


Abstract
Multi-CSAR is a web server that can efficiently and more accurately order and orient the contigs in the assembly of a target genome into larger scaffolds based on multiple reference genomes. Given a target genome and multiple reference genomes, Multi-CSAR first identifies sequence markers shared between the target genome and each reference genome, then utilizes these sequence markers to compute a scaffold for the target genome based on each single reference genome, and finally combines all the single reference-derived scaffolds into a multiple reference-derived scaffold. To run Multi-CSAR, the users need to upload a target genome to be scaffolded and one or more reference genomes in multi-FASTA format. The users can also choose to use the ‘weighting scheme of reference genomes’ for Multi-CSAR to automatically calculate different weights for the reference genomes and choose either ‘NUCmer on nucleotides’ or ‘PROmer on translated amino acids’ for Multi-CSAR to identify sequence markers. In the output page, Multi-CSAR displays its multiple reference-derived scaffold in two graphical representations (i.e. Circos plot and dotplot) for the users to visually validate the correctness of scaffolded contigs and in a tabular representation to further validate the scaffold in detail. Multi-CSAR is available online at http://genome.cs.nthu.edu.tw/Multi-CSAR/.
Graphical Abstract
Graphical Abstract.

Multi-CSAR is a web server that can efficiently and accurately scaffold (i.e. order and orient) the contigs of a target draft genome based on multiple reference genomes.
INTRODUCTION
The reconstruction of complete genomes for the organisms still is a difficult computational task, although the current next generation sequencing (NGS) techniques have been advanced greatly (1,2). Largely due to sequencing errors and large genomic repeats, the sequences produced by current assemblers are just draft genomes that consist of many contigs whose relative positions and orientations along the genome being sequenced are unknown. To further reconstruct chromosome-scale genomes, the contigs of draft genomes usually are required to be ordered and oriented into scaffolds, which are larger gap-containing sequences, so that the gaps between scaffolded contigs can be closed in the subsequent gap-filling process.
In principle, there are a variety of sources of linkage information that can be used to scaffold the contigs of a draft genome, such as long reads, optical maps, Hi-C read pairs and reference genomes (3–5). The scaffolding methods using long reads, optical maps and Hi-C read pairs are considered to be reference-free, because the sources of these linkage information come from the target draft genome to be scaffolded. On the other hand, reference-based scaffolding methods utilize other reference genomes of organisms related to the target genome to guide their scaffolding. Reference-based scaffolders are becoming important because more and more sequenced genomes are available to serve as references (6). Currently, many single reference-based scaffolders have been proposed (7–16). In some situations, however, a single reference genome alone may not be sufficient for a scaffolder to correctly scaffold a target draft genome, especially when the target and reference genomes have a distant phylogenetic relationship or they have undergone some kinds of genome rearrangements. This inspires the development of multiple reference-based scaffolders that can utilize several different but complementary reference genomes to more correctly scaffold the contigs of the target genome. Previously, we have developed a multiple reference-based scaffolder Multi-CSAR (short for ‘Multiple reference-based Contig Scaffolder using Algebraic Rearrangements’) (17), which is an extension of Multi-CAR (18), and have also utilized real biological datasets to demonstrate that Multi-CSAR indeed has better scaffolding performance when compared to other two existing multiple reference-based scaffolders Ragout (19) and MeDuSa (20).
Ragout (19) is a rearrangement-based scaffolder that utilizes synteny blocks between target and multiple reference genomes to scaffold the contigs of the target genome. In the scaffolding process, however, Ragout requires to solve an NP-hard problem on a given phylogenetic tree. For practical usage, Ragout applies a heuristic algorithm to solve this problem approximately. Currently, Ragout has been upgraded into Ragout2 (21) so that it can infer a phylogenetic tree automatically from the target and reference genomes. MeDuSa (20) formulates the scaffolding process as a combinatorial optimization problem, which needs to find a path cover with maximum weight on an edge-weighted graph constructed from the target and reference genomes. However, the problem of finding a maximum-weighted path cover itself is NP-hard and thus MeDuSa applies a 2-approximation algorithm to efficiently find an approximate solution. Multi-CSAR (17) first identifies sequence markers shared between target genome and each of reference genomes, then applies a rearrangement-based algorithm on these sequence markers to compute a single reference-based scaffold for the target genome, and finally uses a graph-based algorithm to combine all the single reference-derived scaffolds together into a multiple reference-derived scaffold. It is worth mentioning that both the algorithms mentioned above in the scaffolding process of Multi-CSAR are solvable in polynomial time.
