


Abstract
VRprofile2 is an updated pipeline that rapidly identifies diverse mobile genetic elements in bacterial genome sequences. Compared with the previous version, three major improvements were made. First, the user-friendly visualization could aid users in investigating the antibiotic resistance gene cassettes in conjunction with various mobile elements in the multiple resistance region with mosaic structure. VRprofile2 could compare the predicted mobile elements to the collected known mobile elements with similar architecture. A new mobilome indicator was proposed to give an overall estimation of the mobilome size in individual bacterial genomes. Second, the relationship between antibiotic resistance genes, mobile elements, and host strains would be efficiently examined with the aid of predicted strain's sequence typing, the incompatibility group and the transferability of plasmids. Finally, the updated back-end database, MobilomeDB2, now collected nearly a thousand active mobile elements retrieved from literature or based on prediction. The pre-computed results of the antibiotic resistance gene-carrying mobile elements of >5500 ESKAPEE genomes were also provided. We expect that VRprofile2 will provide better support for researchers interested in bacterial mobile elements and the dissemination of antibiotic resistance. VRprofile2 is freely available to all users without any login requirement at https://tool2-mml.sjtu.edu.cn/VRprofile.
Graphical Abstract
Graphical Abstract.
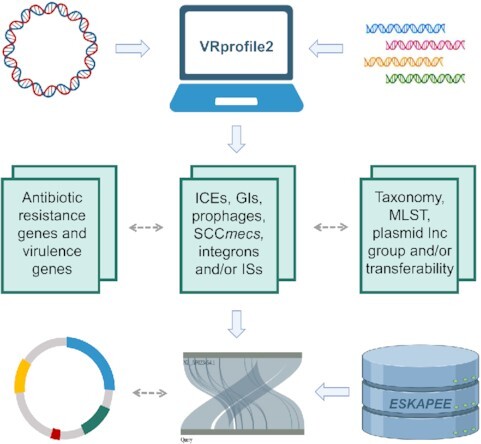
VRprofile2: detection, visualization and comparison of various bacterial MGEs; aid to explore relationships of AGRs, MGEs and host strains; pre-computed mobilome of >5500 genomes of ESKAPEE bacterial group.
INTRODUCTION
The genome sequence of an individual bacterium can be divided into the chromosome backbone, common to the entire species, and a long list of the accessory regions. The latter is termed the ‘mobilome’ (mobile genome) (1,2), which includes a myriad of short IS elements and longer spans of episomal plasmids, transposons, integrons, gene cassettes, prophages, integrative and conjugative elements (ICEs), and genomic islands (GIs). These mobile genetic elements (MGEs) frequently carry antibiotic resistance genes (ARGs). It is a key avenue for the broad distribution of antibiotic resistance in the pathogens with clinical significance, especially for the ESKAPEE bacteria (3). The ESKAPEE group encompasses Gram-positive and Gram-negative species, made up of Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, Enterobacter species and Escherichia coli. The ARG-carrying plasmids, ICEs and other large MGEs often display a mosaic structure composed of multiple modules acquired by complex recombination, insertion, and/or reversion events (4,5). For example, a multiple resistance region contains several antibiotic resistance cassettes in conjunction with various ISs, transposons, as well as integrons and their degenerate elements (6). A recent study revealed that the interaction between the conjugative plasmids and IS elements mediated the ARG transfers (7).
