

Abstract
DAVID is a popular bioinformatics resource system including a web server and web service for functional annotation and enrichment analyses of gene lists. It consists of a comprehensive knowledgebase and a set of functional analysis tools. Here, we report all updates made in 2021. The DAVID Gene system was rebuilt to gain coverage of more organisms, which increased the taxonomy coverage from 17 399 to 55 464. All existing annotation types have been updated, if available, based on the new DAVID Gene system. Compared with the last version, the number of gene-term records for most annotation types within the updated Knowledgebase have significantly increased. Moreover, we have incorporated new annotations in the Knowledgebase including small molecule-gene interactions from PubChem, drug-gene interactions from DrugBank, tissue expression information from the Human Protein Atlas, disease information from DisGeNET, and pathways from WikiPathways and PathBank. Eight of ten subgroups split from Uniprot Keyword annotation were assigned to specific types. Finally, we added a species parameter for uploading a list of gene symbols to minimize the ambiguity between species, which increases the efficiency of the list upload and eliminates confusion for users. These current updates have significantly expanded the Knowledgebase and enhanced the discovery power of DAVID.
Graphical Abstract
Graphical Abstract.
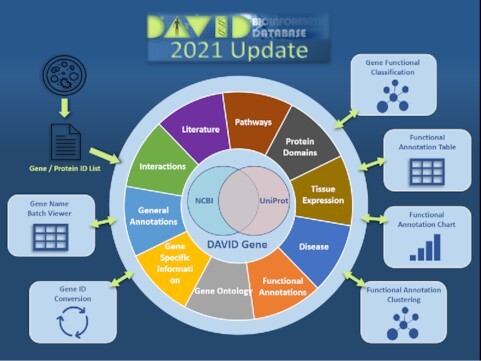
DAVID: a web server for functional enrichment analysis and functional annotation of gene lists (2021 update).
INTRODUCTION
Since nearly complete human genome sequences were published in 2001 (1,2), genome sequences have become available for an exponentially increased number of organisms due to advancement in DNA sequencing technologies (3). Meanwhile, there have been accelerated advancements in computational power, data storage capacity and data transfer speed. Various powerful high-throughput assays (-omics studies) of full genomics scale have been developed for biological and medical research fields using microarrays (4), next-generation sequencing (5), single-cell sequencing (6) and other high-throughput technologies. Large gene lists have been generated from these studies and understanding the biological significance of these gene lists is critical. However, it is a challenging task to interpret these large lists manually. As a result, numerous functional/pathway enrichment analysis applications have been developed since 2002. A review of 68 tools was published in Nucleic Acids Research in 2009 (7), and three new tools were published last year alone (8–10), indicating this is still an active field of development. These tools have played key roles in the functional analyses of large gene lists for various high-throughput biological studies over the last two decades.
DAVID is a popular bioinformatics resource system including a web server and web service for functional enrichment analysis, functional annotation, and ID conversion of gene lists. It has been published in ten papers since 2003 with citations totalling 57 750 as of 14 February 2022 (Supplementary Figure S1), indicating its importance in the scientific community. A total of 1 047 937 gene/protein lists were uploaded to DAVID 6.8 from 216 646 unique IP addresses in 2021. The top uploaded identifier and taxonomy were gene symbol (Supplementary Figure S2) and Homo sapiens (Supplementary Figure S3), respectively.
The first version of DAVID was developed and released in 2003 (11) as a web application version of the standalone software, EASE, which was developed for functional enrichment analysis of large gene lists (12). The central component and foundation of DAVID is the gene-centric DAVID Knowledgebase built upon the DAVID Gene system in 2007. This consists of a single-linkage algorithm used to agglomerate redundant gene/protein identifiers and associated heterogeneous annotation from different databases which are then centralized by DAVID Gene identifiers (13,14) (Figure 1A). Built upon the DAVID Knowledgebase, a set of functional annotation and functional enrichment analysis tools have been developed (14–16) (Figure 1B). There are six tools within DAVID: Functional Annotation Clustering, Functional Annotation Chart and Functional Annotation Table, Gene Functional Classification, Gene ID Conversion and Gene Name Batch Viewer. The detailed description of these tools can be found in the Supplemental Information.
Figure 1.

