


Abstract
The visualization of biological sequences with various functional elements is fundamental for the publication of scientific achievements in the field of molecular and cellular biology. However, due to the limitations of the currently used applications, there are still considerable challenges in the preparation of biological schematic diagrams. Here, we present a professional tool called IBS 2.0 for illustrating the organization of both protein and nucleotide sequences. With the abundant graphical elements provided in IBS 2.0, biological sequences can be easily represented in a concise and clear way. Moreover, we implemented a database visualization module in IBS 2.0, enabling batch visualization of biological sequences from the UniProt and the NCBI RefSeq databases. Furthermore, to increase the design efficiency, a resource platform that allows uploading, retrieval, and browsing of existing biological sequence diagrams has been integrated into IBS 2.0. In addition, a lightweight JS library was developed in IBS 2.0 to assist the visualization of biological sequences in customized web services. To obtain the latest version of IBS 2.0, please visit https://ibs.renlab.org.
Graphical Abstract
Graphical Abstract.
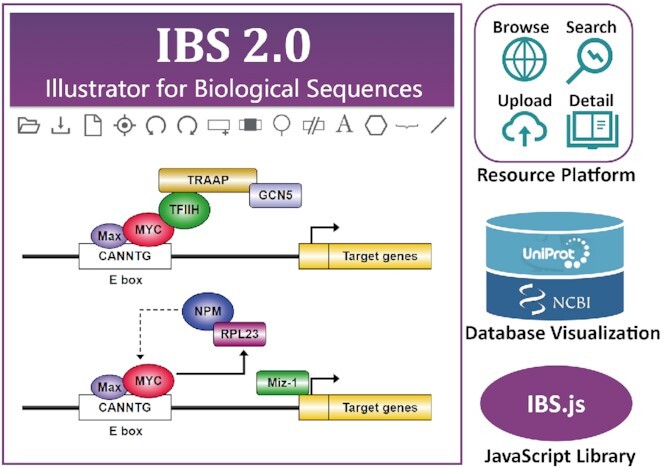
IBS 2.0: an upgraded illustrator for the visualization of biological sequences.
INTRODUCTION
The visualization of functional elements in biological sequences is fundamental when presenting new findings in the field of molecular and cellular biology because it enables the creation of concise and detailed representations of new discoveries for readers. By accurately displaying the organization of biological sequences and presenting other functional elements, such as epigenetic modifications and regulatory factors, in an intuitive way, a schematic diagram can greatly help in explaining the regulatory mechanism of biological macromolecules and further promote the exchange of scientific knowledge. At present, biologists mainly use Microsoft PowerPoint, Adobe Illustrator or Photoshop to draw schematic diagrams for biological sequences. However, these software packages were developed to meet the general needs of image manipulation, and the locations of functional elements cannot be precisely designated in the biological sequences. Therefore, it is difficult to use them to prepare a sequence organization diagram of sufficient accuracy.
Given this situation, our group previously developed a software package called DOG (1) for plotting protein graphs in a step-by-step manner. Later, in 2015, we updated the DOG package and released a new tool called illustrator of biological sequences (IBS) (2) for assisting biologists in drawing publication-quality diagrams of both protein and nucleotide sequences. Although these early versions have successfully helped scientists design fantastic diagrams for presenting their new findings in many published articles, they still have some limitations due to the web technology at that time. For instance, the previous versions were not smooth enough to perform interactive operations, especially for drawing complex diagrams with a large number of elements. Additionally, in the previous version, users could not perform batch visualizations for existing databases and were unable to share their artwork with other researchers.
Therefore, to expand the application scenarios of IBS, we present an upgraded version of IBS with stronger performance and more extensibility. By investigating currently published papers, we collected a set of new functional elements in IBS 2.0, which allows for the use of much more abundant graphical components for representing domain organizations, epigenetic modifications, and regulatory molecules for both protein and nucleotide sequences. In addition, in this update, a database visualization feature was also implemented for the UniProt (3) and NCBI RefSeq (4) databases. Meanwhile, a batch mode was also provided for the visualization of multiple protein or gene entries. In addition, a platform that can share and exchange editable sequence diagrams was implemented in IBS 2.0 to support the uploading, retrieval, and browsing of existing sequence diagrams. To allow an advanced usage of IBS, we have further developed a lightweight and speedy JavaScript library for the users. All the above features are freely available at https://ibs.renlab.org.
