

Abstract
The RNA virus phylum Lenarviricota is composed of the fungi-associated families Narnaviridae and Mitoviridae, the RNA bacteriophage Leviviridae, and the plant and fungi-associated Botourmiaviridae. Members of the Lenarviricota are abundant in most environments and boast remarkable phylogenetic and genomic diversity. As this phylum includes both RNA bacteriophage and fungi- and plant-associated species, the Lenarviricota likely mark a major evolutionary transition between those RNA viruses associated with prokaryotes and eukaryotes. Despite the remarkable expansion of this phylum following metagenomic studies, the phylogenetic relationships among the families within the Lenarviricota remain uncertain. Utilising a large data set of relevant viral sequences, we performed phylogenetic and genomic analyses to resolve the complex evolutionary history within this phylum and identify patterns in the evolution of virus genome organisation. Despite limitations reflecting very high levels of sequence diversity, our phylogenetic analyses suggest that the Leviviridae comprise the basal lineage within the Lenarviricota. Our phylogenetic results also support the construction of a new virus family—the Narliviridae—comprising a set of diverse and phylogenetically distinct species, including a number of uniquely encapsidated viruses. We propose a taxonomic restructuring within the Lenarviricota to better reflect the phylogenetic relationships documented here, with the Botourmiaviridae and Narliviridae combined into the order Ourlivirales, the Narnaviridae remaining in the order Wolframvirales, and these orders combined into the single class, the Amabiliviricetes. In sum, this study provides insights into the complex evolutionary relationships among the diverse families that make up the Lenarviricota.
Keywords: lenarviricota, mitoviridae, metatranscriptomics, phylogenetics, virus taxonomy, genome structure
1. Introduction
The families Narnaviridae and Mitoviridae within the phylum Lenarviricota arguably comprise the simplest of all RNA viruses. They possess very small positive-sense single-stranded genomes (<4,000 nucleotides in length), usually encode a single protein—the RNA-dependent RNA polymerase (RdRp)—and uniquely lack the capsid protein often considered a defining feature of RNA viruses (Hillman and Cai 2013). Although originally discovered in fungal hosts, these families have recently been detected in other microbes, including protists (Akopyants et al. 2016; Grybchuk et al. 2018; Charon et al. 2019; Charon, Murray, and Holmes 2021) and diatoms (Urayama, Takaki, and Nunoura 2016). In addition, some narnaviruses and mitoviruses contain genes additional to the RdRp (Shi et al. 2016; Grybchuk et al. 2018; Charon et al. 2019; Wolf et al. 2020; Charon, Murray, and Holmes 2021).
The Narnaviridae and Mitoviridae were previously classified as two distinct genera (Narnavirus and Mitovirus, respectively) within the family Narnaviridae that could be differentiated by their site of replication. Narnavirusest are restricted to the cell cytosol, while mitoviruses replicate within the cell mitochondria (Hillman and Cai 2013). Accordingly, not only do these families utilise different cell machinery in their replication cycle, but mitoviruses utilise the mitochondrial genetic code, in which the amino acid tryptophan is not only encoded by UGG, but also by UGA that results in a stop codon in the standard genetic code (Cole et al. 2000; Shackelton and Holmes 2008). The function of the UGA codon in the mitoviruses that infect fungi appears to match the bias in the mitochondrial genomes of their hosts (Nibert 2017). These factors, as well as more recent phylogenetic studies (Wolf et al. 2018), particularly utilising data from expansive metagenomic sequencing studies (for example, Shi et al. 2016; Wolf et al. 2020), have led to a taxonomic revision and their current status as two separate families classified into separate orders and classes within the Lenarviricota. Indeed, according to the most recent International Committee on Taxonomy of Viruses (ICTV) release, the Narnaviridae fall within the order Wolframvirales and class Amabiliviricetes, while the Mitoviridae are members of the order Cryppavirales, class Howeltoviricetes (Walker et al. 2020).
Phylogenetic studies have also shown that the Narnaviridae are related to the plant and filamentous-fungi-infecting viruses of the family Botourmiaviridae (Shi et al. 2016; Wolf et al. 2018) that are classified within the order Ourlivirales, class Miaviricetes of the Lenarviricota (Ayllón et al. 2020; Walker et al. 2020). The Botourmiaviridae were initially classified as a single floating genus—‘Ourmiavirus’—following the discovery of the type species, Ourmia melon virus (Ayllón et al. 2020). Ourmiaviruses, of which there are currently only three, are plant-infecting, capsidated, RNA viruses, whose seemingly chimeric genomes are arranged as three segments that encode a narnavirus-like RdRp, a picornavirus-like capsid protein, and a tombusvirus-like movement protein (Rastgou et al. 2009). The other genera currently placed within the Botourmiaviridae—Botoulivirus, Penoulivirus, Magoulivirus, Rhizoulivirus, and Scleroulivirus, each named after the fungal species in which they were discovered—have much smaller and simpler genomes than the ourmiaviruses, ranging between 2 kb and 3.4 kb in length, and only encode an RdRp (Donaire, Rozas, and Ayllón 2016; Marzano et al. 2016; Illana et al. 2017; Nerva et al. 2019). These genera also differ in host range, infecting filamentous fungi as opposed to plants (Ayllón et al. 2020).
Based on previous phylogenetic analyses of the RdRp, the Narnaviridae, Botourmiaviridae, and Mitoviridae have been proposed as related to the bacteriophage-associated Leviviridae (Shi et al. 2016; Wolf et al. 2018). Members of the Leviviridae infect gram-negative bacteria including Enterobacter, Acinetobacter, Caulobacter, and Pseudomonas (King et al. 2012). Leviviruses are widespread and abundant in a range of environments, particularly animal faeces and sediment (Chen et al. 2021). Like the Narnaviridae and Mitoviridae, the Leviviridae are unenveloped and possess very small genomes (<4.3 kb in length). However, while most narnaviruses, botourmiaviruses, and mitoviruses are only composed of an RdRp, the Leviviridae genome is more complex and encodes a capsid protein, a maturation protein, and in some cases, a lysis protein. A read-through protein that extends the capsid protein through the suppression of the terminal UGA codon is also found in some cases (King et al. 2012).
