


Abstract
Protein-ligand blind docking is a powerful method for exploring the binding sites of receptors and the corresponding binding poses of ligands. It has seen wide applications in pharmaceutical and biological researches. Previously, we proposed a blind docking server, CB-Dock, which has been under heavy use (over 200 submissions per day) by researchers worldwide since 2019. Here, we substantially improved the docking method by combining CB-Dock with our template-based docking engine to enhance the accuracy in binding site identification and binding pose prediction. In the benchmark tests, it yielded the success rate of ∼85% for binding pose prediction (RMSD < 2.0 Å), which outperformed original CB-Dock and most popular blind docking tools. This updated docking server, named CB-Dock2, reconfigured the input and output web interfaces, together with a highly automatic docking pipeline, making it a particularly efficient and easy-to-use tool for the bioinformatics and cheminformatics communities. The web server is freely available at https://cadd.labshare.cn/cb-dock2/.
Graphical Abstract
Graphical Abstract.
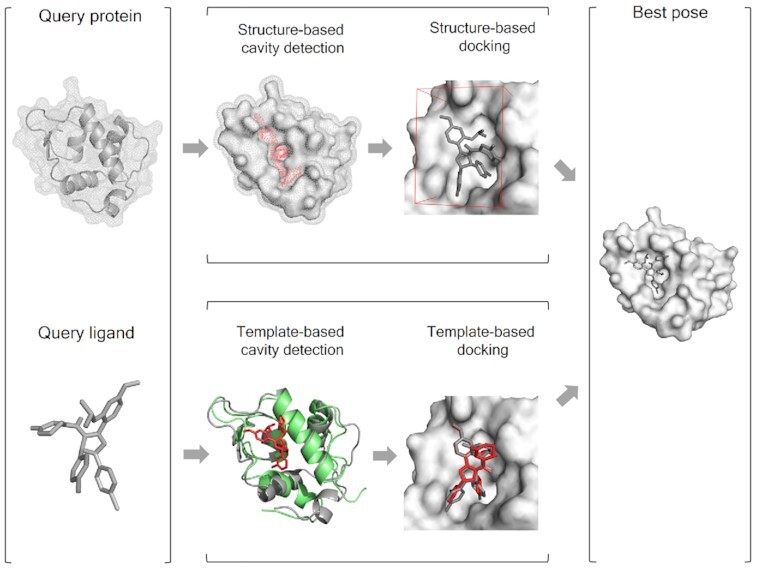
CB-Dock2 integrates the structure-based and template-based blind docking algorithms.
INTRODUCTION
Predicting interactions between proteins and small molecules plays key roles in deciphering a wide variety of biological processes and is crucial to understanding protein functions as well as leveraging drug development (1,2). A powerful approach for this purpose is protein–ligand blind docking, which identifies the binding regions of a protein, and simultaneously predicts the binding pose of a molecule (3,4). Recently, there is an increasingly urgent need of blind docking for the reason that massive protein structures have been determined by AlphaFold2 (5) or RoseTTAFold (6), opening the opportunities to explore new target therapies (7,8). The state-of-the-art blind docking methods such as SwissDock (9), COACH-D (10), EDock (11), MTiAutoDock (12) etc. have been extensively used in exploring potential binding sites or ligand-binding poses.
CB-Dock is a protein–ligand blind docking server developed by our lab (13). It employed our protein-surface-curvature-based cavity detection approach (CurPocket) (13,14) to guide the molecular docking with AutoDock Vina (version 1.1.2) (15,16). Since the original release in 2019, CB-Dock webserver has seen over 200 task submissions worldwide per day, and numerous researchers have used CB-Dock for exploring the binding properties of the compounds as well as the molecular mechanism. For instance, Alvarez et al. discovered a protein functional region by detecting a noncharged binding pocket and docking with acetic acid using CB-Dock (17). Singh et al. utilized the CurPocket algorithm in CB-Dock to predict the binding sites for curcumin on Vibrio cholerae cytolysin (VCC) (18). Particularly, CB-Dock has been broadly used in the study of COVID-19 therapeutic agents (19–24).
The extensive exploitation of CB-Dock can be attributed to the following advantages. (i) Quick result acquisition: the average task time is about one minute, which is suitable for real-time analysis. (ii) High-accuracy prediction: it showed 16%∼30% improvement in terms of docking success rate compared with other blind docking methods in our benchmark (13). (iii) Easy-to-use web interface: it provides interactive and intuitive visualization, which lowers the technical threshold for users. (iv) Exploratory capabilities for docking: it provides centers, sizes and volumes of the predicted cavities to facilitate the migration usage with other molecular docking tools.
