

Abstract
Porcine Parvovirus Type 1 (PPV1) contributes to important losses in the swine industry worldwide. During a PPV1 infection, embryos and fetuses are targeted, resulting in stillbirth, mummification, embryonic death, and infertility (SMEDI syndrome). Even though vaccination is common in gilts and sows, strains mainly belonging to the 27a-like group have been spreading in Europe since early 2000s, resulting in SMEDI problems and requiring in-depth studies into the molecular epidemiology and vaccination efficacy of commercial vaccines. Here, we show that PPV1 has evolved since 1855 [1737, 1933] at a rate of 4.71 × 10−5 nucleotide substitutions per site per year. Extensive sequencing allowed evaluating and reassessing the current PPV1 VP1-based classifications, providing evidence for the existence of four relevant phylogenetic groups. While most European strains belong to the PPV1a (G1) or PPV1b (G2 or 27a-like) group, most Asian and American G2 strains and some European strains were divided into virulent PPV1c (e.g. NADL-8) and attenuated PPV1d (e.g. NADL-2) groups. The increase in the swine population, vaccination degree, and health management (vaccination and biosafety) influenced the spread of PPV1. The reactivity of anti-PPV1 antibodies from sows vaccinated with Porcilis© Parvo, Eryseng© Parvo, or ReproCyc© ParvoFLEX against different PPV1 field strains was the highest upon vaccination with ReproCyc© ParvoFLEX, followed by Eryseng© Parvo, and Porcilis© Parvo. Our findings contribute to the evaluation of the immunogenicity of existing vaccines and support the development of new vaccine candidates. Finally, the potential roles of cluster-specific hallmark amino acids in elevated pathogenicity and viral entry are discussed.
Keywords: molecular phylodynamics, BEAST, viral classification, vaccination, evolution
Introduction
Porcine Parvovirus Type 1 (PPV1) has been circulating in swine herds, resulting in reproductive losses in the swine industry since the 1960s. Since 1967, it has been associated with reproductive failure and is considered as one of the primary agents of stillbirth, mummification, embryonic death, and infertility (SMEDI) (Mayr and Mahnel 1964; Streck and Truyen 2020). During a PPV1 infection, embryos and fetuses of susceptible sows may become infected, which is not surprising as parvoviruses are known to replicate in dividing cells (S-phase) (Cotmore and Tattersall 2007). No treatment is available against PPV1 infection, thus strict biosecurity measures and vaccination have become general practice. The first commercial inactivated vaccines, based on the cell-culture-attenuated NADL-2 strain, became available in the 1980s (Suzuki and Fujisaki 1976; Joo and Johnson 1977; Mengeling et al. 1979; Streck et al. 2013). Since then, several other vaccines have been developed. Despite significant vaccination efforts and improved health management in the swine industry, the number of PPV1-positive SMEDI cases increased from 0.5 per cent in 2017 up to 9.2 per cent in 2019 in Belgium (Activiteitenrapport 2020 Veepeiler Varken; DGZ Vlaanderen). Similarly, in Denmark, less than 5 per cent of the submitted fetuses tested positive for PPV1 until 2015, but from 2015 to 2021 an average of 17.4 per cent of the submitted fetuses tested positive (figures kindly provided by the Danish Pig Research Center). As described by Zimmerman and colleagues in 2006, a new antigenic PPV1 variant, designated 27a, was detected in Germany in the early 2000s. With distinct capsid amino acids, a new phylogenetic cluster was identified (Zimmermann et al. 2006). Moreover, infections and vaccination with heterologous Genotype 2 strains (e.g. NADL-2 and 143a) did not result in cross-neutralizing antibodies against this 27a strain (Zeeuw et al. 2007). In Europe, a predominance of 27a-like PPV1 strains was observed, suggesting that the appearance of 27a-like strains is the result of viral adaptation that occurred over the last 10–30 years due to vaccination pressure in a partially immune swine population (Cadar et al. 2012; Streck and Truyen 2020).
PPV1 is a virus belonging to the family Parvoviridae, showing the typical genomic characteristics of a small single-stranded non-enveloped DNA virus. Its genome (approximately 5 kb) is built up of two terminal hairpins and two major open reading frames (ORFs), including non-structural (NS1, NS2, and NS3) and structural proteins (VP1, VP2, and VP3), which originate from alternative splicing events (Bergeron, Hébert, and Tijssen 1996). Over the last decades, the genetic diversity of the PPV1 was studied extensively by looking at its (partial) VP1/2 or NS1 sequence, which allowed to establish genetic classification systems resulting in either two (Zimmermann et al. 2006), three (Ren et al. 2013), four (Oh et al. 2017), six (Cadar et al. 2012), or seven (Streck, Canal, and Truyen 2015) distinct PPV1 clades. The evolutionary rate of the capsid genes was estimated to be in the range of RNA viruses (10–4 nucleotide substitutions per site per year; (Streck et al. 2011; Cadar et al. 2012; Ren et al. 2013)). Increased sequencing efforts allowed a more accurate estimation of 3.3 × 10−5 nucleotide substitutions per site per year, which is higher than the evolutionary rate of its non-structural genes and lower than that of prototypic RNA viruses (Oh et al. 2017). The surface proteins assemble into a compact icosahedral capsid consisting of sixty identical VP1/VP2 copies, represented in a 1:5 ratio (Fig. 1A–B) (Simpson et al. 2002). These twelve VP1 molecules play an important role in particle assembly, DNA packaging, and infectivity, mediated through a phospholipase 2 (PLA2) domain and linear nuclear localization motifs (Fig. 1D) (Tullis, Burger, and Pintel 1993; Cotmore and Tattersall 2007). Each of three VP1/2 copies assemble into a trimer, resulting in an extrusion at the three-fold axis and a two-fold dimple (Fig. 1). While the three-fold extrusions serve as the main protein receptors and neutralizing antibody binding sites, the two-fold dimple determines tissue specificity and oligosaccharide recognition (Fig. 1D). Specific VP1 amino acid changes (Thr214, Gly378, and Pro436) have been associated with host tropism in tissue cultures, immune response, and hemagglutination activity based on studies with the attenuated NADL-2 strain. However, no clear link with altered antigenicity has been observed so far (Bergeron, Hébert, and Tijssen 1996; Cotmore and Tattersall 2007; Streck, Canal, and Truyen 2015).
Figure 1.

Comprehensive overview of the PPV1 virion. (A) Overall PPV1 capsid structure showing five-fold axes (left) and three- and two-fold (black arrows) axes (right) when centered on the pore and VP2 trimer, respectively. Capsid extrusions, dimples, and five-fold pores are colored in shades of red, gray, and dark gray, respectively; (B) Intersection of pore and VP2-trimer-centered capsid structures, showing a hypothesized VP1 peptide representation (1:5 ratio) at pore interior. Black arrows represent the two-fold dimple between two adjacent VP2 trimers; (C) VP2 trimer representation from side (left) and front (right) orientation; (D) Single VP1/VP2 structure in surface (left) and cartoon (right) representation, highlighting the four major loops (shades of red) and hypothesized VP1 peptide at its 5ʹ end (green). This additional VP1 peptide harbors three linear nuclear localization motifs (LNCM; light green) and a unique PLA2 domain (PLA2; blue). All representations are based on the 1K3V VP2 structure (Tsao et al. 1991; Simpson et al. 2002).
This work focuses on studying the molecular epidemiology of PPV1 strains that are available in genetic databases, supplemented with eighty-seven newly sequenced strains, including strains from Denmark (n = 64), France (n = 7), The Netherlands (n = 7), Germany (n = 4), Belgium (n = 2), the USA (n = 2), and Great Britain (n = 1). The reactivity analyses of antisera obtained from pigs after experimental vaccination with three commercially available vaccines against historical and current PPV1 strains from different clades were also completed.