As reflected by the narnaviruses and mitoviruses, the earliest discovered viruses within the phylum Lenarviricota were defined phenotypically and distinguished by host specificity. However, following the rise of metagenomic sequence data, they now comprise only a small subset of the newly expanded Lenarviricota. Additionally, many of these metagenomic sequences were derived from vertebrate (Mahar et al. 2020; Wille et al. 2020), invertebrate (Shi et al. 2016; Le Lay et al. 2020), and environmental samples (including soils, sediments, and water) (Starr et al. 2019; Wolf et al. 2020; Chen et al. 2021), such that their true host organisms have not been determined. Hence, a large proportion of the viruses within the Lenarviricota have been only defined phylogenetically.
Because of its very broad host range, the phylum Lenarviricota is of significance for understanding the evolutionary transition between RNA bacteriophage and eukaryote-infecting RNA viruses. Critically, however, the evolutionary relationships among the Narnaviridae, Mitoviridae, Leviviridae, and Botourmiaviridae remain uncertain, particularly as the level of sequence divergence among them—as little as only 5 per cent pairwise sequence similarity—introduces significant challenges when constructing reliable sequence alignments and hence phylogenetic trees (Holmes and Duchêne 2019). It has been proposed that the Lenarviricota had a levivirus-like ancestor that lost its capsid protein before giving rise to the mitoviruses (Krupovic and Koonin 2017). This evolutionary transition from bacteriophage to eukaryote-infecting RNA viruses is hypothesised to have occurred during an endosymbiotic event potentially over 1.45 billion years ago in which the α-proteobacteria became intracellular symbionts (Martin and Mentel 2010), after which these mitochondrial viruses escaped to the cell cytosol and became what are now known as the narnaviruses (Wolf et al. 2018). Wolf et al. (2018) further suggest that the Mitoviridae gave rise to the plant-infecting ourmiaviruses alongside the Narnaviridae, making these sister clades with a mitovirus-like common ancestor.
Using a large data set of relevant viruses, we attempted to resolve the evolutionary relationships, and hence transitions, among the diverse virus families that comprise the phylum Lenarviricota. In addition, we provide insights into the complex phylogeny of the Narnaviridae and Botourmiaviridae, identifying a large clade of diverse but distinct species previously classified as ‘narna-like’ viruses, some of which are encapsidated and which we propose might be considered a new family that we tentatively call the Narliviridae. Based on the phylogenetic patterns and genomic structures observed in this study, we also propose a taxonomic restructuring of these three families into the singular class Amabiliviricetes.
2. Methods
2.1. Data collection and processing
We analysed a database comprising 442 meta-transcriptomic libraries from soil, sediment, and animal faecal samples collected, sequenced, and assembled as described previously (Chen et al. 2021). Briefly, 442 RNA-sequencing libraries were generated from samples taken across a wide range of environments and geographical regions in China. These environments included forests, farmland, desert, water environments and sediments, and animal faeces. Total RNA was extracted using the RNeasy® PowerSoil® Total RNA Kit (Qiagen), and each library was sequenced on the Illumina HiSeq X10 platform. The resulting reads were adaptor and quality-trimmed using Trimmomatic (Bolger, Lohse, and Usadel 2014) and assembled de novo using MEGAHIT (Li et al. 2015).
To identify viral hits the assembled contigs were compared to a database, curated in 2019, of Riboviria RdRp sequences available on GenBank using DIAMOND BLASTX (Buchfink, Xie, and Huson 2015). RdRp sequences were obtained by searching the National Center for Biotechnology Information (NCBI) non-redundant (nr) protein database for ‘RdRp’ and ‘RNA dependent RNA polymerase’ entries using Entrez Programming Utilities (https://www.ncbi.nlm.nih.gov/books/NBK25501). All contigs returning a match to a viral RdRp sequence were then run against the nr protein database using DIAMOND BLASTX (Buchfink, Xie, and Huson 2015) with a more sensitive e-value threshold of 1 × 10−5 to exclude false-positives. Those contigs returning a positive hit to a viral RdRp sequence and over 1,000 nucleotides in length were considered likely to be bona fide viral sequences and selected for further analysis, particularly expansive comparisons with other members of the Lenarviricota.
2.2. Sequence alignment and phylogenetic analysis
Contigs that had DIAMOND BLASTX (Buchfink, Xie, and Huson 2015) hits to members of the Mitoviridae and Narnaviridae and were over 1,000 nucleotides in length were imported into Geneious Prime (v2019.1.1). Sequences with multiple stop codons were translated using the mitochondrial genetic code and checked to ensure they resembled a viral RdRp; namely, the presence of conserved A (-DX4D-), B, and C (-GDD-) amino acid motifs in the palm domain of the RdRp (Jácome et al. 2015). These novel viruses were then aligned using Multiple Alignment using Fast Fourier Transform (MAFFT) (v7.450) (Katoh and Standley 2013) with reference RdRp sequences from the Lenarviricota (1,292 reference sequences). Five additional sequence alignments were constructed comprising the novel virus sequences, the Lenarviricota reference sequences, and established members of each of the following families that served as outgroups to root the phylogenies and hence infer the direction of evolutionary change: the Astroviridae (42 sequences), the Partitiviridae-Picobirnaviridae clade (321 sequences), Picornaviridae (176 sequences), Potyviridae (230 sequences), and Tombusviridae (233 sequences). These outgroups were chosen based on their phylogenetic proximity from a large-scale RdRp phylogeny (Wolf et al. 2018). A midpoint-rooted phylogenetic tree was also inferred. Reference sequences, a large proportion of which were described recently (Chen et al. 2021), were obtained by searching the NCBI nr protein database for relevant family and genera names within the Lenarviricota, as well as for the prefixes of all families (i.e. ‘narna’, ‘mito’, ‘levi’, and ‘ourmia’) to include unclassified sequences that contained ‘-like’ in their names. Reference sequence lists were checked manually to ensure that the top hit of each potentially novel virus was included.
The resultant amino acid sequence alignments were trimmed using trimAL (v1.4.1) with conservation thresholds between 3 and 8 per cent (Capella-Gutierrez, Silla-Martinez, and Gabaldon 2009) to remove any ambiguously aligned regions and retain only the most conserved 500–680 amino acid positions. The best-fit amino acid substitution model was determined using ModelFinder (Kalyaanamoorthy et al. 2017) and found to be the Dayhoff model in all cases (although topologically equivalent phylogenies were produced using the Le-Gascual model; not shown). Maximum likelihood phylogenetic trees were then estimated on these data employing 1,000 Shimodaira-Hasegawa-like approximate likelihood ratio test (SH-aLRT) replicates in IQ-TREE (v1.6.12) (Nguyen et al. 2015). Three smaller ‘sub-trees’ were generated using the same method, the first only utilising the Amabiliviricetes (562 sequences), with the second and third based on an alignment of only the Mitoviridae (562 sequences) and Leviviridae (464 sequences), respectively. The unrooted Lenarviricota tree was visualised in FigTree (v1.4.4). All other trees were visualised in R (v.4.1.0) using the packages ape (v5.5) (Paradis and Schliep 2019) and ggtree (v3.0.2) (Yu et al. 2017).
