


Abstract
Recent advances in protein structural modelling have enabled the accurate prediction of the holo 3D structures of almost any protein, however protein function is intrinsically linked to the interactions it makes. While a number of computational approaches have been proposed to explore potential biological interactions, they have been limited to specific interactions, and have not been readily accessible for non-experts or use in bioinformatics pipelines. Here we present CSM-Potential, a geometric deep learning approach to identify regions of a protein surface that are likely to mediate protein-protein and protein–ligand interactions in order to provide a link between 3D structure and biological function. Our method has shown robust performance, outperforming existing methods for both predictive tasks. By assessing the performance of CSM-Potential on independent blind tests, we show that our method was able to achieve ROC AUC values of up to 0.81 for the identification of potential protein-protein binding sites, and up to 0.96 accuracy on biological ligand classification. Our method is freely available as a user-friendly and easy-to-use web server and API at http://biosig.unimelb.edu.au/csm_potential.
Graphical Abstract
Graphical Abstract.
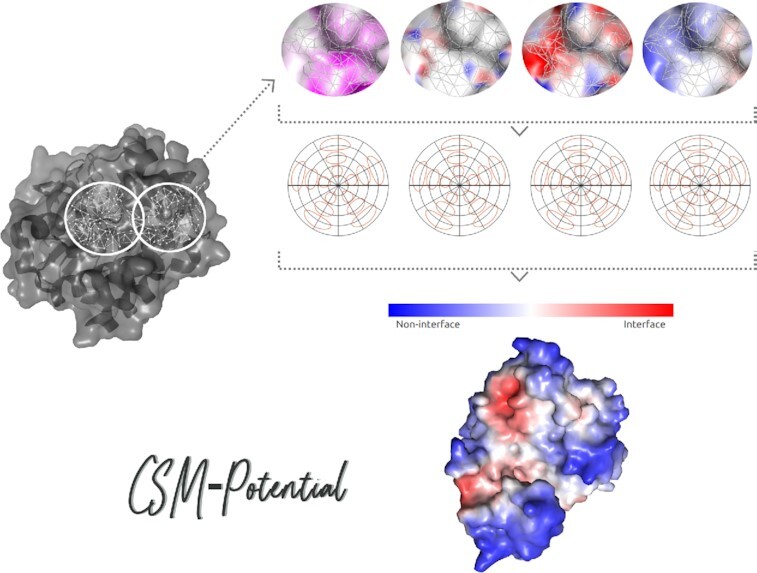
CSM-Potential.
INTRODUCTION
Recent breakthroughs in protein structure prediction by AlphaFold (1,2) and RosettaFold (3), have led to a large proportion of the entire proteome for many organisms, now available in the AlphaFold database (4). These structures, however, lack crucial interactions, which are important for understanding protein function. Consequently, the availability of structural data has prompted an increasing demand for tools that can use these 3D models to help identify key biologically important interacting regions, which is crucial for a better understanding of their biological functions.
Significant efforts have been made towards identification of interacting sites on proteins. For prediction of PPI binding sites previous methods have ranged from those using only information about the protein sequence as input (5,6), to more complex methods, which use a combination of outputs from neural network architectures in combination with changes in relative solvent accessibility upon complexation in a consensus prediction (7). Other approaches have focused on identification of binding sites more likely to participate in interactions with ligands by exploring structural features for classification of pocket regions (8–10), representing an attractive opportunity for elucidating the mechanisms by which these compounds interact with proteins and potentially leading to the design of more safe and effective drugs. More recently, a geometric deep learning framework has been proposed (11) to extract interaction fingerprints from protein surfaces which may then be applied to tasks such as identification of PPI binding sites and binding pocket classification. Despite the diversity of methods currently available, in general these have been developed solely for exploring a specific interaction type, are computationally intensive, and not user friendly.
In this study, we present CSM-Potential, a geometric deep learning approach to identify areas of a protein surface that are likely to participate in protein-protein interactions (PPIs) and protein–ligand interactions (Figure 1). Our method is implemented as an easy-to-use and freely available webserver and an Application Programming Interface (API) to facilitate integration with existing bioinformatics pipelines at http://biosig.unimelb.edu.au/csm_potential.
