

Abstract
Recent studies have revealed that the immune system plays a critical role in various physiological processes beyond its classical pathogen control activity. Even under a sterile condition, various cells and tissues can utilize the immune system to meet a specific demand for proper physiological functions. Particularly, a strong link between immunity and metabolism has been identified. Studies have identified the reciprocal regulation between these two systems. For example, immune signals can regulate metabolism, and metabolism (cellular or systemic) can regulate immunity. In this review, we will summarize recent findings on this reciprocal regulation between immunity and metabolism, and discuss potential biological rules behind this interaction with integrative perspectives.
Keywords: Immunity, Immunometabolism, Inflammation, Metabolism, Stress
INTRODUCTION
The immune system is a specialized system that can protect our body from various pathogens. It can be generally categorized into two systems: an innate immune system and an adaptive immune system. Each system expresses its own receptors to recognize various pathogens. The innate immune system utilizes various pattern recognition receptors (PRRs) including Toll-like receptors, Nod-like receptors, and C-type lectin receptors to recognize specific pathogen-associated molecular patterns (PAMPs). Adaptive immune cells such as T cells and B cells express different types of antigen receptors. Individual T cells and B cells express their unique receptors through genetic recombination, thereby increasing the diversity of their antigen receptors. Through these receptors, adaptive immune cells can recognize various pathogens. These two layers of innate and adaptive immunity can provide strong protection against a variety of pathogenic invasions.
Recent research has revealed that the immune system does more than just controlling pathogens. Even without an infection, the immune system can induce sterile inflammatory responses. This non-canonical function is currently a subject of many discussions. What is suggested through these discussions is that the classical pathogen-killing role of the immune system is just a part of the entire function of the immune system. In this line, efforts are being made to understand the immune system’s role with a comprehensive perspective from the viewpoint of physiological homeostasis (1, 2).
Many properties of immunity allow the immune system to play a crucial role in homeostatic functions of our body beyond pathogen killing. For example, immune cells such as phagocytes can uptake and clean various materials. This property not only can be utilized to kill bacteria, but also can be exploited to remove dying or dead cells for homeostasis. Moreover, the same signaling for cell death in the context of infection can be used for apoptosis under other cellular stresses. Innate immunity can be activated with damage-associated molecular pattern (DAMP) besides PAMP with the same PRR signaling to maintain homeostasis (3). Adaptive immune receptors recognizing various pathogenic molecules can also be utilized to detect mutant proteins that malignant cells make. This idea has become a foundation for cancer immunology and immunotherapy. Various cytokines that immune cells make can have a pleiotropic effect on the function of non-immune cells not only in the context of infection, but also in other stressors. These properties of immune system provide a strong basis for the discussion of immune response beyond pathogen control.
STRESS AND INFLAMMATION
Cellular, tissue-level, and organismal stressors have been linked to inflammation. Various cellular stressors can elicit inflammatory responses even without an infection. For example, genotoxic stress with radiation can cause interferon responses (4). Micronuclei formed under genotoxic stress and cell cycle progression can be sensed by pattern-recognition receptor cyclic GMP–AMP synthase (cGAS) to induce inflammatory responses (4). Endoplasmic reticulum (ER) stress can also induce inflammatory responses. Unfolded protein response is triggered by ER stress, which involves the activation of several transmembrane receptors such as activating transcription factor (ATF) 6, protein kinase RNA-like ER kinase (PERK), and inositol-requiring enzyme 1 (IRE1) α. IRE1α can bind TNF receptor associated factor (TRAF) 2 to the ER membrane once it is activated, triggering inflammatory responses via the nuclear factor kappa B (NF-κB) pathway (5). A recent study has found that two members of the NLR family of PRRs, nucleotide-binding oligomerization domain (NOD) 1 and NOD2, are significant mediators of ER stress-induced inflammation (6). Other studies have found that lysosomal stress can induce inflammation via NOD-like receptor family pyrin domain containing 3 (NLRP3) inflammasome activation. Crystalline substances such as silica, alum, and cholesterol can provoke lysosomal destabilization and rupture and finally lead to NLRP3 activation (7, 8). Other cellular stressors such as mitochondrial stress and oxidative stress can also be sensed by various cellular pathways and lead to NLRP3 inflammasome activation (9). The fundamental biological rationale behind this phenomenon has been discussed in detail elsewhere (1, 10, 11), and it is beyond the scope of this review. However, it is obvious that inflammation induced by cellular stress is beneficial for the survival of an organism either by removing the stressors or by promoting adaptation to the stressors. These reports also strongly support the notion that properties of the immune system can be utilized to regulate other body functions beyond pathogen control.
METABOLISM AND IMMUNITY
Recent reports have revealed a strong link between metabolic regulation and immunity. Cellular and systemic metabolism can greatly affect immune function. Immune responses can also significantly influence metabolism. However, the precise mechanism of their interaction and biological rules behind such interaction remain unclear. Here we will discuss recent findings on this subject.
Type 1 immunity and metabolism
Metabolic regulation of immune function: Traditionally, immune responses have been categorized based on CD4 T helper (Th) cell responses. For example, naïve CD4 T cells can be differentiated into various types of Th cells such as Th1, Th2, Th17, and regulatory T cells (Treg). Each Th cells express their own transcriptional factor and secrete their unique cytokines. Th1 cells express T-bet and produce interferon (IFN)-γ. Th2 cells express GATA3 and secrete IL-4 and IL-13. Th17 cells express RORγt and secrete IL-17. Treg cells express FOXP3 and secrete IL-10. These unique cytokines of each lineage can suppress the expression of other transcriptional factors so that differentiation toward each lineage is stably maintained. Recent discovery of innate lymphoid cells (ILC) has shown that these immune responses are not exclusive to Th cells. The use of an overarching name such as type 1, type 2, and type 3 (type 17) immunity has been suggested.
