


Abstract
5NosoAE is a webserver that can be used for nosocomial bacterial analysis including the identification of similar strains based on antimicrobial resistance profiles (antibiogram) and the spatiotemporal distribution visualization and phylogenetic analysis of identified strains with similar antibiograms. The extensive use of antibiotics has caused many pathogenic bacteria to develop multiple drug resistance, resulting in clinical infection treatment challenges and posing a major threat to global public health. Relevant studies have investigated the key determinants of antimicrobial resistance in the whole-genome sequence of bacteria. However, a web server is currently not available for performing large-scale strain searches according to antimicrobial resistance profiles and visualizing epidemiological information including the spatiotemporal distribution, antibiogram heatmap, and phylogeny of identified strains. Here, we implemented these functions in the new server, referred to as 5NosoAE. This server accepts the genome sequence file in the FASTA format of five nosocomial bacteria, namely Acinetobacter baumannii, Pseudomonas aeruginosa, Klebsiella pneumoniae, Enterococcus faecium and Staphylococcus aureus for query. All visualizations are implemented in JavaScript and PHP. This server will be useful for physicians and epidemiologists involved in research on infectious disease. The 5NosoAE platform is available at https://nosoae.imst.nsysu.edu.tw.
Graphical Abstract
Graphical Abstract.
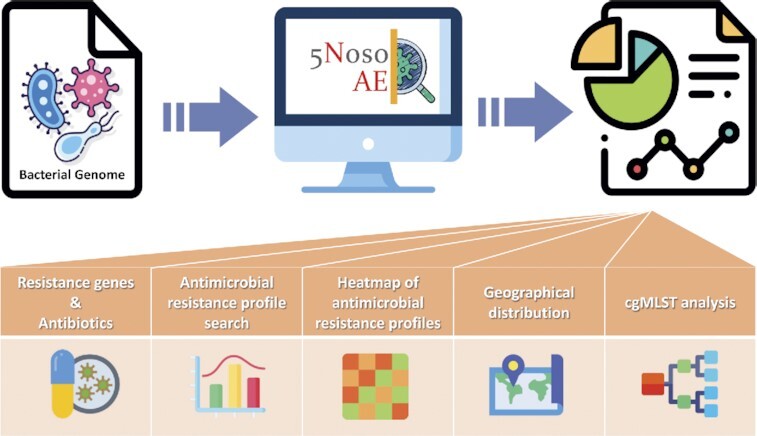
5NosoAE is a web-based tool that allows users to perform large-scale strain searches according to antimicrobial resistance profiles and visualize epidemiological information including the spatiotemporal distribution, antibiogram heatmap, and phylogeny of identified strains.
INTRODUCTION
The extensive use of various antibiotics in clinical practice and even agriculture and livestock breeding has led to the development of various drug-resistant strains, with some strains even being resistant to multiple antibiotics. These drug-resistant bacteria pose a considerable threat to global public health. In inpatients with relatively weak immunity, infection with drug-resistant bacteria may prolong the course of hospitalization and increase the cost and burden of medical care. Moreover, drug-resistant bacteria can cause nosocomial infections in the event of inappropriate infection control, exposing more hospitalized patients to risk. Common multidrug-resistant nosocomial bacteria are carbapenem-resistant Acinetobacter baumannii (1,2), Pseudomonas aeruginosa (3,4), Klebsiella pneumoniae (5,6), vancomycin-resistant Enterococcus faecium (7,8) and Staphylococcus aureus (9,10), all of which pose a considerable threat to inpatients’ lives. Currently, most of the related studies on drug resistance have focused on mechanisms underlying the development of epidemic diseases caused by last-line antibiotic-resistant strains (11,12), and molecular testing methods for the rapid detection of drug resistance (13,14). The advent of next-generation sequencing has enabled the use of the entire bacterial genome for the analysis of drug-resistant strains that cause epidemic diseases, the study of mechanisms underlying drug resistance, and the development of rapid molecular diagnostics (15,16).
Because of the limitations of existing tools, an integrated platform that enables the visualization of drug-resistant bacteria that cause nosocomial infections on a global scale should be constructed. Currently, the Comprehensive Antibiotic Resistance Database (CARD, https://card.mcmaster.ca/) (17) is the most reliable database available for antibiotic resistance gene searches using the Resistance Gene Identifier (RGI) program. The results are presented as a pie chart divided into the three levels of ‘perfect’, ‘strict’ and ‘loose’ for comparing among antibiotic resistance genes in the database. Another popular drug resistance analysis tool is ResFinder (18), developed by the Danish Institute of Technology, which is used for the detection and comparison of antibiotic resistance genes as well as the prediction of antibiotic resistance, and the results are presented in a tabular form. The aforementioned two tools are mainly used by laboratory researchers who are less familiar with command-line operations in the form of website services (RGI and ResFinder can also be downloaded and executed on a local server). NCBI AMRFinder (19) can be downloaded on a local server and is used for large-scale drug resistance gene detection; however, its operation threshold is relatively high and not suitable for general public health laboratory researchers. PATRIC (20) is a large-scale microbiological research integration website that provides microbiological experimental data and analysis tools for download. Pathogenwatch (21,22) is a visualization platform that enables users to analyze drug resistance and query similar strain locations distributed globally for several species. The main purpose of Pathogenwatch is to collect genomes uploaded by users from across the globe for the long-term monitoring of the drug resistance spectrum and the analysis of nosocomial bacteria. BacWGSTdb (23) is a comprehensive whole-genome sequence typing and source tracking tool and database that contains information on 20 microorganisms. Users can upload a single genome to perform 7-gene multilocus sequence typing (MLST) and core genome MLST (cgMLST) and predict antimicrobial resistance and virulence genes. In addition, BacWGSTdb can be used to perform multiple genome analysis. Databases such as BLDB (24) and CBMAR (25) are used for the detection of single-antibiotic resistance genes, and services such as ITEGRALL (26) and ISfinder (27) are used to search specific drug-resistance-related fragments.