Updated DAVID Gene System. (A) Integrated Knowledgebase. The DAVID Gene system was rebuilt based on the NCBI Entrez Gene system and Uniprot Knowledgebase. The categories with new annotation types are coloured green. (B) Analytical Tools. The six DAVID analytical tools were connected to the updated Knowledgebase for comprehensive functional annotation and enrichment analysis of gene/protein lists.
Originally, DAVID was accessible through a web interface and URL-based API. A Web service (DAVID-WS) including four sample clients (Java, Perl, Python and MATLAB) was developed in 2012 to support programmatic access and facilitate automated analysis tasks (17) and an R client was developed by Fresno and Fernandez (18).
Here, we report the major updates made to DAVID in 2021. They have significantly expanded the DAVID knowledgebase, increased gene list upload efficiency, and improved the user experience.
NEW DEVELOPMENTS IN DAVID IN 2021
The major data sources and documentation used in the update can be found in Supplemental Information.
New DAVID gene system
Between 2007 and 2016, the DAVID Knowledgebase had a centralized gene identifier (DAVID Gene ID) to agglomerate redundant gene/protein IDs and associated heterogeneous annotation contents from different annotation databases (13,14). With improved cross-referencing between NCBI, EMBL-EBI and DDBJ, NCBI’s Entrez Gene ID had similar coverage for annotations and identifiers, compared to the DAVID Gene ID in 2016. Therefore, NCBI’s Entrez Gene ID was instituted as the central identifier in DAVID v6.8. However, due to the NCBI Prokaryotic RefSeq Genome Re-annotation Project and subsequent retirement of numerous Prokaryotic Entrez Gene IDs, the DAVID Knowledgebase lost coverage for many Prokaryotic species. Additionally, DAVID did not cover species outside of the core set from the Ensembl project. To overcome these issues, we rebuilt the DAVID Gene system based on the Entrez Gene System and the Uniprot Knowledgebase, merging these knowledgebases, where appropriate, with preference given to NCBI annotations: Uniprot IDs are merged with Entrez Genes stepwise based on specific overlapping gene/protein and taxonomy identifiers. After each step, only unmerged Uniprot IDs proceed to the next round. After all merging steps, the remaining unmerged Uniprot IDs from SwissProt or TrEMBL, if part of a Uniprot Reference Proteome, are assigned a unique DAVID Gene ID outside of the range of Entrez Gene, while the merged identifiers are represented by the Entrez Gene IDs in the DAVID Gene System. All unmerged Entrez Gene IDs are also included in DAVID Gene System (Supplementary Figure S4). The new system has improved overall annotation coverage and increased the number of included taxonomies from 17 399 to 55 464 with the number of bacteria increasing from 2 369 to 12 151 (5-fold) (Figure 2A). Entrez Gene identifiers covered in the knowledgebase have increased from 15 280 218 to 35 202 173 (2.3-fold), Uniprot identifiers from 7 617 882 to 75 375 241 (9.9-fold) and Ensembl Gene identifiers from 756 929 to 43 280 249 (57-fold) (Figure 2B).
Figure 2.

Significant Expansion of the DAVID Knowledgebase. (A) DAVID Taxonomy Coverage. The number of taxonomies in DAVID has increased significantly for all kingdoms due to the increased coverage resulting from the new DAVID Gene system. (B) Major Gene Identifier Type Counts and (C) Significant Increase in Gene-Term Records for Default DAVID Annotation Types. Significant increases in gene-term records are due to both increased annotation from the source databases as well as the increased coverage of the new DAVID Gene System.
Knowledgebase update
Based on the new DAVID Gene System, all existing annotation types in the DAVID Knowledgebase have been updated, if available. Compared with DAVID v6.8, the numbers of gene-term records have increased for 73 out of 78 existing annotation types that have been updated with 58 of them doubling at the minimum (Figure 2C, Supplemental Data 1). The remaining five updated annotation types have a decrease in the number of records (Supplemental Data 1). To understand these numbers, we investigated three annotation types with the greatest decreases: 85.34% decrease for MINT protein interactions, 52.39% decrease for DIP protein interactions and 11.74% decrease for CGNC Gene identifiers. We were not able to obtain updated protein interaction data from DIP directly, however, the data can be obtained via the iMEX consortium (19) together with data for IntAct and MINT. MINT offers a download of their current data which mirrored what we obtained through the iMEX, validating the data from iMEX and suggesting that the differences observed between the two versions of DAVID for DIP and MINT are due to the curation efforts of the iMEX consortium which should improve the quality of these annotation types in the updated DAVID Knowledgebase. Upon comparing differences between CGNC Gene identifiers, we found that a total of 12 545 CGNC IDs within DAVID v6.8 no longer exist in the current CGNC database while the DAVID 2021 update included 8 948 additional CGNC IDs thereby effectively increasing the coverage of valid CGNC Gene identifiers by 49.45% instead of 11.74% decrease seen in the record count (Supplementary Figure S5).
