali DM, Ansari SS, Zepp M, Knapp‐Mohammady M, Berger MR (2019) Optineurin downregulation induces endoplasmic reticulum stress, chaperone‐mediated autophagy, and apoptosis in pancreatic cancer cells. Cell Death Discov 5: 128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaravadi R, Kimmelman AC, White E (2016) Recent insights into the function of autophagy in cancer. Genes Dev 30: 1913–1930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaravadi RK, Kimmelman AC, Debnath J (2019) Targeting autophagy in cancer: recent advances and future directions. Cancer Discov 9: 1167–1181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews LP, Yano H, Vignali DAA (2019) Inhibitory receptors and ligands beyond PD‐1, PD‐L1 and CTLA‐4: breakthroughs or backups. Nat Immunol 20: 1425–1434 [DOI] [PubMed] [Google Scholar]
- Arias E, Cuervo AM (2020) Pros and cons of chaperone‐mediated autophagy in cancer biology. Trends Endocrinol Metab 31: 53–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baginska J, Viry E, Berchem G, Poli A, Noman MZ, van Moer K, Medves S, Zimmer J, Oudin A, Niclou SP et al (2013) Granzyme B degradation by autophagy decreases tumor cell susceptibility to natural killer‐mediated lysis under hypoxia. Proc Natl Acad Sci USA 110: 17450–17455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellodi C, Lidonnici MR, Hamilton A, Helgason GV, Soliera AR, Ronchetti M, Galavotti S, Young KW, Selmi T, Yacobi R et al (2009) Targeting autophagy potentiates tyrosine kinase inhibitor‐induced cell death in Philadelphia chromosome‐positive cells, including primary CML stem cells. J Clin Invest 119: 1109–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellot G, Garcia‐Medina R, Gounon P, Chiche J, Roux D, Pouysségur J, Mazure NM (2009) Hypoxia‐induced autophagy is mediated through hypoxia‐inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol 29: 2570–2581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T (2005) p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin‐induced cell death. J Cell Biol 171: 603–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boone BA, Bahary N, Zureikat AH, Moser AJ, Normolle DP, Wu W‐C, Singhi AD, Bao P, Bartlett DL, Liotta LA et al (2015) Safety and fgb. Ann Surg Oncol 22: 4402–4410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman‐Colin C, Xia B, Bunting S, Klijn C, Drost R, Bouwman P, Fineman L, Chen X, Culhane AC, Cai H et al (2013) Palb2 synergizes with Trp53 to suppress mammary tumor formation in a model of inherited breast cancer. Proc Natl Acad Sci USA 110: 8632–8637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun DA, Bakouny Z, Hirsch L, Flippot R, Van Allen EM, Wu CJ, Choueiri TK (2021) Beyond conventional immune‐checkpoint inhibition – novel immunotherapies for renal cell carcinoma. Nat Rev Clin Oncol 18: 199–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briceño E, Reyes S, Sotelo J (2003) Therapy of glioblastoma multiforme improved by the antimutagenic chloroquine. Neurosurg Focus 14: e3 [DOI] [PubMed] [Google Scholar]
- Bryant KL, Stalnecker CA, Zeitouni D, Klomp JE, Peng S, Tikunov AP, Gunda V, Pierobon M, Waters AM, George SD et al (2019) Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat Med 25: 628–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catalano M, D'Alessandro G, Lepore F, Corazzari M, Caldarola S, Valacca C, Faienza F, Esposito V, Limatola C, Cecconi F et al (2015) Autophagy induction impairs migration and invasion by reversing EMT in glioblastoma cells. Mol Oncol 9: 1612–1625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaanine AH, Kohlbrenner E, Gamb SI, Guenzel AJ, Klaus K, Fayyaz AU, Nair KS, Hajjar RJ, Redfield MM (2016) FOXO3a regulates BNIP3 and modulates mitochondrial calcium, dynamics, and function in cardiac stress. Am J Physiol Heart Circ Physiol 311: H1540–H1559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JL, David J, Cook‐Spaeth D, Casey S, Cohen D, Selvendiran K, Bekaii‐Saab T, Hays JL (2017) Autophagy induction results in enhanced anoikis resistance in models of peritoneal disease. Mol Cancer Res 15: 26–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen N, Eritja N, Lock R, Debnath J (2013) Autophagy restricts proliferation driven by oncogenic phosphatidylinositol 3‐kinase in three‐dimensional culture. Oncogene 32: 2543–2554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Dorn GW (2013) PINK1‐phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 340: 471–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chourasia AH, Tracy K, Frankenberger C, Boland ML, Sharifi MN, Drake LE, Sachleben JR, Asara JM, Locasale JW, Karczmar GS et al (2015) Mitophagy defects arising from BNip3 loss promote mammary tumor progression to metastasis. EMBO Rep 16: 1145–1163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu J, Fu Y, Xu J, Zheng X, Gu Q, Luo X, Dai QI, Zhang S, Liu P, Hong L et al (2018) ATG4B inhibitor FMK‐9a induces autophagy independent on its enzyme inhibition. Arch Biochem Biophys 644: 29–36 [DOI] [PubMed] [Google Scholar]
- Cicchini M, Chakrabarti R, Kongara S, Price S, Nahar R, Lozy F, Zhong H, Vazquez A, Kang Y, Karantza V (2014) Autophagy regulator BECN1 suppresses mammary tumorigenesis driven by WNT1 activation and following parity. Autophagy 10: 2036–2052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuervo AM, Dice JF (2000) Regulation of lamp2a levels in the lysosomal membrane. Traffic 1: 570–583 [DOI] [PubMed] [Google Scholar]
- Cunha LD, Yang M, Carter R, Guy C, Harris L, Crawford JC, Quarato G, Boada‐Romero E, Kalkavan H, Johnson MDL et al (2018) LC3‐associated phagocytosis in myeloid cells promotes tumor immune tolerance. Cell 175: 429–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W et al (2002) Mutations of the BRAF gene in human cancer. Nature 417: 949–954 [DOI] [PubMed] [Google Scholar]
- Delaney JR, Patel CB, Bapat J, Jones CM, Ramos‐Zapatero M, Ortell KK, Tanios R, Haghighiabyaneh M, Axelrod J, DeStefano JW et al (2020) Autophagy gene haploinsufficiency drives chromosome instability, increases migration, and promotes early ovarian tumors. PLoS Genet 16: e1008558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng J, Thennavan A, Dolgalev I, Chen T, Li J, Marzio A, Poirier JT, Peng DH, Bulatovic M, Mukhopadhyay S et al (2021) ULK1 inhibition overcomes compromised antigen presentation and restores antitumor immunity in LKB1 mutant lung cancer. Nat Cancer 2: 503–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dengjel J, Schoor O, Fischer R, Reich M, Kraus M, Müller M, Kreymborg K, Altenberend F, Brandenburg J, Kalbacher H et al (2005) Autophagy promotes MHC class II presentation of peptides from intracellular source proteins. Proc Natl Acad Sci USA 102: 7922–7927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVorkin L, Pavey N, Carleton G, Comber A, Ho C, Lim J, McNamara E, Huang H, Kim P, Zacharias LG et al (2019) Autophagy regulation of metabolism is required for CD8+ T cell anti‐tumor immunity. Cell Rep 27: 502–513 [DOI] [PubMed] [Google Scholar]