The aforementioned platforms are well developed for phylogenetic analysis and antimicrobial resistance gene prediction. For example, BacWGSTdb is a comprehensive database that can be used for conducting epidemiological research based on the pathogenomic comparison and prediction. However, for the investigation and surveillance of the spread of certain critical antibiotic resistance patterns, a platform that can perform the large-scale comparison of antibiotic resistance profiles must be developed. In addition, to study the spread of antimicrobial resistance, the global distribution of strains with similar drug resistance, the timespan of strain isolation, and the source of strain isolation should be determined; moreover, whether strains with similar drug resistance are closely related and whether drug resistance is spread across strains must be investigated. Therefore, an integrated visualization platform is required.
In this study, we developed a standardized method, 5NosoAE, for generating drug resistance profiles and a method for calculating the similarity of strain resistance by using drug resistance profiles. The results indicated that this method enabled the rapid comparison of drug resistance profiles to identify strains with similar drug resistance, and the visualization provided more intuitive epidemiological data. This platform will be useful for the monitoring and clinical treatment of drug-resistant nosocomial bacteria.
METHOD AND IMPLEMENTATION
Figure 1 presents a schematic workflow of 5NosoAE. The 5NosoAE server contains data on the five most crucial nosocomial pathogens (Figure 1A) and can be used for nosocomial bacterial resistance investigation and epidemiology surveys. Users can upload a query genome sequence or enter an NCBI Assembly ID (Figure 1B). The server first identifies the taxonomy of organisms by searching the SILVA (28) ribosomal RNA gene database and then detects antibiotic resistance genes by searching the resfinder_db database through ResFinder strategies (18) and mapping them to the corresponding antibiotics (Figure 1C). The largest evidence score (SEV) of the corresponding resistance genes is used to represent the intensity of the drug resistance of an isolate for each antibiotic to construct antimicrobial resistance profiles for the query isolate and the database of five nosocomial pathogens (Figure 1D). The similarity of antimicrobial resistance profiles is determined using the similarity score (SPRO), and the scores are then fit to a generalized extreme value (GEV) distribution (29) to determine statistical significance (Figure 1E).
Figure 1.

Schematic workflow of the 5NosoAE server.
JavaScript libraries were used for the visualization of antimicrobial resistance profiles and geographical distribution (Figure 1F). The cgMLST analysis in the 5NosoAE server is performed using our PGAdb-builder (30,31), where Python and Perl scripts are used to integrate the analysis pipeline. The webpage was constructed using HTML and PHP. This web server runs on a Linux system with 2.30-GHz Intel Xeon processors consisting of 48 cores.
Collection of the whole-genome sequences of five nosocomial bacterial isolates
Genome sequences in the 5NosoAE server contain data on the five most common nosocomial pathogens, namely A. baumannii, P. aeruginosa, K. pneumoniae, E. faecium and S. aureus. The genome sequences and reads were downloaded from the NCBI Assembly and Sequence Read Archive (SRA) databases, respectively (Figure 1A). To collect high-quality genome sequences, we applied the following filters in the NCBI Assembly database: latest version file, exclude anomalous, and taxonomy check. Furthermore, we used the following filters in the NCBI SRA database: genome, paired end, Illumina, and sequencing depth between 80× and 500×. Prefetch and fasterq-dump, the components of the SRA toolkit, were used to download the SRA file (in the sra format) and convert it to the FASTQ format. Subsequently, the FASTQ reads were assembled into contigs by using SKESA (32). Table 1 lists the detailed organisms and number of isolates included in 5NosoAE. Each genome entry was mapped to the NCBI BioSample database to obtain information on biological source materials used in experimental assays such as the collection date, geographical location, and host.
Table 1.
Name of organisms and genome data distribution by source of database
| Organism | Assemblya | SRAb | Total |
|---|---|---|---|
| Acinetobacter baumannii | 9208 | 1574 | 10 782 |
| Pseudomonas aeruginosa | 8331 | 5626 | 13 957 |
| Klebsiella pneumoniae | 14 766 | 9631 | 24 397 |
| Enterococcus faecium | 11 729 | 4506 | 16 235 |
| Staphylococcus aureus | 25 000 | 17 016 | 42 016 |
| Total | 69 034 | 38 353 | 107 387 |
aAssembly filter criteria: latest version file, exclude anomalous, and taxonomy check.
bSRA filter criteria: genome, paired end, Illumina and sequencing depth between 80× and 500× .