However, Multi-CSAR was developed as a stand-alone application that required the users to install the software of Multi-CSAR on their local computers running with Linux and then execute Multi-CSAR via a command-line interface. In fact, such a requirement for executing Multi-CSAR is inconvenient for those users that are not fully familiar or comfortable with the Linux system and its operation through a command line interface. In this work, therefore, we have further developed Multi-CSAR into a web server so that it can provide the users with an easy-to-operate interface to run Multi-CSAR. In addition, the web server of Multi-CSAR outputs its scaffolding result in two graphical representations using Circos plot and dotplot, both of which allow the users to visually validate the correctness of scaffolded contigs, and in a tabular representation, which allows the users to further validate the scaffold in detail. Our experimental results on some real biological datasets have also shown that Multi-CSAR still has better scaffolding performance when compared to Ragout2.
MATERIALS AND METHODS
The Multi-CSAR web server was developed according to a heuristic approach described briefly as follows. (i) Identify those sequence markers that are shared between the given target genome and each of the given reference genomes. (ii) Utilize these sequence markers to compute the scaffold of the target genome based on each single reference genome. (iii) Combine all the single reference-derived scaffolds together into a multiple reference-derived scaffold. In the following, we describe some details about the program implementation of the Multi-CSAR web server.
The sequence markers mentioned above are similar genomic segments between two biological sequences. The web server of Multi-CSAR applies either NUCmer or PROmer on the target and reference genomes to identify their sequence markers. Both NUCmer and PROmer are sequence aligners from MUMmer’s package (22). Their main difference is that NUCmer detects the sequence markers directly on input DNA sequences, while PROmer finds them on the six-frame protein translation of the input DNA sequences. Furthermore, Multi-CSAR utilizes the identified sequence markers to calculate a weight for each reference genome for measuring its significance in the process of computing multiple reference-based scaffold. The weight of a reference genome with respect to the target genome is the sum of the sequence length times the percent identity for all sequence markers. In principle, the more similar a reference genome is to the target genome, the more weight it receives.
The Multi-CSAR web server applies CSAR (15) to derive the scaffold of the target genome based on each single reference genome, where CSAR is an efficient and accurate single reference-based scaffolder we previously developed based on a near-linear time rearrangement-based scaffolding algorithm (23). The web server of Multi-CSAR continues to construct an edge-weighted contig adjacency graph by using all single reference-derived scaffolds. In the contig adjacency graph, each contig in the target genome is represented by two vertices and there is an edge between any two vertices if they come from the different contigs. Moreover, an edge is said to be supported by a reference genome if the contigs represented by its two vertices are distinct and occur continuously in a single reference-derived scaffold. If an edge is supported by several reference genomes at the same time, then this edge receives a weight equal to the sum of the weights of all the supporting reference genomes. However, if an edge is not supported by any reference genome, then it has a weight of zero. After that, the Multi-CSAR web server computes a maximum weighted perfect matching from the constructed contig adjacency graph by using Blossom V (24), and finally derives a multiple reference-based scaffold of the target genome from this maximum weight perfect matching. For more details about the algorithm of Multi-CSAR, we refer the readers to our previous study (17).
WEB INTERFACE AND USAGE
The Multi-CSAR web server offers an easy-to-operate interface (see Figure 1) to the users. To run the web server of Multi-CSAR, the users first need to upload a target genome and one or more reference genomes in multi-FASTA format. If needed, the users can click the ‘plus’ (respectively, ‘minus’) button to add (respectively, remove) a reference genome field. Currently, many assembled genomes of organisms are maintained in the NCBI Assembly database (https://www.ncbi.nlm.nih.gov/assembly) (25), from which the users can find some reference genomes related to the target genome to be scaffolded by searching in the Assembly database directly or by browsing assembled genomes available for a particular organism. Second, the users can use the ‘weighting scheme of reference genomes’ for Multi-CSAR to automatically compute the weights of all the reference genomes. If the weighting scheme is not used, then the weights of all the reference genomes are defaulted to one. Third, the users can choose to use ‘NUCmer on nucleotides’ or ‘PROmer on translated amino acids’ to identify sequence markers between the target genome and each of the reference genomes. Fourth, the users can enter an email address, which is optional, to run the Multi-CSAR web server in a batch way. In the batch way, the users will be notified of the scaffolding result of Multi-CSAR via email when the submitted job is finished.
