

Abstract
An effective procedure for specific determination of the cap structure at the 5′-terminus of mRNA and for isolation of the corresponding full-length cDNA has been developed. The procedure involves covalent attachment of an oligonucleotide template extender to the 5′-cap structure of mRNA followed by RT–PCR using M-MLV SuperScript II reverse transcriptase. In the course of reverse transcription, the enzyme ‘jumps over’ the cap structure and includes the sequence complementary to the oligonucleotide template extender into the 3′-end of the first cDNA strand. The cap-jumping method was successfully tested using some mammalian cellular mRNAs, genomic RNAs of tobacco mosaic virus (TMV) U1 and the recently isolated crucifer-infecting tobamovirus. Moreover, cDNA products corresponding to the genomic tobamovirus RNA were obtained from total RNA extracted from tobacco plants infected by crucifer-infecting tobamovirus or tobacco mosaic virus. Using the cap-jumping method, we have shown for the first time that genomic crucifer-infecting tobamovirus (crTMV) RNA contains a 5′-cap structure. This improved method can be recommended for the construction of full-length and 5′-end enriched cDNA libraries, identification of capped RNAs and determination of their 5′-terminal sequences.
INTRODUCTION
The generation of full-length cDNAs from mRNA is a major challenge in biotechnology research. Even with the whole genome sequencing initiatives reaching fruition there is still a great need for reliable methods to construct cDNA libraries. In particular, there is a need to identify 5′-untranslated regions (UTRs), novel regulatory motifs, internal ribosome entry sites, etc. Several protocols have been proposed to construct full-length cDNA libraries, however, most of them are not satisfactory in terms of efficiency and bias in the representation of the cDNAs. In general the 5′-ends of genes are under-represented, especially if a poly(dT) primer is used in first strand cDNA synthesis and RNase H is required for second strand cDNA synthesis.
The majority of eukaryotic cellular and viral messenger RNAs (mRNAs) are blocked at the 5′-end by a cap structure, m7G(5′)ppp(5′)Np, which has important effects on the maturation, translation and stability of mRNA. The cap structure consists of an inverted 7-methylguanosine linked via a 5′–5′ triphosphate bridge to the first transcribed residue, and its presence is indicative of the integrity of the proximal 5′-UTR preceding a mRNA coding region (1,2). Traditionally, for most eukaryotic mRNAs the ribosome scanning model of translation initiation proposes that the 40S ribosomal subunit binds the 5′-cap and moves along the 5′-UTR until it reaches an AUG codon (3). Consequently, the majority of structurally polycistronic RNAs of eukaryotic viruses are functionally monocistronic, i.e. only the first open reading frame is translationally active. The downstream genes of viral genomes are commonly expressed by deployment of so-called subgenomic RNAs (sgRNAs) which are 3′-co-terminal with genomic RNA, but lack one or more 5′-proximal genes (4). The 5′-proximal genes of many viral genomic mRNAs, including the RNA of tobamoviruses, encode a virus-specific RNA-dependent RNA polymerase essential for viral RNA synthesis and capping in the cytoplasm of infected cells. It has been shown that the genomic RNA of tobacco mosaic virus (TMV) and many eukaryotic viruses are capped (5,6), whereas RNAs of some other viruses are uncapped or their 5′-terminus can be blocked in different ways (7).
Many viral mRNAs lack a 5′-terminal cap or a poly(A) tail, or both. For example, RNA of viruses belonging to the Piconaviridae family lack a cap structure, but contain a 5′-terminal viral protein linked by a phosphodiester bond to the extremely long, highly structured 5′-UTR (7). Thus, it is important in virus research to know the type of 5′-terminal structure of viral RNA because it reflects different aspects of the viral life cycle.
As the presence of a cap structure at the 5′-end of eukaryotic mRNAs is a good marker for the 5′-terminal part of the coding region, several methods for isolation of full-length mRNAs based on the use of this structure have been developed to improve the construction of cDNA libraries with more full-length sequences of genes (8–14). Some of these protocols include affinity capture of mRNA using a cap-binding protein coupled to a solid support (8), replacing the cap structure with a synthetic oligonucleotide, which is ligated to the mRNA by RNA ligase (9,10), and the chemical addition of a biotin group to the diol residue of the cap structure (11). Another related procedure is the CapFinder technique (12,13), which relies on the terminal transferase and the template switching activity of SuperScript II reverse transcriptase to generate a specific anchor sequence complementary to a template switching oligonucleotide. The CapSelect technique, which was introduced by Schmidt and Mueller (14), avoids the template switching requirement and exploits the extraordinarily high terminal transferase activity of SuperScript II reverse transcriptase with capped mRNAs in the presence of manganese, followed by controlled ribonucleotide tailing of cDNA ends using terminal deoxynucleotidyl transferase and rATP. A double-stranded DNA adapter is then ligated to the known cDNA end. However, these techniques for enrichment of full-length cDNA libraries are time consuming, involve numerous enzymatic steps and yield variable results.
One of the limiting factors in the classical rapid amplification of 5′-cDNA ends is getting the reverse transcriptase to copy the mRNA of interest completely into the first strand cDNA. Prematurely terminated first strand cDNAs are also tailed by terminal transferase, resulting in large populations of truncated cDNAs. To overcome this, the attachment of an anchor primer to the 5′-end of the mRNA, which is incorporated into the first strand cDNA only when reverse transcription proceeds through the entire length of the mRNA of interest, has been proposed (15–17). Before such a linker can be anchored, the mRNA is dephosphorylated using calf intestinal phosphatase, which acts only on degraded mRNA and leaves the capped, full-length mRNAs unaffected (15). Degraded mRNA is then unable to participate in subsequent ligation reactions. The full-length mRNAs are decapped using tobacco acid pyrophosphatase, which leaves them with a phosphorylated 5′-terminus (16,17). A synthetic oligoribonucleotide is then ligated to the mRNA using T4 RNA ligase. The oligo–mRNA hybrids are reverse transcribed using a gene-specific primer to create first strand cDNA. However, the multiple steps and enzymatic reactions required in this system make it a technically difficult procedure. Problems arise when dealing with mRNA populations and rare transcripts due to the inefficiency of the multiple enzymatic steps.
