

Abstract
The mechanisms underlying T-ALL relapse remain essentially unknown. Multilevel-omics in 38 matched pairs of initial and relapsed T-ALL revealed 18 (47%) type-1 (defined by being derived from the major ancestral clone) and 20 (53%) type-2 relapses (derived from a minor ancestral clone). In both types of relapse, we observed known and novel drivers of multidrug resistance including MDR1 and MVP, NT5C2 and JAK-STAT activators. Patients with type-1 relapses were specifically characterized by IL7R upregulation. In remarkable contrast, type-2 relapses demonstrated (1) enrichment of constitutional cancer predisposition gene mutations, (2) divergent genetic and epigenetic remodeling, and (3) enrichment of somatic hypermutator phenotypes, related to BLM, BUB1B/PMS2 and TP53 mutations. T-ALLs that later progressed to type-2 relapses exhibited a complex subclonal architecture, unexpectedly, already at the time of initial diagnosis. Deconvolution analysis of ATAC-Seq profiles showed that T-ALLs later developing into type-1 relapses resembled a predominant immature thymic T-cell population, whereas T-ALLs developing into type-2 relapses resembled a mixture of normal T-cell precursors. In sum, our analyses revealed fundamentally different mechanisms driving either type-1 or type-2 T-ALL relapse and indicate that differential capacities of disease evolution are already inherent to the molecular setup of the initial leukemia.
Subject terms: Cancer genomics, Cancer genomics, Leukaemia
Introduction
Relapse is the main cause of death from pediatric acute precursor T-cell leukemia (T-ALL) [1, 2], but the underlying mechanisms of disease evolution from initial disease to relapse remain incompletely understood and show a remarkable interpatient heterogeneity. The acquisition of mutations and epigenetic modifications represent important components of this process [3–7]. Specifically, activating mutations of the nucleosidase NT5C2 are acquired by app. 20% of patients at relapse [8–10]. These mutations are predicted to confer chemotherapy resistance but are often found to be subclonal [10]. TP53 mutations occur in-app. 12% of relapses and predict a high risk of treatment failure and fatal outcome [11–13]. Although prevalent at T-ALL relapse [14] and correlated with an increased cumulative incidence of relapse [15], RAS-MAPK pathway-activating mutations were reported both, in clones that are eradicated and in those that emerge at relapse at the same time rendering lymphoblasts resistant against methotrexate, while sensitizing them to vincristine [4]. Similarly, NOTCH1 activating mutations, which enhance proliferation and survival [16] have recently been reported to be frequently acquired at later T-ALL stages thus highlighting the importance of NOTCH1-activation for T-ALL progression [17].
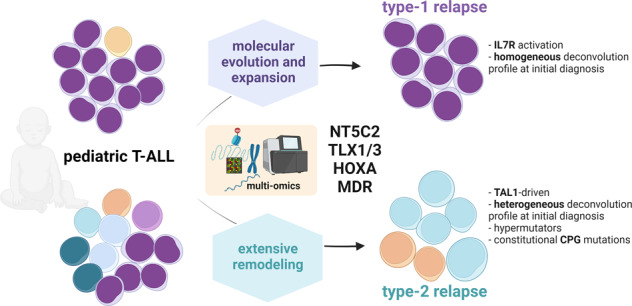
We have previously described two types of T-ALL relapses, characterized either by the clonal evolution of the major clone present at the time of initial presentation (type-1) or by the emergence and evolution of a minor ancestor clone showing a molecular profile that is distinct from the predominant initial clone (type-2) [3].
As cross-sectional genomic and transcriptomic studies failed to identify unifying biological determinants of relapse, we now adopted a longitudinal strategy and performed multi-level omics analyses in 13 matched pairs of initial diagnosis and relapse samples and their corresponding patient-derived xenografts (PDX) models. We extended this sample set by whole-exome sequencing (WES) and methylome analyses in an additional cohort of 25 matched sets of DNA samples obtained from primary patient cells collected at initial diagnosis, remission, and relapse.
Material and methods
Patients
The primary cells were obtained from patients recruited in ALL‐BFM 2000, ALL‐BFM‐2009, CoALL97, CoALL03, CoALL09, and ALL‐REZ BFM 2002 trials or from Schneider Children’s Medical Center of Israel from the time points of first diagnosis, remission, and relapse (Supplementary Table 8). Of the 38 patients described here some details of 13 patients were previously reported [3].
Clinical trials from which samples were used in this analysis had previously received approval from the relevant institutional review boards or ethics committees. Written informed consent had been obtained from all the patients or legal guardians, and the experiments conformed to the principles set out in the WMA Declaration of Helsinki and the Department of Health and Human Services Belmont Report.
Whole-exome and ATAC sequencing
WES and ATAC-Seq were performed as described before [18, 19] - for details see Supplementary Materials.
Analysis of cancer predisposing genes
We generated a list of 227 potential CPG (Suppl. Tab. 9) by combining previously reported tumor suppressors or genes involved in DNA repair [20–24]. Sixty-two remission samples (day 33) from 38 patients who later developed a relapse and from 24 who did not relapse were subjected to WES. Of the constitutional variants (AF ≥30%) we focused exclusively on: stopgain/loss, frameshift InDels, splicesite donor/acceptor in 227 CPGs.
DNA methylation analysis using 450k BeadChip Arrays and Infinium® MethylationEPIC BeadChip Arrays
Bisulfite‐conversion, analysis on the Infinium® Methylation assay and EPIC assay (Illumina) and downstream analysis using the RnBeads software package [25] were performed as reported before [19].
Software and bioinformatical tools
For graphical representation and statistical analyses, GraphPad and R [26] were used. R packages: DESeq2, DNA copy, ggplot2, reshape2, scales.
Graphical abstract was created with BioRender.com.
Results
Clonal selection in type-2 T-ALL relapses is coupled with a high degree of heterogeneity and an increase of mutation load at relapse
Based on the profile of single nucleotide variants (SNV) and insertions/deletions (InDels) (Fig. 1) generated by WES of all 38 patients who were analyzed at the time of initial diagnosis, remission, and relapse we distinguished 18 (47%) patients with type-1 relapses as defined by all clonal mutations with allele frequencies (AF) >30% being preserved at relapse. In 20 (53%) patients with type-2 relapses the major clone present at initial diagnosis was eradicated as defined by a subset of clonal mutations with AF >30% being lost in the relapse [3]. CNA/CN-LOH (copy number alterations/copy-neutral loss of heterozygosity) analyses showed a loss of CNA/CN-LOH profiles detected during initial disease in 9/20 type-2 relapses, confirming the loss of the major clone in these patients (Supplementary Fig. 1). By contrast, no losses of CNA/CN-LOH profiles were observed among type-1 relapses.