2.3. Sequence annotation
To identify possible links between genome structure and the evolutionary patterns within and between the families that comprise the Lenarviricota, we used Prokka (v1.14.5) (Seemann 2014) to annotate the genomes of all available narnavirus and narlivirus sequences, as well as twelve botourmiaviruses, forty mitoviruses, and eighty-nine leviviruses. The representative sequences from the latter groups were chosen based on the phylogenies estimated here to obtain an even distribution across all genera and/or major clades.
3. Results
Our analysis of 442 meta-transcriptomic sequencing libraries from soil, sediment, and animal faeces identified 236 novel mitoviruses utilising the mitochondrial genetic code: that is, when translated under the standard genetic code these viruses contained large numbers of internal UGA stop codons. These novel viruses were aligned with other members of the Lenarviricota (sequences ranging between 326 and 1,913 amino residues in length, final alignment length of 680 amino acids) to generate an RdRp phylogenetic tree from 1,542 viral species (Fig. 1). This unrooted phylogeny clearly displayed a three-way split of similarly high divergence between the Leviviridae, the Mitoviridae, and the class Amabiliviricetes, here defined as comprising the Narnaviridae, Botourmiaviridae, and a large, third clade forming a phylogenetically distinct group that we have provisionally identified as a putative new family—the Narliviridae (Fig. 1). In total, 227 of the 231 sequences comprising the Narliviridae were obtained through metagenomic studies of invertebrates (Shi et al. 2016; François et al. 2019), or soil, sediment, and animal faeces samples (Chen et al. 2021), and classified as ‘narna-like’ viruses at their time of discovery. The remaining four sequences were mechanically isolated from plants (Avgelis, Barba, and Rumbos 1989; Aiton et al. 1988; Lisa et al. 1988) or obtained in a metagenomic study of fungi (Rodríguez-Romero et al. unpublished).
Figure 1.
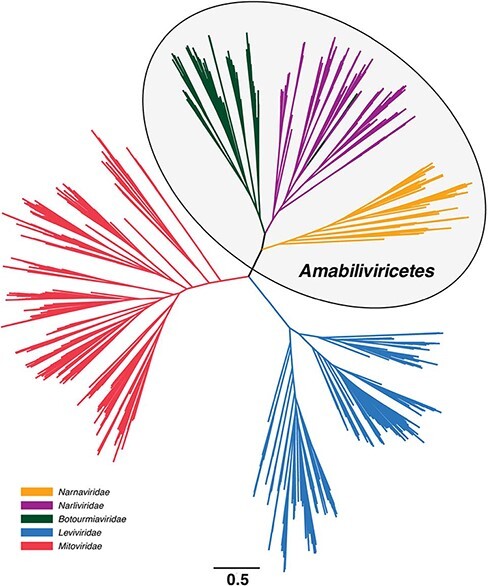
Unrooted phylogeny of the phylum Lenarviricota based on the RdRp domain from 1,542 RNA virus sequences. Branch lengths are scaled according to the number of amino acid substitutions per site, indicated by the scale bar.
We next sought to give this phylogeny an evolutionary directionality from which we could infer the patterns and order of evolutionary transitions in more detail. Accordingly, four virus families and one dual family clade were trialled as potential outgroups based on phylogenetic proximity: the Astroviridae (Fig. 2A), Partitiviridae-Picobirnaviridae (Fig. 2B), Picornaviridae (Fig. 2C), Potyviridae (Fig. 2D), and the Tombusviridae (Fig. 2E). We also estimated a midpoint-rooted Lenarviricota phylogeny (Fig. 2F). Notably, no single tree topology was favoured in all six phylogenies, although in three—those using the Partitiviridae-Picobirnaviridae and Picornaviridae as outgroups as well the midpoint-rooted tree—the Leviviridae fell as the basal group, with the Amabiliviricetes and Mitoviridae then appearing as sister clades (Fig. 2B, C, F). A very different pattern was seen when the Tombusviridae was used as an outgroup: in this case, the Mitoviridae fell as the basal group and the Amabiliviricetes and Leviviridae appeared as sister clades (Fig. 2E). In contrast, when the tree was rooted using the Astroviridae, the Leviviridae and Mitoviridae fell as sister clades to the Amabiliviricetes (Fig. 2A). Finally, when the Potyviridae were used as an outgroup, the Narnaviridae did not appear monophyletic as they did in all other phylogenies, with a group of divergent mitoviruses falling basal to the entire phylum (Fig. 2D). This was the only phylogeny in which each family did not appear as a strictly monophyletic group.
Figure 2.
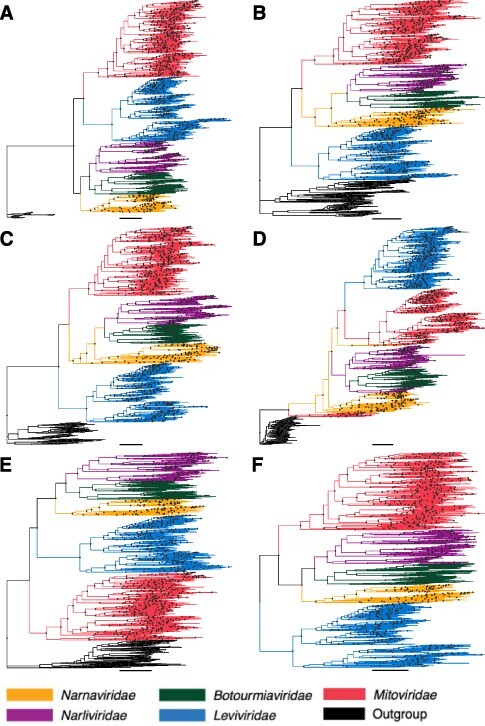
Phylogenies of the phylum Lenarviricota estimated using different groups of RNA viruses as potential outgroups: (A) Astroviridae, (B) Partitiviridae-Picobirnaviridae, (C) Picornaviridae, (D) Potyviridae, and (E) Tombusviridae. Finally, tree (F) is a midpoint-rooted phylogeny with no outgroup. The branch length scale bar represents 0.5 amino acid substitutions per site. Nodes with SH-aLRT support over 80 per cent are marked with circles. Each tree is rooted on its respective outgroup.