As is the nature of publicly available data, source databases for 27 annotation types in DAVID either have not been updated or the data are no longer accessible leading to no or small change for these annotation types (Supplemental Data 2). These legacy databases still contain useful annotation data and were retained except ProDom which has been covered by other sources.
With a new workflow in place, the DAVID Knowledgebase will be updated quarterly, following the UniprotKB update, to keep pace with the constant change in annotation from source databases.
New annotation types added to knowledgebase
We continue to seek annotation from new databases to enrich DAVID. For this update, the new annotations include small molecule-gene interactions from PubChem (20), drug-gene interactions from DrugBank (21), gene–disease associations from DisGeNET (22), pathways from WikiPathways (23) and PathBank (24), and tissue expression data from the Human Protein Atlas database (HPA) (25–27). In addition, UniprotKB Keywords (UP_KW), which constitutes a controlled vocabulary with a hierarchical structure, was grouped into a single annotation type in DAVID v6.8. In this update, we have split UP_KW into ten annotation types based on Uniprot's categorical classification. Among them, ‘Coding sequence diversity’ and ‘Technical term’ were not useful for functional annotation and enrichment analysis and were therefore not included. The remaining eight types have been added to five specific categories. In total, we have added new annotation types to seven Functional Annotations categories in the DAVID Knowledgebase in this update (Figure 1, Supplemental Data 3). It is worthy to note that PubChem and DrugBank annotation types were added to the DAVID Knowledgebase. The addition of these types of interactions makes it possible for users to identify small molecules or drugs that can interact with genes in their lists and help to identify molecules or drugs that can affect the gene/pathway, thus enabling in silico drug discovery.
‘SP_Comment’ within the ‘General Annotations’ category of the Functional Annotation Summary page within DAVID v6.8 contains Uniprot general annotations found in the ‘Comment Section’ of the Uniprot flat files. This annotation type has now been split into subtypes as defined by Uniprot. These have been added to ‘Gene Specific Information’ and are included in the gene report and denoted with the ‘UP_COMMENT’ prefix (Supplemental Data 3)
Updated display of ‘Annotation Summary Results’
Upon starting functional analysis of a gene list in DAVID, the Annotation Summary Results page will display up to ten annotation categories in DAVID v6.8, depending on available annotations associated with the genes in the list. Besides the additional annotation types added to the categories described above, we have renamed or removed some annotation types and annotation categories based on the assessment of the existing annotation types (detailed and colour coded in Supplemental Data 4, summarized in Table S1). Ensembl Gene ID, Entrez Gene ID and Official Gene Symbol do not have functional enrichment analysis value and can be obtained by the Gene ID Conversion tool and accessed within the gene report. Therefore, these types have been removed along with the ‘Main_Accessions’ category under which they were located. ‘SP_COMMENT’ in the ‘General Annotations’ Category has been replaced with ‘UP_COMMENT’ as described above and moved to the gene report. ProDom was removed as described above. ‘SP_COMMENT_TYPE’ in the ‘Functional_Categories’ category did not contain useful annotation and was removed. In addition, we renamed ‘Functional_Categories’ to ‘Functional_Annotations’, ‘CYTOBAND’ to ‘CYTOGENETIC_LOCATION’ and ‘Protein Interactions’ to ‘Interactions’ for consistency and clarity.
Updated gene symbol upload process
Of the 1 047 937 gene/protein lists uploaded to DAVID 6.8 in 2021, the top uploaded gene identifier type was gene symbol (Supplementary Figure S2). Due to the ambiguous nature of gene symbols whereby one symbol may map to multiple species, a gene symbol list uploaded to DAVID would result in the list being mapped to all relevant species and exponentially increasing the number of mappings. This was not only computationally intensive, but also led to confusion for the users as they would need to then select the species and background population accordingly. Many questions from DAVID users arose from this issue. To resolve this issue, we added a parameter allowing users to select their taxonomy of interest before the list is uploaded if ‘OFFICIAL_GENE_SYMBOL’ is selected as the list identifier type (Supplementary Figure S6). By establishing the taxonomy for the list early in process, the gene list upload is more efficient and less confusing for users.