RESULTS
General description of IBS 2.0
The frontend of IBS 2.0 was built using HTML5, CSS, JavaScript and the library SVG.js (https://svgjs.dev/docs/3.0/) for manipulating and animating biological sequences. In addition, the backend was developed based on Java, and all data were stored and managed in MySQL.
The user interface of IBS 2.0 consists of three main functional modules. First, it contains a dual-mode user interface (Figure 1A) that allows biologists to generate their own schematic diagrams for either protein or nucleotide sequences. For user convenience, multiple graphical components, such as polygons, brackets, curves, and polylines, were supported in this module for assisting in the presentation of complicated diagrams. To further facilitate the drawing process, we also implemented a batch visualization module for the automatic drawing of biological sequences retrieved from the UniProt and the NCBI RefSeq databases (Figure 1B). In the new version of IBS, a resource platform (Figure 1C) that collected a wealth of published biological sequence diagrams was established, which enabled the search for existing functional elements for constructing the user's own custom illustrations.
Figure 1.

Overview of the IBS 2.0 server. (A) The main interface of IBS consists of 5 functional parts. (1) The component list panel showing the functional elements drawn in the presented sequence diagram. (2) A toolbox that contains buttons for generating various functional elements in the biological sequences. Some operation commands, such as New, Import, Export, Undo, Redo, Center, Zoom, etc., were also implemented in the toolbox. (3) The interactive interface for drawing biological sequence diagrams. (4) The batch visualization module for the UniProt and RefSeq databases. (5) The interface for setting properties of the selected element. (B) The main pipeline constructed for the batch visualization module. (C) The resource platform in IBS 2.0. Existing biological sequence diagrams can be uploaded, retrieved, and browsed via this module.
Input/output
To design sequence diagrams in IBS 2.0, users can directly drag and drop the graphical components in the main interface. Using the control panel on the right side, the precise position, size, and color of the selected component can be conveniently specified and updated in real-time. In addition, an input file in JSON format is also supported in the recent update. The JSON file records the detailed information of each functional element in the input biological sequences. When uploading this file, IBS 2.0 can automatically parse it and display the biological sequences in the main interface.
To visualize biological sequences from the UniProt and the NCBI RefSeq databases, the accession ID or official gene symbol is needed. For batch visualization, a text file recording the IDs or gene symbols in each line is also supported.
In this updated version, diagrams can be exported as an image file (in SVG, PNG or JPEG format) or a JSON file at any time.
Protein sequence visualization
In IBS 2.0, the protein mode was redesigned with several substantial improvements, such as a multiple sequence presentation, polygonal elements for sequence annotation, and an enhanced marker for presenting functional sites. To further demonstrate the new features of the protein mode, we picked up a striking instance from the published literature and re-illustrated it in Figure 2A.
Figure 2.

Schematic representation of two selected biological sequences. (A) A schematic diagram of the E3 SUMO-protein ligase RanBP2. A RanGAP1-induced activation process is presented on the domain graph. (B) Organization of the fru gene in Drosophila melanogaster. Sex-specific alternative splicing is shown.