Materials and methods
Whole-genome sequencing of PPV1 strains
French (n = 7), Dutch (n = 7), German (n = 4), Belgian (n = 2), American (n = 2), and British (n = 1) PPV1 isolates/strains (sampled between 1963 and 2021) were subjected to whole-genome sequencing at PathoSense BV. In short, nucleic acids were extracted from diagnostic field samples (e.g. 20 per cent w/v suspensions of mummified fetal organs) or cell culture supernatants (NADL-8 USA, 59e/63 GBR, Porcilis© Parvo PPV014, 123456 DEU and 002346-5 USA). The extracted nucleic acids were used in a metagenomics amplification protocol at PathoSense BV prior to long-read nanopore sequencing applying the rapid library preparation (RBK-004; ONT) protocol on a MinION flow cell (R9.4.1; ONT) (Theuns et al. 2018; Bokma & Vereecke et al. 2021). Data were acquired on a GridION (ONT) device, allowing real-time data collection in MinKNOW (v.21.10; ONT) including super-accurate basecalling (guppy_basecaller v.5.0.16; -c dna_r9.4.1_450bps_supac.cfg; ONT) and demultiplexing (guppy_barcoder v.5.0.16; ONT). Raw sequencing reads were quality filtered and taxonomically classified prior to the whole-genome de novo assembly using canu (v2.0.0; (Koren et al. 2017)) and medaka polishing (v1.4.1; ONT). Subsequently, VP1/2/3 sequences were extracted from whole genomes for further analyses. An overview of these strains, their epidemiological relationship, and accompanying accession numbers is available in Supplementary Table S1.
Targeted ORF2 sequencing of Danish PPV1 strains
Total DNA of Danish strains (n = 64; sampled between 2006 and 2021) was extracted manually from cell culture isolates or the primary material (e.g. 20 per cent w/v suspensions of mummified fetal organs) using the QIAamp DNA Mini Kit (QIAGEN, cat. no. 51304) following the manufacturer’s guidelines. The ORF2 encoding the VP1/2/3 genes was amplified by conventional polymerase chain reaction (PCR) using three overlapping primer sets: F2030/R3000; F2562/R3698; and F3570/R4498 (Cadar et al. 2012). The PCR amplification was performed with the AccuPrime Taq DNA High Fidelity (Invitrogen cat. no. 12346-086) as described by the manufacturer using 5 µl of DNA as template. The PCR was run under the following conditions: 2 min 95°C, [35 cycles. 95°C, 15 s; 58°C, 30 s; 72°C, 90 s], 72°C, 10 min; pause 4°C. The PCR products were analyzed by 0.8 per cent agarose gel electrophoresis, and the PCR products of the expected size were purified using Roche’s High Pure PCR product purification kit (cat. no. 11 732 676 001). The samples were prepared for Sanger sequencing at LGC Genomics GmbH, Berlin, with the PCR primers as sequencing primers. The raw sequencing data were assembled against a PPV1 reference sequence using the software CLC main workbench v20.0.4 (QIAGEN). An overview of these strains, their epidemiological relationship, and accompanying accession numbers is available in Supplementary Table S1.
Generation of PPV1 VP1 dataset and evaluation of temporal signal within the dataset
All new VP1/VP2/VP3 sequences (n = 87) were aligned against the available sequences on GenBank (accessed on 21 October 2015) using MAFFT (v.7.310; (Katoh and Standley 2013)). Sequences with incomplete VP1 (<2,190 nt) were removed from the alignment along with sequences showing traces of putative recombination events. The latter was assessed in a preliminary recombination screening using the recombination detection program (RDP) (v.4.99; (Martin et al. 2015)) including the GENECONV, Chimaera, MaxChi, 3Seq, BootScan, SiScan, and RDP approaches. All methods were run using the recommended settings. Sequences showing significant recombinations by three or more methods were removed from the alignment in an iterative way (n = 2; (Sabir et al. 2016)). To evaluate if the PPV1 sampling time window showed sufficient measurable evolution, a Bayesian Evaluation of Temporal Signal (BETS) analysis was performed (Duchene et al. 2020). The BETS analysis was performed for both a strict clock (SC) and a relaxed clock (RC) model with an underlying lognormal distribution, using generalized stepping-stone sampling to accurately estimate each log marginal likelihood (Baele, Lemey, and Suchard 2016). In addition, potential outliers that would render molecular clock estimation difficult were evaluated in TempEst (v.1.5.3; (Rambaut et al. 2016)) after generating a maximum likelihood (ML) phylogenetic tree with IQ-TREE (v.1.6.1; -m GTR + G; (Chernomor, Von Haeseler, and Minh 2016)). No sequences were considered as outliers.
Molecular phylodynamic analyses and generation of a maximum clade credibility tree
Since the BETS and TempEst analyses showed sufficient temporal signal to be present within our dataset, a Bayesian Evolutionary Analysis Sampling Trees (BEAST; v.1.10.4; (Suchard et al. 2018)) analysis was subsequently performed to estimate a time-calibrated phylogeny by employing a GTR + G + I nucleotide substitution model and uncorrelated RC model with an underlying lognormal distribution. A Bayesian Skygrid coalescent model with Hamiltonian Monte Carlo sampling was used to estimate past population dynamics with a cut-off date of 150 years and seventy-five population sizes, respectively (Gill et al. 2013; Baele et al. 2020). To ensure that convergence (Effective Sampling Size > 200) for all studied parameters was reached, log and maximum clade credibility (MCC) tree files of eight independent BEAST runs (500,000,000 iterations each chain and sampling every 50,000th iteration) were combined using LogCombiner (BEAST; v.1.10.4; (Suchard et al. 2018)) and further analyzed in Tracer using a 10 per cent burn-in (v.1.7.2; (Rambaut et al. 2018)), TreeAnnotator (BEAST; v.1.10.4; (Suchard et al. 2018)), and FigTree (v. 1.4.4; https://github.com/rambaut/figtree/). The estimated rates and ancestral dates are represented as mean with accompanying 95 per cent Highest Posterior Density (HPD) interval. To address biological relevance of PPV1 clusters, an ML tree (1,000 bootstraps; GTR + G + I) along with a pairwise amino acid diversity matrix was generated in MEGA (v.11; (Tamura, Stecher, and Kumar 2021)). The latter allowed to construct pairwise identity graphs to assess intra- and inter-cluster pairwise identity frequencies based on MCC tree clusters (Matthijnssens et al. 2008).
Assessing the immunogenicity of commercial PPV1 vaccines
Gilts were vaccinated twice with an interval of 21 or 28 days with either one of three commercial vaccines: octavalent Porcilis© Ery/Parvo/Lepto PPV014 (MSD Animal Health; PPV1a; adj.: dl-alpha-tocoferylacetaat), ReproCyc© bivalent Eryseng© Parvo NADL-2 (HIPRA; PPV1c; adj.: aluminiumhydroxide, DEAE-Dextraan, Ginseng), and ReproCyc© ParvoFLEX 27a subunit vaccine (Boehringer Ingelheim; PPV1b; adj.: Carbopol (ImpranFLEX®; BI Animal Health USA Inc.) as previously described in Noguera et al. (2021). Forty-one or 56 days after vaccination, blood was collected, coagulated in vacutainers, and centrifuged, prior to serum collection. Aliquots of serum were stored at −20°C and subsequently used to quantitate anti-PPV1 antibodies using (1) ImmunoPeroxidase Monolayer Assay (IPMA; (Labarque et al. 2000)), (2) Hemagglutinin Inhibition (HI; (Joo, Donaldson‐Wood, and Johnson 1976; Vannier et al. 1986)), and (3) Serum Neutralization (SN) assays. For the latter, a protocol was adapted from Martínez-Lobo et al. (2011) (Martínez-Lobo et al. 2011). In short, a two-fold serial dilution of serum was carried out in 96-well tissue culture plates with Minimal Essential Medium (MEM) (Gibco: cat. no. 21090-055) supplemented with 5 per cent Fetal Bovine Serum (FBS) (Sigma: cat. no. F7524), 1 per cent Pen/strep/L-Glutamine (Gibco, cat. no. 10378-016), and 1 per cent nonessential amino acids (Sigma: cat. no. M7145-100ml). The virus isolates were diluted to contain 2 × 103 TCID50 ml−1. Fifty microliters of virus suspension or MEM (negative control) was added to 50 µl of each serum dilution and mixed well by pipetting. The mixtures were incubated for 1 h at 37°C. Following incubation, 50 µl of PK15 cell suspension (300,000 cells ml−1) were added to each well and left at 37°C, 5 per cent CO2 for 5 days. In each plate, known positive (PPV1 839-7-2-90 DNK 1976 and hyperimmune serum as described below) and negative serum controls were included. All serum samples were heat-inactivated at 56°C for 30 min before use and were analyzed in duplicate. To investigate the serum-neutralizing abilities, the plates were fixed in absolute ethanol, followed by IPMA staining of the cells as described previously (Markussen and Have 1992; Bøtner, Nielsen, and Bille-Hansen 1994). PPV1 hyperimmune serum (1:200 diluted), retrieved from experimental vaccinated/challenged pigs and Anti-Pig IgG-peroxidase (1:500 diluted; Sigma: cat. no. A5670) was used as primary and secondary antibody, respectively. The plates were read using an inverted light microscope. When cells were found positively stained against PPV1, the corresponding serum dilution was considered not to have PPV1-neutralizing abilities. In all assays, a spectrum of PPV1 field strains was used, including NADL-8 (USA 1976; PPV1d), 839-3-2-83 (Denmark, 1976; PPV1d), 839-7-2-90 (Denmark, 1976; PPV1a), 59e/63 (UK, 1963; PPV1d), 002346-5 USA (USA, 2010; undefined), 19V196 (Belgium, 2019; PPV1b), 8468-1 (Denmark, 2017; PPV1b), and 123456 (DEU, NA; PPV1b) to compare antibody levels between sera. Statistical significance was determined using an ordinary two-way analysis of variance (ANOVA) test, followed by a Tukey’s multiple comparisons test with computation of individual variances for each comparison (n = 3 per sample). All analyses were performed in GraphPad Prism (v.9).
