


Abstract
Protein structure exhibits greater complexity and diversity than DNA structure, and usually affects the interpretation of the function, interactions and biological annotations. Reduced amino acid alphabets (Raaa) exhibit a powerful ability to decrease protein complexity and identify functional conserved regions, which motivated us to create RaacFold. The RaacFold provides 687 reduced amino acid clusters (Raac) based on 58 reduction methods and offers three analysis tools: Protein Analysis, Align Analysis, and Multi Analysis. The Protein Analysis and Align Analysis provide reduced representations of sequence-structure according to physicochemical similarities and computational biology strategies. With the simplified representations, the protein structure can be viewed more concise and clearer to capture biological insight than the unreduced structure. Thus, the design of artificial protein will be more convenient, and redundant interference is avoided. In addition, Multi Analysis allows users to explore biophysical variation and conservation in the evolution of protein structure and function. This supplies important information for the identification and exploration of the nonhomologous functions of paralogs. Simultaneously, RaacFold provides powerful 2D and 3D rendering performance with advanced parameters for sequences, structures, and related annotations. RaacFold is freely available at http://bioinfor.imu.edu.cn/raacfold.
Graphical Abstract
Graphical Abstract.
Natural amino acids with similar properties can be simplified and represented by a smaller group of reduced amino acids, thereby having sufficient capacity for capturing conservative features and biological function hidden in structure noise signals.
INTRODUCTION
Proteins are essential to organisms. Decoding their structures can promote the exploration of function and molecular mechanisms in depth. The standard alphabet of protein is usually composed of 20 natural amino acids, which are shared from prokaryotes to eukaryotes (1). These amino acids determine the structure and function of proteins. However, whether there is a minimal amino acid alphabet for modelling protein structure remains to be determined. In addition, the frequency of amino acids and the size of the alphabet are not constant in the process of evolution. The alphabet used in the earliest protein synthesis is closely related to the origin and early evolution of the genetic code (2). Therefore, it is important to consider how many types of amino acids are required to form a biologically active protein. Furthermore, could we design a functional protein with a clear structure that contains fewer than 20 types of amino acids? Can a simplified alphabet reveal a more unambiguous path to protein evolution? Answering these questions will provide a guideline for the design and synthesis of proteins.
In the past few decades, considerable experimental effort has been made to reduce the standard alphabet (3–5). In 1967, Morita et al. reported the presence of many α-helices in soluble random polypeptides are only comprised of three types of amino acids, including Ala, Glu and Lys (6). In 1997, Riddle et al. suggested that five or more types of residues seem necessary for a foldable protein. They successfully reconstructed the well-ordered SH3 domain with five types of amino acids (7). Reducing the alphabet can produce an active catalyst, which supports the notion that primordial enzymes possess low amino acid diversity. It may be applied to synthesize enzymes with novel structures and functions by combinatorial engineering strategies (8). Recently, an alphabet size of 13 amino acids was used to reconstruct an ancestral nucleoside kinase with stable catalytic activity (1). In computational biology, researchers are seeking to simplify the amino acid alphabet, such as by replacing the generalized global model of the observed amino acid substitution model (PAM and BLOSUM), which has been used for particular protein families (9–11). In 1999, Wang and Wang proposed the first knowledge-based theoretical algorithm for Raac by using residue–residue statistical potentials (12). Subsequently, more reduced schemes were published and demonstrated that the reduced alphabets provided sufficient ability for capturing virtual protein folds (8,13,14). The prospective studies of Raac have provided considerable insight into the early life alphabet, phylogenetic inference, and protein design.
Recently, the emergence of AlphaFold largely extended the protein structure data (15), and the demands for analysing these data are increasing rapidly. In recent decades, excellent structure visualization tools have been proposed, including JSmol (jsmol.sf.net), 3Dmol (3dmol.csb.pitt.edu), NGL Viewer (16), etc. These tools can render sequences or structures based on natural amino acid alphabet. However, they lack the functional connection between sequence and structure. As the most popular viewer, Sehnal et al. provided a simple mapping relationship but involves very poor knowledge of amino acids (17), which may obscure the homology of the protein or underestimate the functional consistency of the paralogs. Amino acid residues are the principal contributor to the protein function (18). And the size of the amino acid alphabet directly determines the complexity and computational handicap of protein analysis. The natural alphabet is harder to interpret in protein function inference than reduced alphabets with a smaller size, which also greatly affect the graphical representation of proteins.