Figure 1.

CSM-Potential methodology workflow. Experimental data on PPIs and biological ligands were extracted from the curated databases followed by interface identification based on the 3D structure. These were then used to generate molecular surfaces from which geometrical and physicochemical properties are calculated. Protein surfaces were then decomposed into overlapping patches based on a geodesic radius of 12 Å, and used in combination with features calculated on the previous step to generate embeddings from learnable Gaussian Kernels. Finally, neural networks were trained to predict PPI binding sites and to classify biological ligand binding sites.
MATERIALS AND METHODS
Data sets
Experimentally characterised structures of PPI complexes were extracted from the PRISM database of non-redundant PPIs (12) for a total of 8,466 proteins. In addition, 3,536 transient interactions were taken from PDBBind (13), SabDab antibody:antigen database (14) and the ZDock benchmark set (15). Proteins were then clustered at a 30% sequence identity using CD-HIT (16) and one representative member from each cluster was chosen, resulting in 3,362 unique proteins. A pairwise matrix of all TM scores for these proteins was then computed followed by hierarchical clustering, via scikit-learn AgglomerativeClustering method, to split our dataset into training and test sets with 3004 and 358 proteins, respectively.
The database used in this work for biological ligand classification was compiled based on all structures cofactor-binding proteins available in the Protein Data Bank (PDB) (17) (accessed on 16 October 2018), where any of the following seven chemical compounds was present: Adenosine diphosphate (ADP), Coenzyme A (CoA), Flavin adenine dinucleotide (FAD), heme (HEM), Nicotinamide adenine dinucleotide (NAD), Nicotinamide adenine dinucleotide phosphate (NADP) or S-adenosyl methionine (SAM). These have been selected due to their large of structure availability in the PDB and for comparison purposes. Details for each ligand, including their chemical structure, are summarised in Table S1 and Figure S1 in the Supplementary data. This resulted in 1,853 ADP structures, 490 CoA, 2,020 FAD, 4,448 HEM, 1,269 NAD, 1,212 NAP and 393 SAM. Proteins were then clustered based on their sequence identity according to the PDB pre-computed sequence clusters, where two proteins were considered to be similar (near-identical) if the associated clusters of both proteins were the same. The final dataset used for biological ligand classification comprises 1468 structures, which were randomly split into training (72%), validation (8%) and testing sets (20%).
The approach used for curating structures for both predictive tasks, PPI binding site prediction and biological ligand classification, follows the protocol described in previous work (11).
Geometric deep learning neural network
In this study, we apply geometric deep learning to the molecular surface of proteins in order to identify regions more likely to participate in interaction with other proteins and with biologically relevant ligands. For both predictive tasks, we trained end-to-end neural networks using the MaSIF framework (11), which decomposes protein surfaces into overlapping patches based on a geodesic radius, and then uses these in combination with geometric and chemical features to generate embeddings from learnable Gaussian Kernels (18). Here, we expanded this approach by capturing a larger geodesic radius size for patch extraction of 12 Å, and combining this with our well established graph-based signatures (19), which have been extensively applied to investigate the role of genetic mutations on protein function (20–22) and small molecule toxicity (23). These are calculated directly from the protein structure for each residue at the surface and then used to extract distance patterns between atoms characterised by their pharmacophores and compiled in signatures as cumulative distributions.
For the predictive model aiming to identify PPI interaction sites, as the number of non-interface points was usually much larger than the number of interface points, during training our neural network, a random number of non-interface points was selected until an equal number of positive and negative samples was achieved.
Definition of interacting interfaces
PPI interacting interface was defined based on the change in solvent accessible surface area (SASA) on interaction. Here, we consider as part of the interface the region of the surface that becomes inaccessible to solvent molecules upon complex formation. This was done by comparing the difference in SASA, at the residue level, between the individual protomers (unbound protein) and within the complex as follow (24):
 |
where A and B are two proteins participating in a pairwise interaction.