As presented in previous studies (5,6), the SUMO E3 ligase RanBP2 and the Ran GTPase activating protein (RanGAP1) are known to form a stable complex during the cell cycle, which indicated a correlation between the sumoylation of proteins and RanGTP hydrolysis. To further demonstrate the synergistic mechanism of these two processes, a detailed complex of RanBP2, sumoylated RanGAP1 and Ubc9 was summarized in Flotho's review (7). The intricate structure of the RanBP2/RanGAP1*SUMO1/Ubc9 complex was picked up and redrawn in Figure 2A using IBS 2.0. In Figure 2A, several FG repeats are represented as dashed lines, while two internal repeats (IR1 and IR2), along with two RanGTP binding domains (RB) are marked as functional domains in the protein sequence. The complex of sumoylated RanGAP1 and Ubc9 can steadily bind to the IR1 domain in RanBP2 and activate the SUMO E3 ligase activity of the IR2 domain. Furthermore, several post-translational modifications, such as phosphorylation and sumoylation, were also found in RanBP2. A serine phosphorylation site at position 3207(8,9) and a lysine sumoylation site at position 2592 (10) are illustrated as examples. The characterization of the above interactions reveals that RanGTP hydrolysis is directly coupled with the catalytic activity of sumoylation. As a major update in the protein mode, a variety of drawing elements, such as polylines, polygons, and information texts, were integrated into IBS 2.0. Benefiting from such a wealth of drawing elements, the above complex sequence structures could be visualized in a concise and clear way. With those drawing elements, the interacting proteins, as well as their annotation notes, can be conveniently marked on the biological sequences, which will be a great help for researchers in interpreting novel regulatory models. In addition, the cutlines were allowed to draw in the protein sequence when representing functional domains that are distant from the ‘core’ regions. In some cases, multiple posttranslational modifications will appear in a single protein. To display different modification sites or functional sites in one protein, IBS 2.0 now provides site markers with different shapes and different colors for users, as shown in Figure 2A.
In conclusion, we suggest that improvements in the protein mode will be a useful feature for experimentalists, allowing them to illustrate their artwork with ease. With the abundant drawing elements provided in our software, more expressive schematic diagrams can be prepared from IBS 2.0.
Nucleotide sequence visualization
A wider variety of functional elements were implemented in the nucleotide mode, which allows users to prepare nucleotide diagrams with distinct drawing styles, than in the protein mode. In nucleotide mode, a ‘Domain’ button is provided for adding functional elements in nucleotide diagrams. Similarly, the ‘Site’ button is designed for representing functional motifs or mutation sites, while a ‘Note’ button is designed for additional annotations. In IBS 2.0, we designed various polygons, such as rectangles, circles, arrows and cylinders, for diversified diagramming. To make it more convenient for users to manage and organize the drawing elements, all polygons can be discretionary zooming and dragging. In addition, when drawing a long sequence of a nucleotide diagram, the functional elements within this sequence may be quite distant from each other. If we illustrate such nucleotide sequence on a real scale, the generated diagram may appear unaesthetic. To avoid such a scenario, we developed the ‘cutline’ element to help users omit inconsequential sequences when representing functional domains that are distant from the ‘core’ regions.
Figure 2B is an example application of our newly developed nucleotide-mode user interface. In 2005, Demir et al. identified a fruitless (fru) gene in Drosophila melanogaster (11). The fru gene is important for the sex-determination cascade and controls male courtship behavior by the establishment and development of a male-specific neuronal circuitry. The genomic organization was reillustrated in Figure 2B with the sex-specific domain, the dimerization domain (BTB), the connecting region and the DNA-binding domains (zinc-figures) marked in different polygons. As mentioned in the original literature, the fru gene is spliced differently in males and females. In Figure 2B, the sex-specific alternative spliced transcripts are shown. Transcripts from the P2–P4 promoters are not sex-specifically spliced and encode a set of common Fru isoforms that control the development of both sexes. However, the transcripts initiated from the distal P1 promoter are sex-specifically spliced under the control of the sex determination factors Tra and Tra-2. In males, the S exon is spliced at its default male-specific donor site, while a splicing event at a more 3′ donor site is found in females and leads to a block translation of the following transcripts. In this example, the polygonal elements provided in nucleotide mode were extensively used. The polygonal elements can be set as different colors and different shapes, which may provide users with unconstrained drawing operations. Additionally, several polyline elements were developed in nucleotide mode. The color and the thickness of the polylines can be varied to represent more diversified content in functional genes, such as different transcriptional directions or various alternative splicing events. Therefore, with the help of these drawing elements, the complex genomic schematic diagram can be clearly and succinctly represented.
For published artwork containing the above abundant polygonal and polyline elements, the organization of a nucleotide sequence can be explicitly interpreted, and visually appealing diagrams will effectively draw attention from readers. This is of great importance for the presentation of scientific achievements in published literature. Taken together, we propose that IBS 2.0 could be a great help for molecular and cellular experimentalists, allowing them to present the nucleotide organization in a more efficient, aesthetic, and concise manner.