Generation of the antimicrobial resistance profile
The antimicrobial resistance profile for the query isolate and the database of the five nosocomial pathogens were generated by detecting resistance genes and mapping their corresponding antibiotics by using ResFinder and resfinder_db (18) (Figure 1C and D). The antimicrobial resistance profile included 91 antibiotics belonging to 24 classes (Supplementary Table S1). The largest evidence score (SEV) of the corresponding resistance genes was used to represent the intensity of the drug resistance of an isolate for each antibiotic to construct the antimicrobial resistance profile. The evidence score (SEV) was calculated as follows:
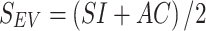 |
(1) |
where SI is the sequence identity and AC is the alignment coverage between the query and subject resistance genes.
Antimicrobial resistance profile comparison and statistical significance
The profile similarity score (SPRO) was calculated by evaluating the root mean square distance (RMSD) between two antimicrobial resistance profiles (Figure 1E). The SPRO was calculated as follows:
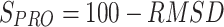 |
(2) |
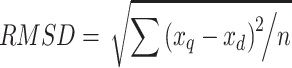 |
(3) |
where xq is the SEV score for each antibiotic of the query genome, xd is the SEV score for each antibiotic of the database, n is the total number of antibiotics in an antimicrobial resistance profile, and RMSD is the measure of the average distance between the profiles.
When compared, the similarity scores of the profiles followed the extreme value distribution. To determine the statistical significance of a database search, all similarity scores for each query were fit to a GEV distribution (29). The GEV distribution is a family of continuous probability distributions that combine the Gumbel, Fréchet, and Weibull distributions. The GEV is parameterized with location and scale parameters, μ and σ, and a shape parameter, k. The cumulative distribution function (CDF) of the GEV distribution is defined as
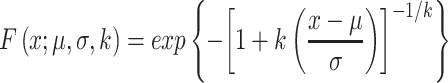 |
(4) |
Finally, the P-value is given by the complementary CDF, which is defined as
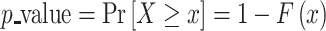 |
(5) |
Core genome multilocus sequence typing
The 5NosoAE server can be used to perform cgMLST analysis to obtain a high-resolution phylogenetic relatedness tree for the top 20 isolates with similar antimicrobial resistance profiles. The cgMLST schemes for the five nosocomial bacterial species were downloaded from the cgMLST nomenclature server (https://www.cgmlst.org). The typing schemes comprised 2390, 3867, 2358, 1423 and 1861 loci for A. baumannii, P. aeruginosa, K. pneumoniae, E. faecium and S. aureus, respectively. The construction of the core genome database and the profiling of alleles were performed using the PGAdb-builder (30,31). A genetic relatedness tree was constructed from the established allelic sequence by using the clustering algorithm of the neighbor-joining method in the PHYLIP v3.6 program (33). Bootstrap values in a dendrogram were calculated using the ETE3 toolkit (34) (Figure 1F).
WEBSERVER
Input format
The 5NosoAE webserver accepts two types of input (Figure 2A): users can upload a genome sequence file in the FASTA format or enter an NCBI Assembly ID. For a genome sequence with a size of ∼5MB, ∼8 min are required for taxonomy identification, resistance gene detection, antibiogram and cgMLST profile construction and comparison, and statistical significance evaluation. However, for a longer genome, more than 20 min in run time is required. Therefore, users are encouraged to enter their e-mail address, and a notification is sent by e-mail when the analysis is complete.
Figure 2.