Type 1 immune responses are traditionally well studied in the context of anti-viral or intracellular pathogen control. Active cytokine players in this axis include IFNs and inflammatory cytokines such as IL-1β and TNF-α. Previous studies have found that the metabolic state is highly linked to proper induction of type 1 immune responses in various immune cells. For example, it is found that polarization of macrophages is accompanied by metabolic reprogramming. When macrophages are treated with lipopolysaccharide (LPS) or IFN-γ, glycolysis is dramatically up-regulated while other metabolic pathways such as tricarbo-xylic cycle (TCA), fatty acid oxidation (FAO), and glutaminolysis are weakened (12, 13). Inhibition of these metabolic alterations leads to the reduction of classical macrophage M1 activation and function (12, 13). Other cytokines such as IFN-β and IL-1β can also increase glucose uptake and glycolysis in macrophages, which augments the inflammatory function of the macrophages (14-16). Metabolic and immune functional changes are tightly intertwined together, so it is not easy to determine which comes first. However, it is clear that enhancement of glycolytic pathway is necessary for the downstream function such as inflammatory cytokine and nitric oxide production in macrophages (Fig. 1A) (17). In contrast to the role of glycolysis, enhancement of oxidative phosphorylation (OXPHOS) in macrophage can lead to reduced inflammatory responses (Fig. 1A) (18). Metabolic alteration in tricarboxylic acid (TCA) cycle also affects macrophage function. Citrate, a component of the TCA cycle, can be catalyzed to itaconate by the enzyme encoded by immune responsive gene 1 (IRG1) (19). Itaconate shows an anti-inflammatory effect by inhibiting the activity of succinate dehydrogenase (SDH) (20). This induction of itaconate and accumulation of succinate has been identified as a hallmark in M1 activation (21). Succinate can stabilize hypoxia-inducible factor (HIF)-1α, thereby increasing the production of IL-1β (22). This is in the line with previous reports showing that M1 macrophage activation is dependent on HIF-1α rather than Myc (12, 23). Recent studies also found that itaconate performs its anti-inflammatory action via activation of nuclear factor erythroid 2-related factor 2 (Nrf2), a transcription factor for anti-oxidant and anti-inflammatory function (24, 25). Fatty acid synthesis (FAS) is also important in the differentiation and function of macrophages (26). In this study, primary lipid class switch from cholesterol to phosphatidylcholine is found during the differentiation of monocyte to macrophages. Inhibition of this process leads to the dysfunction of organelle development and phagocytic function of macrophage (26).
Fig. 1.
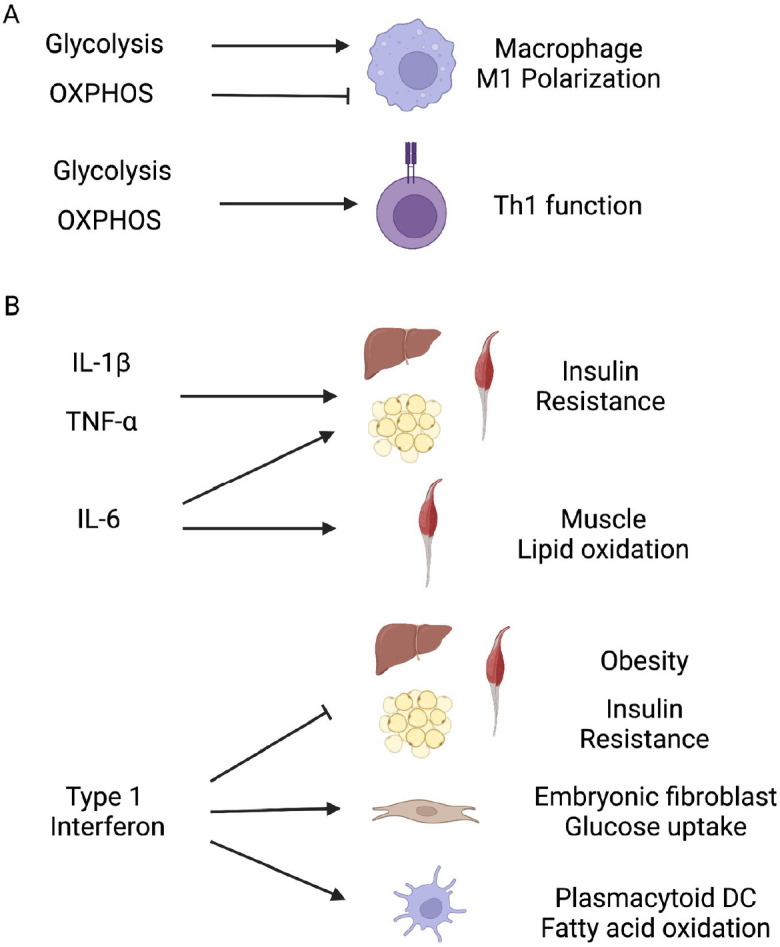
Type 1 immunity and metabolism. (A) Metabolic regulation of type 1 immune function. Glycolysis promotes and OXPHOS inhibits M1 macrophage polarization. Both glycolysis and OXPHOS metabolic pathway (aerobic glycolysis) support Th1 differentiation and function. (B) Type 1 immune regulation of metabolic function. Inflammatory cytokines such as IL-1β, TNF-α, and IL-6 provoke systemic insulin resistance. IL-6 induce lipid oxidation in muscles. Type 1 interferon inhibits insulin resistance and obesity. Type 1 interferon promotes glucose uptake in embryonic fibroblast and fatty acid oxidation in plasmacytoid DC.
