


Abstract
Millions of transcriptome samples were generated by the Library of Integrated Network-based Cellular Signatures (LINCS) program. When these data are processed into searchable signatures along with signatures extracted from Genotype-Tissue Expression (GTEx) and Gene Expression Omnibus (GEO), connections between drugs, genes, pathways and diseases can be illuminated. SigCom LINCS is a webserver that serves over a million gene expression signatures processed, analyzed, and visualized from LINCS, GTEx, and GEO. SigCom LINCS is built with Signature Commons, a cloud-agnostic skeleton Data Commons with a focus on serving searchable signatures. SigCom LINCS provides a rapid signature similarity search for mimickers and reversers given sets of up and down genes, a gene set, a single gene, or any search term. Additionally, users of SigCom LINCS can perform a metadata search to find and analyze subsets of signatures and find information about genes and drugs. SigCom LINCS is findable, accessible, interoperable, and reusable (FAIR) with metadata linked to standard ontologies and vocabularies. In addition, all the data and signatures within SigCom LINCS are available via a well-documented API. In summary, SigCom LINCS, available at https://maayanlab.cloud/sigcom-lincs, is a rich webserver resource for accelerating drug and target discovery in systems pharmacology.
Graphical Abstract
Graphical Abstract.
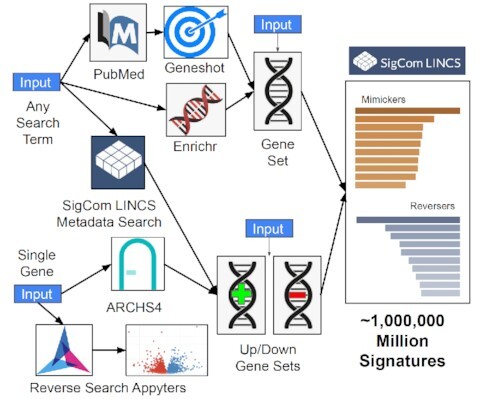
SigCom LINCS user interface workflow map. SigCom LINCS has several entry points to query the database for mimicking and reversing signatures. Users can submit any search term. The search term can be converted to a gene set using the Geneshot or the Enrichr API or used to retrieve SigCom LINCS signatures. Users of SigCom LINCS can also start with a single human gene. The single human gene can be converted into up and down gene sets using co-expression data from ARCHS4 or submitted for reverse search using two Appyters. Users of SigCom LINCS can also submit a gene set or up and down gene sets for signature search.
INTRODUCTION
Following the publication of the human genome sequence (1), genome-wide gene expression profiling with cDNA microarrays became a common tool for molecular and cell biologists. It was then proposed to develop a database of drug-induced gene expression signatures as a reference resource for finding matching signatures between user-submitted up and down genes, and complete signatures, from hundreds of drugs in the reference database. A signature in this context is defined as the differential expression of genes between two conditions, a control condition and a perturbation condition. The differential expression of the genes between the two conditions is computed, and the signature is the ranked list of genes based on their change between the two conditions. It is possible to expand this definition to the top genes that are mostly upregulated, and the top genes downregulated, as two related sets of genes that define the signature. Importantly, differential expression computation can be achieved by different methods that may result in different ranks and sets. The idea of constructing a database of gene expression signatures was first implemented for yeast (2) and later for Mammalia (3–5). This Connectivity Mapping concept (6,7) was popularized by the application of the gene set enrichment analysis method (GSEA) (8) and later by the establishment of the original Connectivity Map (CMAP) (6). The original CMAP database, developed by researchers from the Broad Institute, hosts ∼7000 signatures created by the treatment of four human cancer cell lines with most FDA-approved drugs and a few preclinical compounds. The four cancer cell lines for the original CMAP were profiled with Affymetrix cDNA microarrays before and after drug treatment in different concentrations and where gene expression was measured after 6 h. This Connectivity Mapping approach facilitated early-stage drug discovery by avoiding the need for knowing the exact drug target. Many subsequent publications used the original Connectivity Map (CMAP) resource to identify drugs for repurposing and other applications.
The success and promise of the Connectivity Mapping concept, and the CMAP resource, prompted the NIH to establish the Library of Integrated Network-based Cellular Signatures (LINCS) Common Fund program (9). During the first phase of LINCS, which lasted four years, technology development and data analysis centers were funded. In Phase II of the LINCS program, six data and signature generation centers (DSGCs) and one data coordination and integration center (DCIC) were established. Overall, the LINCS program has generated an extensive collection of perturbation-response signatures over the course of its program, which lasted 10 years (2011–2021). Expanding the original CMAP, the LINCS program employed > 20 assays to catalog the cellular responses of different model cellular systems across a wide range of chemical, genetic, microenvironment, disease, and other perturbations.
The most reused resource produced by the LINCS program is the data generated by the L1000 assay. The L1000 assay is a low-cost, high-throughput, gene expression profiling technology (10). While the L1000 assay directly measures a reduced representation of the transcriptome, the rest of the transcriptome is computationally inferred with an extrapolation algorithm. The advantage of the L1000 assay is that it can be performed in high throughput compared with RNA-seq or microarrays. Five levels of L1000 data are available for download from the CLUE platform (clue.io). Level 3 is the normalized gene expression profiles where the rows are genes, and the columns are samples. Level 5 data are gene expression signatures computed from the Level 3 data. So far, ∼3 million samples were generated (Level 3), and these samples were used to compute ∼1 million signatures (Level 5). Such a digital resource is highly valuable for drug and target discovery and drug repurposing.
Aside from LINCS, publicly available transcriptomics data has significantly expanded over the past decade. The Genotype-Tissue Expression (GTEx) consortium provides a comprehensive resource that serves gene expression data collected from 54 tissue sites of post-mortem donors (11). The latest GTEx release contains RNA-seq samples with limited publicly available metadata that includes tissue of origin, age range, sex, and cause of death. Creating signatures from the GTEx where the younger age group is compared with older ones can provide insights into tissue-specific age-related genes and the biological processes of aging. The most comprehensive and diverse resource for publicly available gene expression data is the Gene Expression Omnibus (GEO) (12). This rapidly growing resource provides transcriptomics data at the sample level, and there is an opportunity to process these data into gene expression signatures to better enable data integration and reuse (13,14). Generating signatures from GEO is challenging because of the diversity of platforms, poor labeling of samples as control and perturbation, diverse options for processing pipelines, non-uniform parameter settings, non-standardized thresholds, and inconsistent data normalization. Several efforts attempted to develop gene expression signatures from GEO. For example, a previous manual effort utilized the participants from a massive open online course (MOOC) on Coursera to crowdsource the labeling of samples for processing microarray data into signatures (13). Other efforts such as MARQ (15), GESgnExt (16), DrugSig (17), iLINCS (18), GREIN (19), ARCHS4 (14), GENEVA (20), GEN3VA (21), GEMMA (22), ExpressionBlast (23), SEEK (24), ExpressionAtlas (25) and NFFinder (26) attempted to automatically mine signatures from GEO and serve these for search through Python and R libraries or web-based search engines. However, most of these resources have limitations that include low coverage of available signatures, slow search algorithms, lack of current availability and continual updating, and poor user interface design.
This article outlines the development of SigCom LINCS, a webserver search engine for gene expression signatures that processes, analyses, and visualizes over one million signatures extracted from LINCS, GEO and GTEx. SigCom LINCS is constructed on the backbone of Signature Commons, an original open-source generic Data Commons template that can be used to host metadata and signatures for other projects. Signature Commons is FAIR compliant; this means that it has facilities for machine readable metadata that is linked to community standard ontologies and dictionaries, well documented open API, and assessment of FAIRness of datasets with FAIRshake (27). Other skeleton data portals options exist, for example, GEN3 (28), CAVATICA (29), cBioPortal (30), DERIVA (31), iRODS (32) and Globus (33). These other Data Commons are geared towards hosting patient protected data and are more elaborate than Signature Commons. Signature Commons can be considered a light-weight Data Commons template that brings together raw and processed data with an intuitive interactive interface that enables users to conduct data discovery tasks without any prior experience, training, or computer programming skills.