Earlier, Carninci et. al. (11) reported a chemical method for specific biotinylation of the cap structure either before or after cDNA synthesis by chemical coupling to the cis-diol groups of the cap structure and at the 3′-end of mRNA. Upon RNase I digestion only mRNA–cDNA hybrids are protected, which allows the 3′-biotin to be eliminated and any non-full-length cDNAs to be excluded from capture by streptavidin-coated magnetic beads. Recently, a method for 5′-capped mRNA isolation has been proposed, which combines the advantages of having an anchor oligonucleotide at the 5′-end of mRNA with the efficiency of a chemical ligation approach (18). The method includes specific coupling of a 3′-amino oligonucleotide tag (particularly the 3′-phosphorohydrazide of a deoxyribooligonucleotide) to the oxidized 5′-terminal 3′,5′-diol group of the capped mRNA with preliminary modification of a diol group at the 3′-end of the mRNA by ligation of a nucleoside 3′,5′-diphosphate in the presence of T4 RNA ligase. The tagged mRNA was used in reverse transcription to obtain single-stranded cDNA followed by cDNA amplification by PCR using a set of two primers. One of the primers represents an oligodeoxyribonucleotide with a sequence corresponding to the ligated tag, while the other primer has a specific sequence complementary to the 3′-region of the tested mRNA.
In this paper, we describe an improved procedure for the construction of full-length and 5′-enriched cDNA libraries as well as for specific detection of the mRNA 5′-cap structure in RNAs. This ‘cap-jumping’ method was tested using some mammalian cellular mRNAs, full-length genomic RNAs isolated from purified preparations of TMV (strain U1), which is a typical member of the Tobamovirus genus, and the recently characterized crucifer-infecting tobamovirus (crTMV) (19). Also, total RNA preparations isolated from HeLa cells and crTMV-infected tobacco plants were used for cap detection on selected cellular and viral RNAs.
MATERIALS AND METHODS
Pure capped rabbit α- and β-globin mRNA transcripts (∼650 bp) and SuperScript II reverse transcriptase were purchased from Gibco BRL, M-MLV reverse transcriptase was purchased from Fermentas and pCR2.1 TOPO vector was purchased from Invitrogen. Escherichia coli strains DH5α and SURE were used for cloning of the recombinant constructs. Oligonucleotides were either purchased from Dharmacon Research or synthesized using an ABI 381A synthesizer. The sequences of the PCR primers are shown in Table 1. Plasmid TMV304 with full-length cDNA of the TMV (U1) genome under control of the SP6 promoter was kindly provided by Dr W. O. Dawson (University of Florida, Lake Alfred, FL). A full-length genomic cDNA clone of crTMV was obtained by Dr P. A. Ivanov (Moscow State University, Moscow, Russia; unpublished results). Inoculation of Nicotiana tabacum cv. Samsun plants with viruses, virus isolation and RNA extraction from virus particles were performed as described by Dorokhov et al. (19). Total RNA preparations from crTMV- or TMV-infected tobacco was obtained by the method of phenol deproteinization as described (20,21). The GeneBee program package (22) was used for RNA secondary structure prediction. Sequences of cDNAs were obtained using an ABI Prism 377. Electrophoresis of PCR products was performed in 2% agarose gels with staining with ethidium bromide.
Table 1. Sequences of oligodeoxyribonucleotide primers used in PCR experiments.
| Primer | Sequence | Primer position on template mRNA | |
|---|---|---|---|
| (5′) | (3′) | ||
| Tag-1 | GCCAGTTTAAACGAT | ||
| Tag-2 | CUAAUACGACUCACUAUAGGG | ||
| Ω | ATATGAATTCGTATTTTTACAACAATTACC | TMV (1–20) | |
| U1-Sph | ATATCCATGGTCTCGAACGTCCAGGTTGGGC | TMV (449–461) | |
| K5 | ATAAGAATTCGTTTTAGTTTTATTGCAACAA | crTMV (1–21) | |
| 2PM | CTCAAGCGATCGAAAGCCA | crTMV (323–339) | |
| α-R-5′ | GGTCCAGTCCGACTGAG | α-Globin (9–25) | |
| α-R-3′ | CTCACTCAGACTTTATTC | α-Globin (530–547) | |
| β-R-5′ | CTTTTGACACAACTGTG | β-Globin (8–24) | |
| β-R-3′ | CCAGAAGTCAGATGCTC | β-Globin (546–563) | |
| G-5′ | TTAGCACCCCTGGCCAAGG | GAPDH (529–547) | |
| G-3′ | CTTACTCCTTGGAGGCCATG | GAPDH (1050–1069) |
Synthesis of the 3′-modified oligonucleotide template extender (OTE)
A deoxyribooligonucleotide containing a 3′-ribo unit or ribooligonucleotide (0.1 µmol) was dissolved in 0.5 ml of water and its 3′-terminal cis-glycol group oxidized with 0.1 M NaIO4 (100 µl) for 15 min at room temperature to generate a dialdehyde unit (23,24). The excess NaIO4 was reduced with 100 µl of 0.2 M sodium hypophosphite for 20 min.