Expansion of gene report
The DAVID Gene Report contains the annotations specific to a given gene available in the DAVID Knowledgebase and is accessible from all DAVID tools. A detailed description can be found in the Supplemental Information. In this update, when available, the following new annotations associated with the DAVID Gene have been added to the gene report: Uniprot identifiers, PubChem (small molecule-gene interactions), DrugBank (drug-gene interactions), Entrez Gene Summary and 27 types of UP_Comment annotations split from the previous SP_Comment (Table S2).
Documentation update and user support
To better serve the community, we have updated the documentation of DAVID including all technical documents and the FAQ. We have also improved the navigation of the DAVID webpages with optimized pull-down menus to make access more efficient. The latest improvements and news concerning DAVID are now included on the home page. Users are actively supported by e-mail communication and as of 24 July 2020, we have re-implemented the DAVID forum where DAVID users can actively communicate with us and each other. The forum is closely monitored by the DAVID Team and questions, when not answered by other users, are addressed promptly.
DISCUSSION
In this update, we have rebuilt the DAVID Gene system to expand annotation and taxonomy coverage. The DAVID Knowledgebase was updated for all annotation types based on the new DAVID Gene system if source database updates were available. Out of 78 updated annotation types, most have significantly increased numbers of gene-term records with only five annotation types decreasing in the number of gene-term records (Supplemental Data 1) including MINT, DIP and CGNC annotation types having the greatest decreases. Analysis shows that a number of the IDs in DAVID v6.8 for these three annotation types have become invalid due to curation, leading to the decrease of the number of records. In fact, CGNC has 49.45% increase of valid IDs in the updated Knowledgebase compared with DAVID v6.8 (Supplementary Figure S5). The number of bacterial taxonomies has increased 5-fold and the number of Ensembl Gene ID records increased 57-fold in the updated DAVID Knowledgebase, indicating the update successfully recovered and expanded coverage of Prokaryotic species and significantly extended coverage of the species from the Ensembl project. With the 2021 update, the DAVID Knowledgebase has Ensembl Gene Id coverage for 90 out of 101 Ensembl plant species, compared with no coverage in DAVID v6.8 (Supplementary Figure S7). These changes highlight both the improvement in the coverage for Ensembl and the need for a mechanism to pull in more high-quality annotations from the Ensembl project in the next update.
Furthermore, we have added new annotation types, including small molecule–gene interactions (PubChem) and drug–gene interactions (DrugBank), allowing the user to identify small-molecules and drugs interacting with the genes in their study and could facilitate in silico drug discovery.
In addition, we have optimized the Annotation Summary Results page: adding the new annotation types and removing redundant or low value ones. Importantly, we have split the Uniprot Keyword annotation type in ‘Functional Annotations’ into more specific subtypes to allow for more specificity in functional enrichment analyses (Supplemental Data 4). The gene report page has been enriched in the update with more available gene specific information in the DAVID Knowledgebase, including the newly added small molecule-gene and drug-gene interaction data (Table S2). Furthermore, a species selection parameter has been added to increase the efficiency and eliminate the confusion for users uploading gene lists containing gene symbols. Finally, new mechanisms are in place to facilitate the efficient update of the new DAVID Gene system and the connected annotation in the Knowledgebase. The DAVID Knowledgebase will be updated quarterly, following the UniprotKB update, to keep the data current.
In conclusion, this update significantly expands the Knowledgebase and enhances the discovery power of DAVID while hopefully improving the user experience. The addition of small molecule–gene and drug–gene interactions to DAVID opens the door for additional discovery and the new update mechanisms developed allows DAVID to stay current and expand in the future.
DATA AVAILABILITY
The DAVID 2021 update is freely available on the DAVID server https://david.ncifcrf.gov/.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank the following colleagues at Frederick National Laboratory for Cancer Research for their support: Dr Lei Xu in LHRI for the active monitoring of the DAVID forum, Mr. Shawn Steward and Mr. Bill Wilton, Clinical Services Program, ADRD and Dr Douglas O’Neal, HPC Systems Administration Group, ITOG for IT support, and Dr Jack Collins, Dr Uma Mudunuri and Mr. Henri Tuthill in the Advanced Biomedical Computational Science, BIDS, for database support. We also thank all previous DAVID Team members for their efforts to develop such an enduring and useful bioinformatic resource. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government.