RESULTS
The user interface of SigCom LINCS
The user interface of SigCom LINCS is divided into several sections: Search, Concierge, UMAPs, Download, API, Help and About. The Search tab enables users to search for mimickers and reversers across over one million LINCS, GTEx, and GEO gene expression signatures collected by L1000, cDNA microarrays, and RNA-seq assays (Supplementary Table S1). SigCom LINCS entry points for signature search include inputting sets of up- and down-genes, a single gene-set, single genes, and any search term(s) (Figure 1). The up/down sets and the single gene set inputs return mimicking and reversing signatures from the various collections of available signatures. The single gene inputs can be converted into up/down sets based on RNA-seq gene–gene co-expression data matrix retrieved from ARCHS4 (14). Alternatively, single gene inputs can be queried with two Appyters (34) that identify signatures where the gene is maximally up- or down-regulated using the same data served by SigCom LINCS. The term search is converted into a gene set using Geneshot's (35) or Enrichr's (36) APIs. A search term can also be used to identify signatures within the SigCom LINCS database.
Figure 1.

SigCom LINCS user interface workflow map. SigCom LINCS has several entry points to query the database for mimicking and reversing signatures. Users can submit any search term. The search term can be converted to a gene set using the Geneshot API, or the Enrichr API, or used to retrieve SigCom LINCS signatures. Users of SigCom LINCS can also start with a single human gene. The single human gene can be converted into up and down gene sets using co-expression data from ARCHS4 or submitted for reverse search using two Appyters. Users of SigCom LINCS can also submit a gene set or up and down gene sets for signature search.
SigCom LINCS supports the entry of gene IDs in different formats including Entrez, HNGC, dbSNP, and ENSEMBL. Variants and ENSEMBL IDs are resolved using the services myvariant.info (37), mygene.info (38) and BioMart (39). Once genes are entered into the input text boxes for single gene sets or up/down sets, there is an option to validate the gene names against all human genes registered in SigCom LINCS (Supplementary Figure S1). The validation function colors the input genes based on their validation status with suggestions for synonyms and corrections, as well as resolution of variant IDs to their respective closest genes using the myvariant.info API (37). Alternatively, users can enter a single gene name or a variant ID in an adjacent text box (Supplementary Figure S2A). Once a valid human gene name, or a valid variant ID, is entered, the gene will be converted into a signature that will populate the up/down text boxes. This signature is created based on the co-expression correlation of the gene with other human genes. This functionality is achieved using the RNA-seq co-expression gene-gene similarity matrix taken from ARCHS4 (14). Using a checkbox, the user can toggle between the up/down gene set input form to the single gene set input form. The single gene set input form provides the ability to identify signatures that maximally up- or down-regulate the expression of a gene set. A search bar is located below the single gene set input form to enable users to access annotated gene sets retrieved from Enrichr (36) or create a gene set from any search term using Geneshot (35). After the user enters a search term in this search bar and presses submit, a PubMed search is invoked. The returned PubMed IDs are converted into a gene set based on co-mentions using the Geneshot API (35). The resultant gene set is then loaded into the SigCom LINCS single gene set input form (Supplementary Figure S2B).
Once the user presses the Search button, SigCom LINCS initially displays the results as columns of bar charts of the top mimickers and reversers for several categories of signatures (Supplementary Figure S3). Expanding the results for each category invokes an alternative, more focused, view with the top matching signatures as bar charts and more detailed results listed in two tables. Users can search for signatures within those tables with a dedicated search input form element on top of each table. The complete contents of the search results for each category are made available for download as tab-separated value (TSV) files (Supplementary Figure S4). The top matching signatures can also be viewed as heatmaps. In these plots, the rows are highly ranked input genes, and the columns are the top 10 matching signatures. The plots are visualized with Clustergrammer (40), which provides interactivity such as zooming, panning, sorting, clustering and filtering (Supplementary Figure S5). This feature assists the user with finding information about the top-ranked genes that are specific to the perturbagen effects.
The Metadata Search interface of SigCom LINCS enables users to search for signatures using any search term, or a combination of search terms. Such terms can be an assay, a cell line, or a drug such as dexamethasone shown in the example (Supplementary Figure S6). The search can be further refined with filters provided on the right side of the search results. Consensus analysis is provided for metadata search results that return fewer than 50 signatures (Supplementary Figure S7). This consensus analysis is performed by piping the selected signature into an Appyter (34). The Appyter produces a report that provides insights on the most common mimickers and reversers among the collection of input signatures. Returned matching signatures are available for download as either a full rank file, or as a gene matrix transpose (GMT) file with the top up- and down-regulated genes. These top up- or down-regulated genes can also be used as input for Signature Search with SigCom LINCS, as well as submitted for enrichment analysis with Enrichr (36) (Supplementary Figure S8). The metadata search results are decorated with the FAIRshake (27) insignia. This insignia represents the FAIR assessment results of each dataset. More details about these FAIR assessments are discussed below. Clicking on the metadata search results redirects users to the metadata dedicated landing pages created for each signature (Supplementary Figure S9).
The Gene Search interface of SigCom LINCS enables users to search for signatures that maximally up- or down-regulate the expression of the queried gene. These searches are performed via two Appyters: the GEO Reverse Search Appyter, and the RNA-seq-like Reverse Search Appyter. These two Appyters visualize the results of the signature search as volcano plots where each point in the plot represents a signature (Supplementary Figure S10). The results from the SigCom LINCS Gene Search Appyters are also provided in tables with ranked signatures. Details about the processing of the GEO and L1000 signature data underlying the Gene Search are provided below under the sections ‘Shaping the L1000 data into RNA-seq-like with Deep Learning’ and ‘RNA-seq gene expression signatures automatically extracted from GEO’. The Fetch Gene Set interface of SigCom LINCS enables users to fetch annotated gene sets from Enrichr (36) or from co-mentions of genes in the literature with any search term using the Geneshot API (35). The results from the search are displayed as bar graphs as well as downloadable tables (Supplementary Figures S3 and S4). Users are also able to view the input gene set by clicking on a button.
Uniquely computing signatures from the LINCS L1000 data
The processed L1000 data in SigCom LINCS consists of uniquely processed Level 3 and Level 5 data not available from the CLUE platform. To process signatures for SigCom LINCS, the Level 3 L1000 data was first downloaded from CLUE on 2 June 2021. Sets of replicate perturbation profiles were identified based on matching metadata for timepoint, dosage, perturbagen, detection plate, and well IDs from the Level 3 metadata. Replicate profiles were then used to compute Level 5 gene expression signatures for each perturbation using the Characteristic Direction (CD) method (41).
To provide a global view of the computed L1000 signatures, we visualized all normalized CD signatures with Uniform Manifold Approximation and Projection (UMAP) (42) (Figure 2A). Signatures are colored by their perturbation type. Besides a UMAP for all L1000 perturbations, UMAP plots that visualize only the chemical perturbations (Figure 2B) and the CRISPR perturbations (Figure 2C) are available. These plots show that some perturbations are cell type-specific and some cell type agnostic. The chemical perturbation plot shows that signatures are clusters by known MOAs, suggesting that for some MOAs the signatures can be predictive about the MOAs of small molecules without previously annotated MOAs. Under the UMAPs tab, SigCom LINCS contains both static and interactive UMAP visualizations for each cell line. These UMAP visualizations are separated into chemical and CRISPR perturbations.
Figure 2.

Global View of L1000 Signatures (A) L1000 signatures are visualized on a 2D space using UMAP with each signature colored by its perturbation type. (B) UMAP plot of the chemical perturbagen signatures colored by the mode of action of the small molecules. (C) UMAP of CRISPR KO signatures colored by cell line.
Benchmarking the LINCS L1000 signatures computed with the CD method
Although the CD method was previously shown to produce high-quality gene expression signatures from L1000 data in past publications (43,44), since then, the L1000 data has undergone a substantial update. Accordingly, we re-benchmarked the CD method against three other differential gene expression analysis methods: fold change, limma (45), and the moderated z-score (MODZ) method used to compute the Level 5 L1000 signatures by the producers of the L1000 data (10). A total of 1218 L1000 signatures for 44 different transcription factors (TFs) targeted by CRISPR knockout perturbations were computed using each of the above differential gene expression methods. For each signature, differentially expressed genes were ranked by the absolute value that quantified the level of differential expression. We then compared these weighted and unweighted ranks to known target genes of the respective TFs based on published ChIP-seq data (Figure 3). Each TF that was knocked out for generating the LINCS L1000 expression samples was matched with the same TF that was also profiled for its targets by ChIP-seq experiments performed by the ENCODE project (46) or by small-scale studies published in the literature and stored in the ChEA database (47). Both weighted and unweighted random walks were computed for each of the signatures and averaged across all TFs and their respective L1000 signatures. In general, the CD method produces the highest peaks in both the unweighted and weighted random walks, suggesting that the CD method best extracts and ranks the most relevant differentially expressed genes. In the unweighted walks, the peak for the CD signatures shows that the CD method recovers more target genes than the other methods without taking expression values into account (Figure 3A, B). The weighted random walks show higher peaks for all methods, possibly because the weighted expression values of the target genes provide additional information than only using the rank (Figure 3C, D). The higher, rounder peaks in the weighted walks also suggest that the MODZ, limma, and CD methods can all sufficiently detect differential expression of the ‘correct’ genes. In contrast, the fold change method performs close to what is expected for a random function. In summary, we decided to provide the L1000 signatures that were uniquely computed with the CD method in SigCom LINCS for use by the community.