Sodium acetate (0.5 M, pH 4.0) was added to the modified OTE obtained as described above to 50 mM final concentration, followed by 300 µl of 20 mM ethylenediamine (or 1,4-phenylenediamine) dihydrochloride. The mixture was incubated with 150 µl of 0.2 M NaCNBH3 in acetonitrile for 1 h at 20°C. Then, the reaction mixture was diluted with water to a volume of 1.5 ml and the amino-modified oligonucleotide was isolated by gel filtration on a Pharmacia NAP-25 column.
Ligation of the OTE to the cap structure
RNA (1–2 µl, 1 µg/µl) was mixed with 14 µl of water and 1 µl of 1 M sodium acetate (pH 5.5). Then, 4 µl of a 0.1 M solution of sodium periodate was added. The reaction mixture was allowed to stand for 1 h and terminated by addition of 0.1 M sodium hypophosphite (5 µl) for 10 min.
Water (23 µl) and 1 M sodium acetate buffer pH 4.5 (1 µl) were added to the 5′,3′-oxidized mRNA and it was treated with 20 µl of 20 mM ethylenediamine dihydrochloride for 15 min at room temperature. Then, 0.2 M sodium cyanoborohydride (10 µl) was added and the reaction mixture was allowed to stand for 2 h. The nucleotide material was precipitated with 2% lithium perchlorate solution in acetone (400 µl) at –20°C for 15 min and isolated by centrifugation at 10 000 g for 5 min. The pellet was washed with acetone (2 × 500 µl) and dissolved in 50 µl of 0.02 M sodium acetate buffer, pH 4.5. A 3′-dialdehyde modified oligonucleotide (2 µl, 30 pmol/µl) was added to the solution of aminated mRNA and, after incubation for 15 min at room temperature, 0.2 M sodium cyanoborohydride (10 µl) was added. The reaction mixture was allowed to stand for 2 h. The nucleic acid material was precipitated with 2% lithium perchlorate solution in acetone (400 µl) at –20°C for 15 min and isolated by centrifugation at 10 000 g for 5 min. The pellet was washed with acetone (2 × 500 µl) and dissolved in 30 µl of 0.4 M NaCl. Then, 10% aqueous cetyltrimethylammonium bromide (CTAB) (3 µl) was added to a final concentration of 1% and the reaction mixture was incubated for 15 min at room temperature. The pellet was isolated by centrifugation and resuspended in 35 µl of 0.4 M NaCl, 1% CTAB. After keeping at room temperature for 15 min and centrifugation for 5 min, the pellet of tagged RNA obtained was dissolved in 1.2 M NaCl (30 µl). Then, it was precipitated with 80 µl of ethanol at –20°C for 16 h. The pellet was isolated by centrifugation at 10 000 g for 5 min and washed with 500 µl of 70% ethanol. The tagged RNA was dissolved in 25 µl of water.
Water (23 µl), 1 M sodium acetate buffer pH 4.5 (1 µl) and the 3′-aminated OTE (2 µl, 30 pmol/µl) were added to the solution of 5′,3′-oxidized mRNA obtained as described above and the reaction mixture incubated for 15 min at room temperature. Then, 0.2 M sodium cyanoborohydride (10 µl) was added and the reaction mixture was allowed to stand for 2 h. The nucleic acid material was precipitated with 2% lithium perchlorate solution in acetone (400 µl) at –20°C for 15 min and isolated by centrifugation at 10 000 g for 5 min. The pellet was washed with acetone (2 × 500 µl) and dissolved in 30 µl of 0.4 M NaCl. The following operations were performed as above.
Reverse transcription and PCR procedures in solution
Tagged RNA (12 µl, 0.5 µg) and a specific 3′-end primer (1 µl of a 25 µM solution) were mixed with 4 µl of a buffer containing 250 mM Tris–HCl, pH 8.3, 375 mM KCl and 15 mM MgCl2 (standard conditions) or 2 µl of the same buffer (low salt conditions) and heated at 95°C for 30 s, followed by cooling on ice. Then, DTT was added to a final concentration of 1 mM together with 2 µl of dNTPs (10 mM) and 0.5 µl of RNasin (Life Technologies). After incubation with 0.5 µl (100 U) of SuperScript II reverse transcriptase for 1 h at 42°C, 40 mM MnCl2 (1 µl) was added and the reaction mixture was incubated for an additional 15 min at 42°C. Then, cDNA was amplified using Taq DNA polymerase (1 µl, 5 U; Sigma) and two sets of the specific primers: 3′ + 5′ or 3′ + Tag (1 µl of a 25 µM solution) (Table 1). PCR was performed using a MiniCycler (MJ Research) and consisted of 30 cycles of denaturation at 94°C for 1 min, annealing at 50°C for 1 min and extension at 72°C for 1 min.