There are many publicly available sources for predicting bacterial MGEs, such as the PLSDB database for plasmids (8), PHASTER tool for prophages (9), ICEberg database for ICEs (10), PAIDB database for pathogenicity islands (11), IslandViewer tool for GIs (12), TnCentral database for transposons (13) and ISfinder database for IS elements (14). They focus on the individual types of MGEs. Several prediction pipelines covering diverse MGE types were recently proposed, such as the MobileElementFinder (15) and MGEfinder (16). MobileElementFinder employs simple BLAST searches against the collected known IS, transposon, and ICE datasets. MGEfinder identifies integrative MGEs from short-read sequencing data but requires bioinformatics to operate its local version. In 2017, we developed the open-access web server VRprofile that allows rapid detection of virulence and antibiotic resistance genes and their flanking MGEs (17). It uses the homologous searches with the combination of the hit co-localization approach to detect gene clusters related to MGEs. However, these tools cannot meet the needs of visualizing and comparing the various mobile elements and their flanking ARGs. To preferably predict the transferability of putative MGEs and to investigate the transmission of antibiotic resistance and virulence factors carried by the bacterial MGEs, it is necessary to obtain the whole picture of mobilome and interpret their interactions, especially in multiple resistance regions, which have a widespread distribution in the clinical isolates of ESKAPEE.
Here, we report the release of VRprofile version 2.0, which offers three major enhancements: (i) graphical representation of the multiple resistance regions with mosaic structures and comparison to the known MGEs with similar architecture; (ii) aid to explore the relationships of antibiotic resistance genes, mobile elements, and host strains; (iii) pre-computed mobilome of >5500 ESKAPEE bacterial genomes. We expect that VRprofile2 will provide better support for researchers interested in bacterial diverse mobile elements and the dissemination of antibiotic resistance.
MATERIALS AND METHODS
Detection and visualization of the MGE regions
The VRprofile2 provides a rapid identification pipeline for various MGEs in the chromosomal or plasmid sequences, including the prophage, ICE, island-like region, IS element, integron and SCCmec (staphylococcal chromosomal cassettes mec) (Supplementary Figure S1). Compared to the original version, the update pipeline newly integrated the following modules: the prediction of integron using IntegronFinder (18), the detection of SCCmec using SCCmecFinder (19), and the BLASTp searches of the transposase (TnpA) and resolvase (TnpR) for the transposons retrieved from the TnCentral (13) and TnRegistry (20) databases. The cluster of ISs/transposons (15) and the flanking ARGs were also detected with an interval <5 kb, which is often present in the multiple resistance region with the complex mosaic structure in a resistance plasmid (7,13,21).
As shown in Supplementary Figure S1, VRprofile2 has different preprocessing modules for the different uploaded sequences. (i) For the chromosomal sequence, the strain typing is done using the script mlst (https://github.com/tseemann/mlst) that employs the PubMLST typing schemes (22). (ii) For the plasmid genome sequence, the plasmid incompatibility group is classified using the tool ABRicate (https://github.com/tseemann/abricate) with an inbuilt database of PlasmidFinder (23). The transferability of the plasmid is predicted as conjugative or mobilizable by using the tool oriTfinder (24). (iii) For the assembled contigs of a bacterial genome or the metagenomic contigs assembled from long reads, the tool viralVerify was integrated into the VRprofile2 pipeline to classify contigs as the fragments of chromosome or plasmid (25). Kraken2 was also employed to determine the taxonomic classification of metagenomic contigs (26).
The VRprofile2 output web page dynamically presents the chromosome or plasmid map with the aid of the AngularPlasmid HTML markup (http://angularplasmid.vixis.com/). And then, an expanded view of the identified MGE region was visualized using SVGene (https://github.com/kblin/svgene), a software that enables in-browser SVG drawings from gene cluster data. These interactive presentations allow the users to conveniently check the genomic coordinates and features of the identified MGE regions and their harboring or flanking ARGs.
Estimation of the mobilome size
To provide an overview of the mobilome of individual strains, we proposed a new indicator, mV (mobilome value), which is estimated by dividing the mobilome size by its whole genome size. The mobilome size is the sum of MGE lengths identified by VRprofile2, irrespective of their types. There are two points to be considered in the mV estimation: (i) for two completely overlapping MGE regions, the length of the larger one is counted while that of the smaller one is omitted. (ii) for two partially overlapping MGE regions, the length of the merged region is counted. In addition, another value, mAV, is employed to examine the mobile elements that harbor ARGs or the ISs/transposons flanked by ARGs with an interval <5 kb. It is calculated by dividing the size of the ARG-associated mobilome by its whole genome size.