The function of T cells and ILCs can also be greatly affected by the regulation of their metabolic pathways during activation (27). When these cells are in a naïve stage, they show quiescent metabolic status which produces ATP from a minimal carbon source because anabolic processes are not required (28-30). In contrast, activated T cells or ILCs can increase their overall metabolic fluxes toward both glycolysis and OXPHOS (31-34). For example, T cell receptor (TCR)-mediated signaling can induce the expression of enzymes involved in glycolysis and glutaminolysis and transporters for glucose and amino acids uptake (35, 36). The increase of these metabolic pathways is required to supply metabolic building blocks for cells to proliferate, differentiate, and release cytokines (37-39). In addition to these general effects, metabolic function can specifically influence Th1 differentiation and function. For example, when aerobic glycolysis is blocked during T cell activation, production of IFN-γ is markedly compromised (Fig. 1A) (40). The intense glycolysis is important to prevent moonlighting action of GAPDH, a key enzyme of glycolysis, which can inhibit the translation of IFN-γ under inhibition of glycolysis (40-42). Metabolic status and Th differentiation are also well described, particularly in the context of mammalian target of rapamycin (mTOR) signaling, which is reviewed elsewhere (43). As reviewed, it is obvious aerobic glycolysis can support Th1 function via the activation of mTOR complex (mTORC) 1 and 2 (43).
Immune regulation of metabolic function: Type 1 immune response can significantly affect metabolism. Upon M1 activation of macrophage, IFN-γ can suppress mTORC 1 and inhibit the translation of anti-inflammatory repressors such as HES1 (44). This metabolic reprogramming can potentiate macrophage activation (44). Increased fatty acid uptake and lipid synthesis but decreased lipolysis has been found upon activation of macrophages with ligands of TLRs (45). On the other hand, alternatively activated M2 macrophages show increased OXPHOS with intact TCA cycle without showing a significant induction of glycolytic activity (13, 21, 46, 47). At the cellular level, type 1 interferon can increase fatty acid oxidation and OXPHOS in plasmacytoid dendritic cells (pDC) (Fig. 1B) (48). This metabolic change of FAO with type 1 interferon is partly dependent on peroxisome proliferator-activated receptor (PPAR) α (48). De novo synthesis of long-chain fatty acid and cholesterol is reduced in bone marrow-derived macrophages (BMDMs) stimulated with IFN-β while lipid import is increased, resulting in an overall increase of fatty acid and cholesterol in cells (49). A study with mouse embryonic fibroblast (MEF) has shown that type 1 interferon IFN-β can reduce AMP-activated protein kinase (AMPK) phosphorylation accompanied by an increase of intracellular ATP (14). In the study, IFN-β induced glucose uptake by translocation of GLUT4 to the cell surface via PI3K-Akt signaling (Fig. 1B) (14). Other studies have found immune-metabolic reciprocal regulation at the level of cell-cell interaction. Monocytes infiltrating neuroblastoma can become M1 macrophages, which release IL-1β and TNF-α. Through p38/ERK signaling, macrophage IL-1β and TNF-α can promote tumor arginase 2 expression and alter arginine metabolism, leading to neuroblastoma cell proliferation (50).
Type 1 immune response can also regulate systemic metabolism. Studies over the past few decades have shown that inflammation is closely linked to various metabolic diseases. In addition, inflammation has been suggested to be a major source of pathology in various metabolic diseases. For example, pro-inflammatory immune cells accumulate in adipose tissues under obesity. Various inflammatory cytokines such as IL-1β, IL-6, and TNF-α produced from these immune cells can directly cause insulin resistance (Fig. 1B) (51-59). However, specific signaling affected by these cytokines can be context-dependent, particularly in the case of pleotropic IL-6. Thus, further studies are required to have a definite answer. Type 1 immune response also has broad effects on other metabolic pathways beyond glucose metabolism. Type 1 interferon signaling via inter-feron receptor interferon alpha and beta receptor subunit 1 (Ifnar1) in adipose tissues plays a significant role in the development of metabolic diseases. Mice without type 1 interferon signaling in adipose tissues develop obesity, glucose intolerance, and insulin resistance with high fat diet challenges (Fig. 1B) (60). Pleiotropic IL-6 modulates fat metabolism in muscles, enhances fatty acid oxidation rates, and reduce lipogenic effects of insulin (Fig. 1B) (61). However, TNF-α shows no effect on fatty acid oxidation, although it can enhance fatty acid incorporation into diacylglyceride, which could contribute TNF-induced insulin resistance in skeletal muscles (61).
Type 2 immunity and metabolism
Metabolic regulation of immune function: Type 2 immunity generally includes responses from immune cells in anti-parasite and allergic responses. Lymphoid cells (such as Th2, ILC2, and IgE+ B cells) and myeloid cells (such as eosinophils, mast cells, and basophils) belong to type 2 immunity. IL-4, IL-5, IL-13, IL-25, and IL-33 are major cytokines in this axis. Studies have also found that metabolic status can greatly influence type 2 immunity. M2 polarization of macrophage is increased by the activation of PPARγ, a master regulator of lipid metabolic pathway (Fig. 2A) (62). PPARγ activation has no effect on M2 marker expression in resting or M1 macrophages. It only has such an effect in monocytes, implying that M2 polarization with PPARγ activation can only be primed in naïve monocytes (62). Another study found that T cells and dendritic cells (DC) undergo development of type 2 immune responses when PPARγ signaling is activated (63). Upregulation of PPARγ signaling by IL-4 and IL-33 in lung-resident CD11b+ DCs can promote the migration of these cells to draining lymph nodes and prime Th2 differentiation (Fig. 2A) (63). Nuclear receptor PPARδ/β also plays an important role in M2 polarization of adipose tissue macrophage and liver residing Kupffer cells (64, 65). Deficiency of PPARδ/β signaling in macrophage results in impairment of glucose tolerance and aggravation of insulin resistance with high-fat diet challenges (Fig. 2A) (64, 65). Lactic acid produced by tumor cells as a by-product of aerobic or anaerobic glycolysis plays a key role in signaling for M2-like polarization of tumor-associated macrophages (Fig. 2A) (66). This effect is mediated by hypoxia-inducible factor 1a (HIF-1α) and arginase 1 (66). Similarly, a recent study found that endothelial lactate promotes M2-like macrophage polarization, which can induce muscle revascularization and regeneration under ischemia (67). Other studies also found that metabolic reprogramming affects germinal center B cell responses. Metabolic reprogramming induced by IL-4 in B cells is crucial for epigenomic activation of Bcl6 expression to promote germinal center (GC) B cell differentiation (68). Another study found that germinal center B cells mainly rely on fatty acid oxidation (but little on glycolysis) for energy generation, which is directly linked to germinal center B cell responses (Fig. 2A) (69).