Residues at the surface of the unbound partners which were not at the surface on the bound complex (solvent inaccessible) were then defined as being part of the PPI interface.
For our final dataset of protein–ligand interactions and following previous work (11), after surface and patch generation, if the center point of a patch was less than 3 Å from an atom any of the seven ligands, the patch was labeled as part of the binding pocket of the corresponding ligand.
WEBSERVER
We have implemented CSM-Potential as a user-friendly and freely available web server (http://biosig.unimelb.edu.au/csm_potential). The server back end is developed using Python via the Flask Framework (version 1.0.2), while the front end was built using Materialize framework version 1.0.0. The web server is hosted on a Linux Server running Apache2.
Input
CSM-Potential can be used in two different ways: for the identification of PPI sites or for the biological classification of pockets on a protein structure. In both cases, users are required to provide a protein structure by either uploading a file in PDB format or by selecting a structure directly from the PDB or AlphaFold databases using the auto-complete input field (Supplementary Figure S2A). By default, CSM-Potential will run predictions on the whole input structure, however, users may indicate specific monomers to be automatically extracted before running the predictions for structures where multiple proteins are present. Furthermore, the biological classification of pockets option requires one extra step before running the predictions, where the user must choose from a list of pockets identified using Ghecom (25) (Supplementary Figure S3A). Finally, for both options, users may provide an email address, which will be later used for notifying the user about the job's results.
Output
For prediction of PPI binding sites (Supplementary Figure S2B), CSM-Potential summarises interface scores for each residue on the input protein structure at a sequence level using the FeatureViewer component (26). In addition, predicted scores are mapped onto the input 3D structure and displayed in an interactive viewer using NGLviewer molecular graphics library (27). Finally, users can download their input protein structure with the predictions annotated on the b-factor column.
For biological ligand classification (Supplementary Figure S3B), the results page shows the input 3D structure with the selected pocket region highlighted by default using NGLviewer and a downloadable table summarising the prediction score for all seven different biological ligands. For each ligand, the predicted score represents the likelihood of binding to the selected pocket. For a given ligand, additional information, such as molecular weight and number of hydrogen acceptors and donors, can be accessed via the Details button on the results table, as well as the ligand depiction built via SmilesDrawer (28) based on its canonical SMILES.
VALIDATION
Performance on training
We evaluated the performance of CSM-Potential to predict PPI binding sites on two different types of cross-validations on our training set. For each cross-validation type, we repeated the experiments five times and reported mean values for each evaluation metric. First we randomly selected 80% of our data for training and the remaining 20% for testing. Performance is reported in terms of ROC AUC values, which were calculated for each protein in the testing set and overall performance is reported after averaging values for all entries. Here, our method achieved an overall ROC AUC of 0.82 on average. Using a similar setup, but varying the proportion of data split between training and testing sets to 50% each, the performance of our approach remained consistent with ROC AUC of 0.79. Additional evaluation metrics, including Matthews correlation coefficient (MCC) and F1 score, have been summarised in Supplementary Table S2, corroborating the robustness of our trained model on CV1 and CV2. The final predictive model was then built using all entries in the training set and evaluated on a blind test set.
To train a predictive model to explore biological ligand binding sites, here we used the training and validation set described in the Materials and Methods section. Performance on the validation set was assessed at each epoch (one forward and backward pass of all entries available in the training set) and used to select the best network after each epoch. Finally, we sampled each pocket 100 times and averaged the resulting 100 predictions to obtain the final prediction, similar to previous work. At the end of our training procedure, our approach achieved a balanced accuracy of up to 0.75 and ROC AUC of 0.87.