Figure 1.

Interface of Multi-CSAR web server.
The Multi-CSAR web server outputs its scaffolding results in four tab pages: (i) input data & parameters, (ii) Circos plot validation, (iii) dotplot validation and (iv) scaffolds of target. In the ‘Input data & parameters’ tab page, Multi-CSAR just displays the sequence information of the input target and reference genomes, the user-specified sequence aligner (either NUCmer or PROmer) to identify sequence markers, and whether the weighting scheme of reference genomes is used or not. By clicking on the links of the target and reference genomes in this tab page, Multi-CSAR will further show their DNA sequences. By clicking on the link ‘Dotplot against target genome’ on each reference genome, Multi-CSAR will display a dotplot for the users to visually inspect sequence markers between the target and reference genomes before scaffolding. The dotplot representation in the Multi-CSAR web server is generated by MUMmerplot in MUMmer’s package (22). In the dotplot (see Figure 2 for an example), the un-scaffolded target genome and the selected reference genome are represented on the y and x axes, respectively, and their contigs and scaffolds are separated by horizontal and vertical dashed lines, respectively. Moreover, each forward (respectively, reverse) sequence marker is shown by a red (respectively, blue) line and its ends are represented by two unfilled circles. The users can sort the contigs in the target genome based on their sizes by clicking on the toggle switch ‘Sort by contig size’. The users can also show or hide the IDs of the contigs and scaffolds by using the toggle switch ‘Show scaffold/contig IDs.’ The format of contig (respectively, scaffold) IDs begins with three-letter prefix CTG (respectively, SCF) followed by an underscore (_) and at least one digit (e.g. CTG_1 and SCF_1). In addition, the users can click the ‘Save as SVG file’ button to download a copy of the dotplot in scalar vector graphics (SVG) format.
Figure 2.

A display of a dotplot between un-scaffolded target genome and a selected reference genome.
In the ‘Circos plot validation’ tab page (see Figure 3 for an example), Multi-CSAR shows its total running time, as well as its scaffolding result displayed in a Circos plot. The Circos plot of a scaffolding result in the Multi-CSAR web server is generated by Circos program (26). In the initial Circos plot, the scaffolds of target genome (displayed in purple) and all the reference genomes (displayed in other colors) are arranged in a circle with the inner links connecting corresponding sequence markers between the target genome and each reference genome. The color of an inner link comes from the reference genome it connects. In the Circos plot, the number of crossing inner links can be viewed as a accuracy measure for a scaffolding result. That is, if the contigs in the target genome are scaffolded well according to a reference genome, the number of crossing inner links between them should be low. For this purpose, the Multi-CSAR web server allows the users to further select any reference genome (by clicking the checkbox next to it) from the top of the tab page to display (by clicking the ‘Display Circos plots’ button) its Circos plot against the scaffolded target genome (see Figure 4 for an example). In this Circos plot, the inner circle displays the sequence markers shared between the target genome and the selected reference genome. As demonstrated in Figure 4, the Circos plot is convenient and helpful for the users to visually validate whether the contigs of the target genome are properly scaffolded according to a reference genome, as well as to visually identify whether there are any genome rearrangements between the scaffolded target and reference genomes. In addition, the Multi-CSAR web server allows the users to download the Circos plot in the SVG format by clicking the ‘Save as SVG file’ button.
Figure 3.

A display of a Circos plot between scaffolded target genome and all reference genomes.
Figure 4.

A display of a Circos plot between scaffolded target genome and a selected reference genome, where the sequence markers are arranged in alternating layers along the two-layer inner circle.