Fig. 2.

Type 2 immunity and metabolism. (A) Metabolic regulation of type 2 immune function. Transcription regulation by PPARγ promotes monocyte M2 polarization and lung CD11b+ DC for Th2 priming. Transcription regulation by PPARδ increases tissue macrophage M2 polarization. Lactic acid also induces tissue macrophage M2 polarization. Fatty acid oxidation promotes germinal B cell reaction. (B) Type 2 immune regulation of metabolic function. IL-4 and its downstream activation of STAT6 inhibit systemic insulin resistance. Type 2 immune activation via IL-33/ILC2 and IL-4/IL-13 responses leads to beige adipose tissue differentiation and lipolysis. IL-13 promotes mitochondrial respiration in muscles.
Immune regulation of metabolic function: Type 2 immunity can significantly affect metabolism. As type 1 and type 2 immunities are mutually exclusive, opposing effect on metabolism has also been identified with type 2 immune response. When type 2 immune responses are blocked with STAT6 deficiency, insulin action is reduced and PPARα-driven oxidative metabolism pro-gram is enhanced (70). On the other hand, activation of STAT6 by IL-4 increases insulin action by blocking the PPARα-regulated food catabolism program and reducing adipose tissue inflammation (Fig. 2B) (70). Moreover, IL-4 can modulate glucose and lipid metabolism by improving glucose tolerance and decreasing adipogenesis via PPARγ down-regulation (71). Furthermore, IL-4 can promote hormone-sensitive lipase translocation, which increases lipolysis in adipocytes (Fig. 2B) (71). Studies have also found that type 2 immunity plays a significant role in cold adaptation and beige fat biogenesis (72). Treatment with IL-33 increases levels of ILC2s in inguinal adipose tissues of mice, which can promote the biogenesis of functional beige fat by stimulating the commitment of PDGFR+ adipocyte pre-cursor cells to the beige adipocyte lineage in an IL-4/13-dependent manner (Fig. 2B) (72). Another study has also found that ILC2s induced by IL-33 can promote beiging of adipose tissues by producing methionine-enkephalin peptides (73). Type 2 immune response also influences muscle endurance exercise and metabolism. Endurance training can enhance a network of mitochondrial and fatty acid oxidation genes, which is dependent on IL-13 signaling (Fig. 2B) (74). IL-13 can activate STAT3 by acting directly on skeletal muscles via its receptor IL-13R1 and boosted mitochondrial respiration (74).
Type 17 and regulatory immunity and metabolism
Metabolic regulation of immune function: Despite the fact that Th17 and Treg have completely distinct functions, they share molecular pathways in differentiation. The unique relationship between Th17 and Treg has been discussed and many molecular mechanisms that control Th17 and Treg balance have been discovered. Interconversion between these two cell types is even possible after differentiation (75, 76). Metabolic context has a significant effect on the differentiation of Th17 and Treg and their cognate functions. In general, inflammatory T cells have a high glycolytic gene expression. They rely on glycolysis to proliferate and survive, whereas Tregs rely on mitochondrial oxidative metabolism to survive and function (77, 78). The HIF-1α-dependent transcriptional pathway is critical for modulating glycolytic activity and influencing Th17 and Treg cell lineage decisions (79). HIF-1α is only expressed in Th17 cells. Its activation is required for mTOR signaling. When the glycolytic pathway is inhibited, Th17 differentiation is blocked, while Treg generation is promoted (Fig. 3A) (79). Another study has found that the fate decision of Treg and Th17 cells is controlled by de novo fatty acid synthesis (80). In the study, inhibition of acetyl-CoA carboxylase 1 (ACC1) decreased the production of Th17 cells but enhanced the development of Treg in humans and mice (Fig. 3A) (80). For their development, Th17 cells rely on de novo fatty acid synthesis driven by ACC1 and the underlying glycolytic-lipogenic metabolic pathway (80). Metabolic alteration can also regulate the balance of Th17 and Treg cells by epigenetic modification. In developing Th17 cells, enhanced transamination primarily catalyzed by glutamate oxaloacetate transaminase 1 (GOT1) results in higher amounts of 2-hydroxyglutarate (81). This 2-hydroxyglutarate can lead to hypermethylation of the Foxp3 gene locus and suppression of Foxp3 transcription, which promotes fate determination toward Th17 cells (Fig. 3A) (81).
Fig. 3.

Type 17 and regulatory immunity and metabolism. (A) Meta-bolic control of type 17 and regulatory immune function. HIF-1α activation and/or glycolysis promotes Th17 and suppresses Treg differentiation. De novo fatty acid synthesis by ACC1 promotes Th17 and suppresses Treg differentiation. 2-hydroxyglutarate by GOT1 also promotes Th17 and suppresses Treg differentiation. (B) Type 17 and regulatory immune control of metabolic function. IL-17 promotes fatty acid synthesis in livers while suppressing adipogenesis. γδ T cells promotes adipose tissue thermogenesis. Treg also supports thermogenesis. Treg inhibits systemic insulin resistance and type 1 interferon can negate this function by depleting Treg.