- Dhingra R, Gang H, Wang Y, Biala AK, Aviv Y, Margulets V, Tee A, Kirshenbaum LA (2013) Bidirectional regulation of nuclear factor‐κB and mammalian target of rapamycin signaling functionally links Bnip3 gene repression and cell survival of ventricular myocytes. Circ Heart Fail 6: 335–343 [DOI] [PubMed] [Google Scholar]
- Dillon BJ, Prieto VG, Curley SA, Ensor CM, Holtsberg FW, Bomalaski JS, Clark MA (2004) Incidence and distribution of argininosuccinate synthetase deficiency in human cancers: a method for identifying cancers sensitive to arginine deprivation. Cancer 100: 826–833 [DOI] [PubMed] [Google Scholar]
- Ding Z‐B, Fu X‐T, Shi Y‐H, Zhou J, Peng Y‐F, Liu W‐R, Shi G‐M, Gao Q, Wang X‐Y, Song K et al (2016) Lamp2a is required for tumor growth and promotes tumor recurrence of hepatocellular carcinoma. Int J Oncol 49: 2367–2376 [DOI] [PubMed] [Google Scholar]
- Dunlop EA, Tee AR (2013) The kinase triad, AMPK, mTORC1 and ULK1, maintains energy and nutrient homoeostasis. Biochem Soc Trans 41: 939–943 [DOI] [PubMed] [Google Scholar]
- Egan D, Chun M, Vamos M, Zou H, Rong J, Miller C, Lou H, Raveendra‐Panickar D, Yang C‐C, Sheffler D et al (2015) Small molecule inhibition of the autophagy kinase ULK1 and identification of ULK1 substrates. Mol Cell 59: 285–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egen JG, Ouyang W, Wu LC (2020) Human anti‐tumor immunity: insights from immunotherapy clinical trials. Immunity 52: 36–54 [DOI] [PubMed] [Google Scholar]
- Eichner LJ, Brun SN, Herzig S, Young NP, Curtis SD, Shackelford DB, Shokhirev MN, Leblanc M, Vera LI, Hutchins A et al (2019) Genetic analysis reveals AMPK is required to support tumor growth in murine Kras‐dependent lung cancer models. Cell Metab 29: 285–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng CH, Wang Z, Tkach D, Toral‐Barza L, Ugwonali S, Liu S, Fitzgerald SL, George E, Frias E, Cochran N et al (2016) Macroautophagy is dispensable for growth of KRAS mutant tumors and chloroquine efficacy. Proc Natl Acad Sci USA 113: 182–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, Tuveson DA (2012) The pancreas cancer microenvironment. Clin Cancer Res 18: 4266–4276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Hong L, Xu J, Zhong G, Gu Q, Gu Q, Guan Y, Zheng X, Dai QI, Luo X et al (2019) Discovery of a small molecule targeting autophagy via ATG4B inhibition and cell death of colorectal cancer cells in vitro and in vivo . Autophagy 15: 295–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara N, Usui T, Ohama T, Sato K (2016) Regulation of Beclin 1 protein phosphorylation and autophagy by protein phosphatase 2A (PP2A) and death‐associated protein kinase 3 (DAPK3). J Biol Chem 291: 10858–10866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- García‐Fernández M, Karras P, Checinska A, Cañón E, Calvo GT, Gómez‐López G, Cifdaloz M, Colmenar A, Espinosa‐Hevia L, Olmeda D et al (2016) Metastatic risk and resistance to BRAF inhibitors in melanoma defined by selective allelic loss of ATG5. Autophagy 12: 1776–1790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- García‐Prat L, Martínez‐Vicente M, Perdiguero E, Ortet L, Rodríguez‐Ubreva J, Rebollo E, Ruiz‐Bonilla V, Gutarra S, Ballestar E, Serrano AL et al (2016) Autophagy maintains stemness by preventing senescence. Nature 529: 37–42 [DOI] [PubMed] [Google Scholar]
- Gautier CA, Kitada T, Shen J (2008) Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc Natl Acad Sci USA 105: 11364–11369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geamănu (Pancă) A, Popa‐Cherecheanu A, Marinescu B, Geamănu C, Voinea L (2014) Retinal toxicity associated with chronic exposure to hydroxychloroquine and its ocular screening. Review. J Med Life 7: 322–326 [PMC free article] [PubMed] [Google Scholar]
- Geisler S, Holmström KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W (2010) PINK1/Parkin‐mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 12: 119–131 [DOI] [PubMed] [Google Scholar]
- Gong C, Bauvy C, Tonelli G, Yue W, Deloménie C, Nicolas V, Zhu Y, Domergue V, Marin‐Esteban V, Tharinger H et al (2013) Beclin 1 and autophagy are required for the tumorigenicity of breast cancer stem‐like/progenitor cells. Oncogene 32: 2261–2272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Y, Zack TI, Morris LGT, Lin K, Hukkelhoven E, Raheja R, Tan I‐L, Turcan S, Veeriah S, Meng S et al (2014) Pan‐cancer genetic analysis identifies PARK2 as a master regulator of G1/S cyclins. Nat Genet 46: 588–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Görgülü K, Diakopoulos KN, Ai J, Schoeps B, Kabacaoglu D, Karpathaki A‐F, Ciecielski KJ, Kaya‐Aksoy E, Ruess DA, Berninger A et al (2019) Levels of the autophagy‐related 5 protein affect progression and metastasis of pancreatic tumors in mice. Gastroenterology 156: 203–217 [DOI] [PubMed] [Google Scholar]
- Gubas A, Dikic I (2022) A guide to the regulation of selective autophagy receptors. FEBS J 289: 75–89 [DOI] [PubMed] [Google Scholar]
- Guo JY, Chen H‐Y, Mathew R, Fan J, Strohecker AM, Karsli‐Uzunbas G, Kamphorst JJ, Chen G, Lemons JMS, Karantza V et al (2011) Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev 25: 460–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JY, Karsli‐Uzunbas G, Mathew R, Aisner SC, Kamphorst JJ, Strohecker AM, Chen G, Price S, Lu W, Teng X et al (2013) Autophagy suppresses progression of K‐ras‐induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev 27: 1447–1461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ (2008) AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell 30: 214–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas NB, Appleman LJ, Stein M, Redlinger M, Wilks M, Xu X, Onorati A, Kalavacharla A, Kim T, Zhen CJ et al (2019) Autophagy inhibition to augment mTOR inhibition: a phase I/II trial of everolimus and hydroxychloroquine in patients with previously treated renal cell carcinoma. Clin Cancer Res 25: 2080–2087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han QI, Deng Y, Chen S, Chen R, Yang M, Zhang Z, Sun X, Wang W, He Y, Wang F et al (2017) Downregulation of ATG5‐dependent macroautophagy by chaperone‐mediated