Solid phase protocol
MGSA4 streptavidin-coated magnetic beads (3 µl) (with a binding capacity of 10 pmol biotinylated oligonucleotide/µl; Seradyne) were washed twice with 30 µl of 250 mM NaCl, 10 mM Tris–HCl, pH 7.5, and suspended in 10 µl of the same buffer. Then, 500 mM NaCl (10 µl) and a solution of the 5′-biotinylated tagged mRNA (10 µl) were added and the mixture was incubated for 30 min at room temperature with mild agitation. The magnetic beads with tagged mRNA were isolated from the mixture by sedimentation on a magnetic stand, washed with 3 × 30 µl of 250 mM NaCl, 10 mM Tris–HCl, pH 7.5, and resuspended in 22 µl of diethylpyrocarbonate (0.1%) treated water. Magnetic beads with tagged mRNA (11 µl) were mixed with oligo(dT) or gene-specific primer (1 µl, 10 pmol) and incubated for 10 min at 70°C. Then, 5× RT buffer (4 µl), RNasin (1 µl), DTT (2 µl) and dNTPs (25 mM, 0.5 µl; Sigma) were added. Reverse transcription was carried out in the presence of SuperScript II enzyme (200 U, 1 µl) for 50 min at 42°C, followed by incubation at 70°C for 10 min to inactivate the enzyme. Amplification of cDNA by RT–PCR was performed using magnetic beads (2 µl) from the reverse transcription experiment in 50 µl of the incubation buffer containing a set of primers: specific 3′ + 5′ primers or 3′ + Tag primers (10 pmol each) (Table 1), dNTPs (0.5 µl, 25 mM) and Taq DNA polymerase (3 U). PCR consisted of 30 cycles of denaturation at 94°C for 0.5 min, annealing at 50°C for 0.5 min and extension at 72°C for 1.5 min. Magnetic beads were removed from the cDNA solution by sedimentation on a magnetic stand.
RESULTS AND DISCUSSION
General outline of the cap-jumping method
Both the 5′- and 3′-ends of full-length mature eukaryotic RNAs contain the same ribonucleotide cis-glycol groups, whereas non-capped mRNAs, for example 5′-deleted mRNAs, have only one cis-glycol group at the 3′-end and a hydroxyl or phosphate (diphosphate or triphosphate) group at the 5′-terminus. This unique structure difference between capped and non-capped mRNAs can be used for discrimination of the two species.
The first step in the cap-jumping method is ligation of a synthetic oligonucleotide to the ends of mRNA using chemical reactions characteristic of the 2′,3′-cis-glycol ribonucleoside group (Fig. 1). Sodium periodate oxidation of the 5′- and 3′-terminal 2′,3′-cis-glycol groups of a capped mRNA generates reactive dialdehyde functions at both mRNA ends. We used two possible ways for addition of an oligonucleotide to the terminal dialdehyde functions of mRNA. In the first variant, the procedure includes generation of amino functions at the ends of mRNA by reductive amination of the dialdehyde groups in the presence of an excess of an aliphatic or aromatic diamine hydrochloride, followed by addition of a previously prepared OTE bearing a 3′-dialdehyde group (Fig. 1, path A). In an alternative procedure, the OTE containing an amino function at the 3′-end is attached to the terminal dialdehyde groups of the modified mRNA using reductive amination in the presence of sodium cyanoborohydride (23,24; Fig. 1, path B). The OTE represents a 3′-modified oligoribonucleotide. As a variant, the OTE can be constructed from deoxyribonucleotides and contain a 3′-terminal ribonucleoside unit.
Figure 1.

Scheme for covalent binding of the OTE to the 5′-terminus of a capped mRNA.
After oligonucleotide attachment to the mRNA, the excess OTE is removed from the RNA by size exclusion chromatography or by precipitation with CTAB in the presence of 0.3 M NaCl. CTAB is a strong cationic detergent that binds nucleic acids to form an insoluble complex. This complex formation is influenced by salt concentration. When the salt concentration is >1 M no complex formation occurs, while below 0.2 M all nucleic acids are efficiently complexed. Between 0.3 and 0.4 M incorporation of small single-stranded nucleic acids into the complex is very inefficient (25,26).
The OTE–mRNA substrate created by chemical ligation is suitable for first strand cDNA synthesis by reverse transcription using a 3′-specific or oligo(dT) primer. A reverse transcriptase ‘reads’ through the cap structure and, using the attached OTE as a template, continues extension to the end. As a result, the sequence complementary to the OTE sequence becomes attached to the 3′-terminus of the first strand cDNA (Fig. 2). Consequently, the first cDNA strand is tagged with a known sequence at its 3′-end, which can subsequently be used to prime second strand synthesis. For the complement of the OTE to be included in the first strand of cDNA it requires a reverse transcriptase which is able to overcome (‘jump over’) the cap structure and continue with first strand cDNA synthesis. It is known that some reverse transcriptases, particularly AMV, HIV and M-MLV, have a terminal transferase activity and are able to add from one to several non-template nucleotides to the 3′-end of a newly synthesized cDNA strand upon reaching the 5′-end of the mRNA template (27,28). These non-template nucleotides could serve as a ‘bridge’ over the cap structure. In the experiments on reverse transcription described in this paper we used M-MLV SuperScript II reverse transcriptase. As shown earlier, this enzyme exhibits high terminal transferase activity (typically it adds 3–4 nt) in the presence of 5′-capped mRNA templates and manganese cations (13), as well as having the ability to switch to a second template (28). We also tested a wild-type M-MLV reverse transcriptase and found that it was able to overcome the cap structure and to reverse transcribe the OTE-I sequence, whereas the HIV and AMV reverse transcriptases did not show positive results in our hands (data not shown).
Figure 2.

Scheme of the cap-jumping approach to full-length double-stranded cDNA synthesis.
Second strand synthesis is achieved using an oligodeoxyribonucleotide primer with a sequence identical to that of the OTE. Full-length cDNA is amplified by PCR using specific primers to the extreme 3′- and 5′-ends. However, to avoid any bias that could be introduced by PCR, it is possible to use restriction enzyme sites encoded by the first and second strand primers to generate overhangs for cloning into a suitable vector.
As a variant, we developed a simple solid phase procedure, which includes ligation of the mRNA to a 5′-biotinylated OTE followed by capture of the biotinylated tagged mRNA on streptavidin-coated magnetic beads. After removal of all non-bound material, the reverse transcription and PCR steps were performed on the solid phase bound mRNA and amplified double-stranded DNA fragments accumulated in solution. After completion of the process, the magnetic beads were separated from the solution of target DNA fragments.