Herein, we present an updated version of CB-Dock, in which multiple new features have been added for both computational methods and web interfaces. It inherits the structure-based cavity detection and docking module and integrates a novel template-based molecular docking module to further enhance the accuracy. Our benchmark tests showed that CB-Dock2 surpassed CB-Dock with over 16% improvement in terms of docking success rate. The details of the updates will be described in the following sections.
CB-DOCK2: OVERVIEW AND NEW FEATURES
Computational pipeline
CB-Dock2 performs highly automatic protein–ligand blind docking by four steps: (i) data input, (ii) data processing, (iii) cavity detection and docking, and (iv) visualization and analysis (Figure 1). The data input includes the PDB file of query protein and the MOL2/SDF/PDB file of query ligand. In this new version, ligands can be drawn manually by using a built-in JSME (25) plug-in. The submitted ligand will be processed by adding hydrogens as well as partial charges, and generated initial 3D conformation by RDKit. CB-Dock2 will check the submitted protein, add the missing side-chain atoms (26,27) and hydrogen atoms, send notices about missing residues in a protein (28) and eliminate the co-crystallized waters as well as other het groups. The cavity detection and docking start with template matching, which searches for known complexes with similar proteins and ligands from the prepared complex database. If any similar complexes are retrieved, CB-Dock2 will use two parallel pipelines, i.e. structure-based and template-based blind docking to perform docking simulation. Among them, the structure-based blind docking pipeline is fully inherited from CB-Dock, and the detailed workflow is described in our publication (13). The template-based blind docking pipeline is powered by our recently developed docking method FitDock (29), which is elaborated in the next section. Each pipeline will produce a list of protein–ligand binding sites as well as binding poses. These results will be integrated by merging the same predicted binding sites and retaining the top scoring binding poses. If no similar complex is retrieved, CB-Dock2 will bypass the template-based blind docking pipeline. The visualization and analysis present the final results, which can be visualized and analyzed by interactive NGL Viewers (30) for 3D structures and 2D sequences, together with abundant information about binding sites, template structures, binding scores, contact residues, docking center, cavity volume, etc. Users can adjust and compare the results in massive forms, and download the results for further off-line analysis.
Figure 1.

The overall pipeline of CB-Dock2. It is constructed by modules of data input, data processing, cavity detection and docking, and visualization and analysis.
New feature: template-based blind docking
As the rapid accumulation of structures in Protein Data Bank (31), docking simulation can be profited by introducing the knowledge of the solved protein–ligand complex structures (32,33). CB-Dock2 not only inherits the structure-based cavity detection and docking module from CB-Dock, but also integrates a template-based molecular docking method FitDock (29) that we developed and published recently. This new module can extract the docking modes from the similar complex structures in the protein–ligand database and transfer to the query protein and ligand, with the assumption that similar ligands result in similar binding modes (32). In our comprehensive benchmark tests, FitDock showed 40–60% improvement in terms of docking success rate and an order of magnitude faster over popular docking methods, if template structures were available (29).
The template structure database used in CB-Dock2 is taken from BioLip (version of 2021.09.15) (34) which is currently the most comprehensive protein–ligand interaction database. After removing the interactions involving ions, peptides, DNA/RNA and the artifact ligands, CB-Dock2 includes 214 506 protein–ligand complex structures. For a given query protein and ligand, CB-Dock2 firstly searches for similar ligands using FP2 fingerprint (35) with a minimum threshold of 0.4. Afterwards, the query protein will be superposed to the corresponding complex structure for FitDock. If none of the template structures were found, CB-Dock2 will only perform the structure-based cavity detection and docking. In addition, when more than one template structure was found, CB-Dock2 will merge cavities from two different templates if they share over 50% binding residues. In the other cases, CB-Dock2 will regard them as two different cavities and perform docking independently.
By taking the advantages of both structure-based and template-based docking, CB-Dock2 showed significant improvement over the original CB-Dock. We performed the same docking test as our previous work using Astex Diverse Set (13). It should be mentioned that 82 in 85 test cases employed template-based docking (see Supplementary Table S1) while the others only performed structure-based docking. The result showed that CB-Dock2 achieved 85.9% success rate (the percentage of top-ranking pose within 2.0 Å root mean squared deviation (RMSD) compared with the crystal structure) in the whole data set, which outperformed 69.4% of CB-Dock or 83.5% of FitDock remarkably (Figure 2A). Compared to the state-of-the-art blind docking servers, such as MTiAutoDock (12), SwissDock (9) and COACH-D (10), CB-Dock2 exhibited at least 16% higher success rate (Figure 2B), which suggests the significance of the update.