autophagy promotes breast cancer cell metastasis. Sci Rep 7: 4759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He R, Wang Z, Cui M, Liu S, Wu W, Chen MO, Wu Y, Qu Y, Lin H, Chen S et al (2021) HIF1A Alleviates compression‐induced apoptosis of nucleus pulposus derived stem cells via upregulating autophagy. Autophagy 17: 3338–3360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horne GA, Stobo J, Kelly C, Mukhopadhyay A, Latif AL, Dixon‐Hughes J, McMahon L, Cony‐Makhoul P, Byrne J, Smith G et al (2020) A randomised phase II trial of hydroxychloroquine and imatinib versus imatinib alone for patients with chronic myeloid leukaemia in major cytogenetic response with residual disease. Leukemia 34: 1775–1786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbi ME, Gilkes DM, Hu H, Kshitiz , Ahmed I, Semenza GL (2014) Cyclin‐dependent kinases regulate lysosomal degradation of hypoxia‐inducible factor 1α to promote cell‐cycle progression. Proc Natl Acad Sci USA 111: E3325–E3334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humpton TJ, Alagesan B, DeNicola GM, Lu D, Yordanov GN, Leonhardt CS, Yao MA, Alagesan P, Zaatari MN, Park Y et al (2019) Oncogenic KRAS induces NIX‐mediated mitophagy to promote pancreatic cancer. Cancer Discov 9: 1268–1287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huo Y, Cai H, Teplova I, Bowman‐Colin C, Chen G, Price S, Barnard N, Ganesan S, Karantza V, White E et al (2013) Autophagy opposes p53‐mediated tumor barrier to facilitate tumorigenesis in a model of PALB2‐associated hereditary breast cancer. Cancer Discov 3: 894–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ianniciello A, Zarou MM, Rattigan KM, Scott M, Dawson A, Dunn K, Brabcova Z, Kalkman ER, Nixon C, Michie AM et al (2021) ULK1 inhibition promotes oxidative stress‐induced differentiation and sensitizes leukemic stem cells to targeted therapy. Sci Transl Med 13: eabd5016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Infante JR, Somer BG, Park JO, Li C‐P, Scheulen ME, Kasubhai SM, Oh D‐Y, Liu Y, Redhu S, Steplewski K et al (2014) A randomised, double‐blind, placebo‐controlled trial of trametinib, an oral MEK inhibitor, in combination with gemcitabine for patients with untreated metastatic adenocarcinoma of the pancreas. Eur J Cancer 50: 2072–2081 [DOI] [PubMed] [Google Scholar]
- Inoki K, Zhu T, Guan KL (2003) TSC2 mediates cellular energy response to control cell growth and survival. Cell 115: 577–590 [DOI] [PubMed] [Google Scholar]
- Ishikawa K, Takenaga K, Akimoto M, Koshikawa N, Yamaguchi A, Imanishi H, Nakada K, Honma Y, Hayashi J‐I (2008) ROS‐generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science 320: 661–664 [DOI] [PubMed] [Google Scholar]
- Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, Jacks T, Tuveson DA (2001) Analysis of lung tumor initiation and progression using conditional expression of oncogenic K‐ras. Genes Dev 15: 3243–3248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jena KK, Kolapalli SP, Mehto S, Nath P, Das B, Sahoo PK, Ahad A, Syed GH, Raghav SK, Senapati S et al (2018) TRIM16 controls assembly and degradation of protein aggregates by modulating the p62‐NRF2 axis and autophagy. EMBO J 37: e98358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jewell JL, Guan KL (2013) Nutrient signaling to mTOR and cell growth. Trends Biochem Sci 38: 233–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, Youle RJ (2010) Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol 191: 933–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen T, Lamark T (2020) Selective Autophagy: ATG8 family proteins, LIR motifs and cargo receptors. J Mol Biol 432: 80–103 [DOI] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T (2000) LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 19: 5720–5728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamphorst JJ, Nofal M, Commisso C, Hackett SR, Lu W, Grabocka E, Vander Heiden MG, Miller G, Drebin JA, Bar‐Sagi D et al (2015) Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res 75: 544–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, Banerjee S, Youle RJ (2014) PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol 205: 143–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karantza‐Wadsworth V, Patel S, Kravchuk O, Chen G, Mathew R, Jin S, White E (2007) Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev 21: 1621–1635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karasic TB, O’Hara MH, Loaiza‐Bonilla A, Reiss KA, Teitelbaum UR, Borazanci E, De Jesus‐Acosta A, Redlinger C, Burrell JA, Laheru DA et al (2019) Effect of gemcitabine and nab‐paclitaxel with or without hydroxychloroquine on patients with advanced pancreatic cancer: a phase 2 randomized clinical trial. JAMA Oncol 5: 993–998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karsli‐Uzunbas G, Guo JY, Price S, Teng X, Laddha SV, Khor S, Kalaany NY, Jacks T, Chan CS, Rabinowitz JD et al (2014) Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov 4: 914–927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsuragi Y, Ichimura Y, Komatsu M (2015) p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J 282: 4672–4678 [DOI] [PubMed] [Google Scholar]
- Kaur S, Singh G, Kaur K (2014) Cancer stem cells: an insight and future perspective. J Cancer Res Ther 10: 846–852 [DOI] [PubMed] [Google Scholar]
- Kazlauskaite A, Kondapalli C, Gourlay R, Campbell DG, Ritorto MS, Hofmann K, Alessi DR, Knebel A, Trost M, Muqit MMK (2014) Parkin is activated by PINK1‐dependent phosphorylation of ubiquitin at Ser65. Biochem J 460: 127–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kepp O, Bezu L, Yamazaki T, Di Virgilio F, Smyth MJ, Kroemer G, Galluzzi L (2021) ATP and cancer immunosurveillance. EMBO J 40: e108130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kim YC, Fang C, Russell RC, Kim JH, Fan W, Liu R, Zhong Q, Guan KL (2013) Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell 152: 290–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B, Guan KL (2011) AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13: 132–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmelman AC, White E (2017) Autophagy and tumor metabolism. Cell Metab 25: 1037–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King KE, Losier TT, Russell RC (2021) Regulation of autophagy enzymes by nutrient signaling. Trends Biochem Sci 46: 687–700 [DOI] [PubMed] [Google Scholar]