In contrast to the procedure described by Merenkova et al. (18), our method uses two possible routes for OTE attachment (Fig. 1), one of which provides the 5′,3′-aminated mRNA, which should be more stable than the 5′,3′-dialdehyde form. Then, we exclude the deactivation of 3′-end mRNA cis-glycol groups step. Ligation of an OTE to the capped mRNA results in its attachment to both mRNA ends. However, the OTE is attached in the correct orientation only at the mRNA 5′-end, whereas at the 3′-end it is attached in the reverse orientation and cannot be used as a template for priming or serve as a primer itself. Therefore, the 3′-terminal OTE does not interfere with any subsequent downstream processes and does not need to be removed. Moreover, for the first time we have developed a simple solid phase modification of the cap-jumping method. It should be noted that all the above-mentioned features greatly simplify the procedure for detection of 5′-capped mRNAs.
Detection of the cap structure at the 5′-ends of cellular mRNAs
As one of our test systems we used purified capped rabbit α- and β-globin mRNA transcripts (∼650 bp) (29,30). Chemically reactive dialdehyde functional groups were generated at both the 5′-cap structure and the 3′-end of the corresponding mRNA (Fig. 1). A 5′-biotinylated oligoribonucleotide GCCAGUUUAAACGAU (OTE-I) was designed to incorporate a PmeI restriction enzyme cleavage site for downstream cloning purposes. It was 3′-modified to carry a dialdehyde (Fig. 1, path A) or aminoethyl (Fig. 1, path B) group as described (23,24) and then attached at the ends of the modified rabbit globin mRNA transcript using reductive amination. The excess OTE-I was removed from the tagged mRNA by size exclusion spin column chromatography. The effectiveness of mRNA modification was estimated in a set of control experiments with a 32P-labeled OTE as 70–80% for the first procedure and 50–70% for the second procedure (data not shown). For comparison, the yield of chemical ligation of the 3′-amino-modified oligonucleotide to mRNA according to previous results (18) was 30–80%. The tagged mRNA was captured on streptavidin-coated magnetic beads and RT–PCR was performed on the bound mRNA using SuperScript II reverse transcriptase and an oligo(dT) primer, followed by PCR with a combination of gene-specific primers to rabbit α- and β-globin and the OTE-I specific primer (Tag-1) (Table 1 and Fig. 3A and B). The amplified fragments were purified by gel electrophoresis (Fig. 3A, lanes 1a and b and 2a and b), cloned into cloning vector pCR2.1 TOPO and for each experiment a set of clones containing the PCR fragments from the same reaction were sequenced. The results obtained (Fig. 4) clearly show that the OTE-I sequence was successfully incorporated into the extreme 5′-end of the sequences of rabbit α- and β-globin mRNA, demonstrating proof of the cap-jumping approach.
Figure 3.
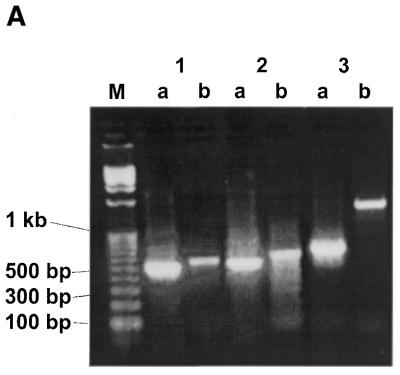

Analysis of the ‘cap jumping’ ability of SuperScript II reverse transcriptase. (A) Agarose gel electrophoresis of PCR products obtained for rabbit α-globin mRNA (1), rabbit β-globin mRNA (2) and GAPDH mRNA (3) with the use of the 5′-biotinylated 15mer OTE-I GCCAGUUUAAACGAU and a solid phase protocol. The appearance of a PCR band with the specific 5′ + 3′ primer sets (1a, 2a and 3a) indicates the presence of tested RNA in the reaction mixture. The appearance of the respective PCR band with the Tag-1 + 3′ (1b, 2b and 3b) primer sets indicates the presence of a cap structure in the RNA. (B) General scheme of these experiments.
Figure 4.

Sequences of the PCR fragments obtained in ‘cap-jumping’ experiments. Nucleotide insertions at the junction of a tag primer with a region that corresponds to the published 5′-mRNA sequence are underlined. For each mRNA, the predominant sequences of nucleotide insertions, which were derived from the analysis of several independent clones, are shown.
Similar experiments were carried out with a population of HeLa cell poly(A)+ mRNAs (31). The tagged HeLa mRNAs were also bound to streptavidin-coated beads and subjected to solid phase RT–PCR using primers specific for glyceraldehyde 3-phosphate dehydrogenase (GAPDH). In this case, the 5′- and 3′-GAPDH gene-specific primers amplified a segment internal to the gene with the 5′-primer annealing ∼600 bp from the published 5′-end of the gene (31). Because of this the Tag-1 + 3′-GAPDH primer pair amplified a band approximately double the size of the band amplified by the 5′- and 3′-GAPDH gene-specific primer pair (Fig. 3A, lanes 3a and b).
These results have demonstrated that SuperScript II reverse transcriptase is able to ‘jump over’ the cap structure together with the unnatural junction of the chemically ligated OTE and continue with first strand cDNA synthesis to the end of the OTE, with addition of one or several nucleotides at the junction point. The sequence procedure revealed that this non-template nucleotide addition is a rather random event. However, for each tested RNA the predominant inserted non-template sequences can be found (Fig. 4). It could be explained as a result of specific interaction of a reverse transcriptase with unique combinations of RNA and OTE sequences or, alternatively, by predominant PCR multiplication of some selected fragments.