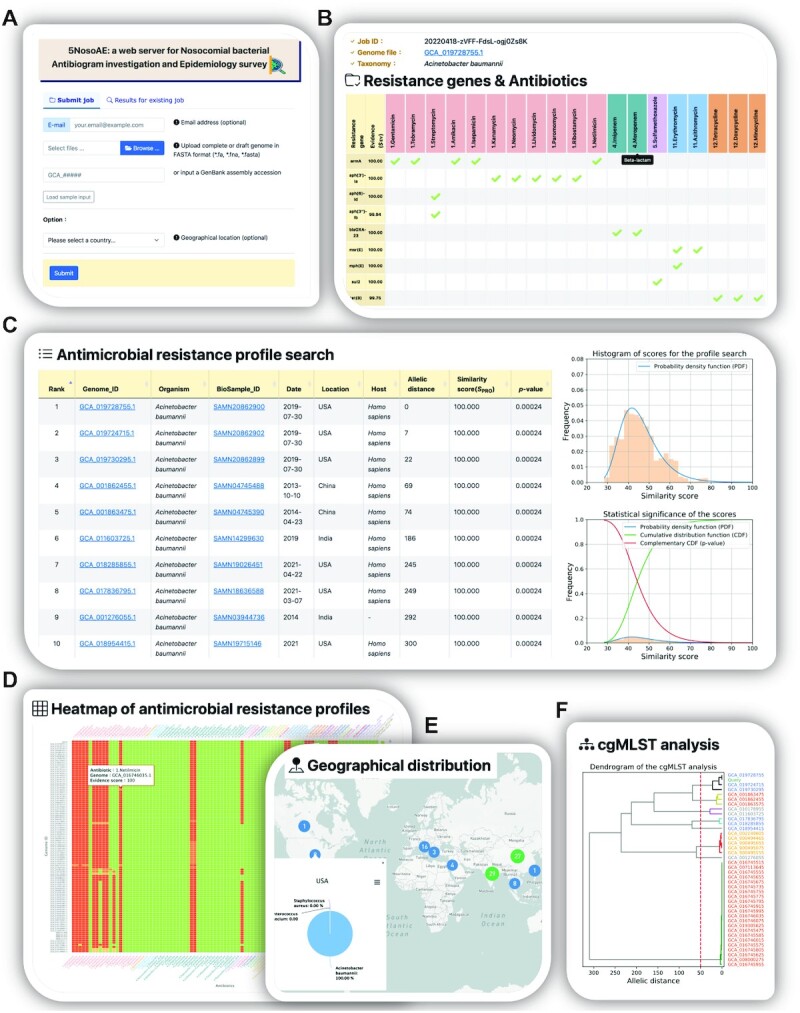
Features of the 5NosoAE web server. (A) Input page of the upload complete or draft genome file in the FASTA format or enter NCBI Assembly ID. (B) Output page of detected resistance genes and their corresponding antibiotics. (C) Results of antimicrobial resistance profile search and statistical significance. (D) Heatmap of antimicrobial resistance profiles. (E) Results of geographical distribution. (F) Results of cgMLST analysis.
Output format
After the analysis, the 5NosoAE server returns five types of results (Figure 2). (1) The server provides information on detected resistance genes, SEV, and their corresponding antibiotics (Figure 2B). Different antibiotic classes are represented as different colors. (2) The analytical results include a table listing the top 100 records of the isolates with similar antimicrobial resistance profiles based on the SPRO score (Figure 2C). Each record includes the genome ID, organism name, BioSample ID, collection date, geographical location, host, allelic distance, SPRO and P-value (Figure 2C). The P-values are used to quantify the statistical significance of the database search. The data can be sorted by clicking on a specific column header and downloaded as a tab-delimited file. (3) The antimicrobial resistance profile of the most similar top 100 isolates is presented as a heatmap (Figure 2D). Color ramps from green to red represent the resistance levels of sensitive (S), intermediate (I) and resistant (R). Antibiotic classes are marked with different colors to help differentiate between various groups. The SEV is shown when the cursor is moved over it. (4) A world map is used to display the geographical distribution of the top 100 isolates (Figure 2E). The pie chart/table is used to indicate the distribution of different organisms with similar antimicrobial resistance profiles in the same geographical location. This helps users to trace the transmission routes of the isolates. (5) The results of the phylogenetic relatedness tree analysis obtained through cgMLST are presented (Figure 2F). The server allows users to download the phylogenetic relatedness tree in the Newick and pdf formats.
EXAMPLE ANALYSIS
To examine the effectiveness of the 5NosoAE server in comparing antimicrobial resistance profiles, a genome sequence of A. baumannii (NCBI Assembly ID: GCA_019728755.1) was used as the query in the example analysis. The strain was isolated from Homo sapiens in 2019 in the United States. The 5NosoAE server first identified its taxonomy as A. baumannii and then detected resistance genes by searching the resfinder_db database and mapping their corresponding antibiotics to generate the antimicrobial resistance profile for the query (Table 2). Nine resistance genes were detected in the genome sequence, and they corresponded to 22 antibiotics distributed in six classes. The results of the antimicrobial resistance profile search are shown in Table 3, where the genome ID, collection date, geographical location, host, allelic distance, SPRO, and p-value are indicated. The 39 isolates had a significantly similar antimicrobial resistance profile with the query (P < 1e−3). The heatmap of antimicrobial resistance profiles is presented in Supplementary Figure S1. Further analysis revealed a time span of >8 years and a spread across four countries (Figure 3A). The identified strains can be grouped by the geographical location and collection date with different leaf colors (Figure 3B). This finding is consistent with the cgMLST analysis, which indicated that the phylogenetic relatedness could be divided into eight groups at an allelic distance cutoff value of 50 (Figure 3B). This finding indicated that these strains have undergone regional evolution. Although their genomes are different (allelic distance of >300), their drug resistance properties are conserved. These strains can be identified using our method, indicating that the 5NosoAE server can be used for conducting drug resistance surveys.
Table 2.