Results
Evaluation of the temporal signal within the PPV1 VP1 dataset
First, the PPV1 VP1 dataset was assessed on potential recombination events using RDP4. Only two sequences (16WS CHN 2013; KM268633 and N2 KOR 2018; MH817779) were removed, showing a recombination signal in three or more used methods. Next, the BETS and TempEst analyses allowed to address the question if sufficient temporal signal was present within the sampling time window of the studied sequences. This analysis strongly supported the presence of a temporal signal within the dataset and opted in favor of an uncorrelated RC model (log Marginal Likelihood Estimation: −7435.86 vs. log MLE: −7455.80 without dates) above a SC model (log MLE: −7461.46 vs. log MLE: −7486.18 without dates). Also, the preliminary TempEst analysis showed a proper root-to-tip plot of the sequences with an estimated evolutionary rate of 2.61 × 10−5 nucleotide substitutions per site per year and estimated time to most recent common ancestor around 1668.
Estimation of PPV1 population dynamics using molecular phylodynamics
Since the preliminary BETS and TempEst analyses showed sufficient support for molecular clock estimation to perform time-calibrated phylogenetic inference, an in-depth BEAST analysis was performed. The phylodynamic analysis with an uncorrelated RC model estimated the evolutionary rate and root age to be around 4.71 × 10−5 nucleotide substitutions per site per year [2.1 × 10−5, 7.6 × 10−5] and 1855 [1737, 1933], respectively. The Bayesian Skygrid coalescent model allowed to assess and visualize the PPV1 population dynamics across years based on the genetic diversity of the PPV1 dataset. As shown in Fig. 2, gradual PPV1 population growth is shown from its founder up to the 1980s, followed by a significant increase in the PPV1 population up to the late 1990s. From then on, a steep decline was observed up to mid-2010s, resulting in a more stabilized population size up to date (2021). Interestingly, the increase in PPV1 population coincided with an increase in the swine population in both Belgium and Denmark.
Figure 2.
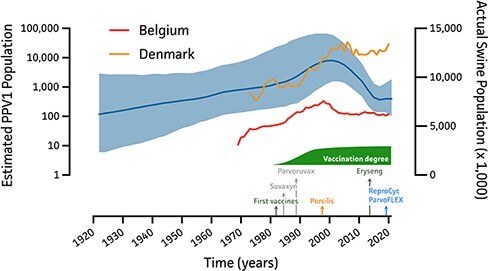
Bayesian Skygrid reconstruction of the PPV1 population in relation to Belgian and Danish swine population. On the left y-axis, the estimated PPV1 population size is shown over a time range of 1921 up to 2021, indicating the mean estimated PPV1 population size as a solid blue line and 95 per cent HPD intervals as blue shade. The right y-axis represents the actual swine population size of Belgium (red) and Denmark (orange) as available from Eurostat (February 2022). At the bottom, the first introductions of PPV1 vaccines in Belgium are shown along with the vaccination degree.
Based on the MCC tree, the Genotype 1 (Zimmerman et al. 2006), Cluster A (Cadar et al. 2012), or European Lineage 2 cluster (Oh et al. 2017) (Fig. 3; orange) showed major divergence between 1942 and 1981, containing the vaccine strains IDT/MSV (DEU, 1972) and Porcilis© Parvo PPV014. Any further divergence of these strains has been limited as only few cases are known since 2020 (DNK = 4 and FRA = 1). A first Genotype 2 divergence was estimated to have occurred around 1903 [1836, 1949], giving rise to Asian Lineage 3 which comprises the attenuated NADL-2 strain (Fig. 3; green). Interestingly, this Asian cluster can still be divided further in the actual Asian Lineage 3 strains (Cluster F; Cadar et al. 2012) and an ‘ancient’ European cluster with the 59e/63 and British Challenge strains from 1963 and 1986, respectively, the Danish 839-3-2-83 strain from 1976, and the German Challenge strain from 2010. For simplicity, this cluster was named Cluster G in compliance with Cadar et al. (2012). Twelve years later, around 1915 [1861, 1953], an additional divergence occurred, resulting in Lineage 4 (Fig. 3; pink). Here again, Lineage 4 shows an additional divergence into an older Cluster E, containing the virulent NADL-8 and American Kresse strains from 1976 and 1985, respectively, and a more recent Asian Cluster H, only comprising Chinese and South-Korean strains from 2003 up to 2018. Again, cluster labeling was done in compliance with Cadar et al. (2012). A final major divergence occurred around 1919 [1868, 1958], resulting in Cluster C and D and Cluster B strains. While Clusters C and D comprise most Romanian PPV1 strains isolated from wild boars, Cluster B is also known as the European Lineage 1 or 27a-like cluster (Fig. 3; blue). The latter highlights the most recent divergence and comprised most of the newly sequenced strains (DNK = 44, NDL = 7, FRA = 6, BEL = 2, and DEU = 2).
Figure 3.

Time-scaled MCC tree of the PPV1 VP1 gene. All PPV1 VP1 sequences are shown indicating the suggested node origins (x-axis). Clusters were determined based on Oh et al. (2017) (colors), Zimmerman et al. (2006) (Genotypes), and Cadar et al. (2012) (Letters). New Clusters G and H are colored in green and magenta, respectively. Relevant vaccine strains and field strains using in IPMA, HI, and SN assays are indicated in bold.
Next, the eight MCC tree-based phylogenetic clusters (A–H) were subjected to an intra- and inter-strain diversity analysis to determine their biological relevance. While amino acid intra-diversity of Cluster A and B strains was higher as compared to inter-diversity values for all other clusters, no overall differences of intra- and inter-cluster diversity for all other clusters were observed (Supplementary Fig. S1 and Supplementary Tables S2 and S3). Interestingly, this analysis highlighted the distinct genetic profile of some strains that were differently classified into one of the MCC tree-based clusters (D: TJ CHN 2012; F: HNAY CHN 2009; and H: VRI-1 KOR 2003). Upon removal of the HNAY CHN 2009 strain from Cluster F, its intra-cluster diversity showed significant differences from all other inter-cluster diversities (Supplementary Fig. S1). Thus, overall, four distinct PPV1 clusters were shown which are thought to be biologically relevant and were renamed as PPV1a, PPV1b, PPV1c, and PPV1d strains, respectively (Fig. 4A, B). It is noteworthy that only for strains 18KWB13 KOR 2018, WB-754 ROM 2011, PS950 FRA 2021, 2003-6 DNK 2021, and 1491-1 DNK 2016 the exact classification to either PPV1b/c could not be determined as they showed ‘mixed/transient’ AA hallmarks, highlighting potential evolutionary traces between PPV1 Clusters b and c.
Figure 4.

Evaluation of cluster divergence based on intra- and inter-cluster amino acid diversities. (A) ML tree (1,000 bootstraps; GTR + G + I) of all PPV1 VP1 sequences highlighting the four biological relevant PPV1 clusters, including PPV1a (G1; orange), PPV1b (G2; blue), PPV1c (G2; green), and PPV1d (G2; pink) strains. Strains used in further analyses are shown in bold; (B–D) Trimer (left) and monomer (right) of the VP2 structure (1K3V) highlighting cluster-specific hallmarks as compared to the PPV1c NADL-8 (USA 1976) strain. Cluster- and subpopulation-specific amino acid changes are shown with solid and dotted lines, respectively. Red and blue circles highlight mutations associated with NADL-2 phenotypic attenuation (Bergeron, Hébert, and Tijssen 1996) and tissue tropism (Simpson et al. 2002), respectively; (E) Overview of new simplified PPV1-VP1-based classification system.