If proteins can be engineered using less than 20 types of amino acid, the mapping between sequence and structure becomes more apparent (13). Using the Raac, we have developed PseKRAAC (19), RAACbook (20), Raaclogo (21). However, there is no tool currently available to analyse the link between protein sequence and structure using Raac. This mapping relation is helpful for uncovering the function of complex protein. This opportunity motivated us to create RaacFold. To the best of our knowledge, this is the first web-based sequence and structure viewer based on Raac. RaacFold can be employed to decrease protein complexity, identify conserved regions of functional domains, and offer new insight into the biomacromolecule interaction.
MATERIALS AND METHODS
Frontend
The frontend visualization is developed using Vue.js web framework. It utilizes latest technologies in HTML5, CSS and JavaScript, which are designed for modern browsers to create a dynamically rendered webserver. The graphic viewers are implemented using D3.js, ngl.js, and three.js. The user request from web interface is sent to Nginx via RESTful API to communicate to the backend server.
Backend
The backend API is implemented in Python using the Django framework. MariaDB is used to store Raac data. The data of Raac is collected from related journal articles on PubMed and is manually curated to create the current database. The database contains 687 Raac, which are generated based on 58 reduction methods. The annotations of protein structure are collected from AlphaFold and PDB databases. The current RaacFold contains 494 948 entries of protein structures from 2951 species covering all of model organisms. The server is hosted on Linux servers with 28 physical cores, which uses the Nginx HTTP as a gateway. After the user uploads protein sequences or Uniprot ids to the database and sets essential parameters, RaacFold temporarily stores the inputs and creates a job with a unique ID. The job is then submitted into a task queue, which will handle the job asynchronously and allocate enough computing resources. Once the job is processed, the frontend will generate the analytical report. RaacFold provides extensive help and example case in navigating website and analysing results.
RESULTS
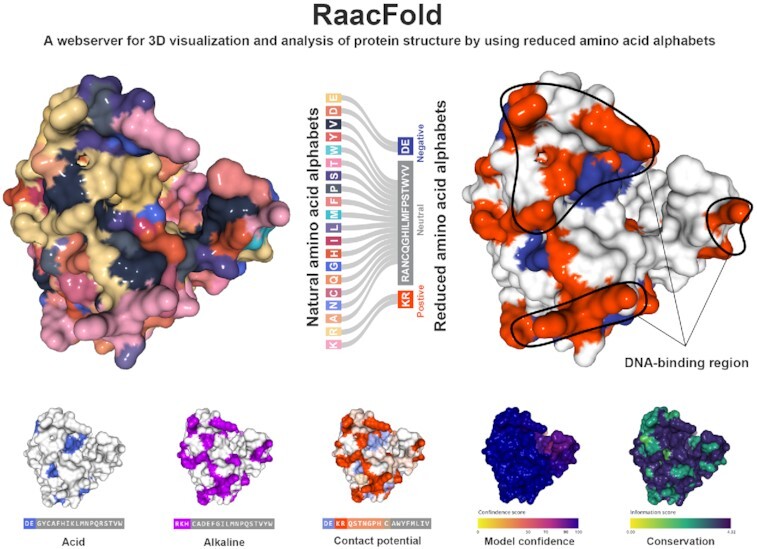
RaacFold contains three analysis tools: Protein Analysis, Align Analysis, and Multi Analysis. All three tools offer detailed analysis of simplified protein sequences and structures. Figure 1A and B shows an overview of RaacFold. In this section, we will describe in more detail of how to use the RaacFold to simplify protein sequences and structures.
Figure 1.

The overview of RaacFold. (A) Input formats for three analysis tools. (B) The framework of database and service. (C) The main window of webserver.
Input data
For Protein Analysis, the input is protein ID/name; for Align Analysis, the inputs are protein sequences containing one or several unaligned sequences in FASTA format. The sequences can either be inputted directly or be uploaded from a FASTA file; for Multi Analysis, the inputs are multiple sequence alignments (MSA) with equal length in FASTA format (Figure 1A).
Raac scheme selection
The desired Raac scheme can be selected from the Raac Selection in the Setting panel on the left (Figure 1C), where the user can select different schemes and clusters. If the user wants to know more about the Raac description, the additional information can be viewed in Information panel on the right of website. This panel contains 11 description items, including the reduction method, description, the authors’ names, source name, PMID, and DOI and other items. If the reduced scheme of interest is not found in the options, the user can also supply their own customized scheme. To do so, the user needs to select ‘custom’ in the Scheme option, and then enter abbreviation of 20 amino acids separated with ‘-’ character. From the custom Raac, RaacFold will generate the corresponding reduced sequence and structure in the Viewers.