Fig. 1. Allele frequency analysis of mutations reveals clonal evolution on the way from initial T-ALL to either type-1 or type-2 relapse.

Scatter plots of allele frequency (AF) of mutations detected at initial diagnosis (INI, x-axis) and relapse (REL, y-axis). Type-1 relapses are defined by all clonal mutations with allele frequencies (AF) > 30% being preserved at relapse; in type-2 relapses, the major clone present at initial diagnosis is eradicated as defined by a subset of clonal mutations with AF > 30% being lost in the relapse. BC = blast content at the time of relapse; dashed red line – 30% allele frequency threshold for clonal (AF ≥ 30%) and subclonal (>30%) mutations; color code: NOTCH1/3 – green, FBXW7 – orange, NRAS/KRAS – red, IL7R/JAK/STAT pathway – yellow, PI3K pathway – violet, ribosomal genes – pink, chromatin modifiers – blue, the remaining mutations are labelled in grey; red frames indicate those patients in whom CNA analysis confirmed type-2 relapse. P4REL – because of the blast content of only 8% the leukemia cells isolated from the relapse, PDX of this patient was used for classification, which demonstrated this relapse to be of type-1; P32 – the mutations lost at relapse had very low coverage; P2 – low blast content at initial diagnosis; the CNA pattern together with the analysis of corresponding PDXs suggests a type-2 relapse.
Whereas leukemias relapsing as type-1 preserved most of the coding mutations (306/337; 91%) detected at initial diagnosis, type-2 leukemias preserved only 51% (181/356; Fig. 2A). Moreover, the number of mutations acquired at the time of type-1 relapse was significantly lower when compared to type-2 (type-1: mean ± SEM: 10.94 ± 1.677 N = 18; vs. type-2: mean ± SEM: 33.05 ± 7.636 N = 20; p = 0.01 (ttest, unpaired)) indicating that type-2 T-ALLs underwent stronger genomic remodeling on the way to relapse (Fig. 2B). A substantial increase in the mutational load in type-2 relapses was particularly evident in three patients: P1, P8, P18 (Fig. 2C), who carried more than 85 coding mutations at the time of relapse. This remarkable accumulation of mutations was likely caused by gains of mutations in the Bloom RecQ helicase (BLM; P18), simultaneous mutations in the mitotic checkpoint serine/threonine-protein kinase (BUB1B) and the mismatch repair endonuclease PMS2 (P8), and in TP53 (P1), respectively. P18 (BLM) and P8 (BUB1B/PMS2) acquired the highest numbers of somatic deletions (Supplementary Fig. 2a), and an analysis of the mutation context suggests a dominant contribution of Cosmic Mutational Signature 6, which is indicative of defective DNA mismatch repair or microsatellite unstable tumors (Supplementary Fig. 2b). Remarkably, the fraction of subclonal mutations of those T-ALLs that later developed into a type-2 relapse was significantly higher already at the time of initial diagnosis than in those T-ALLs that later developed into a type-1 relapse (type-1: 131/337 (39%); type-2: 167/356 (47%), p = 0.0387; Fisher’s exact), a difference that became even more pronounced at the time of relapse (type-1: 153/504 (30%); type-2: 431/842 (51%)); p < 0.0001; Fisher’s exact; Fig. 2D). These findings suggest fundamental biological differences between these types of T-ALLs already at the time of the initial disease, which become even more apparent at the time of relapse. These divergently developing genomic changes are paralleled by a trend towards a longer time to relapse in type-2 than in type-1 patients (p = 0.0896 (Mantel-Cox test); Fig. 2E).
Fig. 2. T-ALL type-1 and type-2 relapses are biologically and clinically distinct.

A Numbers of mutations detected by whole exome sequencing in matched samples collected at initial diagnosis (INI), at relapse (REL) and at both time-points from: 18 T-ALL patients relapsing with type-1 and in 20 T-ALL patients relapsing with type-2. B Boxplot presenting numbers of mutations acquired by T-ALLs relapsing as type-1 and type-2. C Numbers of mutations detected at initial diagnosis plotted against the number of mutations detected at relapse per patient; 3 hypermutator T-ALLs (P1, P8 and P18) carry >85 mutations at relapse; * - the hypermutators were excluded from the correlation analysis. D Proportion of clonal (AF ≥ 30%) and subclonal (AF < 30%) mutations at initial diagnosis and at either type-1 or type-2 relapse. Allele frequencies were not corrected for different blast content in the samples, except for sample collected from P2 at initial diagnosis, where contamination with normal cells was observed (Suppl. Tab. 2). E Kaplan–Meier curve showing leukemia-free survival of T-ALL patients following either type-1 (blue line) or type-2 (red line) relapse. F Recurrent mutations found by WES in the 38 matched pairs of initial (INI) and relapse (REL) T-ALL patients.
We next analyzed the mutation spectrum (Fig. 2F) in the entire group of 38 patients according to relapse type. The most frequent were NOTCH1 mutations (N = 48) found in 21 patients. Of these only patients relapsing with type-1 preserved all the NOTCH1 mutations detected with an AF >30% at the time of initial diagnosis (N = 9 patients). The majority of the lost (11/12) and gained (12/14) NOTCH1 mutations were detected in type-2 relapse (Supplementary Table 1). However, this pattern was also observed for variants of other genes thus not implicating NOTCH1 as a specific driver of either of the types of relapse. Activating mutations of cytosolic 5′-nucleotidase II (NT5C2) occurred in 6/18 type-1 and 8/20 type-2 relapses and in none of the initial diagnosis samples, thus confirming the frequent acquisition of NT5C2 mutations in T-ALL relapses [8–10]. Seven of these mutations were gain-of-function variants at position R367 and two at R238 (for the remaining see Supplementary Tab. 1). Six mutations carried by 5 of these 14 patients (4 type-1 and 1 type-2) were subclonal (AF < 30%). RAS mutations occurred in 4/18 type-1 and in 5/20 type-2 T-ALLs, both at the time of initial and/or relapsed disease. TP53 mutations occurred at relapse in 1/18 type-1 and in 2/20 type-2 T-ALLs. In one of these patients, the TP53 mutation was traced back to the initial diagnosis sample where it was detected with a low AF of 5% (3/58 reads). These findings indicate that the leukemogenic mechanisms that are activated by these mutations are shared between both types of relapses.