VP1 Loop 4 mutations mark the divergence of 27a-like strains
To evaluate the potential impact of VP1-specific mutations on vaccination efficacy, an in-depth analysis of the VP1 protein sequences was performed as compared to the virulent NADL-8 sequence (PPV1d). As shown in Fig. 1D, the VP2 protein is a splicing variant of the VP1 protein, which can be further divided into an internal moiety (white) and four external loops (shades of red). When looking at cluster-specific mutations, a 150-nt sequence unique to VP1 was conserved among all studied strains, only showing a Genotype-1-specific Thr102Ser mutation. All (non-)linear nuclear localization motif sites and the PLA2 catalytic site were also conserved (Supplementary Fig. S2A). While PPV1a (Genotype 1)-specific mutations within the VP2 protein were dispersed over the entire protein, only one mutation (Lys557Asn) was observed in Loop 4 as compared to three unique mutations (Ala564Ser/Glu569Gln/Pro586Thr) for the 27a-like PPV1b group (Figs 1, 4C, D, respectively). Interestingly, when looking at the overall capsid structure, amino acids specific to the PPV1a cluster (Genotype 1; Cluster A) appeared to be present on the lower shoulders of the three-fold axis trimer extrusion or two-fold dimple (Figs 1, 4C; orange). This is in contrast with PPV1b (27a-like Genotype 2; Cluster B) mutations, which are present on top or on the higher shoulders of the three-fold axis trimers (Figs 1, 4D; blue). For PPV1c (Genotype 2; Cluster F) strains only one specific mutation (Thr365Ile) was identified, together with diverse other unique mutations in its subpopulations (Fig. 4E; light green dotted lines). Among these, NADL-2-phenotypic-attenuation-associated mutations were identified, which were mostly located in the two-fold dimple of the virion capsid (Figs 1, 4E; light green and red circles). In agreement with the intra- and inter-strain diversity analyses and the fact that the NADL-8 strain (USA 1976) was used as the reference strain, only some PPV1d strain subsets showed unique hallmarks. For Cluster C and D strains a single unique mutation (Pro586Ala) could be identified (Supplementary Fig. S2C; purple). Also, two unique mutations (Ile295Leu/Ser342Gly) were identified for the new Cluster G, which were located at the inside of the capsid structure (Figs 1, 4E; dark green). No unique markers were identified for Cluster E, except for a Pro586 presented in all but two (Pro586Ala in 18BKWB29 KOR 2018 and Pro586Thr in 18KWB13 KOR 2018) strains. This is the same amino acid as conserved in most of the Genotype 1 strains (Supplementary Fig. S2B).
ReproCyc© ParvoFLEX 27a subunit vaccination results in elevated HI antibody titers
First, VP1 AA sequences of the vaccines (n = 3) and tested field strains (n = 8) were compared as summarized in the pairwise AA identity matrix in Fig. 5A. This showed that the 839-3-2-90 DNK 1976 field strain can be considered homologous to the Porcilis© Parvo vaccine strain. Also, Strains 19V196 BEL 2019, 8468-1 DNK 2017, and 123456 DEU NA were shown to be homologous to the ReproCyc© ParvoFLEX vaccine strain. While all other strains (NADL-8 USA 1976, 839-7-2-83 DNK 1976, and 59e/63 GBR 1963) are considered heterologous strains to all vaccine strains, Strain 002346-5 USA 2010 still showed high AA identity to all PPV1b strains (99.1–99.3 per cent) and the NADL-8 USA 1976 strain (PPV1d; 99.5 per cent). The antisera raised upon vaccination with different commercial vaccines (Porcilis© Ery/Parvo/Lepto (MSD Animal Health; PPV1a), Eryseng© Parvo NADL-2 (HIPRA; PPV1c), and ReproCyc© ParvoFLEX (Boehringer Ingelheim; PPV1b)) were tested with the IPMA and HI assays against the PPV1 field strains NADL-8 (USA 1976; PPV1d), 839-3-2-90 (DNK, 1976; PPV1a), 839-7-2-83 (DNK, 1976; PPV1d), 59e/63 (GBR, 1963; PPV1d), 002346–5 USA (USA, 2010; undefined), 19V196 (BEL, 2019; PPV1b), 8468-1 (DNK, 2017; PPV1b), and 123456 (DEU, NA; PPV1b), allowing to discriminate the total antibody production (IPMA) and those interacting with antigens that potentially link with sugars (e.g. O- and N-linked sialic acids) on red blood cells (RBCs) (Boisvert, Fernandes, and Tijssen 2010). The same antisera were tested with the SN assay against a selection of the PPV1 isolates (839-7-2-90 (DNK, 1976; PPV1a), 002346-5 USA (USA, 2010; undefined), 8468-1 (DNK, 2017; PPV1b), and 123456 (DEU, NA; PPV1b)) to evaluate the production of neutralizing antibodies (Fig. 5A). To allow the proper comparison of resulting antibody titers between different vaccines, Fig. 5B represents differences in mean antibody titers (mean ± 95 per cent confidence interval (CI)) between each of two vaccines for each field strain. Significantly higher IPMA antibody titers were observed with the Porcilis© antisera against the PPV1d NADL-8 (USA 1976) strain (q = 0.0089) as compared to Eryseng© antisera. Also, slightly higher IPMA antibody titers were observed in the ReproCyc© ParvoFLEX antisera, as compared to antisera induced by both other tested vaccines when using the 59e/63 (GBR 1963) strain (Porcilis©q = 0.0170; Eryseng©q = 0.0450). For the German PPV1b strain 123456 DEU (year NA) significantly higher IPMA antibody titers were detected for the ReproCyc© ParvoFLEX versus Eryseng© vaccine (q = 0.0004). When testing for neutralizing antibodies (SN assay) against the Danish PPV1a 839-7-2-90 (1976) strain, both Eryseng© and ReproCyc© ParvoFLEX vaccines showed significantly higher neutralizing antibody titers as compared to the Porcilis© vaccine (q = 0.0066 and q = 0.0017, respectively). Aversely, for all tested PPV1b strains (8468-1 (DNK, 2017) and 123456 (DEU, NA)), significantly higher neutralizing antibody titers were detected in the ReproCyc© ParvoFLEX antisera than in both Eryseng© and Porcilis© Parvo antisera (Fig. 5B and Supplementary Table S4). For the Strain 002346-5 USA (USA, 2010; undefined), only significant higher neutralizing antibodies were detected in ReproCyc© ParvoFLEX antisera as compared to Eryseng© antisera (q = 0.0051). When looking at the HI assay, overall higher HI antibody titers were observed for the ReproCyc© ParvoFLEX antisera against all tested field strains as compared to the other antisera (Fig. 5B–C and Supplementary Table S4). While both other vaccines resulted in lower HI antibody titers, the HI antibody titers against the 002346-5 American strain were elevated for the antisera directed against three tested vaccines. This American strain (2010) is not embedded within the 27a-like cluster but is classified rather as a transient PPV1c/b strain (and Clade C (Cadar et al. 2012)) as presented in Figs 3–4. Strains from both Clusters C and B (PPV1b strains) shared a common ancestor only about 100 years ago [1868, 1958]. Interestingly, the External Loop 4 of the VP1 protein showed a ‘mixed’ genotype with the Pro586Thr mutation, specific for the 27a-like strains, and an Ala564 and Glu569 genotype for all other non-27a-like field strains (Fig. 5D). Loop 2 showed a conserved Gln378, and not the Gln378Glu 27a-like strain hallmark. Also, a unique Thr462Ile mutation was detected within the External Loop 3 at the lower shoulder of the VP2 protein (Supplementary Fig. S2D).
Figure 5.

Immunogenicity of three commercial vaccines. (A) Pairwise AA identity (%) measures of all PPV1 VP1 sequences used in the serological tests are shown in a matrix, highlighting the homo- and heterologous nature of tested field strains in relation to the used commercial vaccines. Antibody titers in the antisera were determined showing (B) PPV1 specific antibodies in the IPMA (left), SN (middle), and HI (right) assays. (C) Multiple comparison plots for pairwise vaccine comparison, showing the mean antibody titer differences and 95 per cent CIs from (B); negative and positive values represent higher antibody titers in Vaccine 1 (left on y-axis) versus Vaccine 2 (right on y-axis), respectively. A value of 0 indicates that no difference in antibody titer was observed. Significance is shown as adjusted P-values based on two-way ANOVA and Tukey testing (0.05–0.01 (*), 0.01–0.001 (**), 0.001–0.0001 (***), and <0.0001 (****)). (D) ML tree of used PPV1 vaccine (bold) and field strains in relation to their VP1-Loop-4-specific mutations.