Figure 3.

L1000 benchmarking. Random walk visualizations of the recovery of transcription factor (TF) target genes for L1000 CRISPR knockdown signatures targeting 44 TFs. The signatures are computed with four different differential expression analysis methods: fold change (FC), moderated Z-score (MODZ), limma, and the characteristic direction (CD). (A) Unweighted random walk comparing ranked genes from L1000 differential expression signatures with ChEA3 TF target gene sets. (B) Unweighted random walk comparing differentially expressed genes to ENCODE TF target gene sets. (C) Weighted walk comparing differentially expressed genes to ChEA3 target gene sets. Weighted increments were determined by the absolute value of the expression value for each gene, normalized to a scale of (0,1). (D) Weighted walk comparing differentially expressed genes to ENCODE target gene sets.
Shaping the L1000 data into RNA-seq-like with Deep Learning
The L1000 assay measures the expression of only 978 genes, and the inferred genes in the Level 3 data provided by CLUE is only a subset of all human coding and non-coding genes (n = 12 327). The lack of full coverage of the genome from those expression profiles prohibits many applications, for example, finding the most potent drugs to up- or down-regulate the expression of genes not reported by the L1000 profiles. To address this limitation, we developed a Deep Learning model that converts L1000 data to RNA-seq-like data. The pipeline takes as input the measured expression levels of the 978 landmark genes from the Level 3 L1000 profiles and outputs 23 614-dimensional RNA-seq-like profiles. The pipeline consists of two steps: the first step is converting L1000 profiles to RNA-seq-like profiles for the landmark genes using a modified version of a CycleGAN (48). The second step extrapolates the inferred 978 genes into full RNA-seq profiles covering the entire genome using a fully connected neural network model trained with RNA-seq data. To benchmark the performance of the 2-step model, data from a collaborative project between LINCS and GTEx was utilized. LINCS and GTEx made publicly available 2929 paired RNA-seq and L1000 profiles collected from the same GTEx tissue samples (GEO accession GSE92743). These paired samples are used to evaluate the performance of the trained model. Pearson's correlation coefficient (PCC) and Root Mean Squared Error (RMSE) were used as the evaluation measures. We proceeded with computing signatures for the RNA-seq-like data using the CD method and made these Level 3 profiles and Level 5 signatures available for download from the SigCom LINCS download page (Supplementary Figure S11).
RNA-seq gene expression signatures automatically extracted from GEO
The Gene Expression Omnibus (GEO) contains the largest and most diverse collection of gene expression data from a wide array of studies and platforms (12). Efforts to uniformly align and curate these publicly available gene expression studies include ARCHS4 (14), Recount (49), Expression Atlas (25) and GEMMA (22). These resources have made publicly available transcriptomics datasets more accessible and reusable. Furthermore, tools such as GEO2R (50), GEO2Enrichr (51), and BioJupies (52) have been developed to assist users to extract signatures from GEO, with the latter utilizing the ARCHS4 resource for access to processed RNA-seq expression data. These tools rely on users to manually annotate the perturbation and control samples that will be used for differential gene expression analysis. This annotation step takes skill, time, and effort. Fully automating signature extraction from GEO studies is desired and was recently attempted to be done at the data level (53). Although samples are labeled during the submission process, labels are not standardized and are often study dependent. To automate the sample annotation process, we first performed term frequency on the tokenized sample terms. Frequent tokenized terms include keywords such as ‘null’, ‘control’, ‘wildtype’ and ‘ctrl’, which were labeled as the control samples, while those samples with frequent terms such as ‘treatment’ or ‘perturbation’ were marked as perturbation samples. A list of frequent terms is provided in Supplementary Table S2. Utilizing only RNA-seq studies that were previously uniformly processed by ARCHS4, we collected and processed signatures from studies that have at least two samples labeled as ‘control’ and two samples labeled as ‘perturbation’. Differential expression analysis was then performed between the two groups using limma (45). Overall, this approach produced 4269 human and 4,216 mouse signatures from 6255 unique GEO series, consisting of 2953 studies with human data, 3275 studies with mouse data and 27 studies with both human and mouse samples. These signatures are made available for search and download from SigCom LINCS.
Microarray gene expression signature from CREEDS
Crowd Extracted Expression of Differential Signatures (CREEDS) was our prior effort to extract gene expression signatures from GEO with the help of participants from a massive open online course (MOOC) we delivered on the Coursera platform (13). Participants of the course extracted gene expression signatures from GEO studies by annotating signatures with the GEO2Enrichr Chrome extension (51). GEO2Enrichr utilizes the Characteristic Direction method (41) to generate up and down gene sets from the annotated studies. This project resulted in 828, 875 and 2176 unique signatures for disease, drug, and single-gene perturbations, respectively. These are made available for download from SigCom LINCS.
Gene expression signatures of aging from GTEx
The GTEx project v8 data release contains gene expression data spanning 49 tissues from 838 individual donors (11). Available metadata for each sample includes the donor age group, the tissue site from which the sample was obtained, and the gender of the subject. To create gene expression signatures from GTEx, comparisons were made at a tissue-specific level, with samples of that tissue obtained from donors in the age of 20–29 group serving as controls, and each of the other age groups serving as cases. Samples were first divided by primary tissue site. For each tissue, genes were filtered using the edgeR (54) package filterByExpr function, using the default minimum count-per-million (CPM) cutoff of 10. Since the number of samples per tissue per age group varies widely, we randomly sampled cases and controls to generate each signature. For each comparison, n samples were chosen from the cases and from the controls, with n being the maximum number of samples from either group such that there is an equal number of cases and controls. Cervix, uterus, and fallopian tube tissues were excluded from the final signature collection because they each were associated with fewer than three total samples in the age of 20–29 control group. 135 total signatures, each of which represents a unique tissue and age group pairing, were computed using the limma-voom R package (55). These signatures are available for download from SigCom LINCS and can be queried using the Signature Search function.
Data access
SigCom LINCS provides access to all the transcriptomics data and signatures via a download page and API. All signatures and other datasets are stored in an S3 bucket using an NIH Science and Technology Research Infrastructure for Discovery, Experimentation and Sustainability (STRIDES) account. Download buttons are also made available from the metadata search result pages. The dedicated download page provides links to the Level 5 full signatures, up and down gene sets stored in GMT files, as well as the predicted L1000 RNA-seq-like profiles all in one place (Supplementary Figure S11). Users of SigCom LINCS can additionally download all other LINCS data sets that are also available from the first published LINCS Data Portal (LDP1) (56). LDP1 contains data packages organized as zipped files with text-based metadata descriptions and tables of data in different formats such as Excel, GCT and text. A dedicated search bar is available for users to find datasets based on key terms such as assay, disease, biological process, cell and organ type, cell line, gene, and drug. The results from such searches can be sorted by size, data level, date, assay, or the resource that generated the data. The web interface of SigCom LINCS is powered by both the metadata API and the data API. These APIs are microservices documented with OpenAPI (57) and are made publicly available for programmatic access. The OpenAPI documentation can be accessed from the API page of SigCom LINCS, and examples are provided in the Help section.
The Signature Commons architecture
SigCom LINCS is deployed on the backbone of Signature Commons, which is a generic skeleton framework developed for quickly deploying light-weight data commons in the cloud (Supplementary Figure S12). The Signature Commons platform is a set of cloud-agnostic REST microservice applications documented with SmartAPI (58) and containerized with Docker (59). Two independent microservices are deployed together to serve a catalog of items accessible via full text search. The items within the Signature Commons database are described as JSON metadata objects. The Signature Commons system automatically provides data repository statistics. In addition to a metadata search engine, Signature Commons also provides a signature search engine. This functionality has real-time querying of gene sets, including enrichment analysis and directional queries applied to full ranked gene signatures. A stateless web interface directly serves the catalog solely through the APIs. Adopting a SmartAPI-microservice-first approach, the APIs provide the same functionality as the web interface, while separation into microservices ensures that the platform can evolve into other kinds of optimized queries.