Constitutional mutations in cancer predisposition genes are enriched in T-ALL patients with a type-2 relapse
Because of the growing evidence for the role of predisposing constitutional variants that contribute to approximately 10% of all pediatric malignancies [20, 21, 27, 28] we analyzed matched remission samples (collected at day 33 of treatment) for inherited variants in 227 known cancer predisposition genes (CPG) compiled from previously reported studies [20, 22–24, 29] and those analyzed by the PCAWG (pan-cancer analysis of whole genomes) network [30]. In 7/20 type-2 patients we detected nine constitutional heterozygous variants (AF ≥30%) predicted to be deleterious or disruptive for splicing (nonsense- frameshift and splice site mutations) in genes associated with DNA repair (CHEK2, ERCC2/3, SPRED1, GTF2H3, RFC3, POLR2L) and in tumor suppressor genes (VHL, FBXW7). Constitutional mutations in these genes are associated with cancer predisposition syndromes [20–22, 24]. By contrast, such constitutional variants were not identified in any of the type-1 patients (type-1 vs. type-2; 0/18 vs. 7/20; p = 0.0087; Fisher's exact test, two-tailed). None of the patients carrying constitutional cancer predisposition mutations exhibited a somatic hypermutator phenotype. In order to assess the role of constitutional CPG mutations (Supplementary Table 8) in conferring a higher risk of relapse we compared the frequency of CPG mutations in remission samples of the relapsing patients with patients who did not develop a relapse. These data did not indicate a significantly difference between these groups. Similarly, a comparison of the frequency of constitutional inactivating mutations in cancer predisposition genes (Supplementary Table 8) between the non-cancer gnomAD cohort (v2.1.1 [31]) and T-ALL patients showed an even distribution (see Supplementary Results for details). These data indicate that CPG mutation shape the evolution of a relapse towards type-2 instead of type-1 but do not increase the overall risk of developing a T-ALL or a T-ALL relapse. We hypothesize that these mutations favor the accumulation of DNA damage under conditions of genotoxic stress such as chemotherapy. Unfortunately, the incompleteness of clinical data and family histories on secondary malignancies precluded a systematic analysis of the association with hereditary cancer predisposition. However, the development of mutational inactivation of a tumor suppressor in a patient with tumor predisposition could be monitored particularly well in P27, who presented with a simultaneous Wilms tumor at the time of initial diagnosis. This patient carried a constitutional previously unknown frameshift mutation of FBXW7 (Met587ArgfsTer41; chr4:152324250). In T-ALL, inactivation of FBXW7 is a common cause for activation of NOTCH1-pathway and other oncogenic clients such as MYC [18]. The sample collected at initial diagnosis of T-ALL exhibited LOH of FBXW7 in addition to the constitutional frameshift mutation (Supplementary Fig. 3), whereas at the time of relapse that occurred unusually late 913 days after the initial diagnosis this T-ALL had acquired a second, somatic missense mutation (c.1033A > G; p.T345A) in proximity to the known hotspot (R347). Samples of the patient’s parents and siblings were not available, nor was a sample representing the synchronous Wilms tumor. We next asked whether constitutional inactivation of FBXW7 might be a common predisposing mechanism in T-ALL and screened 51 remission samples of T-ALL patients in whom we had previously found a somatic FBXW7 mutation [18]. Sanger sequencing of exons 9 and 10 of FBXW7 did not identify constitutional mutations in any of these patients, indicating that constitutional loss of FBXW7 function is not common in pediatric T-ALL.
Transcriptomic and epigenomic profiles of T-ALL are determined by the leukemogenic fusion gene and are preserved between initial diagnosis and relapse
Of the total set of 38 matched pairs of initial diagnosis and relapse, we expanded matched samples of 13 patients in PDX thus enabling us to obtain suitable material (RNA and leukemic cells) for multi-omic analyses (Supplementary Table 2). Of the 12 patients with available mRNA data, 5 were classified as cortical T-ALL, two as mature, two as pre-T, one as pro-T and two as a mixed cortical-T/pre-T (detailed FACS and immunophenotype profiles in Supplementary Table 3). None of the patients was classified as early thymic progenitor (ETP)-ALL, however at the time of diagnosis of some of the patients the high-risk ETP-ALL subtype had not yet been described. Moreover, some expression features resembled those of early thymocytes by high expression of CD34, SPI1 or KIT (Supplementary Fig. 4, Supplementary Table 3). Five of 13 patients developed a type-1 and 8 a type-2 relapse. As previously shown directly, early passages of T-ALL PDXs that were used here preserve the genomic and epigenomic profiles and the subclonal architecture of the primary patients’ leukemias with high fidelity [19, 32]. Based on ectopic expression of known T-ALL drivers, we classified this subset of 13 patients into the following subgroups: TAL1/2 (n = 5), TLX1/3 (n = 3), HOXA (n = 2), NKX2‐4/5 (n = 2), and LMO2 (n = 1; Fig. 3A). Unsupervised learning approaches clustered all 26 samples (obtained at diagnosis and relapse) according to the driving fusion indicating that these are the strongest factors determining the transcriptomic (Fig. 3A), methylation (Supplementary Fig. 5) and chromatin accessibility profiles (Fig. 3B). At the same time, the overall gene expression, methylation, and chromatin accessibility profiles of the initial and relapsed T-ALLs of individual patients were largely retained following the evolution to relapse (Fig. 3; Supplementary Fig. 5). This similarity is also reflected by differential transcriptomic analyses between initial diagnosis and relapse identifying only 0.13% (43/32 529) genes to be significantly up- or downregulated at relapse (DESeq2; padj <0.05; Supplementary Tab. 4a/b). Similarly, analyses by ATAC-Seq revealed significant changes of the chromatin accessibility profile in only 0.53% (557/105 136) of the analyzed ATAC-peaks (DESeq2, padj < 0.05; Supplementary Table 4c/d). Likewise, significant changes on the way to relapse were neither identified in promoter methylation profiles (RnBeads [25]; Supplementary Table 5). The methylation data were further used for classification into either CIMP+ or CIMP– as described previously [33] (Supplementary Fig. 6). These analyses demonstrate that (1) the CIMP status does not change during the transition from initial diagnosis to relapse in majority of the patients (19/23) and (2) does not differ in its distribution between type-1 (5 CIMP–; 6 CIMP+) and type-2 relapses (8 CIMP–; 5 CIMP+). These data indicate that in a cross-sectional or longitudinal approach not considering the type of relapse any relapse-specific alterations in the transcriptomic and epigenomic profiles are masked by the interpatient heterogeneity and by the dominant fusion genes driving the leukemogenesis.
Fig. 3. Unsupervised RNA-Seq and ATAC-Seq principal component analyses of initial diagnosis and relapsed T-ALL cluster samples according to the driver fusion.