Immune regulation of metabolic function: Studies on metabolic alteration by Th17 and Treg immune signaling are also accumulating. These cells and their cognate cytokine signaling have significant effects on the function of various metabolic tissues. In the liver, IL-17 signaling regulates hepatic steatosis (Fig. 3B). Increased IL-17 can promote sterol regulatory element-binding protein 1c (SREBP-1c) and carbohydrate-responsive element-binding protein (ChREBP) transcription while suppressing enoyl-CoA hydratase short chain 1 (ECHS1) and PPARα for the alteration of fatty acid metabolism (82). Mice lacking IL-17 gained more fat mass as they aged, indicating that IL-17 could regulate glucose homeostasis and inhibit adipogenesis (Fig. 3B) (83, 84). This effect is partly dependent on the suppression of adipogenic PPARγ and CCAAT/enhancer binding proteins (C/EBP) α by IL-17 (84). Recent studies have highlighted the role of γδ T cells in metabolic homeostasis and cold adaptation. Visceral adipose tissue resident population of γδ T cells regulate age-dependent Treg expansion and control core body temperature (Fig. 3B) (85). Moreover, γδ T cells in thermogenic adipose tissue play an important role in sympathetic innervation by increasing TGF-β1 production in parenchymal cells, at least in part, through the IL-17 receptor C (IL-17RC) (86). Treg cells re-siding in adipose tissues are depleted in obesity. They can inhibit inflammation and insulin resistance by producing anti-inflammatory IL-10 (Fig. 3B) (87). A recent study has further identified that plasmacytoid dendritic cells that produce IFN-α can provoke the loss of adipose tissue Treg (Fig. 3B) (88). Moreover, a pro-inflammatory state is developed when local Treg cells are removed from brown adipose tissues, which can decrease thermogenic activity (89). Treg is also induced by cold exposure and beta3-adrenergic receptor (ADRB3) stimulation. It can enhance adipose tissue beiging, thermogenesis, and lipolysis, which is mediated by T cell specific STAT6 and Pten signaling (90, 91).
CONCLUSION
Recent reports strongly suggest that the immune system’s traditional pathogen-killing role is only a small part of its overall functions. Studies on sterile inflammation have shown that cellular stress can elicit inflammation which is beneficial for the organism’s survival either by eliminating or promoting adap-tability to stressors. There is also a clear link between metabolic regulation and immunity. We have reviewed how type 1, type 2, type 17, and regulatory immunity are significantly controlled by metabolism and how metabolism is altered by different types of immune responses. Although studies on immunity in systemic metabolism and physiological homeostasis are being accumulated, the discussion on this subject is still stagnant partly due to the lack of a framework that can encompass classical and novel roles of immune responses with homeostatic and evolutionary points of view. Thus, an experiment to test a big idea on this subject is highly required. Emergence of a new perspective may be facilitated by accumulated public OMICS data and systems biology approaches.
ACKNOWLEDGEMENTS
We apologize that we cannot cite all the seminal papers on this subject. This work was supported by the Yonsei Research Fund (2021-22-0049), Yonsei Signature Research Cluster Program of 2022 (2022-22-0013), and the National Research Foundation of Korea (NRF) Ministry of Science, ICT and Future Plan-ning NRF-2021-22-0049.
Footnotes
CONFLICTS OF INTEREST
The authors have no conflicting interests.
REFERENCES
- 1.Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454:428–435. doi: 10.1038/nature07201. [DOI] [PubMed] [Google Scholar]
- 2.Medzhitov R. The spectrum of inflammatory responses. Science. 2021;374:1070–1075. doi: 10.1126/science.abi5200. [DOI] [PubMed] [Google Scholar]
- 3.Roh JS, Sohn DH. damage-associated molecular patterns in inflammatory diseases. Immune Netw. 2018;18:e27. doi: 10.4110/in.2018.18.e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, Greenberg RA. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature. 2017;548:466–470. doi: 10.1038/nature23470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Urano F, Wang X, Bertolotti A, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287:664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- 6.Keestra-Gounder AM, Byndloss MX, Seyffert N, et al. NOD1 and NOD2 signalling links ER stress with inflammation. Nature. 2016;532:394–397. doi: 10.1038/nature17631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duewell P, Kono H, Rayner KJ, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–1361. doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hornung V, Bauernfeind F, Halle A, et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol. 2008;9:847–856. doi: 10.1038/ni.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yabal M, Calleja DJ, Simpson DS, Lawlor KE. Stressing out the mitochondria: mechanistic insights into NLRP3 inflammasome activation. J Leukoc Biol. 2019;105:377–399. doi: 10.1002/JLB.MR0318-124R. [DOI] [PubMed] [Google Scholar]
- 10.Kotas ME, Medzhitov R. Homeostasis, inflammation, and disease susceptibility. Cell. 2015;160:816–827. doi: 10.1016/j.cell.2015.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chovatiya R, Medzhitov R. Stress, inflammation, and defense of homeostasis. Mol Cell. 2014;54:281–288. doi: 10.1016/j.molcel.2014.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu L, Lu Y, Martinez J, et al. Proinflammatory signal suppresses proliferation and shifts macrophage metabolism from Myc-dependent to HIF1α-dependent. Proc Natl Acad Sci U S A. 2016;113:1564–1569. doi: 10.1073/pnas.1518000113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van den Bossche J, Baardman J, de Winther MP. Metabolic characterization of polarized M1 and M2 bone marrow-derived macrophages using real-time extracellular flux analysis. J Vis Exp. 2015;105:53424. doi: 10.3791/53424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burke JD, Platanias LC, Fish EN. Beta interferon regulation of glucose metabolism is PI3K/Akt dependent and important for antiviral activity against coxsackievirus B3. J Virol. 2014;88:3485–3495. doi: 10.1128/JVI.02649-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dror E, Dalmas E, Meier DT, et al. Postprandial macrophage-derived IL-1β stimulates insulin, and both synergistically promote glucose disposal and inflammation. Nat Immunol. 2017;18:283–292. doi: 10.1038/ni.3659. [DOI] [PubMed] [Google Scholar]