Blind test
Here, we compared the results of our PPI binding site predictive model with those reported for MaSIF-site (11) on all 358 entries of our blind-test and three subsets of PPI interactions: transient (59), and large (74) and small (74) hydrophobic interactions. For comparison purposes, results are presented in terms of median ROC AUC per protein. Overall, CSM-Potential achieved a median ROC AUC of 0.90, while MaSIF-site achieved 0.87 ROC AUC. For the three subsets of PPI interactions, CSM-Potential outperformed MaSIF-site for all cases, achieving ROC AUC values ranging from 0.84 to 0.93 (Supplementary Figure S4). Furthermore, here we compared the results of our method with MaSIF-site, SPPIDER (7) and PSIVER (5) on a subset of 53 single-chain transient interactions. SPPIDER AND PSIVER have been previously shown to have top performance in a critical study assessing the robustness of computational predictors of PPI interfaces (29). Here, CSM-Potential achieved the highest performance over all other methods with a ROC AUC of 0.84, while MaSIF-site, SPPIDER and PSIVER achieved 0.81, 0.65 and 0.62, respectively (Figure 2A). Additional evaluation metrics (as median per protein) are shown in Table 1, showing that CSM-Potential and MaSIF-site have a more balanced prediction when differentiating between interface and non-interface residues.
Figure 2.

Performance comparison of CSM-Potential with alternative methods on both predictive tasks: identification of PPI binding sites and biological ligands binding sites classification. Panel A shows overall median ROC AUC values per protein on predictive models for the identification of PPI binding sites, while Panel B summarises performance in terms of ROC curves.
Table 1.
Performance comparison for PPI binding site prediction on a non-redundant blind test
| Method | AUC | MCC | F1 | Sensitivity | Specificity |
|---|---|---|---|---|---|
| CSM-Potential | 0.84 | 0.23 | 0.24 | 0.78 | 0.75 |
| MaSIF-site | 0.81 | 0.21 | 0.20 | 0.75 | 0.73 |
| SPPIDER | 0.65 | 0.11 | 0.19 | 0.25 | 0.86 |
| PSIVER | 0.61 | 0.07 | 0.43 | 0.43 | 0.67 |
For assessing the performance of our predictive model for the classification of biological ligands binding sites, we first investigated how accurate CSM-Potential performed on the blind-test for each specific ligand separately (summarised in Supplementary Figure S5). Accuracies varied from 0.56 for pockets associated with SAM to 0.96 for pockets where HEM is bound. The former may be related to the small number of entries for SAM in our training set (only 23 cofactor-binding proteins), while the latter, in addition to having the highest number of entries available for training of the predictive model (57 cofactor-binding proteins), it also shows a more unique chemical structure when compared with the other ligands (Supplementary Figure S1).
Finally, here we compared the results of our method with three other tools that explore structural features for pocket classification and have shown to perform well in previous study (30): MaSIF-ligand (11), KRIPO (8) and ProBiS (9). Except for MaSIF-ligand, the other three methods have been shown to achieve top performance in a previous study (30). As KRIPO does not support fingerprints for the HEME ligand, this was removed from the comparison. Overall, CSM-Potential achieved ROC AUC of 0.96, which is comparable to MaSIF-ligand with 0.95, and superior to the performance of KRIPO and ProBiS with ROC AUC of 0.93 and 0.90, respectively (Figure 2B). A more recent version of the MaSIF framework has been proposed, namely dMaSIF (31), however we opted to not include it in our comparison given that we were unable to run it locally based on the instructions available.
CONCLUSION
Here, we present CSM-Potential, a webserver that combines geometric deep learning with our graph based signatures for predicting likely binding regions based on protein surface. Our method has similar performance to state of the art methods with robust and accurate predictions on non-redundant blind test sets for identification of PPI binding sites and for the classification of biological ligands binding sites. We believe CSM-Potential will be of great value to the study of protein function prediction for both more experienced and also non-expert users. Our method is freely available as an easy-to-use webserver and API to facilitate large-scale processing and incorporation into analytical pipelines at http://biosig.unimelb.edu.au/csm_potential.
DATA AVAILABILITY
CSM-Potential is freely available (no login or license required) as an easy-to-use webserver and API at http://biosig.unimelb.edu.au/csm_potential. Documentation on how to use the webserver and examples for querying the API using the Python programming language are available at http://biosig.unimelb.edu.au/csm_potential/help. Finally, all the experimental data used to train and evaluate the predictive models described in this work can be accessed at http://biosig.unimelb.edu.au/csm_potential/data.