Unsupervised analysis of (A) the RNA-Seq (26 samples) based on all genes and (B) ATAC-Seq (24 samples) based on all peaks in matched samples of initial diagnosis (circle) and relapse (triangle) collected from 13/12 T-ALL patients (color code). Samples collected from the same patient at different time-points cluster in close vicinity. Most of the variance (RNA-Seq: PC1: 28%, PC2: 14%; ATAC-Seq: PC1: 34%, PC2: 11%) can be explained by aberrant expression of driving fusion genes such as basic helix-loop-helix (bHLH) family members TAL1, TAL2, LIM-only domain (LMO) gene family or the homeobox genes TLX1, TLX3, NKX2-4, NKX2-5; HOXA.
Relapse specific differences at the epigenomic and transcriptomic level develop in type-2 relapses while type-1 relapses resemble the profiles of the initial disease
We next tested whether relapse-specific changes may be unmasked when separately analyzing leukemias that developed into either a type-1 or a type-2 relapse. As detailed above for the entire set of 38 patients, we confirmed in 13 matched pairs of initial diagnosis and relapse expanded in PDX-mice that type-2 relapses acquired significantly more mutations (p = 0.01 (t test, unpaired), Fig. 2A) and are characterized by a higher degree of subclonal complexity than type-1 relapses (Fig. 2D). The analysis of the multi-omics set of 13 patients revealed that the stronger remodeling on the way to type-2 than to type-1 relapses was also reflected by a more complex evolution of epigenetic changes in type-2 relapses. Specifically, when analyzing the methylation profile of the promoters, we observed an almost 10-fold higher mean difference in β value when comparing the initial disease and relapse in type-2 (0.002) with type-1 (0.00034) (p < 0.0001, Chi-square test). When analyzing chromatin accessibility, the number of differentially accessible ATAC-peaks was only 14 (0.013%) in type-1 but 852 (0.81%) in type-2 (p < 0.0001, Chi-square test; Fig. 4A). A similar difference was observed at the transcriptomic level with only 1 gene being differentially expressed in type-1 and 152 genes in type-2 (p < 0.0001, Chi-square test; Fig. 4A, Supplementary Table 6).
Fig. 4. T-ALL type-2 relapses have undergone stronger chromatin remodeling and transcriptional changes than type-1 relapses.

A Differential analyses (DESeq2, adj p < 0.05) of RNA-Seq and ATAC-Seq datasets comparing initial diagnosis and relapse within type-1 and type-2 leukemias. MA plots show log2 fold changes in expression/accessibility in relation to the mean of normalized counts per gene/peak. Significantly differential expressed genes/accessible ATAC-peaks are labeled in red. B Deconvolution analysis of T-ALL samples performed with CIBERSORT trained on the signature of 2823 open chromatin regions selected to distinguish five differentiation stages (DN2, DN3/ISP, DPCD3–/ DPCD3+, CD4+, CD8+) of healthy T-cell precursors as previously reported [35] and applied to decompose T-ALL ATAC-profiles. C Fraction of the dominant population as predicted by deconvolution with CIBERSORT in T-ALLs relapsing either as type-1 or type-2.
We next mapped the T-ALLs to the most closely related maturation stages of thymic T-cell precursors by employing recently reported profiles of chromatin accessibility obtained by ATAC-Seq [19, 34]. Thus, we trained the CIBERSORT algorithm [35] on the signature of 2,823 chromatin regions selected to distinguish five differentiation stages (DN2, DN3/ISP, DPCD3–/ DPCD3+, CD4+, CD8+) of healthy T-cell precursors [34]. The signature regions differentially accessible in maturation stages were enriched for binding motifs of transcription factors that are highly expressed in T-cell precursors, which indicated a match between chromatin accessibility (ATAC-Seq) and gene expression (RNA-Seq). Deconvolution analyses of the T-ALLs obtained at the time of first diagnosis and at relapse revealed that at relapse and even at the time of first diagnosis type-1 leukemias were more homogeneous predominantly resembling a single immature normal T-cell population, whereas the pattern of the type-2 leukemias was more complex resembling a mixture of healthy T-cell precursor populations already at the time of the initial disease (Fig. 4B, p < 0.01, Mann-Whitney test; Fig. 4C). These data show that relapse type-specific differences at the epigenomic and transcriptomic level are already apparent at the time of first diagnosis and that relapse-specific changes largely develop in type-2 relapses, whereas type-1 relapses are essentially identical to the profiles of the initial disease.
Profiles of disease progression and drug resistance in type-1 and type-2 T-ALLs
We next interrogated the epigenomic and transcriptomic data sets of the 13 T-ALLs that could be propagated in xenografted mice for shared and differential profiles in the 5 type-1 and the 8 type-2 T-ALLs (the molecular profiles are summarized in Fig. 5A). We performed differential analyses of the mutation spectrum, RNA-Seq and ATAC-Seq data (DESeq2, p < 0.05) of the initial disease and relapse samples both globally and separately for each of the 13 patients (Suppl. Tab. 7). In 4/13 patients (2 type-1 and 2 type-2) we detected increased mRNA expression (Supplementary Table 4a) of multidrug resistance gene 1 MDR1 (ABCB1) both at the time of initial disease and relapse. In another two patients we observed an at least two-fold upregulation of the MDR1 gene expression at relapse (Fig. 5A; Supplementary Table 4a). MDR1 is known to confer in vitro resistance to many drugs used in the treatment of acute leukemia such as the anthracyclines, vincristine, vinblastine, and methotrexate [36]. Additionally, we identified a relapse-specific 6-fold upregulation of the major vault protein (MVP) gene in two patients whose relapse was classified as type-2. In patient P2 the MVP normalized read count increased from initial disease to relapse from 138 to 800 and in P10 from 409 to 2464 (mean of all patients: 594; median: 416; SD: 493). MVP is involved in nucleocytoplasmic transport and has been reported to be overexpressed in multi-drug resistant solid tumors [37]. These findings, together with the recurrent gain of function mutations of NT5C2 reported previously and shown above, indicate that both, in type-1 and type-2 T-ALL relapses, expression of drug resistance genes contribute to the frequent treatment resistance and poor prognosis. In the set of 13 initial disease/relapse pairs TAL1-driven T-ALLs were exclusively identified in those patients developing type-2 relapses (4/8 type-2 vs. 0/5 type-1; p = 0.1; Fisher's exact test, two-tailed) suggesting that TAL1 may play a specific role in type-2 T-ALL relapses (Fig. 3, Fig. 5), although clearly this trend will have to be validated in a larger number of samples. Functional enrichment analysis of the most recurrently upregulated genes (N > 5) showed enrichment in the regulation of the JAK-STAT cascade. At relapse, 11 of the 13 patients show upregulation of at least one of the SOCS1/2/3, which regulate the JAK-STAT pathway via negative-feedback loop confirming previous reports [38–40] that the activation of this protooncogenic pathway is common in T-ALL. Notably, however, overexpression of the IL7R gene was limited to the 5 patients with type-1 relapses, only in one it was correlated with the presence of an activating IL7R gene mutation. In addition to the overexpression of the IL7R itself, the functional role of this pathway was indicated by the overexpression of other genes involved in its regulation and by the overexpression of MYC, the cell cycle regulator cyclin D2 and the anti-apoptotic member of the Bcl-2 family MCL1 (Supplementary Fig. 7).