Batch visualization of biological sequences from public databases
To facilitate the drawing process, a batch visualization module is also provided in IBS 2.0. A set of database IDs or official gene symbols can be inputted in the text area or uploaded via the file selection box (Figure 3A). After clicking the ‘Search’ button, detailed annotations of the inputted biological sequences were first retrieved from the UniProt or RefSeq database (Figure 3B). In protein mode, the functional domains, repeat regions, coiled coils, motifs, and known post-translational modifications (integrated from the PTMsnp Database (12)) were automatically drawn. In nucleotide mode, the transcript organization, including exon, CDS, 5′-UTR, 3′-UTR, and RNA modification sites (integrated from RMVar (13)), was visualized. Figure 3D and E presents a representative example of the PDZ and LIM domain protein families (PDLIM1, PDLIM2, PDLIM4) and two transcript isoforms of TGFB1. Based on the generated diagram, users can make further manipulations using the aforementioned features provided in IBS 2.0.
Figure 3.

A snapshot of the batch visualization module in IBS 2.0. (A) The data input panel. (B) Retrieval results from the UniProt (left) and RefSeq (right) databases. (C) The main interface of the resource platform. (D) Visualization of the PDZ and LIM domain protein families using the batch visualization module. (E) Visualization of the TGFB1 transcript isoforms using the batch visualization module.
JavaScript library for illustrating biological sequence diagrams
To enhance the scalability and versatility of IBS, we have developed a lightweight and speedy JavaScript library called IBS.js that integrates all functionalities in IBS. Using this JS library, users can easily visualize a biological sequence diagram in their customized web services and make it possible to present functional annotations dynamically during analysis or predictions.
Resource platform for collecting and sharing biological sequence diagrams
By performing an extensive literature survey, we found that many similar elements were drawn in different diagrams. Therefore, establishing a resource platform for collecting existing biological sequence diagrams can further help users search for functional elements of interest and share their artwork with the community, which we believe may be valuable for improving drawing efficacy and quality. To gather an intact set of published biological sequence diagrams, we developed a deep learning model (Supplementary Methods and Figure S1) based on Resnet152 architecture to recognize biological sequence diagrams from published literature. Using this model, we collected and manually reillustrated a total of 347 diagrams in IBS 2.0. For ease of use of these resources, a user-friendly platform that enables users to browse and search the collected sequence diagrams was developed (Figure 3C). In addition, to allow users to share diagrams designed on their own, an upload pipeline was also provided. All the submitted diagrams will be manually reviewed for quality assurance. In this resource platform, users can further edit the collected sequence diagrams using the main interface of IBS 2.0 and access the detailed information, such as the functional annotations, keywords, and associated publications, of each collected diagram.
SUMMARY AND PERSPECTIVES
In this article, we proposed an upgraded version of IBS. By applying the brand-new frontend technology, the performance and user experience were greatly improved. With a dual-mode user interface, experimentalists could produce schematic diagrams of both protein and nucleotide sequences with ease. To provide a drawing environment without restriction, abundant graphical elements, such as polygons, brackets, curves, and polylines, were available. Particularly, a database visualization feature was also implemented in this new version. By retrieving data from the UniProt and the NCBI RefSeq databases, the organization of a given protein and transcript structure can be automatically plotted. The post-translational modifications or RNA modifications on these biological sequences were also annotated in the generated diagram. For user convenience, this feature supports the retrieval of both database IDs and official gene symbols. Meanwhile, a batch mode was also provided for the visualization of multiple protein or gene entries. In addition, through literature research, we found that many diagrams from different published articles may describe similar functions or mechanisms. A platform that can share and exchange editable sequence diagrams may greatly help to improve design efficiency. Therefore, we added a resource platform into IBS 2.0 to support uploading, retrieval, and browsing of existing sequence diagrams. To allow an advanced usage of IBS, we also developed a lightweight and speedy JavaScript library for the users. Using this JS library, one can easily integrate IBS into their web server and dynamically visualize their functional annotations in their analysis or predictions.