Mobilome of ESKAPEE bacteria
The VRprofile2 web server provided the pre-calculated results of the mobilome of the sequenced ESKAPEE bacterial genome. A total of 5,536 completely sequenced genomes of ESKAPEE bacteria were retrieved from GenBank in October 2021, including 235 E. faecium, 1002 S. aureus, 1063 K. pneumoniae, 300 A. baumannii, 359 P. aeruginosa, 207 Enterobacterspp. and 2370 E. coli. The pre-calculated results were listed and visualized on the web pages ‘Browse’, ‘Search’ and ‘Statistics’. On the ‘Browse’ web page, the users can fetch the strain information and the details of identified MGEs. On the ‘Search’ web page, by entering an ARG name, the distribution of the queried ARG in the MGEs of host bacteria can be retrieved (Supplementary Figure S2).
A total of 294 461 MGEs of ESKAPEE were detected, of which 15 650 carry ARGs (Supplementary Table S1). The sequences of these ARG-carrying MGEs were pair-wide aligned using Mash (27) and then grouped using NetworkX with the Mash distances <0.005 (28). The distribution of these obtained ARG-carrying MGE groups among ESKAPEE was examined, and 106 groups were widespread among different MLST of the individual species. These groups were thus labeled as ‘active’.
MobilomeDB2, a collection of active ARG-associated MGEs
The back-end database MobilomeDB2 newly added three datasets related to the active MGEs that are prone to transfer ARGs (Supplementary Table S2). The first dataset, Dataset I, consists of 586 transferable MGEs with experimental supports. It contains 165 conjugative plasmids, 180 ICEs and 241 transposons collected from the literature retrieved from PubMed. The second dataset, Dataset II, was taken from the VRprofile2-predicted active ESKAPEE’s MGEs as defined above. In addition, the third dataset, Dataset III, which contains the publicly available MGE resources, was also integrated into MobilomeDB (Supplementary Table S2). A total of 34 513 plasmids, 65 668 prophage fragments, 1329 pathogenicity islands (PAIs)/antimicrobial resistance islands (REIs), 552 ICEs, and 407 transposons were retrieved from PLSDB, PHASTER, PAIDB, ICEberg, and TnRegistry, respectively. By integrating Mash (27), VRprofile2 can subsequently examine the conserved or rearranged architecture of the predicted MGE regions by comparing them to the MobilomeDB2 datasets.
Implementation of the VRprofile2 server
The VRprofile2 web server is applied to a wide range of bacterial chromosomal and plasmid sequences. It allows users to upload a nucleotide sequence file (in FASTA or GenBank format) containing the completely or partially sequenced bacterial genome or the multiple contigs assembled from the single genome or metagenomic long-read sequencing data. In general, a job can be completed within a minute for a 100-kb plasmid genome sequence or 10 min for a 5-Mb chromosomal sequence. The VRprofile2 server was developed using Python and PHP on a CentOS Linux platform with an Apache Web server. Also, HTML with CSS and bootstrap (http://getbootstrap.com/) were used to generate the web pages. JavaScript and jQuery were used to encounter the dynamic behavior of HTML. Google Chrome is the recommended browser for VRprofile2 for a better user experience.