Supplementary Material
Contributor Information
Carlos H M Rodrigues, Computational Biology and Clinical Informatics, Baker Heart and Diabetes Institute, Melbourne, Victoria, Australia; School of Chemistry and Molecular Biosciences, University of Queensland, Brisbane, Queensland, Australia.
David B Ascher, Computational Biology and Clinical Informatics, Baker Heart and Diabetes Institute, Melbourne, Victoria, Australia; School of Chemistry and Molecular Biosciences, University of Queensland, Brisbane, Queensland, Australia.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Investigator Grant from the National Health and Medical Research Council (NHMRC) of Australia [GNT1174405 to D.B.A.]; Medical Research Council [MR/M026302/1 to D.B.A.]; Victorian Government's Operational Infrastructure Support Program. Funding for open access charge: MRC.
Conflict of interest statement. None declared.
REFERENCES
- 1. Jumper J., Evans R., Pritzel A., Green T., Figurnov M., Ronneberger O., Tunyasuvunakool K., Bates R., Zidek A., Potapenko A.et al.. Highly accurate protein structure prediction with AlphaFold. Nature. 2021; 596:583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Evans R., O’Neill M., Pritzel A., Antropova N., Senior A.W., Green T., Žídek A., Bates R., Blackwell S., Yim J.J.B.. Protein complex prediction with AlphaFold-Multimer. 2021; bioRxiv doi:04 October 2021, preprint: not peer reviewed 10.1101/2021.10.04.463034. [DOI]
- 3. Baek M., DiMaio F., Anishchenko I., Dauparas J., Ovchinnikov S., Lee G.R., Wang J., Cong Q., Kinch L.N., Schaeffer R.D.et al.. Accurate prediction of protein structures and interactions using a three-track neural network. Science. 2021; 373:871–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tunyasuvunakool K., Adler J., Wu Z., Green T., Zielinski M., Zidek A., Bridgland A., Cowie A., Meyer C., Laydon A.et al.. Highly accurate protein structure prediction for the human proteome. Nature. 2021; 596:590–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Murakami Y., Mizuguchi K.. Applying the Naive Bayes classifier with kernel density estimation to the prediction of protein-protein interaction sites. Bioinformatics. 2010; 26:1841–1848. [DOI] [PubMed] [Google Scholar]
- 6. Xue L.C., Dobbs D., Honavar V.. HomPPI: a class of sequence homology based protein-protein interface prediction methods. BMC Bioinformatics. 2011; 12:244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Porollo A., Meller J.. Prediction-based fingerprints of protein-protein interactions. Proteins. 2007; 66:630–645. [DOI] [PubMed] [Google Scholar]
- 8. Ritschel T., Schirris T.J., Russel F.G.. KRIPO - a structure-based pharmacophores approach explains polypharmacological effects. J. Cheminform. 2014; 6:O26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Konc J., Miller B.T., Stular T., Lesnik S., Woodcock H.L., Brooks B.R., Janezic D. ProBiS-CHARMMing: web interface for prediction and optimization of ligands in protein binding sites. J. Chem. Inf. Model. 2015; 55:2308–2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shulman-Peleg A., Nussinov R., Wolfson H.J.. Recognition of functional sites in protein structures. J. Mol. Biol. 2004; 339:607–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gainza P., Sverrisson F., Monti F., Rodola E., Boscaini D., Bronstein M.M., Correia B.E.. Deciphering interaction fingerprints from protein molecular surfaces using geometric deep learning. Nat. Methods. 2020; 17:184–192. [DOI] [PubMed] [Google Scholar]
- 12. Baspinar A., Cukuroglu E., Nussinov R., Keskin O., Gursoy A.. PRISM: a web server and repository for prediction of protein-protein interactions and modeling their 3D complexes. Nucleic Acids Res. 2014; 42:W285–W289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu Z., Li Y., Han L., Li J., Liu J., Zhao Z., Nie W., Liu Y., Wang R.. PDB-wide collection of binding data: current status of the PDBbind database. Bioinformatics. 2015; 31:405–412. [DOI] [PubMed] [Google Scholar]