Results of detected resistance genes and their corresponding antibiotics for the Acinetobacter baumannii isolate (NCBI Assembly ID: GCA_019728755.1)
| Resistance gene | Evidence score (SEV) | 1.Gentamicina | 1.Tobramycin | 1.Streptomycin | 1.Amikacin | 1.Isepamicin | 1.Kanamycin | 1.Neomycin | 1.Lividomycin | 1.Paromomycin | 1.Ribostamycin | 1.Netilmicin | 4.Imipenem | 4.Meropenem | 5.Sulfamethoxazole | 11.Erythromycin | 11.Azithromycin | 12.Tetracycline | 12.Doxycycline | 12.Minocycline | 15.Quinupristin | 15.Pristinamycin_IA | 15.Virginiamycin_S |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| armA | 100 | V | V | V | V | V | |||||||||||||||||
| aph(3')-Ia | 100 | V | V | V | V | V | |||||||||||||||||
| aph( 6 )-Id | 100 | V | |||||||||||||||||||||
| aph(3'')-Ib | 99.94 | V | |||||||||||||||||||||
| blaOXA-23 | 100 | V | V | ||||||||||||||||||||
| msr(E) | 100 | V | V | V | V | V | |||||||||||||||||
| mph(E) | 100 | V | |||||||||||||||||||||
| sul2 | 100 | V | |||||||||||||||||||||
| tet(B) | 99.75 | V | V | V |
aThe front number is used to indicate the class of antibiotics.
Table 3.
Results of the antimicrobial resistance profile search of the Acinetobacter baumannii isolate (NCBI Assembly ID: GCA_019728755.1)
| Rank | Genome ID | Collection date | Geographical location | Host | Allelic distancea | Similarity score (SPRO) | P-value |
|---|---|---|---|---|---|---|---|
| 1 | GCA_019728755.1 | 2019-07-30 | USA | Homo sapiens | 0 | 100.00 | 2.4e-4 |
| 2 | GCA_019724715.1 | 2019-07-30 | USA | Homo sapiens | 7 | 100.00 | 2.4e-4 |
| 3 | GCA_019730295.1 | 2019-07-30 | USA | Homo sapiens | 22 | 100.00 | 2.4e-4 |
| 4 | GCA_001862455.1 | 2013-10-10 | China | Homo sapiens | 69 | 100.00 | 2.4e-4 |
| 5 | GCA_001863475.1 | 2014-04-23 | China | Homo sapiens | 74 | 100.00 | 2.4e-4 |
| 6 | GCA_011603725.1 | 2019 | India | Homo sapiens | 186 | 100.00 | 2.4e-4 |
| 7 | GCA_018285855.1 | 2021-04-22 | USA | Homo sapiens | 245 | 100.00 | 2.4e-4 |
| 8 | GCA_017836795.1 | 2021-03-07 | USA | Homo sapiens | 249 | 100.00 | 2.4e-4 |
| 9 | GCA_001276055.1 | 2014 | India | - | 292 | 100.00 | 2.4e-4 |
| 10 | GCA_018954415.1 | 2021 | USA | Homo sapiens | 300 | 100.00 | 2.4e-4 |
| 11 | GCA_019305625.1 | 2018-10-14 | China | Homo sapiens | 329 | 100.00 | 2.4e-4 |
| 12 | GCA_007113645.1 | 2018-08-13 | China | Homo sapiens | 330 | 100.00 | 2.4e-4 |
| 13 | GCA_016745515.1 | 2019 | China | Homo sapiens | 330 | 100.00 | 2.4e-4 |
| 14 | GCA_016745555.1 | 2019 | China | Homo sapiens | 330 | 100.00 | 2.4e-4 |
| 15 | GCA_016745655.1 | 2019 | China | Homo sapiens | 330 | 100.00 | 2.4e-4 |
| 16 | GCA_016745675.1 | 2019 | China | Homo sapiens | 330 | 100.00 | 2.4e-4 |
| 17 | GCA_016745735.1 | 2019 | China | Homo sapiens | 330 | 100.00 | 2.4e-4 |
| 18 | GCA_016745755.1 | 2019 | China | Homo sapiens | 330 | 100.00 | 2.4e-4 |
| 19 | GCA_016745775.1 | 2019 | China | Homo sapiens | 330 | 100.00 | 2.4e-4 |
| 20 | GCA_016745795.1 | 2019 | China | Homo sapiens | 330 | 100.00 | 2.4e-4 |
| 21 | GCA_016745915.1 | 2019 | China | Homo sapiens | 330 | 100.00 | 2.4e-4 |
| 22 | GCA_016745995.1 | 2019 | China | Homo sapiens | 330 | 100.00 | 2.4e-4 |
| 23 | GCA_016746035.1 | 2019 | China | Homo sapiens | 330 | 100.00 | 2.4e-4 |
| 24 | GCA_016746075.1 | 2019 | China | Homo sapiens | 330 | 100.00 | 2.4e-4 |
| 25 | GCA_016745475.1 | 2019 | China | Homo sapiens | 331 | 100.00 | 2.4e-4 |
| 26 | GCA_016745585.1 | 2019 | China | Homo sapiens | 331 | 100.00 | 2.4e-4 |
| 27 | GCA_016745625.1 | 2019 | China | Homo sapiens | 331 | 100.00 | 2.4e-4 |
| 28 | GCA_016746015.1 | 2019 | China | Homo sapiens | 331 | 100.00 | 2.4e-4 |
| 29 | GCA_008000275.1 | 2018-11-21 | China | - | 332 | 100.00 | 2.4e-4 |
| 30 | GCA_016745575.1 | 2019 | China | Homo sapiens | 332 | 100.00 | 2.4e-4 |
| 31 | GCA_016745805.1 | 2019 | China | Homo sapiens | 332 | 100.00 | 2.4e-4 |