Discussion and conclusions
Here we presented an in-depth molecular epidemiological analysis of all available complete PPV1 VP1/VP2/VP3 sequences, supplemented with the sequences of eighty-seven new PPV1 strains. These new strains comprised sixty-four Danish, seven French, seven Dutch, four German, two Belgian, two American, and one British strains. The largest proportion (38 per cent) of PPV1 samples in our dataset originates from Denmark, which may potentially be a source of sampling bias in our dataset. This is, however, difficult to assess without objective data on how prevalent PPV1 infections are in different countries across the world, i.e. having more sequences from Denmark than from other locations does not necessarily constitute (a form of) sampling bias. Such bias would have clear implications when reconstructing the geographic spread of PPV1 (De Maio et al. 2019), and we have hence refrained from performing such a phylogeographic analysis. Even without performing phylogeographic inference we need to acknowledge that the inferred population size over time may be to a degree driven by the Danish sequences in our dataset. However, this demands a more formal analysis based on objective data—such as case counts over time in different countries—that can then be tested as driving factors of effective population size over time (Gill et al. 2016). To obtain prevalence data, better surveillance systems should be set up in affected countries to regularly analyze and monitor PPV1 strains across the world. Our sequencing efforts also aimed at supplementing our dataset with relevant ancient PPV1 sequences, including the first complete sequence of the highly virulent NADL-8 strain from the USA (1976) and the Porcilis© Ery/Parvo/Lepto PPV014 (MSD Animal Health) vaccine strain, among others. To the authors’ knowledge, this is the first study to show that sufficient temporal signal is present within the PPV1 VP1 sequence data using a BETS analysis (Duchene et al. 2020). Moreover, this analysis allowed to generate preliminary insights into the molecular clock model to be used in subsequent phylodynamic analyses. For the PPV1 VP1 sequence, this was shown to be an uncorrelated RC model, which was previously used in various molecular phylodynamic studies, supporting the power of a preliminary BETS analysis (Streck et al. 2011, 2013; Cadar et al. 2012; Ren et al. 2013; Oh et al. 2017). So far, studies included a limited number of (partial) VP1 sequences, preventing clear and accurate classification and PPV1 population dynamics. Hence, the availability of eighty-seven new complete VP1 sequences enabled to draw more accurate conclusions on the evolutionary rate and root age of PPV1. Our phylodynamic analysis estimated these to be around 4.71 × 10−5 nucleotide substitutions per site per year [2.1 × 10−5; 7.6 × 10−5] and 1855 [1737, 1933], respectively. This is lower than that reported in previous studies (3.02 × 10−4, 2.72 × 10−4, 1.07 × 10−4; (Streck et al. 2011; Cadar et al. 2012; Ren et al. 2013)) and in line with a more recent publication by Oh et al. (2017), estimating the evolutionary rate to be around 3.27 × 10−5 nucleotide substitutions per site per year. This suggests the virus to be evolving at a slower pace than previously thought. This evolutionary rate is still quicker as compared to other DNA viruses (10−9–10−10 nucleotide substitutions per site per year), but slightly slower than RNA viruses (10−3–10−5 nucl. subst. site−1 year−1), which is a characteristic of all autonomous parvoviruses (Streck, Canal, and Truyen 2015). We estimated the most recent common ancestor of PPV1 to have emerged between 284 and 88 years ago, which is in line with previous estimations (250 years) by Ren et al. (2013). Given the lack of samples from before 1964 and the inclusion of several divergent lineages in our dataset (Fig. 3), the inferred time of origin for PPV1 is quite uncertain as indicated by its corresponding 95 per cent HPD. The recent development of novel molecular clock models (e.g. Bletsa et al. 2019; Didelot et al. 2021) may pose an interesting avenue for detailed comparisons on divergence time estimates across the phylogeny, including the root.
Based on the genetic diversity, this extended dataset also allowed to study the past PPV1 effective population size dynamics more accurately using a Bayesian Skygrid reconstruction. This highlighted the steady increase in the PPV1 population over time, followed by a steep increase in the estimated PPV1 population size in the 1980s, reaching its peak around the 2000s. However, this is in contrast with the single available study on the PPV1 population dynamics, which suggested a small steady population, followed by a slow decrease in the PPV1 population from 1980s onward (Streck et al. 2013). It is noteworthy that Streck et al. (2013) only used 739 of the 2,190 VP1 nucleotides from seventy-four samples to estimate the population dynamics. While the authors speculate that this decrease matches the broad implementation of PPV1 vaccination campaigns, our available dataset likely assumes that higher genetic drift was the result of an increased swine population in Belgium and Denmark (Eurostat 2022). This is not surprising, as an increase in swine population requires a higher breeding capacity and thus provides a higher number of infectable hosts for the virus. However, other factors may have had an influence on viral diversity as well, such as improvements in health management, the vaccination degree, and the use of different vaccines with various formulations (e.g. different adjuvants, PPV1 background, and multivalent vaccines). Together, these might explain the observed peak in the 2000s and the subsequent drop in PPV1 population. This is in line with the hypothesis that the antibody pressure causes a lower genetic drift (Streck et al. 2013). The stabilized/constant PPV1 population during the last years suggests that the virus is trying to adapt to persist in the pig population. Again, we would like to emphasize the relevance to set up a surveillance system to regularly analyze and monitor its genetic and amino acid changes in affected countries worldwide.
Ren et al. (2013) found no geographic structures in the phylogeny of PPV1. The current phylogenetic analysis highlights the geographic restriction of some clusters. While PPV1a and PPV1b strains are restricted to Europe, PPV1c is represented by only Asian NADL-2 like strains. In contrast, PPV1d strains show no clear geographic restrictions. Similar trends were previously observed by Oh et al. (2017), resulting in a comparable PPV1 genotypic classification system. Based on our estimated MCC tree, two new distinct clusters were found, which complied most closely with the previously suggested classification of Cadar et al. (2012). Thus, we supplemented their classification system with two new clusters, designated Clusters G and H for an ‘Ancient’ NADL-2-like European Lineage and a more recent Asian Lineage 4, respectively. Even though the MCC tree considers sequence divergence, geography, and temporal data, an intra- and inter-cluster amino acid pairwise identity analysis and ML tree suggest simplifying the current PPV1 VP1-based classification system to four major PPV1 groups. As summarized in Fig. 4B, these PPV1 phylogenetic groups were assigned PPV1a, PPV1b, PPV1c, and PPV1d. To date, this is the most biologically relevant PPV1 classification system that allows the proper classification of the Challenge (UK 1986), Kresse (USA 1985) and VRI-1 (KOR 2003) strains, which was not possible with previous datasets (Streck et al. 2011; Cadar et al. 2012; Ren et al. 2013; Oh et al. 2017).