In the ‘Dotplot validation’ tab page (see Figure 5 for an example), the Multi-CSAR web server displays its scaffolding result by a dotplot between the scaffolded target genome and a selected reference genome (the default is the first reference genome). In fact, the aligned regions of sequence markers should be displayed from the bottom left to the top right in the dotplot (as shown in Figure 5) or from the top left to the bottom right, if the contigs from the target genome are scaffolded perfectly based on the selected reference genome. Showing the scaffolding result in the dotplot display is another way to help the users to visually verify whether the contigs of the target genome are scaffolded properly based on a reference genome. The users can click the ‘Save as SVG file’ button to download the dotplot of a scaffold in the SVG format.
Figure 5.

A display of the ‘Dotplot validation’ tab page.
In the ‘Scaffolds of target’ tab page (see Figure 6 for an example), the Multi-CSAR web server displays its scaffolding result in tabular format, which allows the users to view the scaffolds of the target genome in detail. The scaffolds in the table are sorted according to their sizes, which equals to the sum of contig sizes. For each scaffold, its ordered contigs, as well as their orientations (forward orientation denoted by 0 and reverse orientation by 1), sequences and lengths, are listed in a table. The users can click on the ‘Download scaffolds (.txt)’ and ‘Download scaffolds (.csv)’ buttons to download the scaffolds of the target genome in the tab-delimited text format and comma-delimited CSV format, respectively. In addition, the users can click on the ‘Download sequence’ button to download the scaffold sequences in the multi-FASTA format, in which the contig sequences are separated by 100 Ns if they belong to the same scaffold.
Figure 6.

A display of the ‘Scaffolds of target’ tab page.
EXPERIMENTAL RESULTS
As mentioned before, Ragout (19) has already been upgraded into Ragout2 (21) and therefore we need to re-conduct our previous experiments on a benchmark of five real datasets (see Supplementary Table S1 for their details) for comparing its accuracy performance with those obtained by MeDuSa (version 1.6) and Multi-CSAR. These five testing datasets were originally prepared by Bosi et al. when they studied to develop MeDuSa (20) with each dataset consisting of a target genome to be scaffolded and two or more complete or incomplete reference genomes. For each testing dataset, Bosi et al. (20) also provided a reference order for the contigs of the target genome that can be used a truth standard to evaluate the scaffolding results returned by all the evaluated scaffolders. The metrics used to evaluate the scaffolders include sensitivity, precision, F-score, NGA50, scaffold number and running time. In principle, sensitivity, precision and F-score are all used to estimate the scaffolding accuracy, and both NGA50 and scaffold number are used to estimate the scaffolding contiguity.
Any two consecutive contigs in a scaffolding result are considered as a correct join if they also occur in same order and orientation in the reference order. Moreover, the number of correct joins is called as true positive (denoted by TP) and the number of incorrect joins as false positive (denoted by FP). The sensitivity of a scaffolding result is then defined as TP/P, its precision as TP/(TP + FP), and its 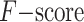 as (2 × sensitivity × precision)/(sensitivity + precision), where P denotes the number of all contig joins in the reference order. Actually, F-score is a balanced measure between sensitivity and precision and F-score is high only when both sensitivity and precision are high.
as (2 × sensitivity × precision)/(sensitivity + precision), where P denotes the number of all contig joins in the reference order. Actually, F-score is a balanced measure between sensitivity and precision and F-score is high only when both sensitivity and precision are high.
All the evaluated scaffolders MeDuSa, Ragout2 and Multi-CSAR were run with their default parameters. Note that unlike its previous version, Ragout2 does not require the users to provide a phylogenetic tree for describing the evolutionary relationships between the input genomes because Ragout2 can infer it automatically. Their average performance results on the five testing datasets are displayed in Table 1 (also see Supplementary Table S2 for detailed scaffolding results), in which sensitivity, precision, F-score are all shown in percentage (%), the size of NGA50 in base pairs (bp), and the running time in minutes. Note that in this experiment, Multi-CSAR was run without using the weighting scheme of reference genomes. As shown in Table 1, Multi-CSAR running with NUCmer gives the best sensitivity, F-score, NGA50 and running time, but still has the second best precision. On the other hand, Multi-CSAR running with PROmer produces the second best sensitivity, F-score, NGA50 and scaffold number, but the third best precision and running time. Ragout2 yields the best precision and scaffold number, but the third best sensitivity and the poorest NGA50 and running time. As for MeDuSa, it obtains the second best running time, but the poorest sensitivity, precision, F-score and scaffold number. Overall speaking, Multi-CSAR has better scaffolding performance than MeDuSa and Ragout2 on the five testing datasets. By the way, the average scaffolding performance of Multi-CSAR on these five testing datasets can be further improved when using with the weighting scheme of reference genomes (see Supplementary Table S3 for its detailed performance results).