| 32 | GCA_016745955.1 | 2019 | China | Homo sapiens | 334 | 100.00 | 2.4e-4 |
| 33 | GCA_900494465.1 | 2016 | Thailand | Homo sapiens | 271 | 99.98 | 2.4e-4 |
| 34 | GCA_002249605.1 | 2016-10-22 | Thailand | Homo sapiens | 273 | 99.98 | 2.4e-4 |
| 35 | GCA_900495075.1 | 2016 | Thailand | Homo sapiens | 273 | 99.98 | 2.4e-4 |
| 36 | GCA_900495055.1 | 2016 | Thailand | Homo sapiens | 274 | 99.98 | 2.4e-4 |
| 37 | GCA_900495155.1 | 2016 | Thailand | Homo sapiens | 276 | 99.98 | 2.4e-4 |
| 38 | GCA_010178955.1 | 2019 | India | Homo sapiens | 204 | 98.43 | 3.1e-4 |
| 39 | GCA_001863575.1 | 2014-08-20 | China | Homo sapiens | 76 | 93.25 | 6.8e-4 |
| 40 | GCA_017349175.1 | 2015 | Malaysia | Homo sapiens | 76 | 89.52 | 1.19e-3 |
| 41 | GCA_017349255.1 | 2015 | Malaysia | Homo sapiens | 76 | 89.52 | 1.19e-3 |
| 42 | GCA_017349095.1 | 2015 | Malaysia | Homo sapiens | 77 | 89.52 | 1.19e-3 |
| 43 | GCA_016484155.1 | 2020-05-14 | USA | Homo sapiens | 82 | 89.52 | 1.19e-3 |
| 44 | GCA_019506555.1 | 2021-07-05 | USA | Homo sapiens | 84 | 89.52 | 1.19e-3 |
| 45 | GCA_011602125.1 | 2019 | India | Homo sapiens | 186 | 89.52 | 1.19e-3 |
| 46 | GCA_002992305.1 | 2015-10-05 | Kuwait | Homo sapiens | 212 | 89.52 | 1.19e-3 |
| 47 | GCA_006937715.1 | 2018-08-26 | China | Homo sapiens | 331 | 89.52 | 1.19e-3 |
| 48 | GCA_011601555.1 | 2019 | India | Homo sapiens | 186 | 89.40 | 1.21e-3 |
| 49 | GCA_011601575.1 | 2019 | India | Homo sapiens | 186 | 89.40 | 1.21e-3 |
| 50 | GCA_011602095.1 | 2019 | India | Homo sapiens | 186 | 89.40 | 1.21e-3 |
aThe allelic distance is calculated by performing cgMLST analysis.
Figure 3.

Analysis of the results of antimicrobial resistance profile search. (A) Stacked histogram of the frequency distribution of years partitioned by country. (B) Dendrogram constructed using cgMLST profiles for the query and 39 hits for Acinetobacter baumannii isolates. Leaf colors indicate the geographical location, which is consistent with the legend of (A). The numbers in the right panel indicate collection years.
In summary, this example indicated that the 5NosoAE server not only provides information on the antimicrobial resistance profile for the query genome but also demonstrates the similarity of the antimicrobial resistance profile, geographical distribution and cgMLST analysis for nosocomial pathogens with similar antibiogram profiles, which can be useful in epidemiological surveys.
CONCLUSION
Herein, we presented the 5NosoAE web server for large-scale antimicrobial resistance profile searching. A unique feature of 5NosoAE is its ability to quantify the difference in antimicrobial resistance among bacterial strains, and this quantification can help identify strains with the same antimicrobial resistance properties. The example demonstrated that the 5NosoAE server is effective for large-scale antimicrobial resistance profile comparison and provides a new option to search strains with the same antimicrobial resistance properties. The 5NosoAE server can be useful for general biologists.
DATA AVAILABILITY
5NosoAE is freely available at https://nosoae.imst.nsysu.edu.tw.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to the Institute of Medical Science and Technology, National Sun Yat-sen University, Taiwan for providing the hardware and software resources.
Author contributions: Conceived and designed the experiments: C.C.C. and Y.Y.L. Performed the experiments and analyzed the data: C.C.C., Y.C.Y. and C.Y.H. Wrote the paper: C.C.C. and Y.Y.L.
Contributor Information
Chih-Chieh Chen, Institute of Medical Science and Technology, National Sun Yat-sen University, Kaohsiung, Taiwan.
Yen-Yi Liu, Department of Public Health, China Medical University, Taichung, Taiwan.
Ya-Chu Yang, Institute of Medical Science and Technology, National Sun Yat-sen University, Kaohsiung, Taiwan.