Main window
The main window displays the analysis results, which contains four resizable panels: the Structure Viewer displays the 3D protein structure; the Sequence Viewer shows the protein sequences and topology information; the Setting panel contains adjustable parameter for customization; the Information panel provides extra analytical results (Figure 1C). The size of each panel can be adjusted by moving the dividing line. It is worth noting that the Sequence Viewer and Structure Viewer are interlinked. By highlighting the amino acid sequences in the Sequence Viewer, the corresponding region of protein structure is also selected. Additionally, the structure can be configured with different aesthetic options in the Setting panel, which allows the user to identify an apparent association between the reduced sequence features and the structures.
Sequence viewer
The Sequence Viewer (Figure 1C) contains natural and reduced amino acid sequences, motif logo, model confidence, sequence conservation, and annotation features (signal peptide, disulfide bond, active site, etc.). The reduced sequence based on the Raac scheme and the natural sequence is located in the first and second row, respectively. The conversion from the natural sequence to the reduced sequence is done by replacing the first letter of every amino acid with the corresponding letter in the reduced cluster. For example, if the 20 natural amino acids are clustered based on the charge scheme (ED-ANCQGILMFPSTWYV-KR), which contains three reduced clusters, they are negative (E), neutral (A), positive (K), respectively. Any amino acid belongs to the first cluster will be replaced with ‘E’ amino acid, and so forth. In Multi Analysis, RaacLogo (21) is also displayed in this viewer. It contains the information entropy used to evaluate the conservation of reduction, where each cluster is represented by a color. The bottom portion of the viewer contains detailed features of sequence annotation that match the corresponding amino acids. And they can be used for the exploration of protein functions. The rendering sequence supplies png, svg, pdf formats for downloading.
Structure viewer
The Structure Viewer displays the rendered protein structure in 3D representation. Various aesthetic options can be selected for customized display, including surfaces and volumes (molecular surface, gaussian volume, etc.), secondary structure (cartoon, ribbon, trace, etc.), and atoms (ball-stick, space fill, points, etc.). For creating visually engaging and interactive structure, the server provides advanced rendering parameters, which contain material, shadow, contour, roughness, etc.
Application
The most outstanding feature of RaacFold is the 3D visualization of the structure with the reduced representations, which reveal essential determinants of protein function. Although the amino acid preferences were shown by simplified alphabet, they cannot link sequence and function directly because functional regions in structure may be dispersed in sequence. For example, after the alphabet was respectively reduced by polarity and secondary structure, G-protein coupled receptor 61 (GPR61) takes several distinct regions of helix, and the 3D structure further shows a large aggregation of nonpolar amino acids, suggesting the hydrophobicity of the transmembrane helices (Figure 2). In 3D visualization, the reduced representations of structure give the spatial relationships of different amino acid clusters, revealing the determinants of amino acid properties on protein function.
Figure 2.

The analysis of the GPR61 protein using reduction schemes. The natural amino acid structure of the G protein is displayed on the left. The reduced protein structures by using polarity and secondary structure are displayed on the right, respectively.
Another powerful feature of RaacFold is the ability to demonstrate functional differentiation of paralogs by matching scattered sites to functional regions. In general, when a residue is replaced by other similar amino acids, a dramatic change in protein activity is unlikely. Conversely, a disruptive change in the properties of residue site, such as a positive charge turning into a negative charge or an acid turning into a base, can lead to functional differentiation of the proteins. Therefore, when the amino acid alphabet of paralogs is reduced by the same method and parameters, the different regions can reflect the generation of nonhomologous functions of sequence and structure to a certain extent. Alkylation repair protein B homolog (ALKBH) and lysine-specific histone demethylase (KDM) belong to the Fe-2OG dioxygenase family, and both have a double stranded beta helix (DSBH) domain as the catalytic core. However, ALKBH2 binds to double-stranded DNA (22), while KDM4B is H3 histone specific (23). When the amino acid alphabet was reduced according to the charge method, the difference in the substrate binding region between ALKBH2 and KDM4B is evident (Figure 3). For ALKBH2, three regions in the sequence clustered around the substrate binding region carry a large amount of positive charges, resulting in strong selectivity for negatively charged nucleic acids rather than H3 histone. In contrast, the catalytic cleft of KDM4B is dominated by electrically neutral amino acids, which is consistent with the characteristics of binding H3 histone.
Figure 3.

The analysis of the ALKBH2 and KDM4B using Charge reduction scheme. The substrate binding area is marked by black lines.