- Kinsey CG, Camolotto SA, Boespflug AM, Guillen KP, Foth M, Truong A, Schuman SS, Shea JE, Seipp MT, Yap JT et al (2019) Protective autophagy elicited by RAF→MEK→ERK inhibition suggests a treatment strategy for RAS‐driven cancers. Nat Med 25: 620–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchner P, Bourdenx M, Madrigal‐Matute J, Tiano S, Diaz A, Bartholdy BA, Will B, Cuervo AM (2019) Proteome‐wide analysis of chaperone‐mediated autophagy targeting motifs. PLoS Biol 17: e3000301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkin V, Rogov VV (2019) A diversity of selective autophagy receptors determines the specificity of the autophagy pathway. Mol Cell 76: 268–285 [DOI] [PubMed] [Google Scholar]
- Klionsky DJ, Petroni G, Amaravadi RK, Baehrecke EH, Ballabio A, Boya P, Bravo‐San Pedro JM, Cadwell K, Cecconi F, Choi AMK et al (2021) Autophagy in major human diseases. EMBO J 40: e108863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kon M, Kiffin R, Koga H, Chapochnick J, Macian F, Varticovski L, Cuervo AM (2011) Chaperone‐mediated autophagy is required for tumor growth. Sci Transl Med 3: 109ra117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, Kimura Y, Tsuchiya H, Yoshihara H, Hirokawa T et al (2014) Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 510: 162–166 [DOI] [PubMed] [Google Scholar]
- Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N (2004) The role of autophagy during the early neonatal starvation period. Nature 432: 1032–1036 [DOI] [PubMed] [Google Scholar]
- Kumar S, Jain A, Choi SW, Duarte P, da Silva G, Allers L, Mudd MH, Peters RS, Anonsen JH, Rusten T‐E et al (2020) Mammalian Atg8‐family proteins are upstream regulators of the lysosomal system by controlling MTOR and TFEB. Autophagy 16: 2305–2306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon M, Berns A (2013) Mouse models for lung cancer. Molecular Oncology 7: 165–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laddha SV, Ganesan S, Chan CS, White E (2014) Mutational landscape of the essential autophagy gene BECN1 in human cancers. Mol Cancer Res 12: 485–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamark T, Svenning S, Johansen T (2017) Regulation of selective autophagy: the p62/SQSTM1 paradigm. Essays Biochem 61: 609–624 [DOI] [PubMed] [Google Scholar]
- Lampert MA, Orogo AM, Najor RH, Hammerling BC, Leon LJ, Wang BJ, Kim T, Sussman MA, Gustafsson ÅB (2019) BNIP3L/NIX and FUNDC1‐mediated mitophagy is required for mitochondrial network remodeling during cardiac progenitor cell differentiation. Autophagy 15: 1182–1198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres‐Cortes J, Minden M, Paterson B, Caligiuri MA, Dick JE (1994) A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 367: 645–648 [DOI] [PubMed] [Google Scholar]
- Lawson KA, Sousa CM, Zhang X, Kim E, Akthar R, Caumanns JJ, Yao Y, Mikolajewicz N, Ross C, Brown KR et al (2020) Functional genomic landscape of cancer‐intrinsic evasion of killing by T cells. Nature 586: 120–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebovitz CB, Robertson AG, Goya R, Jones SJ, Morin RD, Marra MA, Gorski SM (2015) Cross‐cancer profiling of molecular alterations within the human autophagy interaction network. Autophagy 11: 1668–1687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C‐S, Lee LC, Yuan TL, Chakka S, Fellmann C, Lowe SW, Caplen NJ, McCormick F, Luo J (2019) MAP kinase and autophagy pathways cooperate to maintain RAS mutant cancer cell survival. Proc Natl Acad Sci USA 116: 4508–4517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Raffaghello L, Brandhorst S, Safdie FM, Bianchi G, Martin‐Montalvo A, Pistoia V, Wei M, Hwang S, Merlino A et al (2012) Fasting cycles retard growth of tumors and sensitize a range of cancer cell types to chemotherapy. Sci Transl Med 4: 124ra27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Kim J, Nam H‐J, Gao B, Yin P, Qin BO, Yi S‐Y, Ham H, Evans D, Kim S‐H et al (2015) Parkin regulates mitosis and genomic stability through Cdc20/Cdh1. Mol Cell 60: 21–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy JMM, Thompson JC, Griesinger AM, Amani V, Donson AM, Birks DK, Morgan MJ, Mirsky DM, Handler MH, Foreman NK et al (2014) Autophagy inhibition improves chemosensitivity in BRAF(V600E) brain tumors. Cancer Discov 4: 773–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Zhang Y, Cheng X, Yuan H, Zhu S, Liu J, Wen Q, Xie Y, Liu J, Kroemer G et al (2018) PINK1 and PARK2 suppress pancreatic tumorigenesis through control of mitochondrial iron‐mediated immunometabolism. Dev Cell 46: 441–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Shen C, Nakamura E, Ando K, Signoretti S, Beroukhim R, Cowley G, Lizotte P, Liberzon E, Bair S et al (2013) SQSTM1 is a pathogenic target of 5q copy number gains in kidney cancer. Cancer Cell 24: 738–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z‐L, Zhang H‐L, Huang Y, Huang J‐H, Sun P, Zhou N‐N, Chen Y‐H, Mai J, Wang Y, Yu Y et al (2020) Autophagy deficiency promotes triple‐negative breast cancer resistance to T cell‐mediated cytotoxicity by blocking tenascin‐C degradation. Nat Commun 11: 3806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B (1999) Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 402: 672–676 [DOI] [PubMed] [Google Scholar]
- Liu F, Lee JY, Wei H, Tanabe O, Engel JD, Morrison SJ, Guan J‐L (2010) FIP200 is required for the cell‐autonomous maintenance of fetal hematopoietic stem cells. Blood 116: 4806–4814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, He Z, von Rütte T, Yousefi S, Hunger RE, Simon H‐U (2013) Down‐regulation of autophagy‐related protein 5 (ATG5) contributes to the pathogenesis of early‐stage cutaneous melanoma. Sci Transl Med 5: 202ra123 [DOI] [PubMed] [Google Scholar]
- Liu J, Kuang F, Kroemer G, Klionsky DJ, Kang R, Tang D (2020) Autophagy‐dependent ferroptosis: machinery and regulation. Cell Chem Biol 27: 420–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Xia H, Kim M, Xu L, Li Y, Zhang L, Cai YU, Norberg HV, Zhang T, Furuya T et al (2011) Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell 147: 223–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Zhao E, Ilyas G, Lalazar G, Lin Y, Haseeb M, Tanaka KE, Czaja MJ (2015) Impaired macrophage autophagy increases the immune response in obese mice by promoting proinflammatory macrophage polarization. Autophagy 11: 271–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, Ma Q, Zhu C, Wang R, Qi W et al (2012) Mitochondrial outer‐membrane protein FUNDC1 mediates hypoxia‐induced mitophagy in mammalian cells. Nat Cell Biol 14: 177–185 [DOI] [PubMed] [Google Scholar]