Next, our study showed cluster-specific hallmarks highlighting more dispersed mutations to be associated with PPV1a (Genotype 1) strains. In contrast, PPV1b (27a-like Genotype 2)-specific mutations were confined to Loop 4 of VP1/2/3. One of these mutations, Pro586Thr, turned out to be the location with the highest mutational pressure. This is expected as this amino acid is localized on top of the three-fold extrusion and has been confirmed in an in vitro setting before (Fernandes, Boisvert, and Tijssen 2011; Streck et al. 2011). While this mutation was first thought to be associated with increased pathogenicity, this hypothesis was proven wrong using chimeric PPV1 strains in vitro (Fernandes, Boisvert, and Tijssen 2011). Interestingly, during an infection with the 27a strain, more virus replication was detected in fetal organs as compared to another virulent strain 143a (PPV1a) and two vaccine strains IDT/MSV (PPV1a) and NADL-2 (PPV1c) (Wilhelm et al. 2005). While an increased viral fitness and a decreased neutralizing activity were recently reported for 27a strains, the exact sow-to-fetus transfer mechanism has only been hypothesized so far and has also been thought to be the result of transfer of noninfectious viral DNA (Wilhelm et al. 2005; Van Den Born et al. 2020; Streck, Canal, and Truyen 2022). As proposed in previous studies, the PPV1 may use maternal macrophages and/or lymphocytes to cross the placental barrier (Mengeling, Lager, and Vorwald 2000; Wilhelm et al. 2005). The use of macrophages was also suggested for the transplacental spread of porcine reproductive and respiratory syndrome virus-induced reproductive failure (Karniychuk et al. 2011). While PPV1 cell entry is known to happen through terminal α2-3 N- and O-linked sialic acid moieties of glycoproteins on the cell surface, protein receptors (e.g. integrins or transferrin receptors) have been described for parvoviral infections in other mammals (Boisvert, Fernandes, and Tijssen 2010; Tu et al. 2015). Whereas PPV1a and PPV1c hallmark mutations are localized majorly on the lower shoulder of the trimer, PPV1b hallmark mutations were localized on the top and higher shoulder of the trimer. As these hallmarks were determined in relation to the virulent NADL-8 strain (PPV1d), our data show conserved dimple and lower-shoulder trimer amino acid sequences in PPV1b strains. Since this region is known to be important for proper binding to terminal sialic acids, a conserved dimple region might facilitate the cell attachment, followed by conformational change of the virion pore and subsequent VP1 exposure within the endosome (Fig. 1B) (Tullis, Burger, and Pintel 1993; Cotmore and Tattersall 2007; Boisvert, Fernandes, and Tijssen 2010; Tu et al. 2015). Aversely, PPV1a and PPV1c strains have mutations focalized in this sialic-acid-binding region, lowering and/or preventing its efficient cellular entrance, which is in line with its limited persistence and NADL-2-attenuated phenotypes (Suzuki and Fujisaki 1976; Joo and Johnson 1977; Mengeling, Lager, and Vorwald 2000; Jiang et al. 2013). The potential involvement of a protein receptor remains unclear and will require further investigation as mutations on the top or higher shoulder of the trimer might be involved in the receptor binding and/or evasion of neutralizing antibodies and neutralizing antibody binding and not in sialic acid binding (Cotmore and Tattersall 2007). Indeed, upon vaccination with the 27a-based ReproCyc© ParvoFLEX subunit vaccine, transplacental infections are completely abolished by the vaccination-induced immune response (Zeeuw et al. 2007; Jóźwik et al. 2009; Garcia-Morante et al. 2020; Van Den Born et al. 2020).
Finally, we assessed the immunogenicity of three commercial vaccines: octavalent Porcilis© Ery/Parvo/Lepto PPV014 (MSD Animal Health), bivalent Eryseng© Parvo NADL-2 (HIPRA), and the ReproCyc© ParvoFLEX 27a subunit vaccine (Boehringer Ingelheim). Here, the antisera from a previous study by Noguera et al. (2021) were used to determine the antibody reactivity in three assays (IPMA, HI, and SN) against a variety of PPV1 field strains ((NADL-8 (USA 1976), 839-7-2-90 (DNK, 1976), 839-3-2-83 (DNK, 1976), 59e/63 (UK, 1963), 002346-5 USA (USA, 2010), 19V196 (BEL, 2019), 8468-1 (DNK, 2017), and 123456 (DEU, NA)). Our data showed only subtle changes in IPMA antibody titers between the different antisera and tested field strains, showing higher IPMA antibody titers for the Porcilis© Parvo antisera as compared to Eryseng© antisera when tested against the PPV1d NADL-8 (USA 1976) strain. Interestingly, both Eryseng© and ReproCyc© ParvoFLEX antisera showed significantly higher neutralizing antibodies (SN assay) than Porcilis© Parvo anti-serum against the Danish 839-7-2-90 strain (PPV1a). This is surprising as the Porcilis© Parvo PPV014 vaccine strain also belongs to the PPV1a group, which are Genotype 1 strains. This might be a result of a lowered immune response to the Porcilis© Parvo PPV014 vaccine. In addition, significantly higher neutralizing antibody titers against PPV1b strains were present within ReproCyc© ParvoFLEX antisera as compared with antisera induced by other vaccines. This is comparable to in vivo results showing cross-reactive protection of sows vaccinated with the ReproCyc© ParvoFLEX vaccine upon a heterologous PPV1 infection (Garcia-Morante et al. 2020). Results from the HI assay showed more PPV1-specific antibodies to be generated after vaccination with the ReproCyc© ParvoFLEX 27a subunit vaccine than with the two other antisera. The potential effect of different adjuvants used in the vaccines is noteworthy; while the Porcilis© Parvo and Eryseng© Parvo vaccines are adjuvanted with an oil/aluminum, the ReproCyc© ParvoFLEX vaccine is supplemented with Carbopol, a cross-linked polymer of acrylic acid. Vaccines adjuvanted with the latter were shown to be capable of inducing better systemic and mucosal memory responses to non-replicating antigens as compared to oil/aluminum adjuvants (Gasper et al. 2016; Zhang et al. 2018; Garcia-Morante et al. 2019). Upon vaccination with the ReproCyc© ParvoFLEX 27a subunit vaccine, part of the increased antibody levels might be a result of the Carbopol adjuvant. While IPMA assays detect all antibodies, SN detects the antibodies that are involved in the neutralization and blockage of the viral ligands interacting with its receptor(s) on PK15 cells and the HI assay identifies antibodies that are interacting with sugars (e.g. O- and N-linked sialic acids) on the surface of RBCs (Mészáros et al. 2017). Indeed, it has been shown that PPV1 particles enter the cell via oligosaccharide chains with terminal sialic acid residues linked to a common Gal1-4GlcNAc moiety. It is also the interaction of these oligosaccharide chains with the capsid’s two-fold dimple that is trivial for cell tissue tropism rather than the VP1/2 trimer extrusions (Cotmore and Tattersall 2007; Boisvert, Fernandes, and Tijssen 2010). This is also the reason why three two-fold dimple-specific mutations (D528G/H533Q/S586P) were suggested to be important in the cell tropism of the attenuated NADL-2 strain (Bergeron, Hébert, and Tijssen 1996). Interestingly, most PPV1b-specific mutations (Q378E/A564S/E569Q/P586T) are localized on top of or at the shoulders of the trimer extrusion. Hence, elevated HI neutralizing antibodies must be a result of antibodies preventing PPV1 from binding with sugar moieties on RBCs. Whether the PPV1 capsid can bind RBC-specific oligosaccharide moieties with its trimer extrusion is currently unknown. We hypothesize that this is not facilitated as the two-fold dimple is known to interact with terminal sialic acid residues with increased affinity in the function of chain length (Cotmore and Tattersall 2007). Nevertheless, Gln378Glu and Ala564Ser are localized on the shoulder near the two-fold dimple and are part of known antigenic sites as verified by pepscan (Kamstrup et al. 1998). Antibody binding at these positions might result in the steric hindrance of the PPV1 binding to the RBCs in the HI assay. Indeed, Streck and colleagues (2022) identified the amino acids around Position 378 to be a 27a-like neutralizing epitope (Streck, Canal, and Truyen 2022). Moreover, one of the tested field strains (002346-5 USA 2010) showed the overall elevated HI antibody titers in antisera of all three vaccines. Upon further investigation of this PPV1d (Cluster C) strain, a ‘mixed’ genotype could be identified (Cadar et al. 2012). While the strain showed a PPV1b-specific Proline to Threonine mutation on Position 586, a non-PPV1b genotype was present at locations Gln378, Ala564, and Glu569 within Loops 2 and 4 of the VP1 protein. Furthermore, the strain exhibited a unique Thr462Ile mutation within Loop 3, which is located near the G528D mutation, as one of the three mutations involved in NADL-2 phenotypic attenuation (Bergeron, Hébert, and Tijssen 1996). We suggest the increased HI antibody titer to be rather the result of the highly exposed Pro586Thr Loop 4 mutation than the G528D mutation which is on the border of the two-fold dimple. This hypothesis is supported by the fact that neutralizing antibodies are binding to the trimer extrusion and its shoulders, but not to the two-fold dimple (Bergeron, Hébert, and Tijssen 1996; Cotmore and Tattersall 2007; Zeeuw et al. 2007; Streck, Canal, and Truyen 2015). Aversely, epitope mapping and pepscan analyses showed the linear epitopes for major neutralizing antibodies to be located at the N-terminus and Internal Region 4, embedded within the two-fold dimple (Kamstrup et al. 1998; De Souza et al. 2019). Further research will be required to fully assess the potential role of these mutations in the elevated production of HI and neutralizing antibodies. Nevertheless, these findings are interesting since a single strain showed elevated HI antibody titers for all tested antisera. Thus, strains with ‘mixed’ genotypes might be considered for future vaccine development. So far PPV1a (Porcilis© Parvo PPV014), PPV1b (ReproCyc© ParvoFLEX 27a subunit vaccine), and PPV1d (Eryseng© Parvo NADL-2 and Parvoruvax® K-22) strains were used in vaccine development and in vivo trials (Jóźwik et al. 2009; Garcia-Morante et al. 2020; Kiss et al. 2020; Van Den Born et al. 2020; Noguera et al. 2021).