RESULTS AND DISCUSSION
Case Study 1: detection and comparison of a multiple resistance region in the resistance plasmid
VRprofile2 provides a flexible and biologist-friendly web interface consisting of an input page and a result page. In the example shown in Figure 1, the genome sequence of an E. coli M505 plasmid pM505-NDM5 was uploaded with the file in FASTA format (GenBank accession number: AP023220.2) (29). The result page listed the prediction of the incompatibility group (IncFII) and transferability (conjugative) of pM505-NDM5 (Figure 1A). The identified MGE regions were listed and could also be downloaded as an individual text file, while the MGE regions carrying ARGs were highlighted in the table (Figure 1B). Notably, the result page also offered the functional tabs for visualization and further analysis of the AGR-associated MGEs (Figure 1C and D). A carbapenemase gene NDM-5 and eight other ARGs were clustered in a 27-kb region (MGE1 region in Figure 1C), carrying four IS26 elements. Interestingly, an integron that contained three resistance gene cassettes (MGE2 regions in Figure 1C) was located within this multiple resistance region.
Figure 1.

An overview of VRprofile2 outputs using the E. coli M505 plasmid pM505-NDM52 as an example. (A) The predicted incompatibility group and transferability of pM505-NDM5. (B) A table of the identified MGE regions. (C) A circular representation of pM505-NDM52 showing the locations of the identified MGE1 and MGE2 regions (filled with blue) and ARGs (highlighted in red). Four modules related to plasmid conjugation are also marked, including an origin region of transfer (oriT), relaxase gene, type IV coupling protein (T4CP) gene and gene cluster for the type IV secretion system (T4SS) apparatus. (D) An expanded view of the MGE1 region contains four IS26 elements, one integron (MGE2 region), and nine ARGs. (E) A table containing the Mash-facilitated sequence comparison of pM505-NDM52 against the MobilomeDB Dataset II, which contained the active ARG-associated MGE groups of ESKAPEE bacteria. (F) A table containing the sequence comparison of the MGE2 region against the Dataset II.
In addition, the VRprofile2-identified MGE regions can be compared to the known MGEs with similar architecture by using Mash. MobilomeDB2 Database II contains 106 active ARG-associated MGE groups taken from ESKAPEE. Using the whole plasmid pM505-NDM5 as the query to search against Database II, it showed that the skeleton of pM505-NDM5 was highly similar to an active ARG-associated MGE group, which consists of 14 plasmids of similar structure but carrying different ARGs in E. coli and K. pneumoniae (Figure 1E). Remarkably, there was no match for the MGE1 region (an IS and ARG cluster) but massive hits for the MGE2 region (an integron with resistance cassettes) (Figure 1F), indicating that the NDM-5-carrying resistance region in pM505-NDM5 exhibited a unique combination, i.e., a conserved integron located within a new cluster of ISs.
Three other examples with the input of E. coli chromosomal genome sequence, K. pneumoniae plasmid genome sequence (30), and the MinION sequencing metagenomic contigs (31) are available in Supplementary Figures S3, S4 and S5, respectively.
Case Study 2: examination of the relationship between ARGs, plasmids and host strains
VRprofile2 can facilitate an in-depth analysis of the relationships between the host strains with diverse MLSTs, different mobile elements, and various ARGs. The ‘Statistics’ web page of the VRprofile2 web server provides the estimated mobilome sizes of >5500 ESKAPEE bacterial genomes (Figure 2A). The mV values of these ESKAPEE bacteria ranged from 0.01 to 0.36, while the values vary across different species and even within a species.
Figure 2.

VRprofile2-estimated size of the mobilome borne by individual strains of the ESKAPEE group. (A) Bubble chart of the estimated mobilome size. The complete genome sequences of 5,536 ESKAPEE bacteria were retrieved from GenBank. The abscissa axis represents the mV value of individual strains, calculated by dividing the mobilome size by its whole genome size. The bubble size represents the magnitude of the individual strains under analysis. (B) The distribution of MGE types includes the GI, ICE, integron (In), IS, prophage, SCCmec and plasmid. (C) The relationship between the strains (n= 1063), plasmids (n= 3525), and plasmid-carrying ARGs (n= 8924) of Klebsiella pneumoniae. The heat map presents the profile of the plasmids belonging to different incompatibility groups (Inc). The number (log value) of all the identified Inc groups in the single- or multiple-replicon plasmids (Supplementary Table S3) is plotted alongside the map diagonal. The number of the Inc pairs in the individual multiple-replicon plasmids is depicted in the cell. The left bar chart indicates the plasmid number and the proportion in different sequence typing (MLST) strains. For the plasmid containing two replicon regions, the plasmid number of each Inc group was counted separately. The right bar chart denotes the plasmid transferability. The top bar chart presents the number of blaKPC and other carbapenemase genes (see Supplementary Figure S7 for details). The down bar chart shows the number of ARGs and virulence genes.