- Lock R, Kenific CM, Leidal AM, Salas E, Debnath J (2014) Autophagy‐dependent production of secreted factors facilitates oncogenic RAS‐driven invasion. Cancer Discov 4: 466–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lock R, Roy S, Kenific CM, Su JS, Salas E, Ronen SM, Debnath J (2011) Autophagy facilitates glycolysis during Ras‐mediated oncogenic transformation. Mol Biol Cell 22: 165–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losier TT, Akuma M, McKee‐Muir OC, LeBlond ND, Suk Y, Alsaadi RM, Guo Z, Reshke R, Sad S, Campbell‐Valois F‐X et al (2019) AMPK promotes xenophagy through priming of autophagic kinases upon detection of bacterial outer membrane vesicles. Cell Rep 26: 2150–2165 [DOI] [PubMed] [Google Scholar]
- Lv L, Li D, Zhao DI, Lin R, Chu Y, Zhang H, Zha Z, Liu Y, Li ZI, Xu Y et al (2011) Acetylation targets the M2 isoform of pyruvate kinase for degradation through chaperone‐mediated autophagy and promotes tumor growth. Mol Cell 42: 719–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv Q, Wang W, Xue J, Hua F, Mu R, Lin H, Yan J, Lv X, Chen X, Hu Z‐W (2012) DEDD interacts with PI3KC3 to activate autophagy and attenuate epithelial‐mesenchymal transition in human breast cancer. Cancer Res 72: 3238–3250 [DOI] [PubMed] [Google Scholar]
- Lyons A, Coleman M, Riis S, Favre C, O’Flanagan CH, Zhdanov AV, Papkovsky DB, Hursting SD, O’Connor R (2017) Insulin‐like growth factor 1 signaling is essential for mitochondrial biogenesis and mitophagy in cancer cells. J Biol Chem 292: 16983–16998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X‐H, Piao S, Wang D, McAfee QW, Nathanson KL, Lum JJ, Li LZ, Amaravadi RK (2011) Measurements of tumor cell autophagy predict invasiveness, resistance to chemotherapy, and survival in melanoma. Clin Cancer Res 17: 3478–3489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X‐H, Piao S‐F, Dey S, Mcafee Q, Karakousis G, Villanueva J, Hart LS, Levi S, Hu J, Zhang G et al (2014) Targeting ER stress‐induced autophagy overcomes BRAF inhibitor resistance in melanoma. J Clin Invest 124: 1406–1417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macher‐Goeppinger S, Keith M, Hatiboglu G, Hohenfellner M, Schirmacher P, Roth W, Tagscherer KE (2017) Expression and functional characterization of the BNIP3 protein in renal cell carcinomas. Transl Oncol 10: 869–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhavan S, Triplett D, Rao PK (2016) Hydroxychloroquine induced liver injury. Am J Gastroenterol 111: S886 [Google Scholar]
- Maes H, Kuchnio A, Peric A, Moens S, Nys K, De Bock K, Quaegebeur A, Schoors S, Georgiadou M, Wouters J et al (2014) Tumor vessel normalization by chloroquine independent of autophagy. Cancer Cell 26: 190–206 [DOI] [PubMed] [Google Scholar]
- Mancias JD, Kimmelman AC (2016) Mechanisms of selective autophagy in normal physiology and cancer. J Mol Biol 428: 1659–1680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manka D, Spicer Z, Millhorn DE (2005) Bcl‐2/Adenovirus E1B 19 kDa interacting protein‐3 knockdown enables growth of breast cancer metastases in the lung, liver, and bone. Cancer Res 65: 11689–11693 [DOI] [PubMed] [Google Scholar]
- Marsh T, Kenific CM, Suresh D, Gonzalez H, Shamir ER, Mei W, Tankka A, Leidal AM, Kalavacherla S, Woo K et al (2020) Autophagic degradation of NBR1 restricts metastatic outgrowth during mammary tumor progression. Dev Cell 52: 591–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall RS, Hua Z, Mali S, McLoughlin F, Vierstra RD (2019) ATG8‐Binding UIM proteins define a new class of autophagy adaptors and receptors. Cell 177: 766–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin KR, Celano SL, Solitro AR, Gunaydin H, Scott M, O’Hagan RC, Shumway SD, Fuller P, MacKeigan JP (2018) A potent and selective ULK1 inhibitor suppresses autophagy and sensitizes cancer cells to nutrient, Stress. iScience 8: 74–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauthe M, Orhon I, Rocchi C, Zhou X, Luhr M, Hijlkema K‐J, Coppes RP, Engedal N, Mari M, Reggiori F (2018) Chloroquine inhibits autophagic flux by decreasing autophagosome‐lysosome fusion. Autophagy 14: 1435–1455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maycotte P, Aryal S, Cummings CT, Thorburn J, Morgan MJ, Thorburn A (2012) Chloroquine sensitizes breast cancer cells to chemotherapy independent of autophagy. Autophagy 8: 200–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWilliams TG, Prescott AR, Montava‐Garriga L, Ball G, Singh F, Barini E, Muqit MMK, Brooks SP, Ganley IG (2018) Basal mitophagy occurs independently of PINK1 in mouse tissues of high metabolic demand. Cell Metab 27: 439–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellor HR, Harris AL (2007) The role of the hypoxia‐inducible BH3‐only proteins BNIP3 and BNIP3L in cancer. Cancer Metastasis Rev 26: 553–566 [DOI] [PubMed] [Google Scholar]
- Mgrditchian T, Arakelian T, Paggetti J, Noman MZ, Viry E, Moussay E, Van Moer K, Kreis S, Guerin C, Buart S et al (2017) Targeting autophagy inhibits melanoma growth by enhancing NK cells infiltration in a CCL5‐dependent manner. Proc Natl Acad Sci USA 114: E9271–E9279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milan M, Diaferia GR, Natoli G (2021) Tumor cell heterogeneity and its transcriptional bases in pancreatic cancer: a tale of two cell types and their many variants. EMBO J 40: e107206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y (2004) In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 15: 1101–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris SM (2016) Arginine metabolism revisited. J Nutr 146: 2579S–2586S [DOI] [PubMed] [Google Scholar]
- Mortimore GE, Schworer CM (1977) Induction of autophagy by amino‐acid deprivation in perfused rat liver. Nature 270: 174–176 [DOI] [PubMed] [Google Scholar]
- Mulcahy Levy JM, Foreman NK, Thorburn A (2014) Using BRAF(V600E) as a marker of autophagy dependence in pediatric brain tumors. Autophagy 10: 2077–2078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulcahy Levy JM, Zahedi S, Griesinger AM, Morin A, Davies KD, Aisner DL, Kleinschmidt‐DeMasters BK, Fitzwalter BE, Goodall ML, Thorburn J et al (2017) Autophagy inhibition overcomes multiple mechanisms of resistance to BRAF inhibition in brain tumors. Elife 6: e19671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik PP, Birbrair A, Bhutia SK (2019) Mitophagy‐driven metabolic switch reprograms stem cell fate. Cell Mol Life Sci 76: 27–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazio F, Bordi M, Cianfanelli V, Locatelli F, Cecconi F (2019) Autophagy and cancer stem cells: molecular mechanisms and therapeutic applications. Cell Death Differ 26: 690–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noman MZ, Parpal S, Van Moer K, Xiao M, Yu Y, Arakelian T, Viklund J, De Milito A, Hasmim M, Andersson M et al (2020) Inhibition of Vps34 reprograms cold into hot inflamed tumors and improves anti‐PD‐1/PD‐L1 immunotherapy. Sci Adv 6: eaax7881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyfeler B, Eng CH (2016) Revisiting autophagy addiction of tumor cells. Autophagy 12: 1206–1207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver L, Hue E, Priault M, Vallette FM (2012) Basal autophagy decreased during the differentiation of human adult mesenchymal stem cells. Stem Cells Dev 21: 2779–2788 [DOI] [PubMed] [Google Scholar]