SigCom LINCS fast search engine and comparing the Mann–Whitney U test to GSEA
SigCom LINCS computes enrichment scores and P-values given full ranked signatures and gene set libraries using the Mann–Whitney U test (MWU) (60). The test measures the inequality of means for two independent samples. In this case, the average rank of a gene set in a signature is compared to the average rank of a randomly selected gene set. MWU is similar to the Kolmogorov Smirnov test, which is the basis for the more commonly applies signature search algorithm: Gene Set Enrichment Analysis (GSEA) (8). Since the GSEA algorithm is too slow to be applied to hundreds of thousands of signatures, we opted to optimize a fast implementation of the MWU. With such implementation, we can calculate the enrichment of a gene set library against all LINCS signatures in less than one second. The fastest implementations of GSEA, blitzGSEA (61) takes several seconds to compute enrichment scores for a single signature and a gene set library such as Gene Ontology Biological Processes (62). To test whether the MWU test produces comparable results with GSEA as the signature search algorithm of choice, we compared the p-value output produced with MWU to the p-value computed by blitzGSEA for 100 gene sets with varying degrees of significance to a signature. To generate the random gene set ranks, we uniformly sample random rank positions in ranges of the full rank for different sizes of gene sets. MWU and GSEA are producing similar P-value ranks when applied on the same data (R = 0.9869, P-value = 1.896e–79, Supplementary Figure S13). These results strongly suggest that the MWU test, while slightly different from GSEA produces similar ranking of signatures, when used for enrichment analysis in this context.
FAIR assessments
An independent script was devised to process all individual SigCom LINCS signatures and assert several metrics pertinent to the FAIR guiding principles (63). In particular, the following metrics were measured: whether a metadata JSON-schema was present, and whether it was satisfied. The FAIR assessment script also checks for the availability of an associated data generation institution, the presence of an access protocol for accessing the data, the availability of a signature landing page, and the presence and up-to-date validity of several associated ontological identifiers including OBI assay (64), UBERON anatomy (65), MONDO (66), EDAM file type (67), NCBI Taxonomy (68), Cellosaurus Cell Line (69), NCBI Gene Symbol (70) and PubChem (71) to resolve drug names. These per-signature assessment results were assigned scores representing percentage satisfaction. The mean score for each library was computed resulting in a per-library score for each FAIR metric. The scores were registered with FAIRshake (27) and correspond to the individual grid squares in the FAIRshake Insignia that appears on the portal next to each library and dataset (Supplementary Figure S14).
SigCom LINCS signature consensus appyter
The SigCom LINCS metadata signature search returns lists of matching signatures for a text query. For example, a search for a gene name will return all signatures where the gene was knocked down, over-expressed, mutated, or knocked out. Each signature is provided with download links and the ability to submit the signature for analysis with Enrichr (36) as well as to the SigCom LINCS Signature Search as described above. However, it is also desired to perform analyses on a collection of signatures together to compare and combine signatures that share related perturbations and other experimental conditions. To address this type of search, the SigCom LINCS Consensus Appyter was developed. The Appyter accepts collections of up- and down- gene sets and performs signature search on all of them together with the SigCom LINCS signature search API. The matching signatures are ranked using the sum of the z-scores of the up and down gene-sets (z-sum). Signatures with positive scores are labeled as mimickers while signatures with negative scores are considered reversers. The Appyter then constructs a matrix with the top mimickers or reversers as the rows, the input gene-set names as the columns, and the z-sum scores as the data elements within the matrix cells. By default, the consensus signatures are ranked by the sum of z-sum across all the input gene-sets. To ensure that the resulting signatures consistently appear as a hit across several input gene sets, users can define a parameter, min_sigs, to filter out signatures that do not appear in at least a certain number of the input signatures. The default setting for this parameter is set to 2. The top 100 matching signatures are returned as the consensus signatures. A heatmap and an interactive clustergrammer (40) are used to visualize the consensus matrix. Furthermore, the top genes and drugs from the consensus signatures are sent to Enrichr (36) and Drugmonizome (72) for enrichment analysis, respectively. For metadata queries with at most 50 signatures, the web interface of Sigcom LINCS also integrates the Sigcom LINCS Consensus Appyter by providing a button to send the filtered signatures to the consensus Appyter as input.
SUMMARY AND CONCLUSIONS
Here we present SigCom LINCS, a next-generation Data Commons for serving LINCS, GEO and GTEx signatures. While SigCom LINCS improves upon many of the features previously developed to host gene expression signatures created by the LINCS program (Supplementary Table S3), there are many features that are missing. For example, the visualizations of the signatures as scatter plots by SigCom LINCS has less features compared with the scatter plot maps provided by L1000FWD (73) and LINCS Joint Project-Breast Cancer Network Browser (LJP-BCNB) (44). Since SigCom LINCS contains over 1 million signatures, visualizing all signatures as points within an interactive scatter plot is more challenging. In addition, SigCom LINCS does not have yet some of the features provided by other LINCS Data Portals (LDPs), namely LDP1 and LDP2 (74). LDP2 contains more extensive external knowledge about drugs, while LDP1 provides forms for data upload. We opted to exclude these features since they are either no longer needed by the LINCS program (data upload), or difficult to keep current (drug knowledge). Similarly, to SigCom LINCS, the platforms CLUE, L1000FWD, L1000CDS2 and LDP2 host gene expression signatures for search. These applications also have a metadata search engine with filtering options. However, compared with SigCom LINCS, most other websites only host parts of the LINCS data, and have fewer signatures that in SigCom LINCS. SigCom LINCS also has unique features such as starting the analysis with single genes, variants, or annotated gene sets. SigCom LINCS is also easier to navigate, and the search engine is faster.
Besides querying a massive collection of transcriptomics signatures, LINCS data provides the opportunity to study how different layers of biological regulation interact. For example, for the LINCS Joint Project (LJP) (44), transcriptomics data was collected together with cell viability data applied to the same perturbed samples. Another dataset provides matching L1000 samples with matching P100 data (75). In addition, a recent LINCS collaborative project that profiled MCF10A cells, provides multiple layers of omics data collected under the same conditions (76). SigCom LINCS provides the data from the MCF10A project for download. SigCom LINCS also contains signatures created from GTEx and GEO, combining these signatures with the L1000 signatures can lead to many insights and discoveries. The CD method was used for computing signatures from the L1000 datasets because for work described in previous publications, we have found that the CD method better recovers differentially expressed genes in L1000 data more effectively than methods such as limma, which was designed for microarrays (45) and later adapted for RNA-seq. The GEO and GTEx datasets consist of bulk RNA-seq data, and that is why we decided to process them using limma. We could use the CD method to compute signatures for GEO and GTEx but benchmarking such methods for the GEO and GTEx datasets is challenging because there is no easily interpretable global ground truth like we have for the L1000 data.
Other external datasets of high-throughput drug screening, for example, DepMap (77) and CTD2 (78), could be integrated with the expression signatures hosted by SigCom LINCS. The annotated metadata about genes, drugs and cell lines can facilitate such data integration. The gene expression signatures hosted by SigCom LINCS are provided in various forms, and this may facilitate other studies that involve data integration efforts. One area where such data can lead to promising applications is machine learning. Creative methods that used the LINCS L1000 include side-effect predictions (79) and the design of novel compounds for desired effects (80). It is expected that LINCS data will continue to serve as a resource for many other creative applications in the future. One area where the newly published L1000 data can be directly useful is for the identification of drug targets. By combining the L1000 chemical perturbation data together with the L1000 single-gene perturbation CRISPR data, we can identify and prioritize drug targets because small molecules that induce similar but unique effects observed for single gene perturbations directly implicate that the gene product as the target of the matching small molecule. While the future of the L1000 assay is uncertain, the Connectivity Mapping concept is expected to expand. It is expected that new assays will produce Connectivity Maps that will complement the LINCS data, for example, DRUG-seq (81) and RASL-seq (82) developed at Novartis are two new technologies and data collection efforts in this direction. SigCom LINCS and the Signature Commons platform can be repurposed to host and serve data from such future Connectivity Mapping projects. The Signature Commons platform was already used for other projects; for example, as a metadata lake for stem cell related data (83), as a portal for drug-set enrichment analysis (72), as a repository for bioinformatics tools, as well as for mining data and metadata for Lyme disease related projects (84). By utilizing the Signature Commons wireframe, rapid development of future data commons can be facilitated.