We similarly performed a more detailed phylogenetic analysis of the Amabiliviricetes (Fig. 3). This utilised the same narnavirus, narlivirus, and botourmiavirus sequences as above, but with the addition of some members of these families (e.g., the genus Rhizoulivirus and certain divergent narnavirus and narlivirus sequences) that were excluded from the full phylum alignments because their sequences were highly divergent—less than 17 per cent amino acid pairwise identity to even their closest relatives—that they appeared as excessively long branches and negatively impacted the phylogenetic analysis. This analysis revealed three main groups of sequences: (1) the traditional Narnaviridae that occupied the basal position when the tree was midpoint rooted, (2) the Botourmiaviridae, and (3) the newly identified Narliviridae (Fig. 3, Supplementary Fig. S1). Notably, the three plant-infecting members of the genus Ourmiavirus did not fall within the Botourmiaviridae despite being classified in this family. Rather, they grouped with the Narliviridae in every phylogeny (Figs 1–3, Supplementary Fig. S1).
Figure 3.
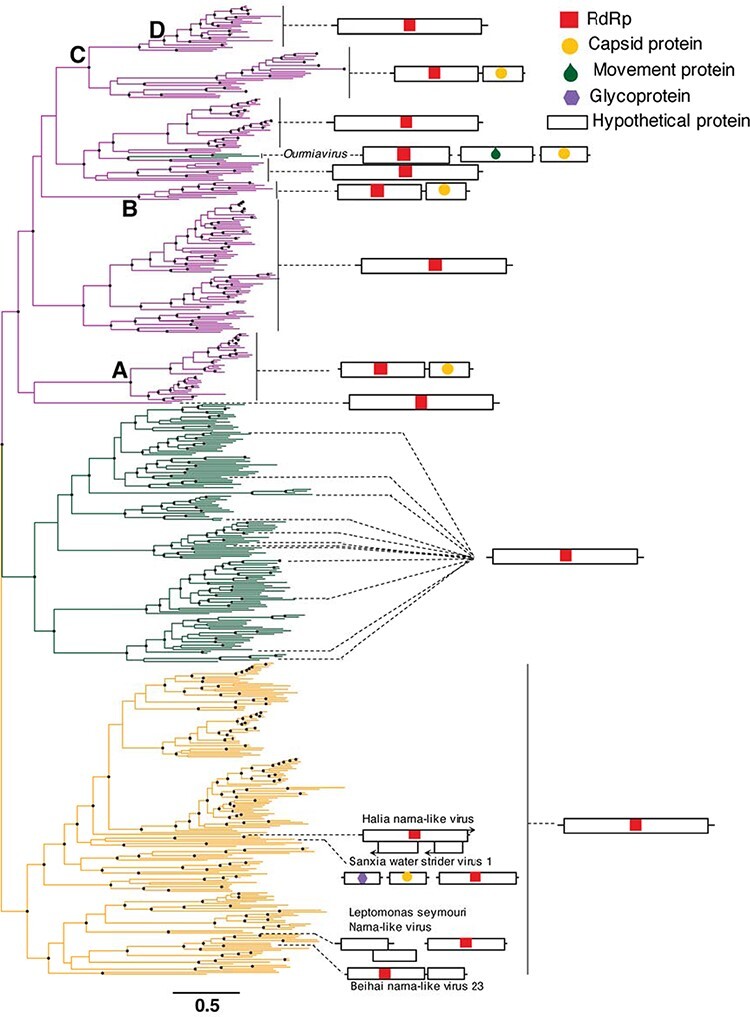
Phylogeny of the Amabiliviricetes based on the RdRp domain from 562 RNA virus sequences. The Narnaviridae occupy the basal position, with Narliviridae (top) and Botourmiaviridae (middle) forming sister clades. General genome organisations for representative species and clades (see Supplementary Figure S1) are shown on the right. ORFs and gene lengths are not drawn to scale. The branch length scale bar represents 0.5 amino acid substitutions per site. Nodes with SH-aLRT support over 80 per cent are marked with circles. The tree is midpoint rooted.
We next annotated the nucleotide sequences of several representative species or clades to identify how well differences in viral genome structure accorded with the overall evolutionary relationships (Figs 3–6). In particular, we carefully annotated the genomes of the Narnaviridae and Narliviridae within the Amabiliviricetes group (Fig. 3), for which the majority of sequences appeared to be complete. As expected, the majority of the traditional Narnaviridae had a single ORF encoding only an RdRp protein. There were four exceptions: Leptomonas seymouri narna-like virus, Sanxia water strider virus 1, Beihai narna-like virus 23, and Halia narna-like virus. These viruses did not group together, nor did they have similar genome structures, such that they comprised four distinct and divergent narnaviruses with unique genome structures. The twelve representative botourmiaviruses and a large majority of the Narliviridae displayed similarly simple genomes to the Narnaviridae, only encoding an RdRp gene (Fig. 3). However, four distinct clades within the Narliviridae contained an additional protein that exhibited 25-82 per cent sequence similarity to viral capsids (Fig. 3). The first clade (Fig. 3, Point A) fell in a basal location within the family, although the associated bootstrap support was low (47.7 per cent) such that the branching position is uncertain. The second and third capsid gains appeared to have evolved more recently (Fig. 3, Points B and C). Most notably, the most recently diverged clade did not contain this additional capsid protein (Fig. 3, Point D), instead reverting to the single RdRp gene, although again with little bootstrap support such that the branching order is uncertain. The fourth occurrence of a capsid protein was within the three ourmiaviruses, each of which had a tri-segmented genome comprising the RdRp, movement protein, and capsid protein, respectively (Fig. 3).
Interestingly, the capsid genes in each of the four occurrences displayed sequence similarity to different sets of other viruses, although usually still with very high levels of divergence (<30 per cent amino acid sequence similarity) (Fig. 4). In the case of the most basal capsid clade (Point A), there was a sequence similarity to the capsid genes of Shahe tombus-like virus 2 and Changjiang narna-like virus 3, as well as to those from some tombusvirus-like and potyvirus-like viruses (Fig. 4). In contrast, the capsid genes at Point B all exhibited sequence similarity to Wenzhou narna-like virus 5. The final group of capsidated viruses (Fig. 4, Point C) had closest matches to Changjiang narna-like virus 2, Hubei narna-like viruses 9 and 10, Hubei tombus-like virus 33, and the nodavirus-like and weivirus-like capsid genes (Fig. 4, Point C). Although the phylogenetic history of these viruses is difficult to infer in places, it is possible that the capsid protein has evolved multiple times independently in the Narliviridae and may have also been lost in one clade.