- 16.Jessop F, Buntyn R, Schwarz B, Wehrly T, Scott D, Bosio CM. Interferon gamma reprograms host mitochondrial metabolism through inhibition of complex II to control intracellular bacterial replication. Infect Immun. 2020;88:e00744–19. doi: 10.1128/IAI.00744-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tan Z, Xie N, Cui H, et al. Pyruvate dehydrogenase kinase 1 participates in macrophage polarization via regulating glucose metabolism. J Immunol. 2015;194:6082–6089. doi: 10.4049/jimmunol.1402469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vats D, Mukundan L, Odegaard JI, et al. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab. 2006;4:13–24. doi: 10.1016/j.cmet.2006.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Michelucci A, Cordes T, Ghelfi J, et al. Immune-responsive gene 1 protein links metabolism to immunity by catalyzing itaconic acid production. Proc Natl Acad Sci U S A. 2013;110:7820–7825. doi: 10.1073/pnas.1218599110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lampropoulou V, Sergushichev A, Bambouskova M, et al. Itaconate links inhibition of succinate dehydrogenase with macrophage metabolic remodeling and regulation of inflammation. Cell Metab. 2016;24:158–166. doi: 10.1016/j.cmet.2016.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jha AK, Huang SC, Sergushichev A, et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity. 2015;42:419–430. doi: 10.1016/j.immuni.2015.02.005. [DOI] [PubMed] [Google Scholar]
- 22.Tannahill GM, Curtis AM, Adamik J, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature. 2013;496:238–242. doi: 10.1038/nature11986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meiser J, Krämer L, Sapcariu SC, et al. Pro-inflammatory macrophages sustain pyruvate oxidation through pyruvate dehydrogenase for the synthesis of itaconate and to enable cytokine expression. J Biol Chem. 2016;291:3932–3946. doi: 10.1074/jbc.M115.676817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mills EL, Ryan DG, Prag HA, et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature. 2018;556:113–117. doi: 10.1038/nature25986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bambouskova M, Gorvel L, Lampropoulou V, et al. Electrophilic properties of itaconate and derivatives regulate the IκBξ-ATF3 inflammatory axis. Nature. 2018;556:501–504. doi: 10.1038/s41586-018-0052-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ecker J, Liebisch G, Englmaier M, Grandl M, Robenek H, Schmitz G. Induction of fatty acid synthesis is a key requirement for phagocytic differentiation of human monocytes. Proc Natl Acad Sci U S A. 2010;107:7817–7822. doi: 10.1073/pnas.0912059107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chapman NM, Chi H. Hallmarks of T-cell exit from quiescence. Cancer Immunol Res. 2018;6:502–508. doi: 10.1158/2326-6066.CIR-17-0605. [DOI] [PubMed] [Google Scholar]
- 28.Rathmell JC, Farkash EA, Gao W, Thompson CB. IL-7 enhances the survival and maintains the size of naive T cells. J Immunol. 2001;167:6869–6876. doi: 10.4049/jimmunol.167.12.6869. [DOI] [PubMed] [Google Scholar]
- 29.Zhang S, Zhang X, Wang K, et al. Newly generated CD4(+) T cells acquire metabolic quiescence after thymic egress. J Immunol. 2018;200:1064–1077. doi: 10.4049/jimmunol.1700721. [DOI] [PubMed] [Google Scholar]
- 30.Miller ML, Mashayekhi M, Chen L, et al. Basal NF-κB controls IL-7 responsiveness of quiescent naïve T cells. Proc Natl Acad Sci U S A. 2014;111:7397–7402. doi: 10.1073/pnas.1315398111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fox CJ, Hammerman PS, Thompson CB. Fuel feeds function: energy metabolism and the T-cell response. Nat Rev Immunol. 2005;5:844–852. doi: 10.1038/nri1710. [DOI] [PubMed] [Google Scholar]
- 32.Loftus RM, Finlay DK. Immunometabolism: cellular metabolism turns immune regulator. J Biol Chem. 2016;291:1–10. doi: 10.1074/jbc.R115.693903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Glick GD, Rossignol R, Lyssiotis CA, et al. Anaple-rotic metabolism of alloreactive T cells provides a metabolic approach to treat graft-versus-host disease. J Pharmacol Exp Ther. 2014;351:298–307. doi: 10.1124/jpet.114.218099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pearce EL, Pearce EJ. Metabolic pathways in immune cell activation and quiescence. Immunity. 2013;38:633–643. doi: 10.1016/j.immuni.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang R, Dillon CP, Shi LZ, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity. 2011;35:871–882. doi: 10.1016/j.immuni.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sinclair LV, Rolf J, Emslie E, Shi YB, Taylor PM, Cantrell DA. Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat Immunol. 2013;14:500–508. doi: 10.1038/ni.2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lochner M, Berod L, Sparwasser T. Fatty acid metabolism in the regulation of T cell function. Trends Immunol. 2015;36:81–91. doi: 10.1016/j.it.2014.12.005. [DOI] [PubMed] [Google Scholar]
- 38.Swamy M, Pathak S, Grzes KM, et al. Glucose and glutamine fuel protein O-GlcNAcylation to control T cell self-renewal and malignancy. Nat Immunol. 2016;17:712–720. doi: 10.1038/ni.3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol. 2011;27:441–464. doi: 10.1146/annurev-cellbio-092910-154237. [DOI] [PubMed] [Google Scholar]