- 14. Dunbar J., Krawczyk K., Leem J., Baker T., Fuchs A., Georges G., Shi J., Deane C.M.. SAbDab: the structural antibody database. Nucleic Acids Res. 2014; 42:D1140–D1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vreven T., Moal I.H., Vangone A., Pierce B.G., Kastritis P.L., Torchala M., Chaleil R., Jimenez-Garcia B., Bates P.A., Fernandez-Recio J.et al.. Updates to the integrated protein-protein interaction benchmarks: docking benchmark version 5 and affinity benchmark version 2. J. Mol. Biol. 2015; 427:3031–3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fu L., Niu B., Zhu Z., Wu S., Li W.. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics. 2012; 28:3150–3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., Shindyalov I.N., Bourne P.E.. The protein data bank. Nucleic Acids Res. 2000; 28:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Monti F., Boscaini D., Masci J., Rodola E., Svoboda J., Bronstein M.M.. Proc. IEEE Conf. Comput. Vis. Pattern Recogn. 2017; 5115–5124. [Google Scholar]
- 19. Pires D.E., de Melo-Minardi R.C., da Silveira C.H., Campos F.F., Meira W. Jr. aCSM: noise-free graph-based signatures to large-scale receptor-based ligand prediction. Bioinformatics. 2013; 29:855–861. [DOI] [PubMed] [Google Scholar]
- 20. Pires D.E., Ascher D.B., Blundell T.L.. mCSM: predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics. 2014; 30:335–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rodrigues C.H.M., Pires D.E.V., Ascher D.B.. mmCSM-PPI: predicting the effects of multiple point mutations on protein-protein interactions. Nucleic Acids Res. 2021; 49:W417–W424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Myung Y., Rodrigues C.H.M., Ascher D.B., Pires D.E.V.. mCSM-AB2: guiding rational antibody design using graph-based signatures. Bioinformatics. 2020; 36:1453–1459. [DOI] [PubMed] [Google Scholar]
- 23. Pires D.E., Blundell T.L., Ascher D.B.. pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015; 58:4066–4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lee B., Richards F.M.. The interpretation of protein structures: estimation of static accessibility. J. Mol. Biol. 1971; 55:379–400. [DOI] [PubMed] [Google Scholar]
- 25. Kawabata T. Detection of multiscale pockets on protein surfaces using mathematical morphology. Proteins. 2010; 78:1195–1211. [DOI] [PubMed] [Google Scholar]
- 26. Garcia L., Yachdav G., Martin M.J.. FeatureViewer, a BioJS component for visualization of position-based annotations in protein sequences. F1000Res. 2014; 3:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rose A.S., Bradley A.R., Valasatava Y., Duarte J.M., Prlic A., Rose P.W.. NGL viewer: web-based molecular graphics for large complexes. Bioinformatics. 2018; 34:3755–3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Probst D., Reymond J.L.. SmilesDrawer: parsing and drawing SMILES-Encoded molecular structures using client-side javascript. J. Chem. Inf. Model. 2018; 58:1–7. [DOI] [PubMed] [Google Scholar]
- 29. Xue L.C., Dobbs D., Bonvin A.M., Honavar V.. Computational prediction of protein interfaces: a review of data driven methods. FEBS Lett. 2015; 589:3516–3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ehrt C., Brinkjost T., Koch O.. A benchmark driven guide to binding site comparison: an exhaustive evaluation using tailor-made data sets (ProSPECCTs). PLoS Comput. Biol. 2018; 14:e1006483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sverrisson F., Feydy J., Correia B.E., Bronstein M.M.. Fast End-to-End Learning on Protein Surfaces. Proceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition. 2021; 2021:15272–15281. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
CSM-Potential is freely available (no login or license required) as an easy-to-use webserver and API at http://biosig.unimelb.edu.au/csm_potential. Documentation on how to use the webserver and examples for querying the API using the Python programming language are available at http://biosig.unimelb.edu.au/csm_potential/help. Finally, all the experimental data used to train and evaluate the predictive models described in this work can be accessed at http://biosig.unimelb.edu.au/csm_potential/data.