Table 1.
Average performance of three evaluated multiple reference-based scaffolders on the five testing datasets
| Scaffolder | Sensitivity | Precision | F-score | NGA50 | #Scaffolds | Time |
|---|---|---|---|---|---|---|
| MeDuSa | 79.7 | 82.7 | 81.1 | 685 150 | 20 | 3.0 |
| Ragout2 | 81.8 | 92.1 | 86.0 | 169 832 | 3 | 22.4 |
| Multi-CSAR (NUCmer) | 89.6 | 90.9 | 90.2 | 1 046 288 | 9 | 1.1 |
| Multi-CSAR (PROmer) | 89.2 | 90.2 | 89.7 | 1 027 066 | 8 | 4.7 |
SUMMARY
In this work, we introduced the Multi-CSAR web server, a multiple reference-based scaffolder, which allows the users to more conveniently scaffold the contigs of a target genome based on multiple reference genomes via an easy-to-operate interface. In particular, the Multi-CSAR web server outputs its scaffolding results in two graphical displays of Circos plot and dotplot, both of which can allow the users to visually validate the correctness of scaffolded contigs. Our experimental results on the testing biological datasets have also shown that Multi-CSAR indeed has better scaffolding performance when compared to other two existing multiple reference-based scaffolders MeDuSa and Ragout2.
Supplementary Material
Contributor Information
Shu-Cheng Liu, Department of Computer Science, National Tsing Hua University, Hsinchu 30013, Taiwan.
Yan-Ru Ju, Department of Computer Science, National Tsing Hua University, Hsinchu 30013, Taiwan.
Chin Lung Lu, Department of Computer Science, National Tsing Hua University, Hsinchu 30013, Taiwan.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Ministry of Science and Technology of Taiwan [MOST110-2221-E-007-083-MY2]. Funding for open access charge: Ministry of Science and Technology of Taiwan.
Conflict of interest statement. None declared.
REFERENCES
- 1. Alkan C., Sajjadian S., Eichler E.E.. Limitations of next-generation genome sequence assembly. Nat. Methods. 2011; 8:61–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. El-Metwally S., Hamza T., Zakaria M., Helmy M.. Next-generation sequence assembly: four stages of data processing and computational challenges. PLoS Comput. Biol. 2013; 9:e1003345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rice E.S., Green R.E.. New approaches for genome assembly and scaffolding. Annu. Rev. Anim. Biosci. 2019; 7:17–40. [DOI] [PubMed] [Google Scholar]
- 4. Ghurye J., Pop M.. Modern technologies and algorithms for scaffolding assembled genomes. PLoS Comput. Biol. 2019; 15:e1006994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Luo J., Wei Y., Lyu M., Wu Z., Liu X., Luo H., Yan C.. A comprehensive review of scaffolding methods in genome assembly. Brief. Bioinform. 2021; 22:bbab033. [DOI] [PubMed] [Google Scholar]
- 6. Mukherjee S., Stamatis D., Bertsch J., Ovchinnikova G., Sundaramurthi J.C., Lee J., Kandimalla M., Chen I.A., Kyrpides N.C., Reddy T.B.K.. Genomes OnLine Database (GOLD) v.8: overview and updates. Nucleic Acids Res. 2021; 49:D723–D733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. van Hijum S.A., Zomer A.L., Kuipers O.P., Kok J.. Projector 2: contig mapping for efficient gap-closure of prokaryotic genome sequence assemblies. Nucleic Acids Res. 2005; 33:W560–W566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Richter D.C., Schuster S.C., Huson D.H.. OSLay: optimal syntenic layout of unfinished assemblies. Bioinformatics. 2007; 23:1573–1579. [DOI] [PubMed] [Google Scholar]