METHODS
Computing the L1000 signatures
The L1000 signatures were computed from the Level 3 L1000 profiles using the characteristic direction (CD) method (41). For each signature, the replicate perturbation profiles were identified based on matching metadata fields for time-point, dosage, perturbagen, detection plate, and well IDs from the Level 3 profile metadata. Each set of perturbation profiles was then compared with all other profiles in the same batch. Batches were identified by the first three terms in the signature ID, consisting of the perturbation group, time-point, and cell line. All computed signatures were divided by perturbation type and compiled into expression tables and rank matrices. The expression tables provide the computed CD differential gene expression coefficients for each signature, while the rank matrices provide the integer gene ranks for each signature as determined by the coefficients. These matrices are stored in GCTx format in S3 and are available for download from the SigCom LINCS download page.
Benchmarking the L1000 data
The CD method was benchmarked against three other differential gene expression analysis methods: fold change, limma (45), and the moderated z-score (MODZ) method (10). For the benchmarks, we examined the recovery of known target genes for 44 different transcription factors (TFs) targeted by a CRISPR knockdown in the L1000 data. These TFs were chosen because they each have corresponding target gene sets in both ENCODE (46) and ChEA (47). All L1000 CRISPR knockdown signatures for a given TF were computed using the CD method, and then each of the three other methods, using the same perturbation and control profiles. All 12 328 genes, including landmark and inferred, were then ranked by the absolute value of the respective expression coefficients: the CD coefficient, the limma logFC value, the MODZ score, and the standard fold change calculation. Comparison gene sets were obtained from both ENCODE and ChEA TF gene set libraries downloaded from Harmonizome (85). The ranked genes for each signature were compared with the corresponding TF target gene sets from each library using both weighted and unweighted random walks to show the deviation from the uniform cumulative distribution function. For the unweighted random walks, the total score increments by 1 when a gene is present in the comparison target gene set. For the weighted random walks, the expression coefficients for all genes in a signature are normalized to a number between (0, 1), and the total score increments by the normalized expression value for a gene present in the comparison gene set. The weighted and unweighted random walks for each method were averaged and plotted for both TF target gene set libraries. The y-axis in each plot measures the deviation from the uniform cumulative distribution, while the x-axis in each plot indicates the rank of each gene, scaled between 0 and 1.
L1000 to RNA-seq transformation pipeline
First, we randomly selected 50 000 L1000 profiles from the LINCS data deposited into the GEO repository (GSE92742). At the same time, we randomly selected 50,000 human RNA-seq samples from ARCHS4 (14). Next, we only retained genes with read counts of at least 10 in at least 2% of the samples. The gene counts were then log2-transformed, and quantile normalized. This left us with RNA-seq profiles with 23,614 genes for each sample. For generating RNA-seq-like profiles from L1000 profiles for the landmark genes, we used the CycleGAN (48) model to convert gene expression values in L1000 space to those in RNA-seq space with unpaired data. Starting with the original architecture of CycleGAN, which uses convolutional neural networks, we modified the model to predict RNA-seq-like profiles as vectors for given L1000 profiles. In the model, there are two generators. One generates RNA-seq profiles from L1000 profiles, and the other generates L1000 profiles from RNA-seq profiles. The first generator takes L1000 profiles as input and outputs RNA-seq profiles. The RNA-seq output by the first generator is used as input to the second generator and the output of the second generator should match the original L1000 profiles. The model also has two discriminators that assess whether a generated sample looks more like it was produced by RNA-seq or L1000. For the model architecture, we use a two-layered fully connected neural network for the generators and discriminators. A learning rate of 0.0002 and the ADAM optimizer are used to train the model. The model is trained over 100 epochs. After training the CycleGAN model, a fully connected neural network model is trained for predicting the expression profile as the full genome RNA-seq space (n = 23 614) given RNA-seq-like profiles in the landmark gene space (n = 978). The model takes the output profiles from the CycleGAN model and predicts the expression of 23 614 genes. This model was trained with another set of 50 000 randomly selected RNA-seq profiles from ARCHS4. Among the 23 614 genes, profiles of the landmark genes were used as input and the full genome profiles were used as target values. The model architecture has four layers, and the activation function is ReLU. The ADAM optimizer (86) was used with a learning rate equal of 0.0002 and batch size of 100. A validation set was used for early stopping with patience set to three epochs. To avoid outputting negative values, ReLU is applied to the output of the model.
Signature Commons architecture
Signature Commons (SigCom) is a cloud-agnostic platform designed to host semi-structured JSON serialized metadata that can be linked with a set or ranked set membership relationships with genes, proteins, drugs, or any other kind of entity. This allows us to deploy instances of SigCom for a variety of purposes, the most recent of which is a drug repurposing hub called Drugmonizome (72) and ReMeDy (83). SigCom is composed of REST microservice APIs documented with SmartAPI. The metadata API provides fast full-text search and field comparison filtering of the metadata, as well as aggregations for statistical summaries. It also performs JSON Schema validation on the JSON serialized entries before ingestion. The data API handles real-time set- and two-sided ranked set-enrichment analysis. A companion web application communicates with these APIs to provide a data portal for querying, browsing, and visualizing data. UI-schemas are JSON serialized entries that define the UI elements of the user interface. This allows for the customization of the Signature Commons interface for a variety of projects. Schemas define the overall look of the SigCom instance. This schema can be extended by adding modular components to the platform, thus extending the functionalities of the interface beyond the original design. All microservices are containerized using Docker, thus ensuring the ease of deployment on any cloud provider. SigCom LINCS is built with the Signature Commons (SigCom) wireframe. The metadata API microservice utilizes TypeORM to communicate with a PostgreSQL database. The database structure is organized in a hierarchy where resources contain libraries, libraries contain signatures, and signatures and entities have a many-to-many relationship. This enables modeling the LINCS datasets as libraries, signatures as signatures, and genes as entities. The data API is written in Java and provides a fast signature similarity search. This is made possible by loading the data matrices from a dedicated S3 bucket and storing them in-memory as hash maps. This improves retrieval time for the signature similarity search. The UI is built using React and Next.js with Material UI as its UI framework. The UI customization can be done by ingesting UI-schemas to the schema table of the database. All the UI-schemas for SigCom LINCS can be accessed from GitHub at https://github.com/MaayanLab/sigcom-lincs/. SigCom LINCS is deployed in AWS and uses Amazon's Relational Database Services (RDS) for its PostgreSQL database and S3 to store the data matrices for signature search.
Signature Commons microservices
The web interface along with each standalone microservice is developed, versioned, and packaged in independent GitHub repositories. A mono-repository using git submodules brings all the components together along with a docker-compose file for complete system deployment and helm chart for deployment to kubernetes directly from the GitHub repository. All relevant components are configurable with operational defaults. After starting the microservices and web interface, the Signature Commons controller can be used to prepare, validate, and ingest data and metadata from several commonly used data formats. After data ingestion, the customizable Signature Commons user interface enables real-time browsing, searching, filtering, and enriching of the data.
Preparing the signatures for UMAP visualization
The landmark genes of the computed CD signatures were z-score normalized on the sample axis such that each gene's coefficient is normally distributed across all signatures. UMAP (42) was then applied to the complete signature matrix. The 50 nearest neighbors and a minimum distance of 0.05 were chosen after several trials of UMAP hyper-parameter combinations. Embeddings are generally stable across runs, and the structures observed are largely preserved.
Signature search
SigCom LINCS utilizes Signature Commons’ signature search functionality to query signatures that reverse or mimic the input of up and down gene sets. Matrices that represent the gene rankings of the L1000 signatures are stored as HashMaps by the data API in random access memory (RAM). HashMaps are data structures designed to efficiently search for key-value pairs, and this implementation drastically cuts the fetch times of the gene rankings by UUIDs, thus improving the speed of the search. The Mann–Whitney U test (60) is performed separately for the up and down gene sets to obtain z-scores. The z-score determines if the genes of an input gene set are mostly positioned on the top or bottom ranks of a signature. Reversers are signatures where the input up genes are more enriched in the bottom of the ranks of a database signature, while the down genes are ranked on top. Mimickers, on the other hand, have the up genes ranked on top and the down genes ranked at the bottom. The matching signatures are then ranked based on the sum of the z-scores of the up and down gene sets (z-sum), with positive scoring z-sum labeled as mimickers and negative ones as reversers.