Chu-Yi Hsu, Institute of Medical Science and Technology, National Sun Yat-sen University, Kaohsiung, Taiwan.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
‘Academic Summit Program’ of Ministry of Science and Technology [MOST 108-2221-E-110-061-MY2 and 110-2221-E-110-032-MY3]; NSYSU-KMU joint research project [NSYSUKMU 109-P007 and 110-P011]; ‘Rapid Screening Research Center for Toxicology and Biomedicine of Higher Education Enhancement Project’ of National Sun Yat-sen University and Ministry of Education, Taiwan. Funding for open access charge: ‘Academic Summit Program’ of Ministry of Science and Technology [MOST 110-2221-E-110-032-MY3].
Conflict of interest statement. None declared.
REFERENCES
- 1. Lotsch F., Albiger B., Monnet D.L., Struelens M.J., Seifert H., Kohlenberg A.European Antimicrobial Resistance Genes Surveillance Network carbapenem-resistant Acinetobacter baumannii capacity survey, g. and group, E.U.-N.c.-r.A.b.c.s. . Epidemiological situation, laboratory capacity and preparedness for carbapenem-resistant Acinetobacter baumannii in Europe, 2019. Euro Surveill. 2020; 25:2001735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Piperaki E.T., Tzouvelekis L.S., Miriagou V., Daikos G.L.. Carbapenem-resistant Acinetobacter baumannii: in pursuit of an effective treatment. Clin. Microbiol. Infect. 2019; 25:951–957. [DOI] [PubMed] [Google Scholar]
- 3. Buehrle D.J., Shields R.K., Clarke L.G., Potoski B.A., Clancy C.J., Nguyen M.H.. Carbapenem-Resistant pseudomonas aeruginosa bacteremia: risk factors for mortality and microbiologic treatment failure. Antimicrob. Agents Chemother. 2017; 61:e01243-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Esposito F., Cardoso B., Fontana H., Fuga B., Cardenas-Arias A., Moura Q., Fuentes-Castillo D., Lincopan N.. Genomic analysis of carbapenem-resistant pseudomonas aeruginosa isolated from urban rivers confirms spread of clone sequence type 277 carrying broad resistome and virulome beyond the hospital. Frontiers in Microbiology. 2021; 12:701921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fang L., Xu H., Ren X., Li X., Ma X., Zhou H., Hong G., Liang X.. Epidemiology and risk factors for carbapenem-resistant klebsiella pneumoniae and subsequent MALDI-TOF MS as a tool to cluster KPC-2-producing Klebsiella pneumoniae, a retrospective study. Front. Cell. Infect. Microbiol. 2020; 10:462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ernst C.M., Braxton J.R., Rodriguez-Osorio C.A., Zagieboylo A.P., Li L., Pironti A., Manson A.L., Nair A.V., Benson M., Cummins K.et al.. Adaptive evolution of virulence and persistence in carbapenem-resistant klebsiella pneumoniae. Nat. Med. 2020; 26:705–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cetinkaya Y., Falk P., Mayhall C.G.. Vancomycin-resistant enterococci. Clin. Microbiol. Rev. 2000; 13:686–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Davis E., Hicks L., Ali I., Salzman E., Wang J., Snitkin E., Gibson K., Cassone M., Mody L., Foxman B.. Epidemiology of vancomycin-resistant enterococcus faecium and Enterococcus faecalis colonization in nursing facilities. Open Forum Infect. Dis. 2020; 7:ofz553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cong Y., Yang S., Rao X.. Vancomycin resistant staphylococcus aureus infections: a review of case updating and clinical features. J. Adv. Res. 2020; 21:169–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shariati A., Dadashi M., Moghadam M.T., van Belkum A., Yaslianifard S., Darban-Sarokhalil D.. Global prevalence and distribution of vancomycin resistant, vancomycin intermediate and heterogeneously vancomycin intermediate staphylococcus aureus clinical isolates: a systematic review and meta-analysis. Sci. Rep. 2020; 10:12689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kumarasamy K.K., Toleman M.A., Walsh T.R., Bagaria J., Butt F., Balakrishnan R., Chaudhary U., Doumith M., Giske C.G., Irfan S.et al.. Emergence of a new antibiotic resistance mechanism in india, pakistan, and the UK: a molecular, biological, and epidemiological study. Lancet Infect. Dis. 2010; 10:597–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Peterson E., Kaur P.. Antibiotic resistance mechanisms in bacteria: relationships between resistance determinants of antibiotic producers, environmental bacteria, and clinical pathogens. Front. Microbiol. 2018; 9:2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Avershina E., Shapovalova V., Shipulin G.. Fighting antibiotic resistance in hospital-acquired infections: current state and emerging technologies in disease prevention, diagnostics and therapy. Front. Microbiol. 2021; 12:707330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Burnham C.-A.D., Leeds J., Nordmann P., O’Grady J., Patel J.. Diagnosing antimicrobial resistance. Nat. Rev. Microbiol. 2017; 15:697–703. [DOI] [PubMed] [Google Scholar]