Figure 4.
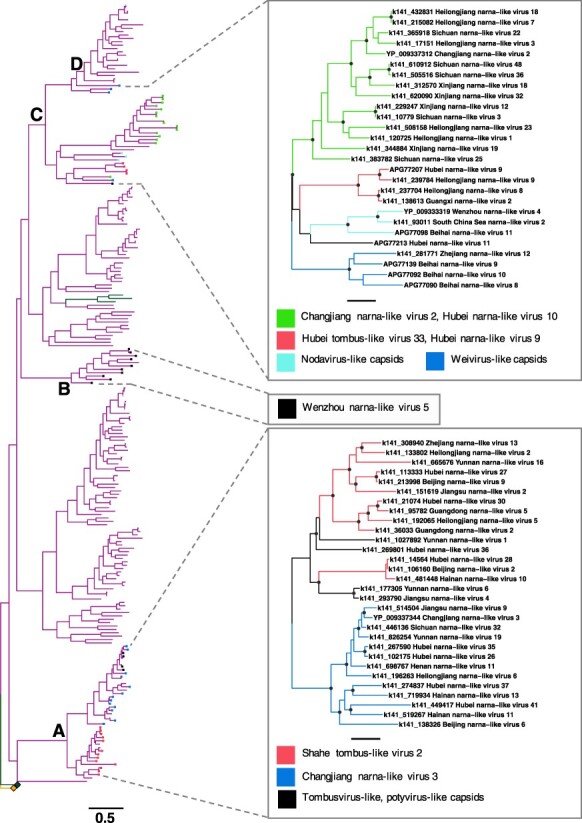
Phylogeny of the Narliviridae (left) within the class Amabiliviricetes based on the RdRp domain. Collapsed clades - Botourmiaviridae (upper) and Narnaviridae (lower) - are shown as squares. Phylogenies estimated using the capsid protein sequences are shown in boxes on the right. Tip colours in the RdRp phylogeny and branch colours in the capsid phylogenies represent the closest capsid protein Blastx hits. The branch length scale bar represents 0.5 amino acid substitutions per site. Nodes with SH-aLRT support over 80 per cent are marked with circles in capsid protein phylogenies. The trees are midpoint rooted.
We similarly annotated genomes within the Mitoviridae and Leviviridae. Overall, thirty-six of the forty representative species within the Mitoviridae contained a single ORF encoding an RdRp (Fig. 5). The four mitoviruses containing additional genes—Daimones mito-like virus, Aiolos mito-like virus, Asopus mito-like virus, and Proteus mito-like virus—were all associated with microalgae, and the latter three displayed ambigrammatic genomes (Charon, Murray, and Holmes 2021); that is, genomes that contain a long, uninterrupted ORF spanning a large proportion of the reverse complement genome (DeRisi et al. 2019) (Fig. 5, Supplementary Fig. S2). In contrast, the majority of Leviviridae genomes contained three genes encoding a maturation protein, a capsid protein, and the viral RdRp (Fig. 6). In several species, a levivirus or levi-like virus lysis protein was also identified. Notably, the leviviruses encoding a lysis protein did not form a single monophyletic group, although they were only present in one of the two lineages within the phylogeny of this family (Fig. 6). Finally, one small group of four leviviruses contained a read-through protein alongside its maturation protein, capsid protein, and RdRp (Fig. 6).
Figure 5.
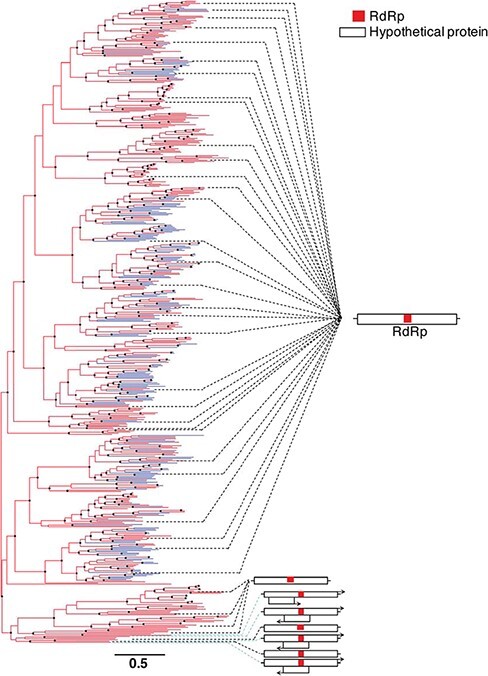
Phylogeny of the family Mitoviridae based on the RdRp domain from 562 RNA virus sequences. General genome organisations for representative species (see Supplementary Figure S2) are shown on the right. ORFs and gene lengths are not drawn to scale. The branch length scale bar represents 0.5 amino acid substitutions per site. Terminal branches coloured differently represent putative novel mitoviruses. Nodes with SH-aLRT support over 80 per cent are marked with circles.
Figure 6.
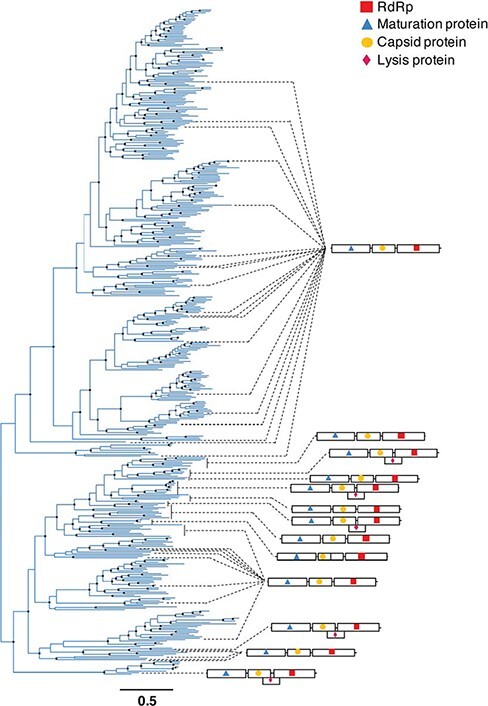
Phylogeny of the family Leviviridae based on the RdRp domain from 464 RNA virus sequences. General genome organisations for representative species and clades (see Supplementary Figure S3) are shown on the right. ORFs and gene lengths are not drawn to scale. The branch length scale bar represents 0.5 amino acid substitutions per site. Nodes with SH-aLRT support over 80 per cent are marked with circles.