- 40.Chang CH, Curtis JD, Maggi LB, Jr, et al. Post-transcriptional control of T cell effector function by aerobic glycolysis. Cell. 2013;153:1239–1251. doi: 10.1016/j.cell.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nagy E, Henics T, Eckert M, Miseta A, Lightowlers RN, Kellermayer M. Identification of the NAD(+)-binding fold of glyceraldehyde-3-phosphate dehydrogenase as a novel RNA-binding domain. Biochem Biophys Res Commun. 2000;275:253–260. doi: 10.1006/bbrc.2000.3246. [DOI] [PubMed] [Google Scholar]
- 42.Boukouris AE, Zervopoulos SD, Michelakis ED. Metabolic enzymes moonlighting in the nucleus: metabo-lic regulation of gene transcription. Trends Biochem Sci. 2016;41:712–730. doi: 10.1016/j.tibs.2016.05.013. [DOI] [PubMed] [Google Scholar]
- 43.Chi H. Regulation and function of mTOR signalling in T cell fate decisions. Nat Rev Immunol. 2012;12:325–338. doi: 10.1038/nri3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Su X, Yu Y, Zhong Y, et al. Interferon-γ regulates cellular metabolism and mRNA translation to potentiate macrophage activation. Nat Immunol. 2015;16:838–849. doi: 10.1038/ni.3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Feingold KR, Shigenaga JK, Kazemi MR, et al. Me-chanisms of triglyceride accumulation in activated macrophages. J Leukoc Biol. 2012;92:829–839. doi: 10.1189/jlb.1111537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rodríguez-Prados JC, Través PG, Cuenca J, et al. Substrate fate in activated macrophages: a comparison between innate, classic, and alternative activation. J Immunol. 2010;185:605–614. doi: 10.4049/jimmunol.0901698. [DOI] [PubMed] [Google Scholar]
- 47.Huang SC, Smith AM, Everts B, et al. Metabolic reprogramming mediated by the mTORC2-IRF4 signaling axis is essential for macrophage alternative activation. Im-munity. 2016;45:817–830. doi: 10.1016/j.immuni.2016.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu D, Sanin DE, Everts B, et al. Type 1 interferons induce changes in core metabolism that are critical for immune function. Immunity. 2016;44:1325–1336. doi: 10.1016/j.immuni.2016.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.York AG, Williams KJ, Argus JP, et al. Limiting cholesterol biosynthetic flux spontaneously engages type I IFN signaling. Cell. 2015;163:1716–1729. doi: 10.1016/j.cell.2015.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fultang L, Gamble LD, Gneo L, et al. macrophage-derived IL1β and TNFα regulate arginine metabo-lism in neuroblastoma. Cancer Res. 2019;79:611–624. doi: 10.1158/0008-5472.CAN-18-2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jager J, Grémeaux T, Cormont M, Le Marchand-Brustel Y, Tanti JF. Interleukin-1beta-induced insulin resistance in adipocytes through down-regulation of insulin receptor substrate-1 expression. Endocrinology. 2007;148:241–251. doi: 10.1210/en.2006-0692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lagathu C, Yvan-Charvet L, Bastard JP, et al. Long-term treatment with interleukin-1beta induces insulin resistance in murine and human adipocytes. Diabetologia. 2006;49:2162–2173. doi: 10.1007/s00125-006-0335-z. [DOI] [PubMed] [Google Scholar]
- 53.Gao D, Madi M, Ding C, et al. Interleukin-1β mediates macrophage-induced impairment of insulin signaling in human primary adipocytes. Am J Physiol Endo-crinol Metab. 2014;307:E289–E304. doi: 10.1152/ajpendo.00430.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Senn JJ, Klover PJ, Nowak IA, Mooney RA. Interleukin-6 induces cellular insulin resistance in hepato-cytes. Diabetes. 2002;51:3391–3399. doi: 10.2337/diabetes.51.12.3391. [DOI] [PubMed] [Google Scholar]
- 55.Kim H-J, Higashimori T, Park SY, et al. Differential effects of interleukin-6 and -10 on skeletal muscle and liver insulin action in vivo. Diabetes. 2004;53:1060–1067. doi: 10.2337/diabetes.53.4.1060. [DOI] [PubMed] [Google Scholar]
- 56.Cai D, Yuan M, Frantz DF, et al. Local and systemic insulin resistance resulting from hepatic activation of IKK-β and NF-κB. Nat Med. 2005;11:183–190. doi: 10.1038/nm1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hotamisligil GS, Arner P, Caro JF, Atkinson RL, Spiegel-man BM. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J Clin Invest. 1995;95:2409–2415. doi: 10.1172/JCI117936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259:87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- 59.Uysal KT, Wiesbrock SM, Marino MW, Hotamisligil GS. Protection from obesity-induced insulin resistance in mice lacking TNF-α function. Nature. 1997;389:610–614. doi: 10.1038/39335. [DOI] [PubMed] [Google Scholar]
- 60.Wieser V, Adolph TE, Grander C, et al. Adipose type I interferon signalling protects against metabolic dysfunction. Gut. 2018;67:157–165. doi: 10.1136/gutjnl-2016-313155. [DOI] [PubMed] [Google Scholar]
- 61.Bruce CR, Dyck DJ. Cytokine regulation of skeletal muscle fatty acid metabolism: effect of interleu-kin-6 and tumor necrosis factor-alpha. Am J Physiol Endo-crinol Metab. 2004;287:E616–E621. doi: 10.1152/ajpendo.00150.2004. [DOI] [PubMed] [Google Scholar]
- 62.Bouhlel MA, Derudas B, Rigamonti E, et al. PPAR-gamma activation primes human monocytes into alterna-tive M2 macrophages with anti-inflammatory properties. Cell Metab. 2007;6:137–143. doi: 10.1016/j.cmet.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 63.Nobs SP, Natali S, Pohlmeier L, et al. PPARγ in dendritic cells and T cells drives pathogenic type-2 effec-tor responses in lung inflammation. J Exp Med. 2017;214:3015–3035. doi: 10.1084/jem.20162069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kang K, Reilly SM, Karabacak V, et al. Adipocyte-derived Th2 cytokines and myeloid PPARδ regulate ma-crophage polarization and insulin sensitivity. Cell Metab. 2008;7:485–495. doi: 10.1016/j.cmet.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Odegaard JI, Ricardo-Gonzalez RR, Red Eagle A, et al. Alternative M2 activation of kupffer cells by PPARδ Ameliorates obesity-induced insulin resistance. Cell Metab. 2008;7:496–507. doi: 10.1016/j.cmet.2008.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Colegio OR, Chu NQ, Szabo AL, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. 2014;513:559–563. doi: 10.1038/nature13490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang J, Muri J, Fitzgerald G, et al. Endothelial lactate controls muscle regeneration from ischemia by inducing M2-like macrophage polarization. Cell Metab. 2020;31:1136–1153.e1137. doi: 10.1016/j.cmet.2020.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Haniuda K, Fukao S, Kitamura D. Metabolic reprogramming induces germinal center B cell differentiation through Bcl6 locus remodeling. Cell Rep. 2020;33:108333. doi: 10.1016/j.celrep.2020.108333. [DOI] [PubMed] [Google Scholar]