Figure 2.

The overall performance of CB-Dock2 on Astex Diverse Set. (A) The success rates of the top-ranking binding modes achieved by CB-Dock, FitDock and CB-Dock2 respectively. (B) The success rates of the top-ranking binding modes achieved by MTiAutoDock, SwissDock, COACH-D and CB-Dock2, respectively. COACH-D1 and COACH-D2 refers to that the best pose generated by COACH-D is selected based on c-score and the docking score of AutoDock Vina, respectively.
New feature: the reconfiguration of input and output interfaces
The web interface was redesigned to provide more useful information and intuitive guidance. Firstly, we added a new function for users to perform cavity detection independently (Figure 3). It will illustrate the predicted binding regions in an interactive 3D viewer, in which the cavities can be selected manually for structure-based or template-based docking (Figure 3B, D). Particularly, the residues at the binding regions are highlighted in a sequence panel to facilitate the identification of binding sites. This function can help the users focus their investigation on any known binding pockets. Secondly, we updated the input interface (Figure 4A) and added a molecule editor, powered by JSME (25), to facilitate the input of ligands (Figure 4B). It allows users to upload query ligands by providing SMILES code or by drawing 2D structure in the JSME window. The ligand uploaded can also be previewed and modified in the window, which is convenient for the comparison studies. Thirdly, the docking result page provides plentiful interactive operations for the online analysis (Figure 4D). For instance, it provides a list of interaction residues with the distance threshold defined by the CASP (the sum of the van der Waals radii of the involved atoms plus a tolerance of 0.5Å) (36), enabling users to obtain contact residues more conveniently. And it also exhibits the results of structure-based and template-based docking to facilitate the neck-to-neck comparison. Fourthly, we added more parameter settings to improve the extensibility of CB-Dock2 and enrich the user experience, including modification of the number of cavities and uploading customized complex structures for template-based docking simulations.
Figure 3.

Output interfaces of cavity detection. (A) The cavities detected by analyzing the concave regions on the solvent accessible surface of the query protein. (B) The docking results after clicking the button of ‘BlindDock’. (C) The cavities detected based on homologus templates. (D) The docking results after clicking the button of ‘Template-based Docking’.
Figure 4.

The input and output interfaces of blind docking. (A) The panel for submission of query protein. (B) The panel for submission of query ligand. (C) The job list and status of the submissions. (D) The interactive visualization and meta-analysis for the blind docking results. Users can select and highlight the docking pose for each cavity.
Other optimization
A drawback in CB-Dock is that the cavity detection approach (CurPocket) is extremely time consuming when the number of residues in the query protein is >2000. To address this issue, we optimized the program of calculating protein-surface curvature and enabled rapid processing of the ultra-large proteins. The test results show that the updated method speeds up to 4–5 times faster and can finish in 50s for a 2500-residue protein (Supplementary Figure S1).
CONCLUSIONS AND FUTURE PERSPECTIVES
CB-Dock2 inherits the popular features of the original version and is improved by integrating a template-based blind docking module, which empowers users to obtain potential binding sites and binding modes by referring to known protein–ligand structure information. The reconfiguration of the user interface allows CB-Dock2 to have more options for sophisticated and diverse data submission, and more convenient visualization of the results. The additional ligand drawing interface and the upload module for user-defined template complex, together with the newly integrated FitDock, empower CB-Dock2 to be particularly convenient for drug design and optimization beyond cavity detection and blind docking.
Despite the enhanced features of CB-Dock2, there are still some shortcomings that need to be further addressed in our subsequent work. For instance, the current version cannot distinguish between the asymmetric unit and the biological assembly for the user-uploaded protein structures, which may result in artificial binding cavities. And it does not support fixing the missing residues, optimizing the protein structures and flexibility of receptors, which may be crucial for molecular docking. More importantly, the docking engines should be continuously improved. Hopefully, CB-Dock2 will benefit from our efforts and users’ feedback to become increasingly strong and prevalent.
DATA AVAILABILITY
The CB-Dock2 web server is publicly available at https://cadd.labshare.cn/cb-dock2/.
Supplementary Material
ACKNOWLEDGEMENTS
The authors wish to thank Prof. Yang Zhang of the University of Michigan for invaluable discussion. The authors also wish to thank Dr Maximilian Grimm and Tingfeng Chen of Sichuan University for their assistance in designing the website.