Detection of the cap structure at the 5′-terminus of viral RNAs
To examine the effectiveness of the cap-jumping approach, the determination of capped and uncapped forms of genomic RNAs from two tobamoviruses was undertaken. The naturally capped genomic RNA was isolated from purified preparations of TMV U1, and a synthetic transcript of the full-length TMV U1 clone TMV304 cDNA (32) was used as its uncapped counterpart. Furthermore, genomic RNA isolated from purified preparations of another tobamovirus (crTMV) (19) was used to reveal the presence of a cap structure in the virion RNA of this virus. The uncapped synthetic transcript obtained from the full-length cDNA clone of crTMV was taken as a negative control. In these cases, the 3′-aminated 21mer OTE-II CUAAUACGACUCACUAUAGGG, which contains a sequence homologous to the T7 polymerase promoter, was used for chemical ligation to the ends of viral RNAs. The 20mer oligodeoxynucleotide homologous to OTE-II (Tag-2) was used as a tag-specific direct (5′) primer in PCR with a tagged viral RNA–cDNA heteroduplex (Table 1). The specific primers Ω and U1-Sph were used as the TMV U1 RNA-specific direct and reverse primers, respectively. Oligonucleotides K5 and 2PM (reverse primer) were used in RT–PCR as the crTMV RNA-specific 5′- and 3′-primers.
Analysis of the RT–PCR reaction products by agarose gel electrophoresis revealed the formation of PCR product in the presence of Tag-2 and the corresponding reverse primers in the case of the naturally capped TMV U1 RNA (Fig. 5), supporting the validity of the ‘cap-jumping’ approach for cap detection. At the same time, no discrete bands were obtained in reaction mixtures with Tag-2, 3′-specific primers and uncapped full-length transcripts of TMV U1 (TMV304) and of crTMV RNA. The presence of discrete products synthesized from the same uncapped templates with the TMV U1-specific (Ω + U1-Sph) or crTMV RNA-specific (K5 + 2PM) primers indicated that the 5′-proximal regions of both viral RNA transcripts were intact (Fig. 5, lanes 3 and 8). As expected, the PCR products obtained with viral RNA-specific primer pairs [Ω + U1-Sph and K5 + 2PM, specific for the TMV U1 and crTMV RNAs, respectively] (Fig. 5, lanes 1 and 6) were slightly shorter than the products obtained with the Tag-2 forward primer in combination with the specific reverse primer (Fig. 5, lanes 2 and 7). Moreover, the band mobilities corresponded to the expected chain lengths (about 460 and 340 bp, respectively) of the PCR products synthesized from uncapped, and hence lacking the 5′-Tag-2 sequence, viral RNAs.
Figure 5.

Analysis of PCR products obtained in the detection of 5′-cap structures in viral RNAs by the ‘cap-jumping’ method with the use of the following templates: RNA-tagged genomic TMV RNA (U1 strain) (lanes 1 and 2), RNA-tagged uncapped RNA transcript of the complete cDNA clone of TMV (lanes 3 and 4), the complete cDNA clone of TMV (lane 5), RNA-tagged genomic crTMV RNA (lanes 6, and 7), RNA-tagged uncapped RNA transcript of the complete cDNA clone of crTMV (lanes 8 and 9), the complete cDNA clone of crTMV (lane 10), RNA-tagged genomic crTMV RNA in total RNA of crTMV-infected tobacco (ribosome fraction) (lanes 11 and 12). M, molecular markers.
To reduce the amplification of non-specific products, we optimized the conditions of the reverse transcription reaction; in particular, we reduced the salt concentration and 2-fold diluted standard buffer was used in these experiments. The necessity to optimize the reverse transcription conditions for genomic crTMV could be due to the presence of a computer-predicted, potentially stable hairpin structure at the 5′-proximal position of crTMV RNA. In contrast to crTMV, the analogous region of TMV U1 genomic RNA is less structured (data not shown).
Consistent with the uncapped nature of full-length crTMV RNA synthetic transcripts, no discrete PCR product was obtained in reactions with Tag-2 and crTMV RNA-specific reverse primer 2PM (Fig. 5, lanes 4 and 9). In contrast, a PCR product was invariably obtained with the natural virion crTMV RNA, which clearly indicated the presence of a cap structure at its 5′-terminus. Sequencing of the cloned PCR fragment confirmed that the genomic crTMV RNA was capped (Fig. 4).
The feasibility of the ‘cap-jumping’ method for cap detection on a selected mRNA species existing in a mixture with other RNAs was examined. The total RNAs isolated from tobacco leaves systemically infected with crTMV (or TMV U1) were subjected to the procedure described above. Reverse transcription was performed with the crTMV RNA-specific primer 2PM (Fig. 5, lane 11) (or with the TMV U1-specific primer U1-Sph). The cap-tagging oligonucleotide-specific Tag-2 and 2PM (Fig. 5, lane 12) (or Tag-2 and U1-Sph) primers were used for PCR amplification. Electrophoretic analysis of the PCR products clearly indicated that the method described can be readily used for detection of a cap structure on a selected mRNA of a multi-component RNA mixture.