Fig. 5. Models of disease progression and drug resistance in type-1 and type-2 T-ALLs are revealed by multi-omics analyses.

A Molecular characteristics of initial diagnosis (INI) and relapse (REL) of matched samples from 13 pediatric T-ALL patients characterized by RNA-Seq (log 2 transformed read counts; read count values of 0 were not transformed; black frame indicates that fused-transcripts were detected), WES (SNVs/InDels), CNAs in targeted T-ALL specific genes and low coverage whole genome sequencing (large CNAs). Displayed are the most recurrent changes and specific hallmarks of each leukemia. B Biological features and potential mechanisms of relapse shared by T-ALLs relapsing either as type-1 or as type-2, and those specific to each relapse type. The font is scaled according to the frequency of the feature.
Discussion
Because most intensive chemotherapy regimens in pediatric T-cell leukemia have reached the limit of tolerability, current research efforts are focusing on stratifying treatment intensity according to risk. While several genetic parameters determining risk are known for precursor B-ALLs [41], risk stratification of T-ALL is still largely based on the kinetics of treatment response [42]. Furthermore, there are currently no established specific molecular targets in T-ALL which can be exploited for therapy. Patients with T-ALL are therefore likely to particularly benefit from a detailed understanding of the biology of the disease and the mechanisms that govern relapse.
We have previously reported that T-ALL relapses can be classified as either type-1, characterized by the major clone present at the time of initial disease evolving by acquiring additional mutations, or type-2, which is characterized by the major clone of the initial disease having been eradicated and the relapse having evolved from a minor ancestral clone [3]. Approximately half of the T-ALL patients who suffer from a relapse can be classified as either type-1 or type-2, respectively. We show here that on the path from initial disease to type-1 relapse, T-ALLs tend to remain stable in their mutational spectrum, promoter methylation, chromatin accessibility and gene expression profile. In contrast, type-2 T-ALL relapses are characterized by intensive remodeling as indicated by an increase in mutational load, changes in DNA methylation, chromatin accessibility and in gene expression. It is important to note that these differences only became apparent when patients were classified according to their disease evolution during progression, whereas they remained masked when analyzing type-1 and type-2 T-ALL relapses together. It has been an unexpected finding, that fundamental differences in genomic, epigenomic and transcriptomic profiles between T-ALLs that will relapse as either type-1 or type-2 are already apparent at the time of first diagnosis. Specifically, T-ALLs later evolving into type-1 relapses show a more homogeneous subclonal architecture and chromatin maturation profiles. An important finding of this study is that constitutional mutations in tumor suppressor genes and in genes involved in DNA repair likely contribute to the genetic instability and accumulation of mutations of leukemias that develop into type-2, but not type-1 relapses. While the analysis of probands reported not to develop malignancies or of T-ALL patients who did not develop a relapse showed that the carrier status for these mutations is neither associated with a significantly increased risk of developing a T-ALL nor of a T-ALL relapse, these mutations implicated in cancer predisposition, likely shape the type of relapse following exposition to leukemogenic stimuli and to genotoxic treatment. Type-1 relapses are characterized by early progression and occur in leukemias that already at the time of initial diagnosis are equipped with mechanisms driving treatment resistance. This is particularly apparent in three patients (P13, P25, and P32), who not only preserved almost all the mutations detected at initial diagnosis but did not acquire additional mutations at the time of relapse. Type-2 relapses, by contrast tend to develop later, the leukemia requiring more extensive genetic and epigenetic remodeling of a subclone of the initial disease, a process that appears to be facilitated by constitutional mutations in cancer predisposition genes. It must be noted that the genomic analyses performed here do not fully explain all differences between T-ALL at diagnosis and at relapse. Specifically, some of the type-2 relapses display a substantially different spectrum of variants when compared to the samples obtained at the time of initial diagnosis. By contrast, these samples do not show equally substantial differences at the epigenomic and transcriptomic level. This observation suggests that many variants may not or only marginally contribute to the driving of the leukemia. Future single cell analyses may provide insight into the specific role of some variants to the biology of the leukemia. With the analysis of a larger number of matched leukemia pairs, we expect that our classification may be updated to a version with more subtypes, for example possibly specifying those that have neither gained or lost mutations during the transition from diagnosis to relapse.
The type-specific and longitudinal analysis of individual patients we performed here revealed shared and type-specific profiles that develop during the transition from initial disease to relapse (Fig. 5B). Both types of relapse share the recurrent acquisition of NT5C2 mutations at relapse, therefore confirming previous reports implicating this mutation as a common relapse-specific mechanism of drug resistance [8–10]. In addition to NT5C2, drug resistance has likely been induced in two type-1 and in two type-2 patients with high expression of MDR1. Overexpression of this gene has previously been reported to cause resistance to drugs used in the treatment of acute leukemia via ATP-dependent drug efflux [36]. At the time of relapse, two further patients exhibited an increased expression of the MVP mRNA, a gene that is implicated in drug resistance in solid tumors [43, 44] and which may thus play a previously unrecognized role in T-ALL. Taken together, these data indicate that the activity of multidrug resistance genes likely contributes to the poor prognosis of both types of T-ALL relapses.