Supplementary Material
Acknowledgements
This work was carried out as a collaborative project between the Belgian Laboratory of Virology, Danish Veterinary Clinical Microbiology, and Boehringer Ingelheim. Carine Boone is acknowledged for her excellent technical work.
Contributor Information
Nick Vereecke, Laboratory of Virology, Faculty of Veterinary Medicine, Ghent University, Merelbeke 9280, Belgium; PathoSense BV, Lier 2500, Belgium.
Lise Kirstine Kvisgaard, Veterinary Clinical Microbiology, Department of Veterinary and Animal Sciences, University of Copenhagen, Copenhagen 1050, Denmark.
Guy Baele, Department of Microbiology, Immunology and Transplantation, Rega Institute, KU Leuven, Leuven 3000, Belgium.
Carine Boone, Laboratory of Virology, Faculty of Veterinary Medicine, Ghent University, Merelbeke 9280, Belgium.
Marius Kunze, Boehringer Ingelheim Vetmedica GmbH, Binger Str. 173, Ingelheim am Rhein 55216, Germany.
Lars Erik Larsen, Veterinary Clinical Microbiology, Department of Veterinary and Animal Sciences, University of Copenhagen, Copenhagen 1050, Denmark.
Sebastiaan Theuns, PathoSense BV, Lier 2500, Belgium.
Hans Nauwynck, Laboratory of Virology, Faculty of Veterinary Medicine, Ghent University, Merelbeke 9280, Belgium.
Supplementary data
Supplementary data are available at Virus Evolution online.
Funding
Flemish Agency for Innovation and Entrepreneurship (Baekeland Mandate HBC.2020.2889) to N.V.; Internal Funds KU Leuven (Grant No. C14/18/094) to G.B.; Research Foundation - Flanders (‘Fonds voor Wetenschappelijk Onderzoek - Vlaanderen’ G0E1420N, G098321N) to G.B.
Conflict of interest:
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication. S.T. and H.N. are co-founders and co-owners of PathoSense..
Data availability
All data are available in figures and supplementary files.
References
- Baele G. et al. (2020) ‘Hamiltonian Monte Carlo Sampling to Estimate Past Population Dynamics Using the Skygrid Coalescent Model in a Bayesian Phylogenetics Framework’, Wellcome Open Research, 5: 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baele G., Lemey P., and Suchard M. A. (2016) ‘Genealogical Working Distributions for Bayesian Model Testing with Phylogenetic Uncertainty’, Systematic Biology, 65: 250–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergeron J., Hébert B., and Tijssen P. (1996) ‘Genome Organization of the Kresse Strain of Porcine Parvovirus: Identification of the Allotropic Determinant and Comparison with Those of NADL-2 and Field Isolates’, Journal of Virology, 70: 2508–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bletsa M., et al. (2019) ‘Divergence dating using mixed effects clock modelling: An application to HIV-1’, Virus Evol., 5: 1–11.doi: 10.1093/ve/vez036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boisvert M., Fernandes S., and Tijssen P. (2010) ‘Multiple Pathways Involved in Porcine Parvovirus Cellular Entry and Trafficking toward the Nucleus’, Journal of Virology, 84: 7782–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokma J. et al. (2021) ‘Evaluation of Nanopore Sequencing as a Diagnostic Tool for the Rapid Identification of Mycoplasma bovis from Individual and Pooled Respiratory Tract Samples’, Journal of Clinical Microbiology, 59: e0111021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bøtner A., Nielsen J., and Bille-Hansen V. (1994) ‘Isolation of Porcine Reproductive and Respiratory Syndrome (PRRS) Virus in a Danish Swine Herd and Experimental Infection of Pregnant Gilts with the Virus’, Veterinary Microbiology, 40: 351–60. [DOI] [PubMed] [Google Scholar]
- Cadar D. et al. (2012) ‘Phylogeny and Evolutionary Genetics of Porcine Parvovirus in Wild Boars’, Infection, Genetics and Evolution, 12: 1163–71. [DOI] [PubMed] [Google Scholar]
- Chernomor O., Von Haeseler A., and Minh B. Q. (2016) ‘Terrace Aware Data Structure for Phylogenomic Inference from Supermatrices’, Systematic Biology, 65: 997–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotmore S. F., and Tattersall P. (2007) ‘Parvoviral Host Range and Cell Entry Mechanisms’, Advances in Virus Research, 70: 183–232. [DOI] [PubMed] [Google Scholar]
- De Maio N. et al. (2019) ‘Comparison of Long-read Sequencing Technologies in the Hybrid Assembly of Complex Bacterial Genomes’, Microbial Genomics, 5: 1–22.doi: 10.1099/mgen.0.000294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Souza A. R. et al. (2019) ‘Porcine Parvovirus VP1/VP2 on a Time Series Epitope Mapping: Exploring the Effects of High Hydrostatic Pressure on the Immune Recognition of Antigens’, Virology Journal, 16: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didelot X., Siveroni I., and Volz E.M. (2021) ‘Additive Uncorrelated Relaxed Clock Models for the Dating of Genomic Epidemiology Phylogenies’, Molecular Biology and Evolution, 38: 307–17.doi: 10.1093/molbev/msaa193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchene S. et al. (2020) ‘Bayesian Evaluation of Temporal Signal in Measurably Evolving Populations’, Molecular Biology and Evolution, 37: 3363–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes S., Boisvert M., and Tijssen P. (2011) ‘Genetic Elements in the VP Region of Porcine Parvovirus are Critical to Replication Efficiency in Cell Culture’, Journal of Virology, 85: 3025–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Morante B. et al. (2020) ‘Duration of Immunity against Heterologous Porcine Parvovirus 1 Challenge in Gilts Immunized with a Novel Subunit Vaccine Based on the Viral Protein 2’, BMC Veterinary Research, 16: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2019) ‘Field Evaluation of the Safety and Compatibility of a Combined Vaccine against Porcine Parvovirus 1 and Porcine Reproductive and Respiratory Syndrome Virus in Breeding Animals’, Porcine Health Management, 5: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasper D. J. et al. (2016) ‘Effective Respiratory CD8 T-Cell Immunity to Influenza Virus Induced by Intranasal Carbomer-Lecithin-Adjuvanted Non-replicating Vaccines’, PLOS Pathogens, 12: 1–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill M. S. et al. (2016) ‘Understanding past Population Dynamics: Bayesian Coalescent-Based Modeling with Covariates’, Systematic Biology, 65: 1041–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2013) ‘Improving Bayesian Population Dynamics Inference: A Coalescent-based Model for Multiple Loci’, Molecular Biology and Evolution, 30: 713–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W. et al. (2013) ‘RNA-guided Editing of Bacterial Genomes Using CRISPR-Cas Systems’, Nature Biotechnology, 31: 233–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo H. S., Donaldson-Wood C. R., and Johnson R. H. (1976) ‘A Standardised Haemagglutination Inhibition Test for Porcine Parvovirus Antibody’, Australian Veterinary Journal, 52: 422–4. [DOI] [PubMed] [Google Scholar]
- Joo H. S., and Johnson R. H. (1977) ‘Serological Responses in Pigs Vaccinated with Inactivated Porcine Parvovirus’, Australian Veterinary Journal, 53: 550–2. [DOI] [PubMed] [Google Scholar]
- Jóźwik A. et al. (2009) ‘Vaccination against Porcine Parvovirus Protects against Disease, but Does Not Prevent Infection and Virus Shedding after Challenge Infection with a Heterologous Virus Strain’, Journal of General Virology, 90: 2437–41. [DOI] [PubMed] [Google Scholar]
- Kamstrup S. et al. (1998) ‘Mapping the Antigenic Structure of Porcine Parvovirus at the Level of Peptides’, Virus Research, 53: 163–73. [DOI] [PubMed] [Google Scholar]
- Karniychuk U. U. et al. (2011) ‘Porcine Reproductive and Respiratory Syndrome Virus (PRRSV) Causes Apoptosis during Its Replication in Fetal Implantation Sites’, Microbial Pathogenesis, 51: 194–202. [DOI] [PubMed] [Google Scholar]