DISCUSSION
The complexity of the natural amino acid alphabet may obscure the identification of conservative features in the protein sequence and structure. Natural amino acids with similar biochemical properties, atomic arrangements, or frequency distributions can be represented by a smaller group of reduced alphabets, thereby having sufficient capacity for capturing conservative features hidden in noise signals (5). Based on reduced amino acid alphabets, we have developed the first Raac webserver PseKRAAC for feature extraction of protein sequences (19) and the comprehensive Raac database RaacBook for machine learning of protein function prediction (20). Subsequently, the Raac method was further applied to RaacLogo, a motif analysis service for simplifying protein sequence (21).
Currently, RaacFold is the first integrated analysis platform of protein sequence and structure that exhibits powerful ability in conservation, evolution, function, and mechanistic analysis. The performance of RaacFold depends on many factors. The selection of the Raac scheme and the quality of the submitted sequence are the two most critical factors. According to the deposit reduction information provided by RaacFold webserver, users need to choose the appropriate scheme to obtain the best result. For the Multi Analysis tool, due to background noise existing in the multiple sequences, the conservative signal and display quality may be worse. Therefore, we suggest users to select the input sequences by multiple sequence alignment (MSA), which includes enough conserved regions. If the users need a high-quality reduced structure, we recommend they employ more Raac schemes and advanced structure rendering options.
RaacFold shows better sensitivity in decreasing protein complexity and capturing the conservative features hidden in the noise signals. For computational and experimental biologists, it has the potential to become an integral part of protein analysis, which include homology detection, evolutionary inference, and function prediction. Furthermore, this webserver may provide new insight into the protein design in synthetic biology. In future, the RaacFold will be continuously maintained with user feedback, and keep pace with the constant change from Alphafold/PDB databases.
DATA AVAILABILITY
The webserver is available at http://bioinfor.imu.edu.cn/raacfold.
ACKNOWLEDGEMENTS
We would like to thank the anonymous reviewers and Chao Chen, member of the Inner Mongolia Intelligent Union Big Data Academy, for his valuable feedback and suggestions. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Contributor Information
Lei Zheng, State key Laboratory of Reproductive Regulation and Breeding of Grassland Livestock, College of Life Sciences, Inner Mongolia University, Hohhot 010070, China.
Dongyang Liu, Photosynthesis Research Center, Key Laboratory of Photobiology, Institute of Botany, Chinese Academy of Sciences, Beijing 100093, China; University of Chinese Academy of Sciences, Beijing 100049, China.
Yuan Alex Li, WorkFusion Inc, New York, NY 10005, USA.
Siqi Yang, State key Laboratory of Reproductive Regulation and Breeding of Grassland Livestock, College of Life Sciences, Inner Mongolia University, Hohhot 010070, China.
Yuchao Liang, State key Laboratory of Reproductive Regulation and Breeding of Grassland Livestock, College of Life Sciences, Inner Mongolia University, Hohhot 010070, China.
Yongqiang Xing, The Inner Mongolia Key Laboratory of Functional Genome Bioinformatics, School of Life Science and Technology, Inner Mongolia University of Science and Technology, Baotou 014010, China; Department of Biological Sciences, Center for Systems Biology, the University of Texas at Dallas, Richardson, TX 75080-3021, USA.
Yongchun Zuo, State key Laboratory of Reproductive Regulation and Breeding of Grassland Livestock, College of Life Sciences, Inner Mongolia University, Hohhot 010070, China.
FUNDING
National Nature Scientific Foundation of China [62061034, 62171241, 61861036, 62161039]; key technology research program of Inner Mongolia Autonomous Region [2021GG0398]; Science and Technology Major Project of Inner Mongolia Autonomous Region of China to the State Key Laboratory of Reproductive Regulation and Breeding of Grassland Livestock [2019ZD031]. Funding for open access charge: National Nature Scientific Foundation of China [62061034, 62171241, 61861036, 62161039].
Conflict of interest statement. None declared.