Notes
Present address: Ming Hao, RNAimmune, 401 Professional Drive #280, Gaithersburg, MD 20879, USA.
Present address: Xiaoli Jiao, RNAimmune, 401 Professional Drive #280, Gaithersburg, MD 20879, USA.
Contributor Information
Brad T Sherman, Laboratory of Human Retrovirology and Immunoinformatics, Applied and Developmental Research Directorate, Frederick National Laboratory for Cancer Research, Frederick, MD 21702, USA.
Ming Hao, Laboratory of Human Retrovirology and Immunoinformatics, Applied and Developmental Research Directorate, Frederick National Laboratory for Cancer Research, Frederick, MD 21702, USA.
Ju Qiu, Laboratory of Human Retrovirology and Immunoinformatics, Applied and Developmental Research Directorate, Frederick National Laboratory for Cancer Research, Frederick, MD 21702, USA.
Xiaoli Jiao, Laboratory of Human Retrovirology and Immunoinformatics, Applied and Developmental Research Directorate, Frederick National Laboratory for Cancer Research, Frederick, MD 21702, USA.
Michael W Baseler, Clinical Services Program, Applied and Developmental Research Directorate, Frederick National Laboratory for Cancer Research, Frederick, MD, 21702, USA.
H Clifford Lane, Laboratory of Immunoregulation, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD 20892, USA.
Tomozumi Imamichi, Laboratory of Human Retrovirology and Immunoinformatics, Applied and Developmental Research Directorate, Frederick National Laboratory for Cancer Research, Frederick, MD 21702, USA.
Weizhong Chang, Laboratory of Human Retrovirology and Immunoinformatics, Applied and Developmental Research Directorate, Frederick National Laboratory for Cancer Research, Frederick, MD 21702, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Cancer Institute, National Institutes of Health [HHSN261200800001E]; National Institute of Allergy and Infectious Disease (in part). Funding for open access charge: National Cancer Institute, National Institutes of Health [HHSN261200800001E].
Conflict of interest statement. None declared.
REFERENCES
- 1. Lander E.S., Linton L.M., Birren B., Nusbaum C., Zody M.C., Baldwin J., Devon K., Dewar K., Doyle M., FitzHugh W.et al.. Initial sequencing and analysis of the human genome. Nature. 2001; 409:860–921. [DOI] [PubMed] [Google Scholar]
- 2. Venter J.C., Adams M.D., Myers E.W., Li P.W., Mural R.J., Sutton G.G., Smith H.O., Yandell M., Evans C.A., Holt R.A.et al.. The sequence of the human genome. Science. 2001; 291:1304–1351. [DOI] [PubMed] [Google Scholar]
- 3. Schuster S.C. Next-generation sequencing transforms today's biology. Nat. Methods. 2008; 5:16–18. [DOI] [PubMed] [Google Scholar]
- 4. Cuzin M. DNA chips: a new tool for genetic analysis and diagnostics. Transfus Clin. Biol. 2001; 8:291–296. [DOI] [PubMed] [Google Scholar]
- 5. Morozova O., Marra M.A.. Applications of next-generation sequencing technologies in functional genomics. Genomics. 2008; 92:255–264. [DOI] [PubMed] [Google Scholar]
- 6. Wang Y., Navin N.E.. Advances and applications of single-cell sequencing technologies. Mol. Cell. 2015; 58:598–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Huang da W., Sherman B.T., Lempicki R.A.. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009; 37:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hellstern M., Ma J., Yue K., Shojaie A.. netgsa: Fast computation and interactive visualization for topology-based pathway enrichment analysis. PLoS Comput. Biol. 2021; 17:e1008979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Manzini S., Busnelli M., Colombo A., Franchi E., Grossano P., Chiesa G.. reString: an open-source Python software to perform automatic functional enrichment retrieval, results aggregation and data visualization. Sci. Rep. 2021; 11:23458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Thanati F., Karatzas E., Baltoumas F.A., Stravopodis D.J., Eliopoulos A.G., Pavlopoulos G.A.. FLAME: a web tool for functional and literature enrichment analysis of multiple gene lists. Biology (Basel). 2021; 10:665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dennis G., Sherman B.T., Hosack D.A., Yang J., Gao W., Lane H.C., Lempicki R.A. DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 2003; 4:P3. [PubMed] [Google Scholar]