- 9. Rissman A.I., Mau B., Biehl B.S., Darling A.E., Glasner J.D., Perna N.T.. Reordering contigs of draft genomes using the Mauve Aligner. Bioinformatics. 2009; 25:2071–2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Husemann P., Stoye J.. r2cat: synteny plots and comparative assembly. Bioinformatics. 2010; 26:570–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Alonge M., Soyk S., Ramakrishnan S., Wang X., Goodwin S., Sedlazeck F.J., Lippman Z.B., Schatz M.C.. RaGOO: fast and accurate reference-guided scaffolding of draft genomes. Genome Biol. 2019; 20:224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Munoz A., Zheng C., Zhu Q., Albert V.A., Rounsley S., Sankoff D.. Scaffold filling, contig fusion and comparative gene order inference. BMC Bioinformatics. 2010; 11:304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dias Z., Dias U., Setubal J.C.. SIS: a program to generate draft genome sequence scaffolds for prokaryotes. BMC Bioinformatics. 2012; 13:96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lu C.L., Chen K.-T., Huang S.-Y., Chiu H.-T.. CAR: contig assembly of prokaryotic draft genomes using rearrangements. BMC Bioinformatics. 2014; 15:381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen K.-T., Liu C.-L., Huang S.-H., Shen H.-T., Shieh Y.-K., Chiu H.-T., Lu C.L.. CSAR: a contig scaffolding tool using algebraic rearrangements. Bioinformatics. 2018; 34:109–111. [DOI] [PubMed] [Google Scholar]
- 16. Chen K.-T., Lu C.L.. CSAR-web: a web server of contig scaffolding using algebraic rearrangements. Nucleic Acids Res. 2018; 46:W55–W59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen K.-T., Shen H.-T., Lu C.L.. Multi-CSAR: a multiple reference-based contig scaffolder using algebraic rearrangements. BMC Syst. Biol. 2018; 12:139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chen K.-T., Chen C.-J., Shen H.-T., Liu C.-L., Huang S.-H., Lu C.L.. Multi-CAR: a tool of contig scaffolding using multiple references. BMC Bioinformatics. 2016; 17:469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kolmogorov M., Raney B., Paten B., Pham S.. Ragout-a reference-assisted assembly tool for bacterial genomes. Bioinformatics. 2014; 30:i302–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bosi E., Donati B., Galardini M., Brunetti S., Sagot M.F., Lio P., Crescenzi P., Fani R., Fondi M.. MeDuSa: a multi-draft based scaffolder. Bioinformatics. 2015; 31:2443–2451. [DOI] [PubMed] [Google Scholar]
- 21. Kolmogorov M., Armstrong J., Raney B.J., Streeter I., Dunn M., Yang F., Odom D., Flicek P., Keane T.M., Thybert D.et al.. Chromosome assembly of large and complex genomes using multiple references. Genome Res. 2018; 28:1720–1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kurtz S., Phillippy A., Delcher A.L., Smoot M., Shumway M., Antonescu C., Salzberg S.L.. Versatile and open software for comparing large genomes. Genome Biol. 2004; 5:R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lu C.L. An efficient algorithm for the contig ordering problem under algebraic rearrangement distance. J. Comput. Biol. 2015; 22:975–987. [DOI] [PubMed] [Google Scholar]
- 24. Kolmogorov V. Blossom V: a new implementation of a minimum cost perfect matching algorithm. Math. Program. Comput. 2009; 1:43–67. [Google Scholar]
- 25. Kitts P.A., Church D.M., Thibaud-Nissen F., Choi J., Hem V., Sapojnikov V., Smith R.G., Tatusova T., Xiang C., Zherikov A.et al.. Assembly: a resource for assembled genomes at NCBI. Nucleic Acids Res. 2016; 44:D73–D80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Krzywinski M., Schein J., Birol I., Connors J., Gascoyne R., Horsman D., Jones S.J., Marra M.A.. Circos: an information aesthetic for comparative genomics. Genome Res. 2009; 19:1639–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.