When considering type-specific differences, overexpression of IL7R emerged as a characteristic feature of type-1 leukemias, remarkably both at the time of relapse and already at the time of first diagnosis. While only one of the patients analyzed here carries an activating IL7R mutation, we observed a possibly compensatory upregulation of components of the IL7R-JAK-STAT pathway: its ligand HGF, IL7R itself and of suppressors of cytokine signaling (SOCS1, SOCS2, SOCS3), which regulate the pathway via a negative-feedback loop. Suppressors of cytokine signaling were previously identified as part of a stemness-related molecular signature signifying unfavorable outcome in acute myeloid and lymphoblastic leukemia [45]. Gain-of-function mutations in interleukin-7 receptor-α conferring cytokine-independent growth of progenitor lymphoid cells were first described by Shochat and colleagues [46, 47] and shown to be involved in human T-cell leukemogenesis [48] and drug resistance in T-ALL [49]. Although analyses in the UK ALL2003 trial showed that an activation of IL7R-JAK did not confer an adverse prognosis in T-ALL [50], in vitro analysis of steroid resistance showed an association of IL7R-JAK-STAT mutations with a strong activation of the downstream oncogenic MEK-ERK and AKT pathways; thereby inducing a robust antiapoptotic response by upregulating MCL1 and BCLXL expression [49]. Moreover, recent studies in pediatric B-cell precursor (BCP) acute lymphoblastic leukemia linked high expression of IL7R with CNS infiltration and relapse [51]. Previous reports suggest that overexpression of IL7R and the activation of downstream targets may represent therapeutic targets of either specific antibodies or small molecule inhibitors such as ruxolitinib [51, 52]. However, overexpression alone of molecular targets has recently emerged not to reliably predict treatment response by targeted therapy in pediatric malignancies [53]. Irrespective of its potential role as a molecular target, the results reported here implicate activation of the IL7R pathway as a leukemogenic event that preferentially occurs in type-1 T-ALL.
In conclusion, the data presented here show that the analysis of relapse-specific mechanisms in T-ALL is substantially facilitated by analyzing type-1 and type-2 relapses separately. Specifically, this work shows shared and type-specific profiles that are already apparent at the time of diagnosis. These profiles include the frequent upregulation of the IL7R pathway in type-1 and the enrichment of constitutional cancer predisposition genes and hypermutator phenotypes in type-2 relapses.
Supplementary information
Acknowledgements
We thank all the patients who participated in the study and their families. We thank EMBL IT Services and EMBL GeneCore for technical support. José Carreras Leukämie‐Stiftung (José Carreras Leukämie‐Stiftung e.V.). Bundesministerium für Bildung und Forschung (BMBF) [TRANSCALL— part of the ERA‐NET on “Translational Cancer Research” (TRANSCAN)]. Dietmar Hopp Stiftung. Manfred Lautenschläger Stiftung. Tour der Hoffnung Bensheim. Tour der Hoffnung Gießen.
Author contributions
PR‐P designed the experiments, performed the bioinformatic analyses, and wrote the manuscript; JBK designed the research, contributed to the interpretation of the results, and wrote the manuscript; TR performed bioinformatic analyses, aided in interpreting the results and wrote the manuscript; BE‐U performed the ATAC‐Seq experiments and their analysis and contributed to the writing of the manuscript; BB established the PDX models and contributed to the interpretation of the results; VF established the PDX models and provided the data; YA performed bioinformatic analysis of the methylation results and contributed to their interpretation; MZ provided the patients’ clinical data and their analysis; MH performed the experiments; CKL-D performed the methylation experiments; NN provided the samples and the clinical data; RK provided the remission samples and the clinical data; MSt, MSc, GC, GE, RK‐S, CE and SA provided patients samples and data for the analyses; SP contributed to the analysis of the results and to the writing of the manuscript; MUM contributed to the analysis of the results and to the writing of the manuscript; J‐PB supervised the establishment of the PDX models and contributed to the design of the research; JOK supervised data analysis and interpretation; and AEK designed the research, supervised the project, and wrote the manuscript. All authors reviewed and contributed to the final manuscript.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Data availability
Sequence data are deposited at the European Genome‐phenome Archive (http://www.ebi.ac.uk/ega/) under accession number EGAS00001003248. Scripts for the plots and tables are available in a repository (https://github.com/tobiasrausch/t-all).
Code availability
The code for RNA & ATAC alignments, peak calling, read counting etc. is available on GitHub (https://github.com/tobiasrausch/ATACseq, https://github.com/tobiasrausch/RNAseq). For further information see Suppl. Materials.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Paulina Richter-Pechańska, Joachim B. Kunz, Tobias Rausch, Büşra Erarslan-Uysal.
Contributor Information
Jan O. Korbel, Email: korbel@embl.de
Andreas E. Kulozik, Email: andreas.kulozik@med.uni-heidelberg.de
Supplementary information
The online version contains supplementary material available at 10.1038/s41375-022-01587-0.
References
- 1.Bhojwani D, Pui CH Relapsed childhood acute lymphoblastic leukaemia. Lancet Oncol 2013;14. 10.1016/S1470-2045(12)70580-6. [DOI] [PubMed]
- 2.Schrappe M, Valsecchi MG, Bartram CR, Schrauder A, Panzer-Grümayer R, Möricke A, et al. Late MRD response determines relapse risk overall and in subsets of childhood T-cell ALL: Results of the AIEOP-BFM-ALL 2000 study. Blood. 2011;118:2077–84. doi: 10.1182/blood-2011-03-338707. [DOI] [PubMed] [Google Scholar]