- 15. Kumburu H.H., Sonda T., van Zwetselaar M., Leekitcharoenphon P., Lukjancenko O., Mmbaga B.T., Alifrangis M., Lund O., Aarestrup F.M., Kibiki G.S.. Using WGS to identify antibiotic resistance genes and predict antimicrobial resistance phenotypes in MDR acinetobacter baumannii in tanzania. J. Antimicrob. Chemother. 2019; 74:1484–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Argimón S., Masim M.A.L., Gayeta J.M., Lagrada M.L., Macaranas P.K.V., Cohen V., Limas M.T., Espiritu H.O., Palarca J.C., Chilam J.et al.. Integrating whole-genome sequencing within the national antimicrobial resistance surveillance program in the philippines. Nat. Commun. 2020; 11:2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Alcock B.P., Raphenya A.R., Lau T.T.Y., Tsang K.K., Bouchard M., Edalatmand A., Huynh W., Nguyen A.V., Cheng A.A., Liu S.et al.. CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2020; 48:D517–D525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bortolaia V., Kaas R.S., Ruppe E., Roberts M.C., Schwarz S., Cattoir V., Philippon A., Allesoe R.L., Rebelo A.R., Florensa A.F.et al.. ResFinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. 2020; 75:3491–3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Feldgarden M., Brover V., Gonzalez-Escalona N., Frye J.G., Haendiges J., Haft D.H., Hoffmann M., Pettengill J.B., Prasad A.B., Tillman G.E.et al.. AMRFinderPlus and the reference gene catalog facilitate examination of the genomic links among antimicrobial resistance, stress response, and virulence. Sci. Rep. 2021; 11:12728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Davis J.J., Wattam A.R., Aziz R.K., Brettin T., Butler R., Butler R.M., Chlenski P., Conrad N., Dickerman A., Dietrich E.M.et al.. The PATRIC bioinformatics resource center: expanding data and analysis capabilities. Nucleic Acids Res. 2020; 48:D606–D612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sánchez-Busó L., Yeats C.A., Taylor B., Goater R.J., Underwood A., Abudahab K., Argimón S., Ma K.C., Mortimer T.D., Golparian D.et al.. A community-driven resource for genomic epidemiology and antimicrobial resistance prediction of neisseria gonorrhoeae at pathogenwatch. Genome Med. 2021; 13:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Argimón S., Yeats C.A., Goater R.J., Abudahab K., Taylor B., Underwood A., Sánchez-Busó L., Wong V.K., Dyson Z.A., Nair S.et al.. A global resource for genomic predictions of antimicrobial resistance and surveillance of salmonella typhi at pathogenwatch. Nat. Commun. 2021; 12:2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Feng Y., Zou S., Chen H., Yu Y., Ruan Z.. BacWGSTdb 2.0: a one-stop repository for bacterial whole-genome sequence typing and source tracking. Nucleic Acids Res. 2021; 49:D644–D650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Naas T., Oueslati S., Bonnin R.A., Dabos M.L., Zavala A., Dortet L., Retailleau P., Iorga B.I.. Beta-lactamase database (BLDB) - structure and function. J. Enzyme Inhib. Med. Chem. 2017; 32:917–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Srivastava A., Singhal N., Goel M., Virdi J.S., Kumar M.. CBMAR: a comprehensive beta-lactamase molecular annotation resource. Database (Oxford). 2014; 2014:bau111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Moura A., Soares M., Pereira C., Leitão N., Henriques I., Correia A.. INTEGRALL: a database and search engine for integrons, integrases and gene cassettes. Bioinformatics. 2009; 25:1096–1098. [DOI] [PubMed] [Google Scholar]
- 27. Siguier P., Perochon J., Lestrade L., Mahillon J., Chandler M.. ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res. 2006; 34:D32–D36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Quast C., Pruesse E., Yilmaz P., Gerken J., Schweer T., Yarza P., Peplies J., Glockner F.O.. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013; 41:D590–D596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hosking J.R.M., Wallis J.R., Wood E.F.. Estimation of the generalized extreme-value distribution by the method of probability-weighted moments. Technometrics. 1985; 27:251–261. [Google Scholar]
- 30. Liu Y.Y., Lin J.W., Chen C.C.. cano-wgMLST_BacCompare: a bacterial genome analysis platform for epidemiological investigation and comparative genomic analysis. Front. Microbiol. 2019; 10:1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu Y.Y., Chiou C.S., Chen C.C.. PGAdb-builder: a web service tool for creating pan-genome allele database for molecular fine typing. Sci. Rep. 2016; 6:36213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Souvorov A., Agarwala R., Lipman D.J.. SKESA: strategic k-mer extension for scrupulous assemblies. Genome Biol. 2018; 19:153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Felsenstein J. Evolutionary trees from DNA sequences: a maximum likelihood approach. J. Mol. Evol. 1981; 17:368–376. [DOI] [PubMed] [Google Scholar]
- 34. Huerta-Cepas J., Serra F., Bork P.. ETE 3: reconstruction, analysis, and visualization of phylogenomic data. Mol. Biol. Evol. 2016; 33:1635–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
5NosoAE is freely available at https://nosoae.imst.nsysu.edu.tw.