Recently, several similar tools have been developed for the visualization of biological sequences. MyDomains (14) is one of the typical representatives. Compared to MyDomains, IBS 2.0 is considerably more powerful and user-friendly in the following aspects. First, MyDomains only supports drawing protein diagrams with up to 6 different shapes and 4 colors, while IBS 2.0 provides 22 types of graphical elements and any desired color for producing both protein and nucleotide sequence diagrams. Second, MyDomains can only plot one protein each time, but IBS 2.0 is free to add sequences without this restriction. Third, in MyDomains, schematic diagrams can only be exported into low-resolution bitmaps. However, in IBS 2.0, artwork can be saved in high-resolution bitmaps and editable vector images, which may be more suitable for producing publication-quality figures. Except for MyDomains, the Pfam (15) and the SMART (16) databases also provided a simple functionality for protein domain visualization. Nevertheless, similar to MyDomains, only one protein was allowed to draw in these kinds of websites at a time. Moreover, unlike IBS 2.0, none of the abovementioned tools are capable of providing an interactive operation for users, therefore causing great inconveniences in the drawing process. In conclusion, we suggest that IBS 2.0 is a useful tool for the community since it allows the researchers to represent their discoveries in a simpler and more comfortable way.
In the near future, we will update IBS as a comprehensive graphical software dedicated to biological research. A number of new features will be added, including the drawing of organelles, metabolic pathways, and cell signaling pathways. Additionally, to allow sharing diagrams between different tools, standard interchange formats, such as the Systems Biology Graphical Notation (17), will be further supported in our feature version. In addition, we will continue to collect as many biological sequence diagrams as possible and integrate them into the resource platform. Other functions will also be added based on suggestions and comments from our users.
DATA AVAILABILITY
IBS is a web-accessible open resource available at https://ibs.renlab.org. The complete tutorial and source code for the JavaScript library IBS.js are freely available at https://ibs.renlab.org/#/documentation.
Supplementary Material
Contributor Information
Yubin Xie, School of Life Sciences, Precision Medicine Institute, the First Affiliated Hospital, Sun Yat-sen University, Guangzhou 510060, China.
Huiqin Li, School of Life Sciences, Precision Medicine Institute, the First Affiliated Hospital, Sun Yat-sen University, Guangzhou 510060, China.
Xiaotong Luo, State Key Laboratory of Oncology in South China, Cancer Center, Collaborative Innovation Center for Cancer Medicine, Sun Yat-sen University, Guangzhou 510060, China.
Hongyu Li, School of Life Sciences, Precision Medicine Institute, the First Affiliated Hospital, Sun Yat-sen University, Guangzhou 510060, China.
Qiuyuan Gao, School of Life Sciences, Precision Medicine Institute, the First Affiliated Hospital, Sun Yat-sen University, Guangzhou 510060, China.
Luowanyue Zhang, School of Life Sciences, Precision Medicine Institute, the First Affiliated Hospital, Sun Yat-sen University, Guangzhou 510060, China.
Yuyan Teng, School of Life Sciences, Precision Medicine Institute, the First Affiliated Hospital, Sun Yat-sen University, Guangzhou 510060, China.
Qi Zhao, State Key Laboratory of Oncology in South China, Cancer Center, Collaborative Innovation Center for Cancer Medicine, Sun Yat-sen University, Guangzhou 510060, China.
Zhixiang Zuo, State Key Laboratory of Oncology in South China, Cancer Center, Collaborative Innovation Center for Cancer Medicine, Sun Yat-sen University, Guangzhou 510060, China.
Jian Ren, School of Life Sciences, Precision Medicine Institute, the First Affiliated Hospital, Sun Yat-sen University, Guangzhou 510060, China; State Key Laboratory of Oncology in South China, Cancer Center, Collaborative Innovation Center for Cancer Medicine, Sun Yat-sen University, Guangzhou 510060, China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Natural Science Foundation of China [31771462, 81772614, U1611261, 31801105, 81802438]; National Key R&D Program of China [2017YFA0106700]; Program for Guangdong Introducing Innovative and Entrepreneurial Teams [2017ZT07S096]; Guangdong Basic and Applied Basic Research Foundation [2020A1515010220]. Funding for open access charge: National Natural Science Foundation of China [31771462].
Conflict of interest statement. None declared.