DATA AVAILABILITY
The SigCom LINCS webserver is available at: https://maayanlab.cloud/sigcom-lincs
The SigCom LINCS source code is available at: https://github.com/MaayanLab/sigcom-lincs
The Signature Commons Data Commons template source code is available at: https://github.com/MaayanLab/signature-commons/
The source code for automatically extracting signatures from GEO is available from: https://github.com/MaayanLab/AutoSigGen
Supplementary Material
Contributor Information
John Erol Evangelista, Department of Pharmacological Sciences, Department of Artificial Intelligence and Human Health, Mount Sinai Center for Bioinformatics, Icahn School of Medicine at Mount Sinai, One Gustave L. Levy Place, Box 1603, New York, NY 10029, USA.
Daniel J B Clarke, Department of Pharmacological Sciences, Department of Artificial Intelligence and Human Health, Mount Sinai Center for Bioinformatics, Icahn School of Medicine at Mount Sinai, One Gustave L. Levy Place, Box 1603, New York, NY 10029, USA.
Zhuorui Xie, Department of Pharmacological Sciences, Department of Artificial Intelligence and Human Health, Mount Sinai Center for Bioinformatics, Icahn School of Medicine at Mount Sinai, One Gustave L. Levy Place, Box 1603, New York, NY 10029, USA.
Alexander Lachmann, Department of Pharmacological Sciences, Department of Artificial Intelligence and Human Health, Mount Sinai Center for Bioinformatics, Icahn School of Medicine at Mount Sinai, One Gustave L. Levy Place, Box 1603, New York, NY 10029, USA.
Minji Jeon, Department of Pharmacological Sciences, Department of Artificial Intelligence and Human Health, Mount Sinai Center for Bioinformatics, Icahn School of Medicine at Mount Sinai, One Gustave L. Levy Place, Box 1603, New York, NY 10029, USA.
Kerwin Chen, Department of Pharmacological Sciences, Department of Artificial Intelligence and Human Health, Mount Sinai Center for Bioinformatics, Icahn School of Medicine at Mount Sinai, One Gustave L. Levy Place, Box 1603, New York, NY 10029, USA.
Kathleen M Jagodnik, Department of Pharmacological Sciences, Department of Artificial Intelligence and Human Health, Mount Sinai Center for Bioinformatics, Icahn School of Medicine at Mount Sinai, One Gustave L. Levy Place, Box 1603, New York, NY 10029, USA.
Sherry L Jenkins, Department of Pharmacological Sciences, Department of Artificial Intelligence and Human Health, Mount Sinai Center for Bioinformatics, Icahn School of Medicine at Mount Sinai, One Gustave L. Levy Place, Box 1603, New York, NY 10029, USA.
Maxim V Kuleshov, Department of Pharmacological Sciences, Department of Artificial Intelligence and Human Health, Mount Sinai Center for Bioinformatics, Icahn School of Medicine at Mount Sinai, One Gustave L. Levy Place, Box 1603, New York, NY 10029, USA.
Megan L Wojciechowicz, Department of Pharmacological Sciences, Department of Artificial Intelligence and Human Health, Mount Sinai Center for Bioinformatics, Icahn School of Medicine at Mount Sinai, One Gustave L. Levy Place, Box 1603, New York, NY 10029, USA.
Stephan C Schürer, Department of Biomedical Informatics, University of Cincinnati College of Medicine, Cincinnati, OH 45267, USA.
Mario Medvedovic, Department of Pharmacology, Miller School of Medicine, University of Miami, Miami, FL 33136, USA.
Avi Ma’ayan, Department of Pharmacological Sciences, Department of Artificial Intelligence and Human Health, Mount Sinai Center for Bioinformatics, Icahn School of Medicine at Mount Sinai, One Gustave L. Levy Place, Box 1603, New York, NY 10029, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [U54HL127624, R01DK131525, OT2OD030160]. Funding for open access charge: NIH [OT2OD030160].
Conflict of interest statement. None declared.
REFERENCES
- 1. Venter J.C., Adams M.D., Myers E.W., Li P.W., Mural R.J., Sutton G.G., Smith H.O., Yandell M., Evans C.A., Holt R.A.. The sequence of the human genome. Science. 2001; 291:1304–1351. [DOI] [PubMed] [Google Scholar]
- 2. Hughes T.R., Marton M.J., Jones A.R., Roberts C.J., Stoughton R., Armour C.D., Bennett H.A., Coffey E., Dai H., He Y.D.. Functional discovery via a compendium of expression profiles. Cell. 2000; 102:109–126. [DOI] [PubMed] [Google Scholar]
- 3. Waring J.F., Jolly R.A., Ciurlionis R., Lum P.Y., Praestgaard J.T., Morfitt D.C., Buratto B., Roberts C., Schadt E., Ulrich R.G.. Clustering of hepatotoxins based on mechanism of toxicity using gene expression profiles. Toxicol. Appl. Pharmacol. 2001; 175:28–42. [DOI] [PubMed] [Google Scholar]
- 4. Gunther E.C., Stone D.J., Gerwien R.W., Bento P., Heyes M.P.. Prediction of clinical drug efficacy by classification of drug-induced genomic expression profiles in vitro. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:9608–9613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Steiner G., Suter L., Boess F., Gasser R., de Vera M.C., Albertini S., Ruepp S.. Discriminating different classes of toxicants by transcript profiling. Environ. Health Perspect. 2004; 112:1236–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lamb J., Crawford E.D., Peck D., Modell J.W., Blat I.C., Wrobel M.J., Lerner J., Brunet J.P., Subramanian A., Ross K.N.et al.. The connectivity map: using gene-expression signatures to connect small molecules, genes, and disease. Science (New York, N.Y.). 2006; 313:1929–1935. [DOI] [PubMed] [Google Scholar]
- 7. Keenan A.B., Wojciechowicz M.L., Wang Z., Jagodnik K.M., Jenkins S.L., Lachmann A., Ma’ayan A.. Connectivity mapping: methods and applications. Annu. Rev. Biomed. Data Sci. 2019; 2:69–92. [Google Scholar]
- 8. Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S.. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Keenan A.B., Jenkins S.L., Jagodnik K.M., Koplev S., He E., Torre D., Wang Z., Dohlman A.B., Silverstein M.C., Lachmann A.et al.. The library of integrated network-based cellular signatures NIH program: system-level cataloging of human cells response to perturbations. Cell Syst. 2018; 6:13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Subramanian A., Narayan R., Corsello S.M., Peck D.D., Natoli T.E., Lu X., Gould J., Davis J.F., Tubelli A.A., Asiedu J.K.et al.. A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell. 2017; 171:1437–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. GTEx Consortium The genotype-tissue expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015; 348:648–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Edgar R., Domrachev M., Lash A.E.. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002; 30:207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang Z., Monteiro C.D., Jagodnik K.M., Fernandez N.F., Gundersen G.W., Rouillard A.D., Jenkins S.L., Feldmann A.S., Hu K.S., McDermott M.G.. Extraction and analysis of signatures from the gene expression omnibus by the crowd. Nat. Commun. 2016; 7:12846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lachmann A., Torre D., Keenan A.B., Jagodnik K.M., Lee H.J., Wang L., Silverstein M.C., Ma’ayan A.. Massive mining of publicly available RNA-seq data from human and mouse. Nat. Commun. 2018; 9:1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vazquez M., Nogales-Cadenas R., Arroyo J., Botï¿ ½as P., Garcï¿ ½a R., Carazo J.M., Tirado F., Pascual-Montano A., Carmona-Saez P.. MARQ: an online tool to mine GEO for experiments with similar or opposite gene expression signatures. Nucleic Acids Res. 2010; 38:W228–W232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yan S., Wong K.-C.. GESgnExt: gene expression signature extraction and meta-analysis on gene expression omnibus. IEEE J. Biomed. Health Inform. 2019; 24:311–318. [DOI] [PubMed] [Google Scholar]