CONCLUSIONS
In this study we have developed an improved method for the detection of 5′-capped mRNAs and isolation of full-length cDNA which requires minimum manipulations. The method was tested for cellular RNAs as well as RNAs isolated from viral particles. This procedure can significantly simplify identification of the 5′-end of a mature mRNA and amplification of a full-length cDNA using the corresponding 5′- and 3′-deoxyribonucleotide primers. Moreover, the specific attachment of an OTE to the 5′-cap structure of mRNA allows one to overcome some problems. Thus, the modification of 3′-OH ends of total mRNA before reverse transcription greatly decreases the level of background priming as a result of the RNA 3′-ends, i.e. random self-priming (cf. 33). The ability to synthesize second strand cDNA without the use of RNase H to generate mRNA primers is essential if longer and rarer transcripts are to be identified, and libraries constructed using this technique would consist of predominantly full-length cDNAs. This can simplify downstream screening of these libraries as there is a greater chance of obtaining a complete sequence of the coding region and also the 5′- and 3′-UTRs. Our experiments have also shown that, in contrast to the SMART PCR procedure (13), the sequence of an OTE, particularly the sequence of its 3′-end, has no influence on the effectiveness of the cap-jumping process.
It should be noted that translation in plants is highly cap dependent and plant mRNAs known to naturally lack a cap structure are viral in origin. TMV, which is a typical member of the Tobamovirus genus, has a positive-stranded RNA genome of 6395 nt that encodes at least four proteins (34). Only the 126 and 183 kDa proteins are translated directly from the genomic TMV RNA, whereas the 30 kDa movement protein and 17.5 kDa coat protein are translated from individual 3′-co-terminal sgRNAs (35–38). It was shown previously that both genomic TMV RNA and sgRNA for TMV coat protein were 5′-capped (6,37,38). Use of the ‘cap-jumping’ method allowed us, for the first time, to show that the 5′-terminus of the genomic crTMV RNA contains a cap structure similar to other TMV RNAs.
Acknowledgments
ACKNOWLEDGEMENTS
We are grateful to Dr W. O. Dawson for plasmid TMV304 coding for the complete infectious cDNA of TMV and to Drs P. A. Ivanov, M. V. Skulachev and E. Skurat for providing the crTMV full-length genomic cDNA clone, viral RNAs, assistance in sequencing and helpful discussions.
REFERENCES
- 1.Shumann S. (1982) Capping enzyme in eukaryotic mRNA synthesis. Prog. Nucleic Acid Res. Mol. Biol., 50, 101–129. [DOI] [PubMed] [Google Scholar]
- 2.Shatkin A.J. and Manley,J.L. (2000) The ends of the affair: capping and polyadenylation. Nature Struct. Biol., 7, 838–842. [DOI] [PubMed] [Google Scholar]
- 3.Kozak M. (1989) The scanning model for translation: an update. J. Mol. Biol., 108, 229–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miller W.A. and Koev,G. (2000) Synthesis of subgenomic RNAs by positive-strand RNA viruses. Virology, 273, 1–8. [DOI] [PubMed] [Google Scholar]
- 5.Keith J. and Fraenkel-Conrat,H. (1975) Tobacco mosaic virus RNA carries 5′-terminal triphosphorylated guanosine blocked by 5′-linked 7-methylguanosine. FEBS Lett., 57, 31–34. [DOI] [PubMed] [Google Scholar]
- 6.Zimmern D. (1975) The 5′ end group of tobacco mosaic virus RNA is m7G5′ppp5′Gp. Nucleic Acids Res., 2, 1189–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pelletier J. and Sonenberg,N. (1988) Internal initiation of translation of eukaryotic mRNA directed by a sequence derived from poliovirus RNA. Nature, 334, 320–325. [DOI] [PubMed] [Google Scholar]
- 8.Edery I., Chu,L.L., Sonenberg,N. and Pelletier,J. (1995) An efficient strategy to isolate full-length cDNAs based on an mRNA cap retention procedure (CAPture). Mol. Cell. Biol., 15, 3363–3371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maruyama K. and Sugano,S. (1994) Oligo-capping: a simple method to replace the cap structure of eukaryotic mRNAs with oligoribonucleotides. Gene, 138, 171–174. [DOI] [PubMed] [Google Scholar]
- 10.Suzuki Y., Yoshitomo-Nakagawa,K., Maruyama,K., Suyama,A. and Sugano,S. (1997) Construction and characterization of a full length-enriched and a 5′-end-enriched cDNA library. Gene, 200, 149–156. [DOI] [PubMed] [Google Scholar]
- 11.Carninci P., Westover,A., Nishiyama,Y., Ohsumi,T., Itoh,M., Nagaoka,S., Sasaki,N., Okazaki,Y., Muramatsu,M., Schneider,C. and Hayashizaki,Y. (1997) High efficiency selection of full-length cDNA by improved biotinylated cap trapper. DNA Res., 4, 61–66. [DOI] [PubMed] [Google Scholar]
- 12.Maleszka R. and Stange,G. (1997) Molecular cloning, by a novel approach, of a cDNA encoding a putative olfactory protein in the labial palps of the moth Cactoblastis cactorum. Gene, 202, 39–43. [DOI] [PubMed] [Google Scholar]
- 13.Chenchik A., Zhu,Y.Y., Diatchenko,L., Li,R., Hill,J. and Siebert,P.D. (1998) Generation and use of high-quality cDNA from small amounts of total RNA by SMART™ PCR. In Siebert,P. and Larrick,J. (eds), Gene Cloning and Analysis by RT-PCR. Biotechniques Books, Natick, MA, pp. 305–319.