- 69.Weisel FJ, Mullett SJ, Elsner RA, et al. Germinal center B cells selectively oxidize fatty acids for energy while conducting minimal glycolysis. Nat Immunol. 2020;21:331–342. doi: 10.1038/s41590-020-0598-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ricardo-Gonzalez RR, Red Eagle A, Odegaard JI, et al. IL-4/STAT6 immune axis regulates peripheral nutrient metabolism and insulin sensitivity. Proc Natl Acad Sci U S A. 2010;107:22617–22622. doi: 10.1073/pnas.1009152108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tsao CH, Shiau MY, Chuang PH, Chang YH, Hwang J. Interleukin-4 regulates lipid metabolism by inhibi-ting adipogenesis and promoting lipolysis. J Lipid Res. 2014;55:385–397. doi: 10.1194/jlr.M041392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lee MW, Odegaard JI, Mukundan L, et al. Activated type 2 innate lymphoid cells regulate beige fat biogenesis. Cell. 2015;160:74–87. doi: 10.1016/j.cell.2014.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Brestoff JR, Kim BS, Saenz SA, et al. Group 2 innate lymphoid cells promote beiging of white adipose tissue and limit obesity. Nature. 2015;519:242–246. doi: 10.1038/nature14115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Knudsen NH, Stanya KJ, Hyde AL, et al. Inter-leukin-13 drives metabolic conditioning of muscle to en-durance exercise. Science. 2020;368:eaat3987. doi: 10.1126/science.aat3987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gagliani N, Amezcua Vesely MC, Iseppon A, et al. Th17 cells transdifferentiate into regulatory T cells during resolution of inflammation. Nature. 2015;523:221–225. doi: 10.1038/nature14452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schenk U, Frascoli M, Proietti M, et al. ATP inhibits the generation and function of regulatory T cells through the activation of purinergic P2X receptors. Sci Signal. 2011;4:ra12. doi: 10.1126/scisignal.2001270. [DOI] [PubMed] [Google Scholar]
- 77.Gerriets VA, Kishton RJ, Nichols AG, et al. Meta-bolic programming and PDHK1 control CD4+ T cell sub-sets and inflammation. J Clin Invest. 2015;125:194–207. doi: 10.1172/JCI76012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Michalek RD, Gerriets VA, Jacobs SR, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic pro-grams are essential for effector and regulatory CD4+ T cell subsets. J Immunol. 2011;186:3299–3303. doi: 10.4049/jimmunol.1003613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shi LZ, Wang R, Huang G, et al. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med. 2011;208:1367–1376. doi: 10.1084/jem.20110278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Berod L, Friedrich C, Nandan A, et al. De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat Med. 2014;20:1327–1333. doi: 10.1038/nm.3704. [DOI] [PubMed] [Google Scholar]
- 81.Xu T, Stewart KM, Wang X, et al. Metabolic control of T(H)17 and induced T(reg) cell balance by an epige-netic mechanism. Nature. 2017;548:228–233. doi: 10.1038/nature23475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shi W, Zhu Q, Gu J, et al. Anti-IL-17 antibody improves hepatic steatosis by suppressing interleukin-17-related fatty acid synthesis and metabolism. Clin Dev Immunol. 2013;2013:253046. doi: 10.1155/2013/253046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zúñiga LA, Shen WJ, Joyce-Shaikh B, et al. IL-17 regulates adipogenesis, glucose homeostasis, and obesity. J Immunol. 2010;185:6947–6959. doi: 10.4049/jimmunol.1001269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ahmed M, Gaffen SL. IL-17 inhibits adipo-genesis in part via C/EBPα, PPARγ and Krüppel-like factors. Cytokine. 2013;61:898–905. doi: 10.1016/j.cyto.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kohlgruber AC, Gal-Oz ST, LaMarche NM, et al. γδ T cells producing interleukin-17A regulate adipose regulatory T cell homeostasis and thermogenesis. Nat Immunol. 2018;19:464–474. doi: 10.1038/s41590-018-0094-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hu B, Jin C, Zeng X, et al. γδ T cells and adipocyte IL-17RC control fat innervation and thermogenesis. Nature. 2020;578:610–614. doi: 10.1038/s41586-020-2028-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Feuerer M, Herrero L, Cipolletta D, et al. Lean, but not obese, fat is enriched for a unique population of re-gulatory T cells that affect metabolic parameters. Nat Med. 2009;15:930–939. doi: 10.1038/nm.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Li C, Wang G, Sivasami P, et al. Interferon-α-pro-ducing plasmacytoid dendritic cells drive the loss of adi-pose tissue regulatory T cells during obesity. Cell Metab. 2021;33:1610–1623.e1615. doi: 10.1016/j.cmet.2021.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Medrikova D, Sijmonsma TP, Sowodniok K, et al. Brown adipose tissue harbors a distinct sub-population of regulatory T cells. PLoS One. 2015;10:e0118534. doi: 10.1371/journal.pone.0118534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kälin S, Becker M, Ott VB, et al. A Stat6/Pten axis links regulatory T cells with adipose tissue function. Cell Metab. 2017;26:475–492.e477. doi: 10.1016/j.cmet.2017.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fang W, Deng Z, Benadjaoud F, Yang D, Yang C, Shi GP. Regulatory T cells promote adipocyte beiging in subcutaneous adipose tissue. FASEB J. 2020;34:9755–9770. doi: 10.1096/fj.201902518R. [DOI] [PubMed] [Google Scholar]