4. Discussion
The phylum Lenarviricota is composed of RNA viruses that likely mark a major evolutionary transition event between RNA bacteriophage and early eukaryote-infecting RNA viruses. Members of this unique phylum are highly diverse, abundant in virtually every environment (Chen et al. 2021), and associated with a broad range of hosts including bacteria, protists, fungi, and plants. Here, we investigate the evolutionary relationships among the diverse families comprising the Lenarviricota, utilising a large data set of relevant viral sequences—including 236 novel mitoviruses identified in this study—the majority of which have been obtained from large-scale metagenomic studies.
A key goal of our study was to resolve the phylogenetic history of the phylum Lenarviricota. Due to a trichotomy (and similar levels of divergence) between the Leviviridae, the Mitoviridae, and the Amabiliviricetes, the exact pattern of ancestor–descendent relationships among these viruses and hence between those viruses infecting prokaryotes and eukaryotes is difficult to determine. In addition, the long branches at the base of each group imply missing phylogenetic diversity that has yet to be identified. To help overcome these major issues in phylogenetic analysis, we rooted the Lenarviricota phylogeny using five different outgroups—the families Astroviridae, a Partitiviridae-Picobirnaviridae clade, Picornaviridae, Potyviridae, and Tombusviridae—as well as estimating a simple midpoint-rooted tree. Notably, however, this analysis did not result in a consistent tree topology. This is most clearly seen in the tree rooted on the Potyviridae, in which neither the Narnaviridae nor the Amabiliviricetes formed monophyletic groups and a group of divergent mitoviruses fell basal to the entire Lenarviricota phylum. Hence, the use of highly divergent outgroups cannot reliably resolve the evolution of the Lenarviricota. Indeed, it is clear that RNA viruses as a whole and likely the Lenarviricota, in particular, are too diverse to align with sufficient reliability to produce a robust phylogeny tree (Edgar 2021), with individual amino acid sites subject to extensive multiple substitution (Holmes and Duchêne 2019).
Despite these limitations, given that the codon bias in Mitoviridae genomes reflects that of their respective fungal hosts (Nibert 2017), the alternative codon usage by members of the Mitoviridae is likely a derived, adaptive function acquired after the origin of organisms containing the mitochondria. This means the mitoviruses are unlikely to be an ancestral group to RNA viruses as a whole as implied in the phylogeny using the Tombusviridae as an outgroup. In addition, the most common and perhaps likely phylogenetic pattern observed in this study (in three of the six phylogenetic trees) suggests that the Leviviridae is the basal lineage within the Lenarviricota, with the Mitoviridae and Amabiliviricetes falling as derived groups. This supports the most popular hypothesis for the evolutionary pathway of this phylum, in which a levivirus-like ancestral virus gave rise to the Mitoviridae that acquired the capacity to replicate in the newly emerged mitochondrion of early eukaryotic organisms (Koonin and Dolja 2014; Wolf et al. 2018).
The Botourmiaviridae were previously considered to be a monophyletic, phylogenetically distinct sister clade to the Narnaviridae (Ayllón et al. 2020). However, the phylogeny of the Narnaviridae, Botourmiaviridae, and sequences classified as ‘narna-like’ at their time of discovery—that we now term as the Narliviridae—has changed considerably with the periodic addition of a huge number of diverse viruses found in invertebrates, soil, and marine samples (Shi et al. 2016; Wolf et al. 2020; Chen et al. 2021). In all phylogenies estimated here using an alignment containing sequences from the Amabiliviricetes, the non-encapsidated, filamentous-fungi-infecting genera within Botourmiaviridae (Botoulivirus, Magoulivirus, Penoulivirus, Rhizoulivirus and Scleroulivirus) remained monophyletic. However, according to our phylogenetic analysis the family no longer includes the plant-infecting ourmiaviruses, which instead appear to have evolved from an entirely different lineage within the Narliviridae. Importantly, this contradicts previous phylogenetic analyses and thus challenges the family’s current taxonomic organisation (Ayllón et al. 2020). Currently, the Narnaviridae and Botourmiaviridae are separated at the class level: the Narnaviridae in the Amabiliviricetes and the Botourmiaviridae in the Miaviricetes (Ayllón et al. 2020; Walker et al. 2020). In contrast, the phylogeny produced in this study suggests the Narnaviridae, Botourmiaviridae, and Narliviridae likely fall within a single taxonomic class. Hence, we propose that the Botourmiaviridae and newly classified Narliviridae should be combined into one order—the Ourlivirales, while the Narnaviridae remain in the order Wolframvirales and that both orders be combined into one class—the Amabiliviricetes. Importantly, this taxonomic distinction is robust to all the phylogenetic trees presented in this paper.
The family Narnaviridae has traditionally been defined as having a remarkably simple genome of a single ORF encoding only the viral RdRp (Hillman and Cai 2013), although some recently identified members of this family appear to contain additional genes and multiple ORFs or ambigrammatic genomes (Shi et al. 2016; Grybchuk et al. 2018; Charon et al. 2019; Chiapello et al. 2020; Wolf et al. 2020; Charon, Murray, and Holmes 2021). Notably, large-scale metagenomic studies have suggested the presence of a capsid protein in assembled sequences resembling narnaviruses (Shi et al. 2016; Wolf et al. 2020), all of which appear to fall within the newly proposed Narliviridae. The genome structures of viruses within the Amabiliviricetes also support the taxonomic distinction between the families Narnaviridae and Narliviridae. While the vast majority of species within the Narnaviridae do indeed have the typical narnavirus genome comprising a single RdRp gene, the Narliviridae appear to have gained a capsid gene at multiple distinct points and lost it at one, suggesting that they may possess more flexible genomes than those of the Narnaviridae. These diverse capsid genes show some sequence similarity to the capsids of tombusviruses, nodaviruses, and sobemoviruses with the picorna-like single jelly-roll fold (Koonin et al. 2008), suggesting frequent and independent instances of horizontal gene transfer between these plant and animal-associated virus families and the Narliviridae. This has been proposed as a mechanism for how ourmiaviruses gained their capsid and movement proteins (Koonin and Dolja 2014). The presence of capsid genes within this family shows that despite these viruses having similarity to the narnavirus RdRp (Shi et al. 2016; Chen et al. 2021), they instead likely comprise a new family with variable genome structures.