There are 3525 plasmids in 1063 K. pneumoniae genomes, representing about 30% of the total mobilome in an individual strain (Figure 2B and Supplementary Figure S6). The relationships between ARGs, plasmids, and host strains are graphically shown in Figure 2C and Supplementary Figure S7 and Table S3. For example, out of all the 3525 plasmids under analysis, 314 plasmids carried 326 blaKPC genes. Notably, 190 blaKPC genes were harbored in 182 IncFII-related plasmids, including 64.3% (117/182) IncFII plasmids in the ST11 strains and 11.5% (21/182) in the ST258 strains.
CONCLUSION
Here, we reported a major update of VRprofile and focused on ARG-carrying mobilome, especially in the ESKAPEE bacteria. The rapid detection, user-friendly visualization and comparison could aid users in investigating the multiple resistance regions and other mosaic regions. With the prediction of the strain MLST, plasmid incompatibility group, and transferability, VRprofile2 could facilitate examining the relationships between ARGs, MGEs and host strains. The newly proposed indicator mV also gives an overall estimation of the mobilome size in an individual bacterial genome. The pre-computed results of the mobilome of >5500 ESKAPEE bacterial genomes are provided. They will be updated and improved regularly from the rapidly growing bacterial genomic data. Ultimately, we propose an updated mobilome detection pipeline to efficiently explore large numbers of mobile elements that play key roles in the rapid dissemination of antibiotic resistance.
Supplementary Material
ACKNOWLEDGEMENTS
The graphical abstract was created with BioRender (BioRender.com).
Contributor Information
Meng Wang, State Key Laboratory of Microbial Metabolism, Joint International Laboratory on Metabolic & Developmental Sciences, School of Life Sciences & Biotechnology, Shanghai Jiao Tong University, Shanghai 200030, China.
Ying-Xian Goh, State Key Laboratory of Microbial Metabolism, Joint International Laboratory on Metabolic & Developmental Sciences, School of Life Sciences & Biotechnology, Shanghai Jiao Tong University, Shanghai 200030, China.
Cui Tai, State Key Laboratory of Microbial Metabolism, Joint International Laboratory on Metabolic & Developmental Sciences, School of Life Sciences & Biotechnology, Shanghai Jiao Tong University, Shanghai 200030, China.
Hui Wang, State Key Laboratory of Pathogens and Biosecurity, Beijing Institute of Microbiology and Epidemiology, Beijing 100071, China.
Zixin Deng, State Key Laboratory of Microbial Metabolism, Joint International Laboratory on Metabolic & Developmental Sciences, School of Life Sciences & Biotechnology, Shanghai Jiao Tong University, Shanghai 200030, China.
Hong-Yu Ou, State Key Laboratory of Microbial Metabolism, Joint International Laboratory on Metabolic & Developmental Sciences, School of Life Sciences & Biotechnology, Shanghai Jiao Tong University, Shanghai 200030, China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Key Research and Development Program of China [2018YFE0102400]; National Natural Science Foundation of China [32070572]; Science and Technology Commission of Shanghai Municipality [19JC1413000 and 19430750600]; Medicine and Engineering Interdisciplinary Research Fund of Shanghai Jiao Tong University [19x190020171]. Funding for open access charge: National Key Research and Development Program of China [2018YFE0102400].
Conflict of interest statement. None declared.