- 3.Kunz JB, Rausch T, Bandapalli OR, Eilers J, Pechanska P, Schuessele S, et al. Pediatric T-cell lymphoblastic leukemia evolves into relapse by clonal selection, acquisition of mutations and promoter hypomethylation. Haematologica. 2015;100:1442–50. doi: 10.3324/haematol.2015.129692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oshima K, Khiabanian H, da Silva-Almeida AC, Tzoneva G, Abate F, Ambesi-Impiombato A, et al. Mutational landscape, clonal evolution patterns, and role of RAS mutations in relapsed acute lymphoblastic leukemia. Proc Natl Acad Sci USA. 2016;113:11306–11. doi: 10.1073/pnas.1608420113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van Vlierberghe P, Ferrando A. The molecular basis of T cell acute lymphoblastic leukemia. J Clin Investig. 2012;122:3398–406. doi: 10.1172/JCI61269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vicente C, Cools J. The origin of relapse in pediatric T-cell acute lymphoblastic leukemia. Haematologica. 2015;100:1373–5. doi: 10.3324/haematol.2015.136077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yadav BD, Samuels AL, Wells JE, Sutton R, Venn NC, Bendak K, et al. Heterogeneity in mechanisms of emergent resistance in pediatric T-cell acute lymphoblastic leukemia. Oncotarget. 2016;7:58728–42. doi: 10.18632/oncotarget.11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tzoneva G, Perez-Garcia A, Carpenter Z, Khiabanian H, Tosello V, Allegretta M, et al. Activating mutations in the NT5C2 nucleotidase gene drive chemotherapy resistance in relapsed ALL. Nat Med. 2013;19:368–71. doi: 10.1038/nm.3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meyer JA, Wang J, Hogan LE, Yang JJ, Dandekar S, Patel JP, et al. Relapse-specific mutations in NT5C2 in childhood acute lymphoblastic leukemia. Nat Genet. 2013;45:290–4. doi: 10.1038/ng.2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barz MJ, Hof J, Groeneveld-Krentz S, Loh JW, Szymansky A, Astrahantseff K, et al. Subclonal NT5C2 mutations are associated with poor outcomes after relapse of pediatric acute lymphoblastic leukemia. Blood. 2020;135:921–33. doi: 10.1182/blood.2019002499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hof J, Krentz S, van Schewick C, Körner G, Shalapour S, Rhein P, et al. Mutations and deletions of the TP53 gene predict nonresponse to treatment and poor outcome in first relapse of childhood acute lymphoblastic leukemia. J Clin Oncol. 2011;29:3185–93. doi: 10.1200/JCO.2011.34.8144. [DOI] [PubMed] [Google Scholar]
- 12.Richter-Pechańska P, Kunz JB, Hof J, Zimmermann M, Rausch T, Bandapalli OR et al. Identification of a genetically defined ultra-high-risk group in relapsed pediatric T-lymphoblastic leukemia. Blood Cancer Journal 2017; 7. 10.1038/bcj.2017.3. [DOI] [PMC free article] [PubMed]
- 13.Hsiao MH, Yu AL, Yeargin J, Ku D, Haas M. Nonhereditary p53 mutations in T-cell acute lymphoblastic leukemia are associated with the relapse phase. Blood. 1994;83:2922–30. doi: 10.1182/blood.V83.10.2922.2922. [DOI] [PubMed] [Google Scholar]
- 14.Irving J, Matheson E, Minto L, Blair H, Case M, Halsey C, et al. Ras pathway mutations are prevalent in relapsed childhood acute lymphoblastic leukemia and confer sensitivity to MEK inhibition. Blood. 2014;124:3420–30. doi: 10.1182/blood-2014-04-531871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gianfelici V, Chiaretti S, Demeyer S, di Giacomo F, Messina M, la Starza R, et al. RNA sequencing unravels the genetics of refractory/relapsed T-cell acute lymphoblastic leukemia. Prognostic and therapeutic implications. Haematologica. 2016;101:941–50. doi: 10.3324/haematol.2015.139410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weng AP, Ferrando AA, Lee W, Morris JP 4th, Silverman LB, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 2004; 306. www.sciencemag.org (Accessed 22 Mar2022). [DOI] [PubMed]
- 17.Single-cell DNA amplicon sequencing reveals clonal heterogeneity and evolution in T-cell acute lymphoblastic leukemia. Mission Bio. https://missionbio.com/resources/publications/alberti-servera-blood-2021/ (Accessed 22 Mar2022). [DOI] [PMC free article] [PubMed]
- 18.Kox C, Zimmermann M, Stanulla M, Leible S, Schrappe M, Ludwig WD, et al. The favorable effect of activating NOTCH1 receptor mutations on long-term outcome in T-ALL patients treated on the ALL–BFM 2000 protocol can be separated from FBXW7 loss of function. Leukemia. 2010;24:2005. doi: 10.1038/leu.2010.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Richter‐Pechańska P, Kunz JB, Bornhauser B, Knebel Doeberitz C, Rausch T, Erarslan‐Uysal B, et al. PDX models recapitulate the genetic and epigenetic landscape of pediatric T‐cell leukemia. EMBO Molecular Medicine 2018; 10. 10.15252/emmm.201809443. [DOI] [PMC free article] [PubMed]
- 20.Zhang J, Walsh MF, Wu G, Edmonson MN, Gruber TA, Easton J, et al. Germline Mutations in Predisposition Genes in Pediatric Cancer. N. Engl J Med. 2015;373:2336–46. doi: 10.1056/NEJMoa1508054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stieglitz E, Loh ML. Genetic predispositions to childhood leukemia. Therapeutic Adv Hematol. 2013;4:270–90. doi: 10.1177/2040620713498161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Topka S, Vijai J, Walsh MF, Jacobs L, Maria A, Villano D, et al. Germline ETV6 Mutations Confer Susceptibility to Acute Lymphoblastic Leukemia and Thrombocytopenia. PLoS Genetics 2015; 11. 10.1371/journal.pgen.1005262. [DOI] [PMC free article] [PubMed]
- 23.Liu Y, Easton J, Shao Y, Maciaszek J, Wang Z, Wilkinson MR, et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat Genet. 2017;49:1211–8. doi: 10.1038/ng.3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rahman N. Realizing the promise of cancer predisposition genes. Nature. 2014;505:302–8. doi: 10.1038/nature12981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Assenov Y, Müller F, Lutsik P, Walter J, Lengauer T, Bock C. Comprehensive analysis of DNA methylation data with RnBeads. Nat Methods. 2014;11:1138–40. doi: 10.1038/nmeth.3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria 2019. https://www.R-project.org/.