Further metagenomic studies will inevitably increase the number of viruses within this phylum, although the identification of potential ‘intermediate’ species alone may not resolve their evolutionary history. Large-scale virus discovery projects are identifying viruses so diverse that even the most conserved regions of their genomes (i.e. the RdRp) are difficult to align with currently available computational tools. Hence, if RNA virus taxonomy continues to increasingly depend on RdRp phylogenies, it is likely to be continually disrupted by the inevitable discovery of diverse viral species. In contrast, protein structures are considerably more conserved than primary sequences (Illergård, Ardell, and Elofsson 2009; Černý et al. 2014), with polymerases exhibiting relatively high levels of conservation reflecting their central function in the viral life cycle. This makes structural analysis an attractive tool for the discovery of highly divergent viruses (Ortiz-Baez et al. 2020). With both the growing availability of structural data and advances in protein modelling (Kelley et al. 2015), it is likely that uncovering the evolutionary history of RNA viruses will rely increasingly on structure-based phylogenies.
Supplementary Material
Contributor Information
Sabrina Sadiq, Sydney Institute for Infectious Diseases, School of Life and Environmental Sciences and School of Medical Sciences, The University of Sydney, Sydney, NSW 2006, Australia.
Yan-Mei Chen, Shanghai Public Health Clinical Center, State Key Laboratory of Genetic Engineering, School of Life Sciences and Human Phenome Institute, Fudan University, Shanghai 200438, China.
Yong-Zhen Zhang, Shanghai Public Health Clinical Center, State Key Laboratory of Genetic Engineering, School of Life Sciences and Human Phenome Institute, Fudan University, Shanghai 200438, China.
Edward C Holmes, Sydney Institute for Infectious Diseases, School of Life and Environmental Sciences and School of Medical Sciences, The University of Sydney, Sydney, NSW 2006, Australia.
Supplementary data
Supplementary data are available at Virus Evolution online.
Funding
Australian Research Council Australian Laureate Fellowship (FL170100022) to E.C.H.; National Natural Science Foundation of China (31930001 and 32130002) to Y.Z.Z.
Conflict of interest:
None declared.
Data availability
Sequence reads are available at the NCBI Sequence Read Archive database under BioProject accession PRJNA716119. Novel viral sequences identified in this study are available in GenBank under the accession numbers ON001450–ON001685.
References
- Aiton M. M. et al. (1988) ‘Two New Cassava Viruses from Africa’, In: 5th International Congress of Plant Pathology, Kyoto, Japan, 43. [Google Scholar]
- Akopyants N. S. et al. (2016) ‘A Narnavirus in the Trypanosomatid Protist Plant Pathogen Phytomonas serpens’, Genome Announcements, 4: e00711–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avgelis A., Barba M., and Rumbos I. (1989) ‘Erirus Cherry Virus, an Unusual Virus Isolated from Cherry with Rasp-Leaf Symptoms in Greece’, Journal of Phytopathology, 126: 51–88. [Google Scholar]
- Ayllón M. Á. et al. (2020) ‘ICTV Virus Taxonomy Profile: Botourmiaviridae’, Journal of General Virology, 101: 454–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger A. M., Lohse M., and Usadel B. (2014) ‘Trimmomatic: A Flexible Trimmer for Illumina Sequence Data’, Bioinformatics, 30: 2114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchfink B., Xie C., and Huson D. H. (2015) ‘Fast and Sensitive Protein Alignment Using DIAMOND’, Nature Methods, 12: 59–60. [DOI] [PubMed] [Google Scholar]
- Capella-Gutierrez S., Silla-Martinez J. M., and Gabaldon T. (2009) ‘trimAl: AtTool for Automated Alignment Trimming in Large-Scale Phylogenetic Analyses’, Bioinformatics, 25: 1972–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Černý J. et al. (2014) ‘Evolution of Tertiary Structure of Viral RNA Dependent Polymerases’, PLoS One, 9: e96070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charon J. et al. (2019) ‘Novel RNA Viruses Associated with Plasmodium vivax in Human Malaria and Leucocytozoon Parasites in Avian Disease’, PLoS Pathogens, 15: e1008216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charon J., Murray S., and Holmes E. C. (2021) ‘Revealing RNA Virus Diversity and Evolution in Unicellular Algae tTanscriptomes’, Virus Evolution, 7: veab070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y. M. et al. (2021) ‘RNA Virome Composition Is Shaped by Sampling Ecotype’, SSRN Electronic Journal.doi: 10.2139/ssrn.3934022. [DOI] [Google Scholar]
- Chiapello M. et al. (2020) ‘Analysis of the Virome Associated to Grapevine Downy Mildew Lesions Reveals New Mycovirus Lineages’, Virus Evolution, 6: veaa058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole T. E. et al. (2000) ‘Detection of an RNA-Dependent RNA Polymerase in Mitochondria from a Mitovirus-Infected Isolate of the Dutch Elm Disease Fungus, Ophiostoma Novo-Ulmi’, Virology, 268: 239–43. [DOI] [PubMed] [Google Scholar]
- DeRisi J. L. et al. (2019) ‘An Exploration of Ambigrammatic Sequences in Narnaviruses’, Scientific Reports, 9: 17982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaire L., Rozas J., and Ayllón M. A. (2016) ‘Molecular Characterization of Botrytis Ourmia-Like Virus, a Mycovirus Close to the Plant Pathogenic Genus Ourmiavirus’, Virology, 489: 158–64. [DOI] [PubMed] [Google Scholar]
- Edgar R. C. (2021) ‘MUSCLE v5 Enables Improved Estimates of Phylogenetic Tree Confidence by Ensemble Bootstrapping’, bioRxiv.doi: 10.1101/2021.06.20.449169. [DOI] [Google Scholar]
- François S. et al. (2019) ‘A New Prevalent Densovirus Discovered in Acari. Insight from Metagenomics in Viral Communities Associated with Two-Spotted Mite (Tetranychus urticae) Populations’, Viruses, 13: 233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grybchuk D. et al. (2018) ‘Viral Discovery and Diversity in Trypanosomatid Protozoa with a Focus on Relatives of the Human Parasite Leishmania’, Proceedings of the National Academy of Sciences USA, 11: 506–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillman B. I., and Cai G. (2013) ‘The Family Narnaviridae’, Advances in Virus Research, 86: 149–76. [DOI] [PubMed] [Google Scholar]