REFERENCES
- 1. Ren J., Wen L., Gao X., Jin C., Xue Y., Yao X.. DOG 1.0: illustrator of protein domain structures. Cell Res. 2009; 19:271–273. [DOI] [PubMed] [Google Scholar]
- 2. Liu W., Xie Y., Ma J., Luo X., Nie P., Zuo Z., Lahrmann U., Zhao Q., Zheng Y., Zhao Y.et al.. IBS: an illustrator for the presentation and visualization of biological sequences. Bioinformatics. 2015; 31:3359–3361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. UniProt C. UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 2021; 49:D480–D489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. O’Leary N.A., Wright M.W., Brister J.R., Ciufo S., Haddad D., McVeigh R., Rajput B., Robbertse B., Smith-White B., Ako-Adjei D.et al.. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016; 44:D733–D745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Reverter D., Lima C.D.. Insights into E3 ligase activity revealed by a SUMO-RanGAP1-Ubc9-Nup358 complex. Nature. 2005; 435:687–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Werner A., Flotho A., Melchior F.. The ranbp2/rangap1*SUMO1/Ubc9 complex is a multisubunit SUMO E3 ligase. Mol. Cell. 2012; 46:287–298. [DOI] [PubMed] [Google Scholar]
- 7. Flotho A., Werner A.. The ranbp2/rangap1*SUMO1/Ubc9 complex: a multisubunit E3 ligase at the intersection of sumoylation and the rangtpase cycle. Nucleus (Austin, Tex.). 2012; 3:429–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Olsen J.V., Vermeulen M., Santamaria A., Kumar C., Miller M.L., Jensen L.J., Gnad F., Cox J., Jensen T.S., Nigg E.A.et al.. Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci. Signal. 2010; 3:ra3. [DOI] [PubMed] [Google Scholar]
- 9. Rigbolt K.T., Prokhorova T.A., Akimov V., Henningsen J., Johansen P.T., Kratchmarova I., Kassem M., Mann M., Olsen J.V., Blagoev B.. System-wide temporal characterization of the proteome and phosphoproteome of human embryonic stem cell differentiation. Sci. Signal. 2011; 4:rs3. [DOI] [PubMed] [Google Scholar]
- 10. Pichler A., Knipscheer P., Saitoh H., Sixma T.K., Melchior F.. The ranbp2 SUMO E3 ligase is neither HECT- nor RING-type. Nat. Struct. Mol. Biol. 2004; 11:984–991. [DOI] [PubMed] [Google Scholar]
- 11. Demir E., Dickson B.J.. fruitless splicing specifies male courtship behavior in Drosophila. Cell. 2005; 121:785–794. [DOI] [PubMed] [Google Scholar]
- 12. Peng D., Li H., Hu B., Zhang H., Chen L., Lin S., Zuo Z., Xue Y., Ren J., Xie Y.. PTMsnp: a web server for the identification of driver mutations that affect protein Post-translational modification. Front. Cell Dev. Biol. 2020; 8:593661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Luo X., Li H., Liang J., Zhao Q., Xie Y., Ren J., Zuo Z.. RMVar: an updated database of functional variants involved in RNA modifications. Nucleic Acids Res. 2021; 49:D1405–D1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sigrist C.J., de Castro E., Cerutti L., Cuche B.A., Hulo N., Bridge A., Bougueleret L., Xenarios I.. New and continuing developments at PROSITE. Nucleic Acids Res. 2013; 41:D344–D347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mistry J., Chuguransky S., Williams L., Qureshi M., Salazar G.A., Sonnhammer E.L.L., Tosatto S.C.E., Paladin L., Raj S., Richardson L.J.et al.. Pfam: the protein families database in 2021. Nucleic Acids Res. 2021; 49:D412–D419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Letunic I., Khedkar S., Bork P.. SMART: recent updates, new developments and status in 2020. Nucleic Acids Res. 2021; 49:D458–D460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Le Novere N., Hucka M., Mi H., Moodie S., Schreiber F., Sorokin A., Demir E., Wegner K., Aladjem M.I., Wimalaratne S.M.et al.. The systems biology graphical notation. Nat. Biotechnol. 2009; 27:735–741. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
IBS is a web-accessible open resource available at https://ibs.renlab.org. The complete tutorial and source code for the JavaScript library IBS.js are freely available at https://ibs.renlab.org/#/documentation.