Contributor Information
Yang Liu, Center of Growth, Metabolism and Aging, Key Laboratory of Bio-Resource and Eco-Environment of Ministry of Education, College of Life Sciences, Sichuan University, Chengdu, China.
Xiaocong Yang, Center of Growth, Metabolism and Aging, Key Laboratory of Bio-Resource and Eco-Environment of Ministry of Education, College of Life Sciences, Sichuan University, Chengdu, China.
Jianhong Gan, Center of Growth, Metabolism and Aging, Key Laboratory of Bio-Resource and Eco-Environment of Ministry of Education, College of Life Sciences, Sichuan University, Chengdu, China.
Shuang Chen, Chengdu Institute of Biology, Chinese Academy of Sciences, Chengdu, China.
Zhi-Xiong Xiao, Center of Growth, Metabolism and Aging, Key Laboratory of Bio-Resource and Eco-Environment of Ministry of Education, College of Life Sciences, Sichuan University, Chengdu, China.
Yang Cao, Center of Growth, Metabolism and Aging, Key Laboratory of Bio-Resource and Eco-Environment of Ministry of Education, College of Life Sciences, Sichuan University, Chengdu, China; Animal Disease Prevention and Food Safety Key Laboratory of Sichuan Province, Microbiology and Metabolic Engineering Key Laboratory of Sichuan Province, Chengdu, China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Natural Science Foundation of China [81973243]. Funding for open access charge: National Natural Science Foundation of China.
Conflict of interest statement. None declared.
REFERENCES
- 1. Salentin S., Schreiber S., Haupt V.J., Adasme M.F., Schroeder M.. PLIP: fully automated protein–ligand interaction profiler. Nucleic Acids Res. 2015; 43:W443–W447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jacob L., Vert J.P.. Protein-ligand interaction prediction: an improved chemogenomics approach. Bioinformatics. 2008; 24:2149–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hassan N.M., Alhossary A.A., Mu Y., Kwoh C.-K.. Protein-ligand blind docking using QuickVina-W with inter-process spatio-temporal integration. Sci. Rep. 2017; 7:15451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hetényi C., van der Spoel D.. Efficient docking of peptides to proteins without prior knowledge of the binding site. Protein Sci. 2009; 11:1729–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tunyasuvunakool K., Adler J., Wu Z., Green T., Zielinski M., Žídek A., Bridgland A., Cowie A., Meyer C., Laydon A.et al.. Highly accurate protein structure prediction for the human proteome. Nature. 2021; 596:590–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Baek M., DiMaio F., Anishchenko I., Dauparas J., Ovchinnikov S., Lee G.R., Wang J., Cong Q., Kinch L.N., Schaeffer R.D.et al.. Accurate prediction of protein structures and interactions using a three-track neural network. Science. 2021; 373:871–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rask-Andersen M., Almén M.S., Schiöth H.B.. Trends in the exploitation of novel drug targets. Nat. Rev. Drug Discov. 2011; 10:579–590. [DOI] [PubMed] [Google Scholar]
- 8. Singh V., Mizrahi V.. Identification and validation of novel drug targets in mycobacterium tuberculosis. Drug Discov. Today. 2017; 22:503–509. [DOI] [PubMed] [Google Scholar]
- 9. Grosdidier A., Zoete V., Michielin O.. SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res. 2011; 39:W270–W7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wu Q., Peng Z., Zhang Y., Yang J.. COACH-D: improved protein–ligand binding sites prediction with refined ligand-binding poses through molecular docking. Nucleic Acids Res. 2018; 46:W438–W442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang W., Bell E.W., Yin M., Zhang Y.. EDock: blind protein–ligand docking by replica-exchange monte carlo simulation. J. Cheminform. 2020; 12:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Labbé C.M., Rey J., Lagorce D., Vavruša M., Becot J., Sperandio O., Villoutreix B.O., Tufféry P., Miteva M.A.. MTiOpenScreen: a web server for structure-based virtual screening. Nucleic Acids Res. 2015; 43:W448–W54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu Y., Grimm M., Dai W-tao, Hou M-chun, Xiao Z.X., Cao Y.. CB-Dock: a web server for cavity detection-guided protein–ligand blind docking. Acta Pharmacol. Sin. 2020; 41:138–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cao Y., Li L.. Improved protein–ligand binding affinity prediction by using a curvature-dependent surface-area model. Bioinformatics. 2014; 30:1674–1680. [DOI] [PubMed] [Google Scholar]