- Holmes E. C., and Duchêne S. (2019) ‘Can Sequence Phylogenies Safely Infer the Origin of the Global Virome?’, mBio, 10: e00289–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illana A. et al. (2017) ‘Molecular Characterization of a Novel ssRNA Ourmia-Like Virus from the Rice Blast Fungus Magnaporthe oryzae’, Archives of Virology, 162: 891–5. [DOI] [PubMed] [Google Scholar]
- Illergård K., Ardell D. H., and Elofsson A. (2009) ‘Structure Is Three to Ten Times More Conserved than Sequence - a Study of Structural Response in Protein Cores’, Proteins: Structure, Function, and Bioinformatics, 77: 499–508. [DOI] [PubMed] [Google Scholar]
- Jácome R. et al. (2015) ‘Structural Analysis of Monomeric RNA-Dependent Polymerases: Evolutionary and Therapeutic Implications’, PLoS One, 10: e0139001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalyaanamoorthy S. et al. (2017) ‘ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates’, Nature Methods, 14: 587–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K., and Standley D. M. (2013) ‘MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability’, Molecular Biology and Evolution, 30: 772–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley L. A. et al. (2015) ‘The Phyre2 Web Portal for Protein Modeling, Prediction and Analysis’, Nature Protocols, 10: 845–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King A. M. Q. et al. (2012) ‘Leviviridae’, Virus Taxonomy, 1: 1035–43. [Google Scholar]
- Koonin E. V., and Dolja V. V. (2014) ‘Virus World as an Evolutionary Network of Viruses and Capsidless Selfish Elements’, Microbiology and Molecular Biology Reviews, 78: 278–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin E. V. et al. (2008) ‘The Big Bang of Picorna-Like Virus Evolution Antedates the Radiation of Eukaryotic Supergroups’, Nature Reviews. Microbiology, 6: 925–39. [DOI] [PubMed] [Google Scholar]
- Krupovic M., and Koonin E. V. (2017) ‘Multiple Origins of Viral Capsid Proteins from Cellular Ancestors’, Proceedings Of the National Academy Of Sciences, 114: 2401–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Lay C. et al. (2020) ‘Unmapped RNA Virus Diversity in Termites and Their Symbionts’, Viruses, 12: 1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D. et al. (2015) ‘MEGAHIT: An Ultra-Fast Single-Node Solution for Large and Complex Metagenomics Assembly via Succinct de Bruijn Graph’, Bioinformatics, 31: 1674–6. [DOI] [PubMed] [Google Scholar]
- Lisa V. et al. (1988) ‘Ourmia Melon Virus, a Virus from Iran with Novel Properties’, Annals of Applied Biology, 112: 291–302. [Google Scholar]
- Mahar J. E. et al. 2020. ‘Comparative Analysis of RNA Virome Composition in Rabbits and Associated Ectoparasites’, Journal of Virology, 94: e02119–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin W. F., and Mentel M. (2010) ‘The Origin of Mitochondria’, Nature Education, 3: 58. [Google Scholar]
- Marzano S. Y. L. et al. (2016) ‘Identification of Diverse Mycoviruses through Metatranscriptomics Characterization of the Viromes of Five Major Fungal Plant Pathogens’, Journal of Virology, 90: 6846–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nerva L. et al. (2019) ‘Isolation, Molecular Characterization and Virome Analysis of Culturable Wood Fungal Endophytes in Esca Symptomatic and Asymptomatic Grapevine Plants’, Environmental Microbiology, 21: 2886–904. [DOI] [PubMed] [Google Scholar]
- Nguyen L.-T. et al. (2015) ‘IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies’, Molecular Biology and Evolution, 32: 268–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nibert M. L. (2017) ‘Mitovirus UGA(Trp) Codon Usage Parallels that of Host Mitochondria’, Virology, 507: 96–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz-Baez A. S. et al. (2020) ‘A Divergent Articulavirus in an Australian Gecko Identified Using Meta-Transcriptomics and Protein Structure Comparisons’, Viruses, 12: 613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis E., and Schliep K. (2019) ‘Ape 5.0: An Environment for Modern Phylogenetics and Evolutionary Analyses in R’, Bioinformatics, 35: 526–8. [DOI] [PubMed] [Google Scholar]
- Rastgou M. et al. (2009) ‘Molecular Characterization of the Plant Virus Genus Ourmiavirus and Evidence of Inter-Kingdom Reassortment of Viral Genome Segments as Its Possible Route of Origin’, Journal of General Virology, 90: 2525–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seemann T. (2014) ‘Prokka: Rapid Prokaryotic Genome Annotation’, Bioinformatics, 30: 2068–6. [DOI] [PubMed] [Google Scholar]
- Shackelton L. A., and Holmes E. C. (2008) ‘The Role of Alternative Genetic Codes in Viral Evolution and Emergence’, Journal of Theoretical Biology, 254: 128–34. [DOI] [PubMed] [Google Scholar]
- Shi M. et al. (2016) ‘Redefining the Invertebrate RNA Virosphere’, Nature, 540: 539–43. [DOI] [PubMed] [Google Scholar]
- Starr E. P. et al. (2019) ‘Metatranscriptomic Reconstruction Reveals RNA Viruses with the Potential to Shape Carbon Cycling in Soil’, Proceedings of the National Academy of Sciences, 116: 25900–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urayama S., Takaki Y., and Nunoura T. (2016) ‘FLDS: A Comprehensive dsRNA Sequencing Method for Intracellular RNA Virus Surveillance’, Microbes and Environments, 31: 33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker P. J. et al. (2020) ‘Changes to Virus Taxonomy and the Statutes Ratified by the International Committee on Taxonomy of Viruses (2020)’, Archives of Virology, 165: 2737–48. [DOI] [PubMed] [Google Scholar]
- Wille M. et al. (2020) ‘Sustained RNA Virome Diversity in Antarctic Penguins and Their Ticks’, The ISME Journal, 14: 1768–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf Y. I. et al. (2018) ‘Origins and Evolution of the Global RNA Virome’, mBio, 9: e02329–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2020) ‘Doubling of the Known Set of RNA Viruses by Metagenomic Analysis of an Aquatic Virome’, Nature Microbiology, 5: 1262–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G. et al. (2017) ‘Ggtree: An R Package for Visualization and Annotation of Phylogenetic Trees with Their Covariates and Other Associated Data’, Methods in Ecology and Evolution, 8: 28–36. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequence reads are available at the NCBI Sequence Read Archive database under BioProject accession PRJNA716119. Novel viral sequences identified in this study are available in GenBank under the accession numbers ON001450–ON001685.