REFERENCES
- 1. Ou H.-Y., Smith R., Lucchini S., Hinton J., Chaudhuri R.R., Pallen M., Barer M.R., Rajakumar K.. ArrayOme: a program for estimating the sizes of microarray-visualized bacterial genomes. Nucleic Acids Res. 2005; 33:e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shao Y., He X., Harrison E.M., Tai C., Ou H.-Y., Rajakumar K., Deng Z.. mGenomeSubtractor: a web-based tool for parallel in silico subtractive hybridization analysis of multiple bacterial genomes. Nucleic Acids Res. 2010; 38:W194–W200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Partridge S.R., Kwong S.M., Firth N., Jensen S.O.. Mobile genetic elements associated with antimicrobial resistance. Clin. Microbiol. Rev. 2018; 31:e0008817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dionisio F., Zilhão R., Gama J.A.. Interactions between plasmids and other mobile genetic elements affect their transmission and persistence. Plasmid. 2019; 102:29–36. [DOI] [PubMed] [Google Scholar]
- 5. Deng Y., Bao X., Ji L., Chen L., Liu J., Miao J., Chen D., Bian H., Li Y., Yu G.. Resistance integrons: class 1, 2 and 3 integrons. Ann. Clin. Microbiol. Antimicrob. 2015; 14:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Harmer C.J., Partridge S.R., Hall R.M.. pDGO100, a type 1 IncC plasmid from 1981 carrying ARI-A and a Tn1696-like transposon in a novel integrating element. Plasmid. 2016; 86:38–45. [DOI] [PubMed] [Google Scholar]
- 7. Che Y., Yang Y., Xu X., Břinda K., Polz M.F., Hanage W.P., Zhang T.. Conjugative plasmids interact with insertion sequences to shape the horizontal transfer of antimicrobial resistance genes. Proc. Natl. Acad. Sci. U.S.A. 2021; 118:e2008731118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schmartz G.P., Hartung A., Hirsch P., Kern F., Fehlmann T., Müller R., Keller A.. PLSDB: advancing a comprehensive database of bacterial plasmids. Nucleic Acids Res. 2022; 50:D273–D278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Arndt D., Grant J.R., Marcu A., Sajed T., Pon A., Liang Y., Wishart D.S.. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016; 44:W16–W21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liu M., Li X., Xie Y., Bi D., Sun J., Li J., Tai C., Deng Z., Ou H.-Y.. ICEberg 2.0: an updated database of bacterial integrative and conjugative elements. Nucleic Acids Res. 2019; 47:D660–D665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yoon S.H., Park Y.-K., Kim J.F.. PAIDB v2.0: exploration and analysis of pathogenicity and resistance islands. Nucleic Acids Res. 2015; 43:D624–D30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bertelli C., Laird M.R., Williams K.P.Simon Fraser University Research Computing Group Simon Fraser University Research Computing Group Lau B.Y., Hoad G., Winsor G.L., Brinkman F.S.L.. IslandViewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017; 45:W30–W35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ross K., Varani A.M., Snesrud E., Huang H., Alvarenga D.O., Zhang J., Wu C., McGann P., Chandler M.. TnCentral: a prokaryotic transposable element database and web portal for transposon analysis. mBio. 2021; 12:e0206021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Siguier P., Perochon J., Lestrade L., Mahillon J., Chandler M.. ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res. 2006; 34:D32–D36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Johansson M.H.K., Bortolaia V., Tansirichaiya S., Aarestrup F.M., Roberts A.P., Petersen T.N.. Detection of mobile genetic elements associated with antibiotic resistance in Salmonellaenterica using a newly developed web tool: MobileElementFinder. J. Antimicrob. Chemother. 2021; 76:101–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Durrant M.G., Li M.M., Siranosian B.A., Montgomery S.B., Bhatt A.S.. A bioinformatic analysis of integrative mobile genetic elements highlights their role in bacterial adaptation. Cell Host & Microbe. 2020; 27:140–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li J., Tai C., Deng Z., Zhong W., He Y., Ou H.-Y.. VRprofile: gene-cluster-detection-based profiling of virulence and antibiotic resistance traits encoded within genome sequences of pathogenic bacteria. Brief. Bioinform. 2018; 19:566–574. [DOI] [PubMed] [Google Scholar]