- Katoh K., and Standley D. M. (2013) ‘MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability’, Molecular Biology and Evolution, 30: 772–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss I. et al. (2020) ‘Vaccine Protection against Experimental Challenge Infection with a PPV-27a Genotype Virus in Pregnant Gilts’, Veterinary Medicine: Research and Reports, 11: 17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren S. et al. (2017) ‘Canu: Scalable and Accurate Long-read Assembly via Adaptive K -mer Weighting and Repeat Separation’, Genome Research, 27: 722–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labarque G. G. et al. (2000) ‘Effect of Cellular Changes and Onset of Humoral Immunity on the Replication of Porcine Reproductive and Respiratory Syndrome Virus in the Lungs of Pigs’, The Journal of General Virology, 81: 1327–34. [DOI] [PubMed] [Google Scholar]
- Markussen N. H., and Have P. (1992) ‘Phocine Distemper Virus Infection in Harp Seals (Phoca groenlandica)’, Marine Mammal Science, 8: 19–26. [Google Scholar]
- Martin D. P. et al. (2015) ‘RDP4: Detection and Analysis of Recombination Patterns in Virus Genomes’, Virus Evolution, 1: 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Lobo F. J. et al. (2011) ‘Porcine Reproductive and Respiratory Syndrome Virus Isolates Differ in Their Susceptibility to Neutralization’, Vaccine, 29: 6928–40. [DOI] [PubMed] [Google Scholar]
- Matthijnssens J. et al. (2008) ‘Full Genome-Based Classification of Rotaviruses Reveals a Common Origin between Human Wa-Like and Porcine Rotavirus Strains and Human DS-1-Like and Bovine Rotavirus Strains’, Journal of Virology, 82: 3204–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr A. and Mahnel H. (1964) ‘Cultivation of Hog Cholera Virus in Pig Kidney Cultures with Cytopathogenic Effect’, Zentralblatt Fur Bakteriologie, Parasitenkunde, Infektionskrankheiten Und Hygiene. 1. Abt. Medizinisch-hygienische Bakteriologie, Virusforschung Und Parasitologie. Originale, 195: 66. [PubMed] [Google Scholar]
- Mengeling W. L. et al. (1979) ‘Efficacy of an Inactivated Virus Vaccine for Prevention of Porcine Parvovirus-induced Reproductive Failure’, American Journal of Veterinary Research, 40: 204–7. [PubMed] [Google Scholar]
- Mengeling W. L., Lager K. M., and Vorwald A. C. (2000) ‘The Effect of Porcine Parvovirus and Porcine Reproductive and Respiratory Syndrome Virus on Porcine Reproductive Performance’, Animal Reproduction Science, 60–61: 199–210. [DOI] [PubMed] [Google Scholar]
- Mészáros I. et al. (2017) ‘Biology of Porcine Parvovirus (Ungulate Parvovirus 1)’, Viruses, 9: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguera M. et al. (2021) ‘Effects of Three Commercial Vaccines against Porcine Parvovirus 1 in Pregnant Gilts’, Vaccine, 39: 3997–4005. [DOI] [PubMed] [Google Scholar]
- Oh W. T. et al. (2017) ‘Perspectives on the Evolution of Porcine Parvovirus’, Viruses, 9: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A. et al. (2018) ‘Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7’, Systematic Biology, 67: 901–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2016) ‘Exploring the Temporal Structure of Heterochronous Sequences Using TempEst (Formerly Path-O-Gen)’, Virus Evolution, 2: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren X. et al. (2013) ‘Phylogeny and Evolution of Porcine Parvovirus’, Virus Research, 178: 392–7. [DOI] [PubMed] [Google Scholar]
- Sabir J. S. M. et al. (2016) ‘Co-circulation of Three Camel Coronavirus Species and Recombination of MERS-CoVs in Saudi Arabia’, Science (80-.), 351: 81–4. [DOI] [PubMed] [Google Scholar]
- Simpson A. A. et al. (2002) ‘The Structure of Porcine Parvovirus: Comparison with Related Viruses’, Journal of Molecular Biology, 315: 1189–98. [DOI] [PubMed] [Google Scholar]
- Streck A. et al. (2011) ‘High Rate of Viral Evolution in the Capsid Protein of Porcine Parvovirus’, Journal of General Virology, 92: 2628–36. [DOI] [PubMed] [Google Scholar]
- Streck A. F., Canal C. W., and Truyen U. (2015) ‘Molecular Epidemiology Evolution of Porcine Parvoviruses’, Infection, Genetics and Evolution, 36: 300–6. [DOI] [PubMed] [Google Scholar]
- ——— (2022) ‘Viral Fitness and Antigenic Determinants of Porcine Parvovirus at the Amino Acid Level of the Capsid Protein’, Journal of Virology, 96: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streck A. F. et al. (2013) ‘Population Dynamics and in Vitro Antibody Pressure of Porcine Parvovirus Indicate a Decrease in Variability’, Journal of General Virology, 94: 2050–5. [DOI] [PubMed] [Google Scholar]
- Streck A. F., and Truyen U. (2020) ‘Porcine Parvovirus’, Current Issues in Molecular Biology, 37: 33–45. [DOI] [PubMed] [Google Scholar]
- Suchard M. A. et al. (2018) ‘Bayesian Phylogenetic and Phylodynamic Data Integration Using BEAST 1.10’, Virus Evolution, 4: 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H., and Fujisaki Y. (1976) ‘Immunizing Effect of Inactivated Porcine Parvovirus Vaccine on Piglets’, National Institute of Animal Health Quarterly, 16. [Google Scholar]
- Tamura K., Stecher G., and Kumar S. (2021) ‘MEGA11: Molecular Evolutionary Genetics Analysis Version 11’, Molecular Biology and Evolution, 38: 3022–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theuns S. et al. (2018) ‘Nanopore Sequencing as a Revolutionary Diagnostic Tool for Porcine Viral Enteric Disease Complexes Identifies Porcine Kobuvirus as an Important Enteric Virus’, Scientific Reports, 8: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsao J. et al. (1991) ‘The Three-dimensional Structure of Canine Parvovirus and Its Functional Implications’, Science (80-.), 251: 1456–64. [DOI] [PubMed] [Google Scholar]
- Tu M. et al. (2015) ‘Role of Capsid Proteins in Parvoviruses Infection’, Virology Journal, 12: 114–21.doi: 10.1186/s12985-015-0344-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tullis G. E., Burger L. R., and Pintel D. J. (1993) ‘The Minor Capsid Protein VP1 of the Autonomous Parvovirus Minute Virus of Mice Is Dispensable for Encapsidation of Progeny Single-stranded DNA but Is Required for Infectivity’, Journal of Virology, 67: 131–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Den Born E. et al. (2020) ‘An Octavalent Vaccine Provides Pregnant Gilts Protection against a Highly Virulent Porcine Parvovirus Strain’, BMC Veterinary Research, 16: 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannier P. et al. (1986) ‘Study of the Efficacy of an Inactivated Virus Vaccine against Porcine Parvovirus’, Annales de Recherches Veterinaires, 17: 425–32. [PubMed] [Google Scholar]
- Wilhelm S. et al. (2005) ‘Tissue Distribution of Two Field Isolates and Two Vaccine Strains of Porcine Parvovirus in Foetal Organs after Experimental Infection of Pregnant Sows as Determined by Real-time PCR’, Journal of Veterinary Medicine Series B, 52: 323–6. [DOI] [PubMed] [Google Scholar]
- Zeeuw E. J. L. et al. (2007) ‘Study of the Virulence and Cross-neutralization Capability of Recent Porcine Parvovirus Field Isolates and Vaccine Viruses in Experimentally Infected Pregnant Gilts’, Journal of General Virology, 88: 420–7. [DOI] [PubMed] [Google Scholar]
- Zhang J. et al. (2018) ‘Evaluation of Carbopol as an Adjuvant on the Effectiveness of Progressive Atrophic Rhinitis Vaccine’, Vaccine, 36: 4477–84. [DOI] [PubMed] [Google Scholar]
- Zimmermann P. et al. (2006) ‘VP1 Sequences of German Porcine Parvovirus Isolates Define Two Genetic Lineages’, Journal of General Virology, 87: 295–301. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available in figures and supplementary files.