- 17. Wu H., Huang J., Zhong Y., Huang Q.. DrugSig: a resource for computational drug repositioning utilizing gene expression signatures. PLoS One. 2017; 12:e0177743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pilarczyk M., Kouril M., Shamsaei B., Vasiliauskas J., Niu W., Mahi N., Zhang L., Clark N., Ren Y., White S.. Connecting omics signatures of diseases, drugs, and mechanisms of actions with iLINCS. 2020; bioRxiv doi:31 October 2019, preprint: not peer reviewed 10.1101/826271. [DOI] [PMC free article] [PubMed]
- 19. Mahi N.A., Najafabadi M.F., Pilarczyk M., Kouril M., Medvedovic M.. GREIN: an interactive web platform for re-analyzing GEO RNA-seq data. Sci. Rep. 2019; 9:7580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tanner S.W., Agarwal P.. Gene vector analysis (Geneva): a unified method to detect differentially-regulated gene sets and similar microarray experiments. BMC Bioinf. 2008; 9:348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gundersen G.W., Jagodnik K.M., Woodland H., Fernandez N.F., Sani K., Dohlman A.B., Ung P.M.-U., Monteiro C.D., Schlessinger A., Ma’ayan A.. GEN3VA: aggregation and analysis of gene expression signatures from related studies. BMC Bioinf. 2016; 17:461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zoubarev A., Hamer K.M., Keshav K.D., McCarthy E.L., Santos J.R.C., Van Rossum T., McDonald C., Hall A., Wan X., Lim R.. Gemma: a resource for the reuse, sharing and meta-analysis of expression profiling data. Bioinformatics. 2012; 28:2272–2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zinman G.E., Naiman S., Kanfi Y., Cohen H., Bar-Joseph Z.. ExpressionBlast: mining large, unstructured expression databases. Nat. Methods. 2013; 10:925–926. [DOI] [PubMed] [Google Scholar]
- 24. Zhu Q., Wong A.K., Krishnan A., Aure M.R., Tadych A., Zhang R., Corney D.C., Greene C.S., Bongo L.A., Kristensen V.N.et al.. Targeted exploration and analysis of large cross-platform human transcriptomic compendia. Nat. Methods. 2015; 12:211–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Papatheodorou I., Fonseca N.A., Keays M., Tang Y.A., Barrera E., Bazant W., Burke M., Füllgrabe A., Fuentes A.M.-P., George N.. Expression Atlas: gene and protein expression across multiple studies and organisms. Nucleic Acids Res. 2018; 46:D246–D251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Setoain J., Franch M., Martínez M., Tabas-Madrid D., Sorzano C.O., Bakker A., Gonzalez-Couto E., Elvira J., Pascual-Montano A.. NFFinder: an online bioinformatics tool for searching similar transcriptomics experiments in the context of drug repositioning. Nucleic Acids Res. 2015; 43:W193–W199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Clarke D.J., Wang L., Jones A., Wojciechowicz M.L., Torre D., Jagodnik K.M., Jenkins S.L., McQuilton P., Flamholz Z., Silverstein M.C.. FAIRshake: toolkit to evaluate the FAIRness of research digital resources. Cell Syst. 2019; 9:417–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hughes L., Grossman R.L., Flamig Z., Prokhorenkov A., Lukowski M., Fitzsimons M., Lichtenberg T., Tang Y.. American Society of Clinical Oncology. 2019; [Google Scholar]
- 29. Raman P., Waanders A.J., Storm P.B., Lilly J.V., Mason J., Heath A.P., Felmeister A.S., Cros A., Zhu Y., Sender L.. gene-15. Cavatica-a pediatric genomic cloud empowering data discovery through the pediatric brain tumor atlas. Neuro-oncol. 2017; 19:iv21. [Google Scholar]
- 30. Gao J., Aksoy B.A., Dogrusoz U., Dresdner G., Gross B., Sumer S.O., Sun Y., Jacobsen A., Sinha R., Larsson E.. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013; 6:pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bugacov A., Czajkowski K., Kesselman C., Kumar A., Schuler R.E., Tangmunarunkit H.. 2017 IEEE 13th International Conference on e-Science (e-Science). 2017; IEEE; 79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rajasekar A., Moore R., Hou C.-Y., Lee C.A., Marciano R., de Torcy A., Wan M., Schroeder W., Chen S.-Y., Gilbert L.. iRODS primer: integrated rule-oriented data system. Synth. Lect. Inform. Concepts Retriev. Serv. 2010; 2: 10.2200/S00233ED1V01Y200912ICR012. [DOI] [Google Scholar]
- 33. Foster I. Globus online: accelerating and democratizing science through cloud-based services. IEEE Internet Comput. 2011; 15:70–73. [Google Scholar]
- 34. Clarke D.J.B., Jeon M., Stein D.J., Moiseyev N., Kropiwnicki E., Dai C., Xie Z., Wojciechowicz M.L., Litz S., Hom J.et al.. Appyters: turning jupyter notebooks into data-driven web apps. Patterns. 2021; 2:100213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lachmann A., Schilder B.M., Wojciechowicz M.L., Torre D., Kuleshov M.V., Keenan A.B., Ma’ayan A.. Geneshot: search engine for ranking genes from arbitrary text queries. Nucleic Acids Res. 2019; 47:W571–W577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kuleshov M.V., Jones M.R., Rouillard A.D., Fernandez N.F., Duan Q., Wang Z., Koplev S., Jenkins S.L., Jagodnik K.M., Lachmann A.et al.. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016; 44:W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Xin J., Mark A., Afrasiabi C., Tsueng G., Juchler M., Gopal N., Stupp G.S., Putman T.E., Ainscough B.J., Griffith O.L.et al.. High-performance web services for querying gene and variant annotation. Genome Biol. 2016; 17:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wu C., Macleod I., Su A.I.. BioGPS and mygene.info: organizing online, gene-centric information. Nucleic Acids Res. 2013; 41:D561–D565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Durinck S., Moreau Y., Kasprzyk A., Davis S., De Moor B., Brazma A., Huber W.. BioMart and bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics. 2005; 21:3439–3440. [DOI] [PubMed] [Google Scholar]
- 40. Fernandez N.F., Gundersen G.W., Rahman A., Grimes M.L., Rikova K., Hornbeck P., Ma’ayan A.. Clustergrammer, a web-based heatmap visualization and analysis tool for high-dimensional biological data. Scientific Data. 2017; 4:170151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Clark N.R., Hu K.S., Feldmann A.S., Kou Y., Chen E.Y., Duan Q., Ma’ayan A.. The characteristic direction: a geometrical approach to identify differentially expressed genes. BMC Bioinf. 2014; 15:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. McInnes L., Healy J., Melville J.. UMAP: uniform manifold approximation and projection for dimension reduction. 2018; arXiv doi:18 September 2020, preprint: not peer reviewedhttps://arxiv.org/abs/1802.03426.