- Palikaras K, Lionaki E, Tavernarakis N (2018) Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat Cell Biol 20: 1013–1022 [DOI] [PubMed] [Google Scholar]
- Parker TM, Henriques V, Beltran A, Nakshatri H, Gogna R (2020) Cell competition and tumor heterogeneity. Semin Cancer Biol 63: 1–10 [DOI] [PubMed] [Google Scholar]
- Parzych KR, Klionsky DJ (2014) An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal 20: 460–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y‐F, Shi Y‐H, Ding Z‐B, Ke A‐W, Gu C‐Y, Hui B, Zhou J, Qiu S‐J, Dai Z, Fan J (2013) Autophagy inhibition suppresses pulmonary metastasis of HCC in mice via impairing anoikis resistance and colonization of HCC cells. Autophagy 9: 2056–2068 [DOI] [PubMed] [Google Scholar]
- Perera RM, Stoykova S, Nicolay BN, Ross KN, Fitamant J, Boukhali M, Lengrand J, Deshpande V, Selig MK, Ferrone CR et al (2015) Transcriptional control of autophagy‐lysosome function drives pancreatic cancer metabolism. Nature 524: 361–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrova V, Mancini M, Agostini M, Knight RA, Annicchiarico‐Petruzzelli M, Barlev NA, Melino G, Amelio I (2015) TAp73 transcriptionally represses BNIP3 expression. Cell Cycle 14: 2484–2493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrocola F, Pol J, Vacchelli E, Rao S, Enot D, Baracco E, Levesque S, Castoldi F, Jacquelot N, Yamazaki T et al (2016) Caloric restriction mimetics enhance anticancer immunosurveillance. Cancer Cell 30: 147–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poillet‐Perez L, Sharp DW, Yang Y, Laddha SV, Ibrahim M, Bommareddy PK, Hu ZS, Vieth J, Haas M, Bosenberg MW et al (2020) Autophagy promotes growth of tumors with high mutational burden by inhibiting a T‐cell immune response. Nat Cancer 1: 923–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poillet‐Perez L, Xie X, Zhan LE, Yang Y, Sharp DW, Hu ZS, Su X, Maganti A, Jiang C, Lu W et al (2018) Autophagy maintains tumour growth through circulating arginine. Nature 563: 569–573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole LP, Bock‐Hughes A, Berardi DE, Macleod KF (2021) ULK1 promotes mitophagy via phosphorylation and stabilization of BNIP3. Sci Rep 11: 20526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porporato P, Payen Valéry L, Pérez‐Escuredo J, De Saedeleer C, Danhier P, Copetti T, Dhup S, Tardy M, Vazeille T, Bouzin C et al (2014) A mitochondrial switch promotes tumor metastasis. Cell Rep 8: 754–766 [DOI] [PubMed] [Google Scholar]
- Qiang L, Zhao B, Ming M, Wang N, He T‐C, Hwang S, Thorburn A, He Y‐Y (2014) Regulation of cell proliferation and migration by p62 through stabilization of Twist1. Proc Natl Acad Sci USA 111: 9241–9246 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Qin W, Li C, Zheng W, Guo Q, Zhang Y, Kang M, Zhang BO, Yang B, Li B, Yang H et al (2015) Inhibition of autophagy promotes metastasis and glycolysis by inducing ROS in gastric cancer cells. Oncotarget 6: 39839–39854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, Rosen J, Eskelinen E‐L, Mizushima N, Ohsumi Y et al (2003) Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest 112: 1809–1820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovich S, Adler L, Yizhak K, Sarver A, Silberman A, Agron S, Stettner N, Sun Q, Brandis A, Helbling D et al (2015) Diversion of aspartate in ASS1‐deficient tumours fosters de novo pyrimidine synthesis. Nature 527: 379–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao S, Tortola L, Perlot T, Wirnsberger G, Novatchkova M, Nitsch R, Sykacek P, Frank L, Schramek D, Komnenovic V et al (2014) A dual role for autophagy in a murine model of lung cancer. Nat Commun 5: 3056 [DOI] [PubMed] [Google Scholar]
- Ravenhill BJ, Boyle KB, von Muhlinen N, Ellison CJ, Masson GR, Otten EG, Foeglein A, Williams R, Randow F (2019) The cargo receptor NDP52 initiates selective autophagy by recruiting the ULK complex to cytosol‐invading bacteria. Mol Cell 74: 320–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravikumar B, Duden R, Rubinsztein DC (2002) Aggregate‐prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum Mol Genet 11: 1107–1117 [DOI] [PubMed] [Google Scholar]
- Ravikumar B, Stewart A, Kita H, Kato K, Duden R, Rubinsztein DC (2003) Raised intracellular glucose concentrations reduce aggregation and cell death caused by mutant huntingtin exon 1 by decreasing mTOR phosphorylation and inducing autophagy. Hum Mol Genet 12: 985–994 [DOI] [PubMed] [Google Scholar]
- Reis LB, Filippi‐Chiela EC, Ashton‐Prolla P, Visioli F, Rosset C (2021) The paradox of autophagy in tuberous sclerosis complex. Genet Mol Biol 44: e20200014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogov VV, Suzuki H, Marinković M, Lang V, Kato R, Kawasaki M, Buljubašić M, Šprung M, Rogova N, Wakatsuki S et al (2017) Phosphorylation of the mitochondrial autophagy receptor Nix enhances its interaction with LC3 proteins. Sci Rep 7: 1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojo de la Vega M, Chapman E, Zhang DD (2018) NRF2 and the hallmarks of cancer. Cancer Cell 34: 21–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronan B, Flamand O, Vescovi L, Dureuil C, Durand L, Fassy F, Bachelot M‐F, Lamberton A, Mathieu M, Bertrand T et al (2014) A highly potent and selective Vps34 inhibitor alters vesicle trafficking and autophagy. Nat Chem Biol 10: 1013–1019 [DOI] [PubMed] [Google Scholar]
- Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N (2015) Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 160: 48–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld MR, Ye X, Supko JG, Desideri S, Grossman SA, Brem S, Mikkelson T, Wang D, Chang YC, Hu J et al (2014) A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy 10: 1359–1368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeldt MT, O’Prey J, Morton JP, Nixon C, MacKay G, Mrowinska A, Au A, Rai TS, Zheng L, Ridgway R et al (2013) p53 status determines the role of autophagy in pancreatic tumour development. Nature 504: 296–300 [DOI] [PubMed] [Google Scholar]