- 14.Schmidt W.M. and Mueller,M.W. (1999) CapSelect: a highly sensitive method for 5′cap-dependent enrichment of full-length cDNA in PCR-mediated analysis of mRNAs. Nucleic Acids Res., 27, e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Volloch V., Schweitzer,B., Zhang,X. and Rits,S. (1991) Identification of negative-strand complements to cytochrome oxidase subunit III RNA in Trypanosoma brucei. Proc. Natl Acad. Sci. USA, 88, 10671–10675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mandl C., Heinz,F., Puchhammer-Stockl,E. and Kunz,C. (1991) Sequencing the termini of capped viral RNA by 5′-3′ ligation and PCR. Biotechniques, 10, 484–486. [PubMed] [Google Scholar]
- 17.Fromont-Racine M., Bertrand,E., Pictet,R. and Grange,T. (1993) A highly sensitive method for mapping the 5′ termini of mRNAs. Nucleic Acids Res., 21, 1683–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Merenkova I. and Dumas Milne Edwards,J.-B. (2000) Method for the specific coupling of the cap of the 5′-end of an mRNA fragment and preparation of mRNA and complete cDNA. US patent no. 6,022,715.
- 19.Dorokhov Yu.L., Ivanov,P.A., Novikov,V.K., Agranovsky,A.A., Morozov,S.Yu., Efimov,V.A., Casper,R. and Atabekov,J.G. (1994) Complete nucleotide sequence and genome organization of a tobamovirus infecting cruciferae plants. FEBS Lett., 350, 5–8. [DOI] [PubMed] [Google Scholar]
- 20.Fraenkel-Conrat H., Singer,B. and Tsugita,A. (1961) Purification of viral RNA by means of bentonite. Virology, 14, 54–58. [DOI] [PubMed] [Google Scholar]
- 21.Leiser R., Ziegler-Graff,V., Reutenauer,A., Herrbach,E., Lemaire,O., Guilley,H., Richards,K. and Jonard,G. (1992) Agroinfection as an alternative to insects for infecting plants with beet western yellows luteovirus. Proc. Natl Acad. Sci. USA, 89, 9136–9140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brodsky L., Ivanov,V., Kalaydzidis,Y., Leontovich,A., Nikolaev,V., Feranchuk,S. Drachev,V. (1995) GeneBee-NET: Internet-based server for analyzing biopolymers structure. Biochemistry, 60, 923–928. [PubMed] [Google Scholar]
- 23.Proudnikov D. and Mirzabekov,A. (1996) Chemical methods of DNA and RNA fluorescent labeling. Nucleic Acids Res., 24, 4535–4542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Efimov V.A., Buryakova,A.A. and Chakhmakhcheva,O.G. (1999) Synthesis of polyacrylamides N-substituted with PNA-like oligonucleotide mimics for molecular diagnostic applications. Nucleic Acids Res., 27, 4416–4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Belyavsky A., Vinogradova,T. and Rajewsky,K. (1989) PCR-based cDNA library construction: general cDNA libraries at the level of a few cells. Nucleic Acids Res., 25, 2919–2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bertioli D.J., Smoker,M., Brown,A.C.P., Jones,M.G.K. and Burrows,P.R. (1994) A method based on PCR for the construction of cDNA libraries and probes from small amounts of tissue. Biotechniques, 16, 1054–1058. [PubMed] [Google Scholar]
- 27.Patel P. and Preston,B. (1994) Marked infidelity of human immunodeficiency virus type 1 reverse transcriptase at RNA and DNA template ends. Proc. Natl Acad. Sci. USA, 91, 549–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kulpa D., Topping,R. and Telesnitsky,A. (1997) Determination of the site of first strand transfer during Moloney murine leukemia virus reverse transcription and identification of strand transfer-associated reverse transcriptase errors. EMBO J., 16, 856–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heindell H., Liu,A., Paddock,G., Studnicka,G. and Salser,W. (1978) The primary sequence of rabbit alpha-globin mRNA. Cell, 15, 43–54. [DOI] [PubMed] [Google Scholar]
- 30.Kafatos F., Efatratiadis,A., Forget,B. and Weissman,S. (1977) Molecular evolution of human and rabbit beta-globin mRNAs. Proc. Natl Acad. Sci. USA, 74, 5618–5622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tokunaga K., Nakamura,Y., Sakata,K., Fujimori,K., Ohkubo,M., Sawada,K. and Sakiyama,S. (1987) Enhanced expression of a glyceraldehyde-3-phosphate dehydrogenase gene in human lung cancers. Cancer Res., 47, 5616–5619. [PubMed] [Google Scholar]
- 32.Culver J.N., Lehto,K., Close,S.M., Hilf,M.E. and Dawson,W.O. (1993) Genomic position affects the expression of tobacco mosaic virus movement protein and coat protein genes. Proc. Natl Acad. Sci. USA, 90, 2055–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Frech B. and Peterhans,E. (1994) RT–PCR: ‘background priming’ during reverse transcription. Nucleic Acids Res., 22, 4342–4343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goelet P., Lomonossoff,G., Butler,P., Akam,M., Gait,M. and Karn,J. (1982) Nucleotide sequence of tobacco mosaic virus RNA. Proc. Natl Acad. Sci. USA, 79, 5818–5822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jackson A., Zaitlin,M., Siegel,A. and Francki,R. (1972) Replication of tobacco mosaic virus. 3. Viral RNA metabolism in separated leaf cells. Virology, 48, 655–665. [DOI] [PubMed] [Google Scholar]
- 36.Hunter T., Hunt,T., Knowland,J. and Zimmern,D. (1976) Messenger RNA for the coat protein of tobacco mosaic virus. Nature, 260, 759–764. [DOI] [PubMed] [Google Scholar]
- 37.Guilley H., Jonard,G., Kukla,B. and Richards,K. (1979) Sequence of 1000 nucleotides at the 3′ end of tobacco mosaic virus RNA. Nucleic Acids Res., 6, 1287–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lewandowski D. and Dawson,W. (2000) Functions of the 126- and 183-kDa proteins of tobacco mosaic virus. Virology, 271, 90–98. [DOI] [PubMed] [Google Scholar]