- 18. Cury J., Jové T., Touchon M., Néron B., Rocha E.P.. Identification and analysis of integrons and cassette arrays in bacterial genomes. Nucleic Acids Res. 2016; 44:4539–4550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kaya H., Hasman H., Larsen J., Stegger M., Johannesen T.B., Allesøe R.L., Lemvigh C.K., Aarestrup F.M., Lund O., Larsen A.R.. SCCmecFinder, a web-based tool for typing of staphylococcal cassette chromosome mec in Staphylococcus aureus using whole-genome sequence data. mSphere. 2018; 3:e0061217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tansirichaiya S., Rahman M.A., Roberts A.P.. The transposon registry. Mob. DNA. 2019; 10:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ellabaan M.M.H., Munck C., Porse A., Imamovic L., Sommer M.O.A.. Forecasting the dissemination of antibiotic resistance genes across bacterial genomes. Nat. Commun. 2021; 12:2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jolley K.A., Bray J.E., Maiden M.C.J.. Open-access bacterial population genomics: BIGSdb software, the pubmlst.org website and their applications. Wellcome Open Res. 2018; 3:124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Carattoli A., Zankari E., García-Fernández A., Voldby Larsen M., Lund O., Villa L., Møller Aarestrup F., Hasman H.. In silico detection and typing of plasmids using plasmidfinder and plasmid multilocus sequence typing. Antimicrob. Agents Chemother. 2014; 58:3895–3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li X., Xie Y., Liu M., Tai C., Sun J., Deng Z., Ou H.-Y.. oriTfinder: a web-based tool for the identification of origin of transfers in DNA sequences of bacterial mobile genetic elements. Nucleic Acids Res. 2018; 46:W229–W234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Antipov D., Raiko M., Lapidus A., Pevzner P.A.. Metaviral SPAdes: assembly of viruses from metagenomic data. Bioinformatics. 2020; 36:4126–4129. [DOI] [PubMed] [Google Scholar]
- 26. Wood D.E., Lu J., Langmead B.. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019; 20:257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ondov B.D., Treangen T.J., Melsted P., Mallonee A.B., Bergman N.H., Koren S., Phillippy A.M.. Mash: fast genome and metagenome distance estimation using minhash. Genome Biol. 2016; 17:132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Matlock W., Chau K.K., AbuOun M., Stubberfield E., Barker L., Kavanagh J., Pickford H., Gilson D., Smith R.P., Gweon H.S.et al.. Genomic network analysis of environmental and livestock F-type plasmid populations. ISME J. 2021; 15:2322–2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sugawara Y., Akeda Y., Hagiya H., Sakamoto N., Takeuchi D., Shanmugakani R.K., Motooka D., Nishi I., Zin K.N., Aye M.M.et al.. Spreading patterns of NDM-producing Enterobacteriaceae in clinical and environmental settings in Yangon, Myanmar. Antimicrob. Agents Chemother. 2019; 63:e01924-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Harmer C.J., Partridge S.R., Hall R.M.. pDGO100, a type 1 IncC plasmid from 1981 carrying ARI-A and a Tn1696-like transposon in a novel integrating element. Plasmid. 2016; 86:38–45. [DOI] [PubMed] [Google Scholar]
- 31. Bertrand D., Shaw J., Kalathiyappan M., Ng A.H.Q., Kumar M.S., Li C., Dvornicic M., Soldo J.P., Koh J.Y., Tong C.et al.. Hybrid metagenomic assembly enables high-resolution analysis of resistance determinants and mobile elements in human microbiomes. Nat. Biotechnol. 2019; 37:937–944. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.