- Rosenfeldt MT, O’Prey J, Lindsay CR, Nixon C, Roth S, Sansom OJ, Ryan KM (2021) Loss of autophagy affects melanoma development in a manner dependent on PTEN status. Cell Death Differ 28: 1437–1439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T, Ichimura Y, Taguchi K, Suzuki T, Mizushima T, Takagi K, Hirose Y, Nagahashi M, Iso T, Fukutomi T et al (2016) p62/Sqstm1 promotes malignancy of HCV‐positive hepatocellular carcinoma through Nrf2‐dependent metabolic reprogramming. Nat Commun 7: 12030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjurjo L, Aran G, Téllez É, Amézaga N, Armengol C, López D, Prats C, Sarrias M‐R (2018) CD5L promotes M2 macrophage polarization through autophagy‐mediated upregulation of ID3. Front Immunol 9: 480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarraf SA, Raman M, Guarani‐Pereira V, Sowa ME, Huttlin EL, Gygi SP, Harper JW (2013) Landscape of the PARKIN‐dependent ubiquitylome in response to mitochondrial depolarization. Nature 496: 372–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarraf SA, Sideris DP, Giagtzoglou N, Ni L, Kankel MW, Sen A, Bochicchio LE, Huang C‐H, Nussenzweig SC, Worley SH et al (2019) PINK1/Parkin influences cell cycle by sequestering TBK1 at damaged mitochondria, inhibiting mitosis. Cell Rep 29: 225–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schadendorf D, Fisher DE, Garbe C, Gershenwald JE, Grob J‐J, Halpern A, Herlyn M, Marchetti MA, McArthur G, Ribas A et al (2015) Melanoma. Nat Rev Dis Primers 1: 15003 [DOI] [PubMed] [Google Scholar]
- Sharifi MN, Mowers EE, Drake LE, Collier C, Chen H, Zamora M, Mui S, Macleod KF (2016) Autophagy promotes focal adhesion disassembly and cell motility of metastatic tumor cells through the direct interaction of paxillin with LC3. Cell Rep 15: 1660–1672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi G, McQuibban GA (2017) The mitochondrial rhomboid protease PARL is regulated by PDK2 to integrate mitochondrial quality control and metabolism. Cell Rep 18: 1458–1472 [DOI] [PubMed] [Google Scholar]
- Smith AG, Macleod KF (2019) Autophagy, cancer stem cells and drug resistance. J Pathol 247: 708–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soria LR, Gurung S, De Sabbata G, Perocheau DP, De Angelis A, Bruno G, Polishchuk E, Paris D, Cuomo P, Motta A et al (2021) Beclin‐1‐mediated activation of autophagy improves proximal and distal urea cycle disorders. EMBO Mol Med 13: e13158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa CM, Biancur DE, Wang X, Halbrook CJ, Sherman MH, Zhang LI, Kremer D, Hwang RF, Witkiewicz AK, Ying H et al (2016) Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 536: 479–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa CM, Kimmelman AC (2014) The complex landscape of pancreatic cancer metabolism. Carcinogenesis 35: 1441–1450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starobinets H, Ye J, Broz M, Barry K, Goldsmith J, Marsh T, Rostker F, Krummel M, Debnath J (2016) Antitumor adaptive immunity remains intact following inhibition of autophagy and antimalarial treatment. J Clin Invest 126: 4417–4429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strohecker AM, Guo JY, Karsli‐Uzunbas G, Price SM, Chen GJ, Mathew R, McMahon M, White E (2013) Autophagy sustains mitochondrial glutamine metabolism and growth of BrafV600E‐driven lung tumors. Cancer Discov 3: 1272–1285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sucker A, Zhao F, Pieper N, Heeke C, Maltaner R, Stadtler N, Real B, Bielefeld N, Howe S, Weide B et al (2017) Acquired IFNγ resistance impairs anti‐tumor immunity and gives rise to T‐cell‐resistant melanoma lesions. Nat Commun 8: 15440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui X, Chen R, Wang Z, Huang Z, Kong N, Zhang M, Han W, Lou F, Yang J, Zhang Q et al (2013) Autophagy and chemotherapy resistance: a promising therapeutic target for cancer treatment. Cell Death Dis 4: e838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, Eishi Y, Hino O, Tanaka K, Mizushima N (2011) Autophagy‐deficient mice develop multiple liver tumors. Genes Dev 25: 795–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian W, Alsaadi R, Guo Z, Kalinina A, Carrier M, Tremblay M‐E, Lacoste B, Lagace D, Russell RC (2020) An antibody for analysis of autophagy induction. Nat Methods 17: 232–239 [DOI] [PubMed] [Google Scholar]
- Toma MI, Wuttig D, Kaiser S, Herr A, Weber T, Zastrow S, Koch R, Meinhardt M, Baretton GB, Wirth MP et al (2013) PARK2 and PACRG are commonly downregulated in clear‐cell renal cell carcinoma and are associated with aggressive disease and poor clinical outcome. Genes Chromosomes Cancer 52: 265–273 [DOI] [PubMed] [Google Scholar]
- Tra T, Gong L, Kao L‐P, Li X‐L, Grandela C, Devenish RJ, Wolvetang E, Prescott M (2011) Autophagy in human embryonic stem cells. PLoS One 6: e27485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turco E, Savova A, Gere F, Ferrari L, Romanov J, Schuschnig M, Martens S (2021) Reconstitution defines the roles of p62, NBR1 and TAX1BP1 in ubiquitin condensate formation and autophagy initiation. Nat Commun 12: 5212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turco E, Witt M, Abert C, Bock‐Bierbaum T, Su M‐Y, Trapannone R, Sztacho M, Danieli A, Shi X, Zaffagnini G et al (2019) FIP200 claw domain binding to p62 promotes autophagosome formation at ubiquitin condensates. Mol Cell 74: 330–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdor R, García‐Bernal D, Riquelme D, Martinez CM, Moraleda JM, Cuervo AM, Macian F, Martinez S (2019) Glioblastoma ablates pericytes antitumor immune function through aberrant up‐regulation of chaperone‐mediated autophagy. Proc Natl Acad Sci USA 116: 20655–20665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vara‐Perez M, Felipe‐Abrio B, Agostinis P (2019) Mitophagy in cancer: a tale of adaptation. Cells 8: E493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas JNS, Wang C, Bunker E, Hao L, Maric D, Schiavo G, Randow F, Youle RJ (2019) Spatiotemporal control of ULK1 activation by NDP52 and TBK1 during selective autophagy. Mol Cell 74: 347–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vázquez P, Arroba AI, Cecconi F, de la Rosa EJ, Boya P, de Pablo F (2012) Atg5 and Ambra1 differentially modulate neurogenesis in neural stem cells. Autophagy 8: 187–199 [DOI] [PubMed] [Google Scholar]