- 15. Trott O., Olson A.J.. AutoDock VINA: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010; 31:455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Eberhardt J., Santos-Martins D., Tillack A.F., Forli S.. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021; 61:3891–3898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Alvarez A.F., Rodríguez C., González-Chávez R., Georgellis D.. The Escherichia coli two-component signal sensor BarA binds protonated acetate via a conserved hydrophobic-binding pocket. J. Biol. Chem. 2021; 297:101383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Singh M., Rupesh N., Pandit S.B., Chattopadhyay K.. Curcumin inhibits membrane-damaging pore-forming function of the β-Barrel pore-forming toxin vibrio cholerae cytolysin. Front. Microbiol. 2022; 12:809782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mishra P.M., Nandi C.K.. Structural decoding of a small molecular inhibitor on the binding of SARS-CoV-2 to the ACE 2 receptor. J. Phys. Chem. B. 2021; 125:8395–8405. [DOI] [PubMed] [Google Scholar]
- 20. Ye X.-W., Deng Y.-L., Zhang X., Liu M.-M., Liu Y., Xie Y.-T., Wan Q., Huang M., Zhang T., Xi J.-H.et al.. Study on the mechanism of treating COVID-19 with SHENQI wan based on network pharmacology. Drug Dev. Ind. Pharm. 2021; 47:1279–1289. [DOI] [PubMed] [Google Scholar]
- 21. Somasekharan S.P., Gleave M.. SARS-CoV-2 nucleocapsid protein interacts with immunoregulators and stress granules and phase separates to form liquid droplets. FEBS Lett. 2021; 595:2872–2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Padhi A.K., Seal A., Khan J.M., Ahamed M., Tripathi T.. Unraveling the mechanism of arbidol binding and inhibition of SARS-CoV-2: insights from atomistic simulations. Eur. J. Pharmacol. 2021; 894:173836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hosseini M., Chen W., Xiao D., Wang C.. Computational molecular docking and virtual screening revealed promising SARS-CoV-2 drugs. Precis. Clin. Med. 2021; 4:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dey D., Borkotoky S., Banerjee M.. In silico identification of tretinoin as a SARS-CoV-2 envelope (E) protein ion channel inhibitor. Comput. Biol. Med. 2020; 127:104063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bienfait B., Ertl P.. JSME: a free molecule editor in JavaScript. J. Cheminform. 2013; 5:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dolinsky T.J., Czodrowski P., Li H., Nielsen J.E., Jensen J.H., Klebe G., Baker N.A.. PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 2007; 35:W522–W525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cao Y., Song L., Miao Z., Hu Y., Tian L., Jiang T.. Improved side-chain modeling by coupling clash-detection guided iterative search with rotamer relaxation. Bioinformatics. 2011; 27:785–790. [DOI] [PubMed] [Google Scholar]
- 28. Liu J.L., Miao Z.C., Li L., Xiao Z.X., Cao Y.. DRSP: a structural database for single residue substitutions in PDB. Prog. Biochem. Biophys. 2016; 43:810–816. [Google Scholar]
- 29. Yang X., Liu Y., Gan J., Xiao Z.-X., Cao Y.. FitDock: protein–ligand docking by template fitting. Brief. Bioinform. 2022; 10.1093/bib/bbac087. [DOI] [PubMed] [Google Scholar]
- 30. Rose A.S., Hildebrand P.W.. NGL viewer: a web application for molecular visualization. Nucleic Acids Res. 2015; 43:W576–W579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Berman H.M. The protein data bank. Nucleic Acids Res. 2000; 28:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Paggi J.M., Belk J.A., Hollingsworth S.A., Villanueva N., Powers A.S., Clark M.J., Chemparathy A.G., Tynan J.E., Lau T.K., Sunahara R.K.et al.. Leveraging nonstructural data to predict structures and affinities of protein–ligand complexes. Proc. Natl. Acad. Sci. U.S.A. 2021; 118:e2112621118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang W., Bell E.W., Yin M., Zhang Y.. EDock: blind protein–ligand docking by replica-exchange monte carlo simulation. J. Cheminform. 2020; 12:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yang J., Roy A., Zhang Y.. BioLiP: a semi-manually curated database for biologically relevant ligand–protein interactions. Nucleic Acids Res. 2012; 41:D1096–D1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. O’Boyle N.M., Banck M., James C.A., Morley C., Vandermeersch T., Hutchison G.R.. Open babel: an open chemical toolbox. J. Cheminform. 2011; 3:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gallo Cassarino T., Bordoli L., Schwede T.. Assessment of ligand binding site predictions in CASP10. Proteins. 2014; 82(Suppl. 2):154–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The CB-Dock2 web server is publicly available at https://cadd.labshare.cn/cb-dock2/.