- 12. Hosack D.A., Dennis G., Sherman B.T., Lane H.C., Lempicki R.A. Identifying biological themes within lists of genes with EASE. Genome Biol. 2003; 4:R70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sherman B.T., Huang da W., Tan Q., Guo Y., Bour S., Liu D., Stephens R., Baseler M.W., Lane H.C., Lempicki R.A.. DAVID knowledgebase: a gene-centered database integrating heterogeneous gene annotation resources to facilitate high-throughput gene functional analysis. BMC Bioinformatics. 2007; 8:426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Huang D.W., Sherman B.T., Tan Q., Kir J., Liu D., Bryant D., Guo Y., Stephens R., Baseler M.W., Lane H.C.et al.. DAVID bioinformatics resources: expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007; 35:W169–W175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Huang da W., Sherman B.T., Stephens R., Baseler M.W., Lane H.C., Lempicki R.A.. DAVID gene ID conversion tool. Bioinformation. 2008; 2:428–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huang D.W., Sherman B.T., Tan Q., Collins J.R., Alvord W.G., Roayaei J., Stephens R., Baseler M.W., Lane H.C., Lempicki R.A.. The DAVID gene functional classification tool: a novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 2007; 8:R183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jiao X., Sherman B.T., Huang da W., Stephens R., Baseler M.W., Lane H.C., Lempicki R.A.. DAVID-WS: a stateful web service to facilitate gene/protein list analysis. Bioinformatics. 2012; 28:1805–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fresno C., Fernández E.A.. RDAVIDWebService: a versatile R interface to DAVID. Bioinformatics. 2013; 29:2810–2811. [DOI] [PubMed] [Google Scholar]
- 19. Orchard S., Kerrien S., Abbani S., Aranda B., Bhate J., Bidwell S., Bridge A., Briganti L., Brinkman F.S., Cesareni G.et al.. Protein interaction data curation: the International Molecular Exchange (IMEx) consortium. Nat. Methods. 2012; 9:345–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kim S., Chen J., Cheng T., Gindulyte A., He J., He S., Li Q., Shoemaker B.A., Thiessen P.A., Yu B.et al.. PubChem 2019 update: improved access to chemical data. Nucleic Acids Res. 2019; 47:D1102–D1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wishart D.S., Feunang Y.D., Guo A.C., Lo E.J., Marcu A., Grant J.R., Sajed T., Johnson D., Li C., Sayeeda Z.et al.. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 2018; 46:D1074–D1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bauer-Mehren A., Rautschka M., Sanz F., Furlong L.I.. DisGeNET: a Cytoscape plugin to visualize, integrate, search and analyze gene-disease networks. Bioinformatics. 2010; 26:2924–2926. [DOI] [PubMed] [Google Scholar]
- 23. Pico A.R., Kelder T., van Iersel M.P., Hanspers K., Conklin B.R., Evelo C.. WikiPathways: pathway editing for the people. PLoS Biol. 2008; 6:e184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wishart D.S., Li C., Marcu A., Badran H., Pon A., Budinski Z., Patron J., Lipton D., Cao X., Oler E.et al.. PathBank: a comprehensive pathway database for model organisms. Nucleic Acids Res. 2020; 48:D470–D478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sjöstedt E., Zhong W., Fagerberg L., Karlsson M., Mitsios N., Adori C., Oksvold P., Edfors F., Limiszewska A., Hikmet F.et al.. An atlas of the protein-coding genes in the human, pig, and mouse brain. Science. 2020; 367:eaay5947. [DOI] [PubMed] [Google Scholar]
- 26. Karlsson M., Zhang C., Méar L., Zhong W., Digre A., Katona B., Sjöstedt E., Butler L., Odeberg J., Dusart P.et al.. A single-cell type transcriptomics map of human tissues. Sci. Adv. 2021; 7:eabh2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Uhlén M., Fagerberg L., Hallström B.M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson Å., Kampf C., Sjöstedt E., Asplund A.et al.. Proteomics. Tissue-based map of the human proteome. Science. 2015; 347:1260419. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The DAVID 2021 update is freely available on the DAVID server https://david.ncifcrf.gov/.