- Vives‐Bauza C, Zhou C, Huang Y, Cui M, de Vries RLA, Kim J, May J, Tocilescu MA, Liu W, Ko HS et al (2010) PINK1‐dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci USA 107: 378–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Burikhanov R, Jayswal R, Weiss HL, Arnold SM, Villano JL, Rangnekar VM (2018) Neoadjuvant administration of hydroxychloroquine in a phase 1 clinical trial induced plasma Par‐4 levels and apoptosis in diverse tumors. Genes Cancer 9: 190–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warr MR, Binnewies M, Flach J, Reynaud D, Garg T, Malhotra R, Debnath J, Passegué E (2013) FOXO3A directs a protective autophagy program in haematopoietic stem cells. Nature 494: 323–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei H, Wang C, Croce CM, Guan J‐L (2014) p62/SQSTM1 synergizes with autophagy for tumor growth in vivo . Genes Dev 28: 1204–1216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei H, Wei S, Gan B, Peng X, Zou W, Guan J‐L (2011) Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis. Genes Dev 25: 1510–1527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijshake T, Zou Z, Chen B, Zhong L, Xiao G, Xie Y, Doench JG, Bennett L, Levine B (2021) Tumor‐suppressor function of Beclin 1 in breast cancer cells requires E‐cadherin. Proc Natl Acad Sci USA 118: e2020478118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolpin BM, Rubinson DA, Wang X, Chan JA, Cleary JM, Enzinger PC, Fuchs CS, McCleary NJ, Meyerhardt JA, Ng K et al (2014) Phase II and pharmacodynamic study of autophagy inhibition using hydroxychloroquine in patients with metastatic pancreatic adenocarcinoma. Oncologist 19: 637–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong P‐M, Feng Y, Wang J, Shi R, Jiang X (2015) Regulation of autophagy by coordinated action of mTORC1 and protein phosphatase 2A. Nat Commun 6: 8048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu K, Lin K, Li X, Yuan X, Xu P, Ni P, Xu D (2020) Redefining tumor‐associated macrophage subpopulations and functions in the tumor microenvironment. Front Immunol 11: 1731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia H‐G, Najafov A, Geng J, Galan‐Acosta L, Han X, Guo Y, Shan B, Zhang Y, Norberg E, Zhang T et al (2015) Degradation of HK2 by chaperone‐mediated autophagy promotes metabolic catastrophe and cell death. J Cell Biol 210: 705–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao B, Heath R, Saiu P, Leiper FC, Leone P, Jing C, Walker PA, Haire L, Eccleston JF, Davis CT et al (2007) Structural basis for AMP binding to mammalian AMP‐activated protein kinase. Nature 449: 496–500 [DOI] [PubMed] [Google Scholar]
- Xie X, Koh JY, Price S, White E, Mehnert JM (2015) Atg7 overcomes senescence and promotes growth of BrafV600E‐driven melanoma. Cancer Discov 5: 410–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K, Venida A, Yano J, Biancur DE, Kakiuchi M, Gupta S, Sohn ASW, Mukhopadhyay S, Lin EY, Parker SJ et al (2020) Autophagy promotes immune evasion of pancreatic cancer by degrading MHC‐I. Nature 581: 100–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang A, Herter‐Sprie G, Zhang H, Lin EY, Biancur D, Wang X, Deng J, Hai J, Yang S, Wong K‐K et al (2018) Autophagy sustains pancreatic cancer growth through both cell‐autonomous and nonautonomous mechanisms. Cancer Discov 8: 276–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang A, Kimmelman AC (2014) Inhibition of autophagy attenuates pancreatic cancer growth independent of TP53/TRP53 status. Autophagy 10: 1683–1684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang A, Rajeshkumar NV, Wang X, Yabuuchi S, Alexander BM, Chu GC, Von Hoff DD, Maitra A, Kimmelman AC (2014) Autophagy is critical for pancreatic tumor growth and progression in tumors with p53 alterations. Cancer Discov 4: 905–913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, Bause A, Li Y, Stommel JM, Dell'Antonio G et al (2011) Pancreatic cancers require autophagy for tumor growth. Genes Dev 25: 717–729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo SK, Wen J, Chen S, Guan J‐L (2016) Autophagy differentially regulates distinct breast cancer stem‐like cells in murine models via EGFR/Stat3 and Tgfβ/Smad signaling. Cancer Res 76: 3397–3410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying H, Dey P, Yao W, Kimmelman AC, Draetta GF, Maitra A, DePinho RA (2016) Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev 30: 355–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young ARJ, Narita M, Ferreira M, Kirschner K, Sadaie M, Darot JFJ, Tavaré S, Arakawa S, Shimizu S, Watt FM et al (2009) Autophagy mediates the mitotic senescence transition. Genes Dev 23: 798–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Luo K, Zhang L, Cheville JC, Lou Z (2010) USP10 regulates p53 localization and stability by deubiquitinating p53. Cell 140: 384–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue Z, Jin S, Yang C, Levine AJ, Heintz N (2003) Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci USA 100: 15077–15082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C‐S, Hawley SA, Zong Y, Li M, Wang Z, Gray A, Ma T, Cui J, Feng J‐W, Zhu M et al (2017) Fructose‐1,6‐bisphosphate and aldolase mediate glucose sensing by AMPK. Nature 548: 112–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Hama Y, Mizushima N (2021) The evolution of autophagy proteins – diversification in eukaryotes and potential ancestors in prokaryotes. J Cell Sci 134: jcs233742 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Morgan MJ, Chen K, Choksi S, Liu Z (2012) Induction of autophagy is essential for monocyte‐macrophage differentiation. Blood 119: 2895–2905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Codogno P, Zhang H (2021) Machinery, regulation and pathophysiological implications of autophagosome maturation. Nat Rev Mol Cell Biol 22: 733–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Yang J, Fan X, Hu S, Zhou F, Dong J, Zhang S, Shang Y, Jiang X, Guo H et al (2016) Chaperone‐mediated autophagy regulates proliferation by targeting RND3 in gastric cancer. Autophagy 12: 515–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Liu J, Fu T, Wu P, Peng C, Gong X, Wang Y, Zhang M, Li Y, Wang Y et al (2021) Phosphorylation regulates the binding of autophagy receptors to FIP200 Claw domain for selective autophagy initiation. Nat Commun 12: 1570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Massen S, Terenzio M, Lang V, Chen‐Lindner S, Eils R, Novak I, Dikic I, Hamacher‐Brady A, Brady NR (2013) Modulation of serines 17 and 24 in the LC3‐interacting region of Bnip3 determines pro‐survival mitophagy versus apoptosis. J Biol Chem 288: 1099–1113 [DOI] [PMC free article] [PubMed] [Google Scholar]