REFERENCES
- 1. Shibue R., Sasamoto T., Shimada M., Zhang B., Yamagishi A., Akanuma S.. Comprehensive reduction of amino acid set in a protein suggests the importance of prebiotic amino acids for stable proteins. Sci. Rep. 2018; 8:1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lu Y., Freeland S.. On the evolution of the standard amino-acid alphabet. Genome Biol. 2006; 7:102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Etchebest C., Benros C., Bornot A., Camproux A.C., de Brevern A.G.. A reduced amino acid alphabet for understanding and designing protein adaptation to mutation. Eur. Biophys. J.: EBJ. 2007; 36:1059–1069. [DOI] [PubMed] [Google Scholar]
- 4. Stephenson J.D., Freeland S.J.. Unearthing the root of amino acid similarity. J. Mol. Evol. 2013; 77:159–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chan H.S. Folding alphabets. Nat. Struct. Biol. 1999; 6:994–996. [DOI] [PubMed] [Google Scholar]
- 6. Morita K., Simons E.R., Blout E.R.. Polypeptides. 53. Water-soluble copolypeptides of L-glutamic acid, L-lysine, and L-alanine. Biopolymers. 1967; 5:259–271. [DOI] [PubMed] [Google Scholar]
- 7. Riddle D.S., Santiago J.V., Bray-Hall S.T., Doshi N., Grantcharova V.P., Yi Q., Baker D.. Functional rapidly folding proteins from simplified amino acid sequences. Nat. Struct. Biol. 1997; 4:805–809. [DOI] [PubMed] [Google Scholar]
- 8. Walter K.U., Vamvaca K., Hilvert D.. An active enzyme constructed from a 9-amino acid alphabet. J. Biol. Chem. 2005; 280:37742–37746. [DOI] [PubMed] [Google Scholar]
- 9. Henikoff S., Henikoff J.G.. Amino acid substitution matrices from protein blocks. Proc. Nat. Acad Sci. U.S.A. 1992; 89:10915–10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vilim R.B., Cunningham R.M., Lu B., Kheradpour P., Stevens F.J.. Fold-specific substitution matrices for protein classification. Bioinformatics. 2004; 20:847–853. [DOI] [PubMed] [Google Scholar]
- 11. Teodorescu O., Galor T., Pillardy J., Elber R.. Enriching the sequence substitution matrix by structural information. Proteins. 2004; 54:41–48. [DOI] [PubMed] [Google Scholar]
- 12. Wang J., Wang W.. A computational approach to simplifying the protein folding alphabet. Nat. Struct. Biol. 1999; 6:1033–1038. [DOI] [PubMed] [Google Scholar]
- 13. Solis A.D. Amino acid alphabet reduction preserves fold information contained in contact interactions in proteins. Proteins. 2015; 83:2198–2216. [DOI] [PubMed] [Google Scholar]
- 14. Murphy L.R., Wallqvist A., Levy R.M.. Simplified amino acid alphabets for protein fold recognition and implications for folding. Protein Eng. 2000; 13:149–152. [DOI] [PubMed] [Google Scholar]
- 15. Jumper J., Evans R., Pritzel A., Green T., Figurnov M., Ronneberger O., Tunyasuvunakool K., Bates R., Žídek A., Potapenko A.et al.. Highly accurate protein structure prediction with alphafold. Nature. 2021; 596:583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rose A.S., Hildebrand P.W.. NGL viewer: a web application for molecular visualization. Nucleic Acids Res. 2015; 43:W576–W579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sehnal D., Bittrich S., Deshpande M., Svobodová R., Berka K., Bazgier V., Velankar S., Burley S.K., Koča J., Rose A.S.. Mol* viewer: modern web app for 3D visualization and analysis of large biomolecular structures. Nucleic Acids Res. 2021; 49:W431–W437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liu D., Li G., Zuo Y.. Function determinants of TET proteins: the arrangements of sequence motifs with specific codes. Brief Bioinform. 2019; 20:1826–1835. [DOI] [PubMed] [Google Scholar]
- 19. Zuo Y., Li Y., Chen Y., Li G., Yan Z., Yang L.. PseKRAAC: a flexible web server for generating pseudo K-tuple reduced amino acids composition. Bioinformatics. 2017; 33:122–124. [DOI] [PubMed] [Google Scholar]
- 20. Zheng L., Huang S., Mu N., Zhang H., Zhang J., Chang Y., Yang L., Zuo Y.. RAACBook: a web server of reduced amino acid alphabet for sequence-dependent inference by using chou's five-step rule. Database (Oxford). 2019; 2019:baz131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zheng L., Liu D., Yang W., Yang L., Zuo Y.. RaacLogo: a new sequence logo generator by using reduced amino acid clusters. Brief. Bioinf. 2021; 22:bbaa096. [DOI] [PubMed] [Google Scholar]
- 22. Xu B., Liu D., Wang Z., Tian R., Zuo Y.. Multi-substrate selectivity based on key loops and non-homologous domains: new insight into ALKBH family. Cell Mol. Life Sci. 2021; 78:129–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang Z., Liu D., Xu B., Tian R., Zuo Y.. Modular arrangements of sequence motifs determine the functional diversity of KDM proteins. Brief. Bioinf. 2021; 22:bbaa215. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The webserver is available at http://bioinfor.imu.edu.cn/raacfold.