- 43. Duan Q., Reid S.P., Clark N.R., Wang Z., Fernandez N.F., Rouillard A.D., Readhead B., Tritsch S.R., Hodos R., Hafner M.. L1000CDS 2: LINCS L1000 characteristic direction signatures search engine. NPJ Syst. Biol. Appl. 2016; 2:16015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Niepel M., Hafner M., Duan Q., Wang Z., Paull E.O., Chung M., Lu X., Stuart J.M., Golub T.R., Subramanian A.et al.. Common and cell-type specific responses to anti-cancer drugs revealed by high throughput transcript profiling. Nat. Commun. 2017; 8:1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Smyth G.K. Bioinformatics and Computational Biology Solutions Using R and Bioconductor. 2005; Springer; 397–420. [Google Scholar]
- 46. ENCODE Project Consortium The ENCODE (ENCyclopedia of DNA elements) project. 2004; 306:636–640. [DOI] [PubMed] [Google Scholar]
- 47. Keenan A.B., Torre D., Lachmann A., Leong A.K., Wojciechowicz M.L., Utti V., Jagodnik K.M., Kropiwnicki E., Wang Z., Ma’ayan A.. ChEA3: transcription factor enrichment analysis by orthogonal omics integration. Nucleic Acids Res. 2019; 47:W212–W224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhu J.-Y., Park T., Isola P., Efros A.A.. Proceedings of the IEEE International Conference on Computer Vision. 2017; 2223–2232. [Google Scholar]
- 49. Wilks C., Zheng S.C., Chen F.Y., Charles R., Solomon B., Ling J.P., Imada E.L., Zhang D., Joseph L., Leek J.T.et al.. recount3: summaries and queries for large-scale RNA-seq expression and splicing. Genome Biol. 2021; 22:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Barrett T., Wilhite S.E., Ledoux P., Evangelista C., Kim I.F., Tomashevsky M., Marshall K.A., Phillippy K.H., Sherman P.M., Holko M.. NCBI GEO: archive for functional genomics data sets—update. Nucleic Acids Res. 2012; 41:D991–D995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gundersen G.W., Jones M.R., Rouillard A.D., Kou Y., Monteiro C.D., Feldmann A.S., Hu K.S., Ma’ayan A.. GEO2Enrichr: browser extension and server app to extract gene sets from GEO and analyze them for biological functions. Bioinformatics. 2015; 31:3060–3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Torre D., Lachmann A., Ma’ayan A.. BioJupies: automated generation of interactive notebooks for RNA-seq data analysis in the cloud. Cell Syst. 2018; 7:556–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kaur N., Oskotsky B., Butte A.J., Hu Z.. Systematic identification of ACE2 expression modulators reveals cardiomyopathy as a risk factor for mortality in COVID-19 patients. Genome Biol. 2022; 23:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Robinson M.D., McCarthy D.J., Smyth G.K.. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010; 26:139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Law C.W., Chen Y., Shi W., Smyth G.K.. voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014; 15:R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Koleti A., Terryn R., Stathias V., Chung C., Cooper D.J., Turner J.P., Vidović D., Forlin M., Kelley T.T., D’Urso A. Data portal for the library of integrated Network-based cellular signatures (LINCS) program: integrated access to diverse large-scale cellular perturbation response data. Nucleic Acids Res. 2018; 46:D558–D566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sferruzza D., Rocheteau J., Attiogbé C., Lanoix A.. International Conference on Web Information Systems and Technologies. 2018; [Google Scholar]
- 58. Zaveri A., Dastgheib S., Wu C., Whetzel T., Verborgh R., Avillach P., Korodi G., Terryn R., Jagodnik K., Assis P.. European Semantic Web Conference. 2017; Springer; 154–169. [Google Scholar]
- 59. Bhat S. Practical Docker with Python. 2018; Springer; 53–89. [Google Scholar]
- 60. Mann H.B., Whitney D.R.. On a test of whether one of two random variables is stochastically larger than the other. Ann. Math. Statist. 1947; 50–60. [Google Scholar]
- 61. Lachmann A., Xie Z., Ma’ayan A.. blitzGSEA: efficient computation of gene set enrichment analysis through gamma distribution approximation. Bioinformatics. 2022; 38:2356–2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. The Gene Ontology Consortium The gene ontology resource: 20 years and still GOing strong. Nucleic Acids Res. 2019; 47:D330–d338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wilkinson M.D., Dumontier M., Aalbersberg I.J., Appleton G., Axton M., Baak A., Blomberg N., Boiten J.W., da Silva Santos L.B., Bourne P.E.et al.. The FAIR guiding principles for scientific data management and stewardship. Scientific Data. 2016; 3:160018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Bandrowski A., Brinkman R., Brochhausen M., Brush M.H., Bug B., Chibucos M.C., Clancy K., Courtot M., Derom D., Dumontier M.. The ontology for biomedical investigations. PLoS One. 2016; 11:e0154556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Mungall C.J., Torniai C., Gkoutos G.V., Lewis S.E., Haendel M.A.. Uberon, an integrative multi-species anatomy ontology. Genome Biol. 2012; 13:R5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Vasilevsky N., Essaid S., Matentzoglu N., Harris N.L., Haendel M., Robinson P., Mungall C.J.. CEUR Workshop Proceedings. 2020; 2807:CEUR-WS. [Google Scholar]
- 67. Ison J., Kalaš M., Jonassen I., Bolser D., Uludag M., McWilliam H., Malone J., Lopez R., Pettifer S., Rice P.. EDAM: an ontology of bioinformatics operations, types of data and identifiers, topics and formats. Bioinformatics. 2013; 29:1325–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Schoch C.L., Ciufo S., Domrachev M., Hotton C.L., Kannan S., Khovanskaya R., Leipe D., Mcveigh R., O’Neill K., Robbertse B.. NCBI taxonomy: a comprehensive update on curation, resources and tools. Database (Oxford). 2020; 2020:baaa062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Bairoch A. The cellosaurus, a cell-line knowledge resource. J. Biomol. Tech. 2018; 29:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Brown G.R., Hem V., Katz K.S., Ovetsky M., Wallin C., Ermolaeva O., Tolstoy I., Tatusova T., Pruitt K.D., Maglott D.R.. Gene: a gene-centered information resource at NCBI. Nucleic Acids Res. 2015; 43:D36–D42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kim S., Chen J., Cheng T., Gindulyte A., He J., He S., Li Q., Shoemaker B.A., Thiessen P.A., Yu B.et al.. PubChem in 2021: new data content and improved web interfaces. Nucleic Acids Res. 2021; 49:D1388–D1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kropiwnicki E., Evangelista J.E., Stein D.J., Clarke D.J.B., Lachmann A., Kuleshov M.V., Jeon M., Jagodnik K.M., Ma’ayan A.. Drugmonizome and drugmonizome-ml: integration and abstraction of small molecule attributes for drug enrichment analysis and machine learning. Database (Oxford). 2021; 2021:baab017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wang Z., Lachmann A., Keenan A.B., Ma’ayan A.. L1000FWD: fireworks visualization of drug-induced transcriptomic signatures. Bioinformatics. 2018; 34:2150–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Stathias V., Turner J., Koleti A., Vidovic D., Cooper D., Fazel-Najafabadi M., Pilarczyk M., Terryn R., Chung C., Umeano A.. LINCS data portal 2.0: next generation access point for perturbation-response signatures. Nucleic Acids Res. 2020; 48:D431–D439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Litichevskiy L., Peckner R., Abelin J.G., Asiedu J.K., Creech A.L., Davis J.F., Davison D., Dunning C.M., Egertson J.D., Egri S.. A library of phosphoproteomic and chromatin signatures for characterizing cellular responses to drug perturbations. Cell Syst. 2018; 6:424–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Gross S.M., Dane M.A., Smith R.L., Devlin K., Mclean I., Derrick D., Mills C., Subramanian K., London A., Torre D. A LINCS microenvironment perturbation resource for integrative assessment of ligand-mediated molecular and phenotypic responses. 2021; bioRxiv doi:09 August 2021, preprint: not peer reviewed 10.1101/2021.08.06.455429. [DOI] [PMC free article] [PubMed]
- 77. Tsherniak A., Vazquez F., Montgomery P.G., Weir B.A., Kryukov G., Cowley G.S., Gill S., Harrington W.F., Pantel S., Krill-Burger J.M.. Defining a cancer dependency map. Cell. 2017; 170:564–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Aksoy B.A., Dančík V., Smith K., Mazerik J.N., Ji Z., Gross B., Nikolova O., Jaber N., Califano A., Schreiber S.L.. CTD2 dashboard: a searchable web interface to connect validated results from the cancer target discovery and development network. Database (Oxford). 2017; 2017:bax054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Wang Z., Clark N.R., Ma’ayan A.. Drug-induced adverse events prediction with the LINCS L1000 data. Bioinformatics. 2016; 32:2338–2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Méndez-Lucio O., Baillif B., Clevert D.-A., Rouquié D., Wichard J.. De novo generation of hit-like molecules from gene expression signatures using artificial intelligence. Nat. Commun. 2020; 11:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Ye C., Ho D.J., Neri M., Yang C., Kulkarni T., Randhawa R., Henault M., Mostacci N., Farmer P., Renner S.. DRUG-seq for miniaturized high-throughput transcriptome profiling in drug discovery. Nat. Commun. 2018; 9:4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Li H., Qiu J., Fu X.D.. RASL-seq for massively parallel and quantitative analysis of gene expression. Curr. Protoc. Mol. Biol. 2012; 98: 10.1002/0471142727.mb0413s98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Borziak K., Parvanova I., Finkelstein J.. ReMeDy: a platform for integrating and sharing published stem cell research data with a focus on iPSC trials. Database. 2021; 2021:baab038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Bobe J.R., Jutras B.L., Horn E.J., Embers M.E., Bailey A., Moritz R.L., Zhang Y., Soloski M.J., Ostfeld R.S., Marconi R.T.. Recent progress in Lyme disease and remaining challenges. Front. Med. 2021; 8:666554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Rouillard A.D., Gundersen G.W., Fernandez N.F., Wang Z., Monteiro C.D., McDermott M.G., Ma’ayan A.. The harmonizome: a collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database (Oxford). 2016; 2016:baw100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Kingma D.P., Ba J.. Adam: a method for stochastic optimization. 2014; arXiv doi:30 January 2017, preprint: not peer reviewedhttps://arxiv.org/abs/1412.6980.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The SigCom LINCS webserver is available at: https://maayanlab.cloud/sigcom-lincs
The SigCom LINCS source code is available at: https://github.com/MaayanLab/sigcom-lincs
The Signature Commons Data Commons template source code is available at: https://github.com/MaayanLab/signature-commons/
The source code for automatically extracting signatures from GEO is available from: https://github.com/MaayanLab/AutoSigGen