- 27.Wong M, Mayoh C, Lau LMS, Khuong-Quang DA, Pinese M, Kumar A, et al. Whole genome, transcriptome and methylome profiling enhances actionable target discovery in high-risk pediatric cancer. Nat Med. 2020;26:1742–53. doi: 10.1038/s41591-020-1072-4. [DOI] [PubMed] [Google Scholar]
- 28.Newman S, Nakitandwe J, Kesserwan CA, Azzato EM, Wheeler DA, Rusch M, et al. Genomes for kids: the scope of pathogenic mutations in pediatric cancer revealed by comprehensive DNA and RNA sequencing. Cancer Disco. 2021;11:3008–35. doi: 10.1158/2159-8290.CD-20-1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gröbner SN, Worst BC, Weischenfeldt J, Buchhalter I, Kleinheinz K, Rudneva VA, et al. The landscape of genomic alterations across childhood cancers. Nature. 2018;555:321–7. doi: 10.1038/nature25480. [DOI] [PubMed] [Google Scholar]
- 30.Rodriguez-Martin B, Alvarez EG, Baez-Ortega A, Zamora J, Supek F, Demeulemeester J, et al. Pan-cancer analysis of whole genomes identifies driver rearrangements promoted by LINE-1 retrotransposition. Nat Genet. 2020;52:306–19. doi: 10.1038/s41588-019-0562-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–43. doi: 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang K, Sanchez-Martin M, Wang X, Knapp KM, Koche R, Vu L, et al. Patient-derived xenotransplants can recapitulate the genetic driver landscape of acute leukemias. Leukemia. 2017;31:151–8. doi: 10.1038/leu.2016.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Borssén M, Palmqvist L, Karrman K, Abrahamsson J, Behrendtz M, Heldrup J, et al. Promoter DNA methylation pattern identifies prognostic subgroups in childhood T-cell acute lymphoblastic leukemia. PLOS ONE. 2013;8:e65373. doi: 10.1371/journal.pone.0065373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Erarslan‐Uysal B, Kunz JB, Rausch T, Richter‐Pechańska P, Belzen IA, Frismantas V et al. Chromatin accessibility landscape of pediatric T‐lymphoblastic leukemia and human T‐cell precursors. EMBO Molecular Medicine 2020; 12. 10.15252/emmm.202012104. [DOI] [PMC free article] [PubMed]
- 35.Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12:453–7. doi: 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Szakács G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discov. 2006;5:219–34. doi: 10.1038/nrd1984. [DOI] [PubMed] [Google Scholar]
- 37.Xiao YS, Zeng D, Liang YK, Wu Y, Li MF, Qi YZ, et al. Major vault protein is a direct target of Notch1 signaling and contributes to chemoresistance in triple-negative breast cancer cells. Cancer Lett. 2019;440-1:156–67. doi: 10.1016/j.canlet.2018.09.031. [DOI] [PubMed] [Google Scholar]
- 38.Steelman LS, Abrams SL, Whelan J, Bertrand FE, Ludwig DE, Bäsecke J, et al. Contributions of the Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and Jak/STAT pathways to leukemia. Leukemia. 2008;22:686–707. doi: 10.1038/leu.2008.26. [DOI] [PubMed] [Google Scholar]
- 39.Vainchenker W, Constantinescu SN. JAK/STAT signaling in hematological malignancies. Oncogene. 2012;32:2601–13. doi: 10.1038/onc.2012.347. [DOI] [PubMed] [Google Scholar]
- 40.Migone TS, Lin JX, Cereseto A, Mulloy JC, O’Shea JJ, Franchini G, et al. Constitutively activated Jak-STAT pathway in T cells transformed with HTLV-I. Science. 1995;269:79–81. doi: 10.1126/science.7604283. [DOI] [PubMed] [Google Scholar]
- 41.Mullighan CG. Molecular genetics of B-precursor acute lymphoblastic leukemia. J Clin Investig. 2012;122:3407–15. doi: 10.1172/JCI61203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Raetz EA, Teachey DT. T-cell acute lymphoblastic leukemia. Hematol: Am Soc Hematol Educ Program. 2016;2016:580. doi: 10.1182/asheducation-2016.1.580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Steiner E, Holzmann K, Pirker C, Elbling L, Micksche M, Sutterlüty H, et al. The major vault protein is responsive to and interferes with interferon-γ-mediated STAT1 signals. J Cell Sci. 2006;119:459–69. doi: 10.1242/jcs.02773. [DOI] [PubMed] [Google Scholar]
- 44.Mossink MH, van Zon A, Scheper RJ, Sonneveld P, Wiemer EAC. Vaults: a ribonucleoprotein particle involved in drug resistance? Oncogene. 2003;22:7458–67. doi: 10.1038/sj.onc.1206947. [DOI] [PubMed] [Google Scholar]
- 45.Vitali C, Bassani C, Chiodoni C, Fellini E, Guarnotta C, Miotti S, et al. SOCS2 controls proliferation and stemness of hematopoietic cells under stress conditions and its deregulation marks unfavorable acute leukemias. Cancer Res. 2015;75:2387–99. doi: 10.1158/0008-5472.CAN-14-3625. [DOI] [PubMed] [Google Scholar]
- 46.Shochat C, Tal N, Bandapalli OR, Palmi C, Ganmore I, te Kronnie G, et al. Gain-of-function mutations in interleukin-7 receptor-α (IL7R) in childhood acute lymphoblastic leukemias. J Exp Med. 2011;208:901–8. doi: 10.1084/jem.20110580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shochat C, Tal N, Gryshkova V, Birger Y, Bandapalli OR, Cazzaniga G, et al. Novel activating mutations lacking cysteine in type i cytokine receptors in acute lymphoblastic leukemia. Blood. 2014;124:106–10. doi: 10.1182/blood-2013-10-529685. [DOI] [PubMed] [Google Scholar]
- 48.Zenatti PP, Ribeiro D, Li W, Zuurbier L, Silva MC, Paganin M, et al. Oncogenic IL7R gain-of-function mutations in childhood T-cell acute lymphoblastic leukemia. Nat Genet. 2011;43:932–41. doi: 10.1038/ng.924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li Y, Buijs-Gladdines JGCAM, Canté-Barrett K, Stubbs AP, Vroegindeweij EM, Smits WK, et al. IL-7 Receptor mutations and steroid resistance in pediatric t cell acute lymphoblastic leukemia: a genome sequencing study. PLoS Medicine 2016; 13. 10.1371/journal.pmed.1002200. [DOI] [PMC free article] [PubMed]
- 50.Vicente C, Schwab C, Broux M, Geerdens E, Degryse S, Demeyer S, et al. Targeted sequencing identifies associations between IL7R-JAK mutations and epigenetic modulators in T-cell acute lymphoblastic leukemia. Haematologica. 2015;100:1301–10. doi: 10.3324/haematol.2015.130179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Alsadeq A, Lenk L, Vadakumchery A, Cousins A, Vokuhl C, Khadour A, et al. IL7R is associated with CNS infiltration and relapse in pediatric B-cell precursor acute lymphoblastic leukemia. Blood. 2018;132:1614–7. doi: 10.1182/blood-2018-04-844209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gianfelici V, Messina M, Paoloni F, Peragine N, Lauretti A, Fedullo AL, et al. IL7R overexpression in adult acute lymphoblastic leukemia is associated to JAK/STAT pathway mutations and identifies patients who could benefit from targeted therapies. Leuk Lymphoma. 2019;60:829–32. doi: 10.1080/10428194.2018.1499906. [DOI] [PubMed] [Google Scholar]
- 53.van Tilburg CM, Pfaff E, Pajtler KW, Langenberg KPS, Fiesel P, Jones BC, et al. The pediatric precision oncology inform registry: clinical outcome and benefit for patients with very high-evidence targets. Cancer Disco. 2021;11:2764–79. doi: 10.1158/2159-8290.CD-21-0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequence data are deposited at the European Genome‐phenome Archive (http://www.ebi.ac.uk/ega/) under accession number EGAS00001003248. Scripts for the plots and tables are available in a repository (https://github.com/tobiasrausch/t-all).
The code for RNA & ATAC alignments, peak calling, read counting etc. is available on GitHub (https://github.com/tobiasrausch/ATACseq, https://github.com/tobiasrausch/RNAseq). For further information see Suppl. Materials.