

Abstract
Pospiviroidae, with their main representative potato spindle tuber viroid (PSTVd), are replicated via a rolling circle mechanism by the host-encoded DNA-dependent RNA polymerase II (pol II). In the first step, the (+)-strand circular viroid is transcribed into a (–)-strand oligomer intermediate. As yet it is not known whether transcription is initiated by promotors at specific start sites or is distributed non-specifically over the whole circle. An in vitro transcription extract was prepared from a non-infected potato cell culture which exhibited transcriptional activity using added circular PSTVd (+)-strand RNA as template. In accordance with pol II activity, transcription could be inhibited by α-amanitin. RT–PCR revealed the existence of at least two different start sites and primer extension identified these as nucleotides A111 and A325. The sequences of the first 7 nt transcribed are very similar, 105GGAGCGA111 and 319GGGGCGA325. GC-boxes are located at a distance of 15 and 16 nt upstream, respectively, in the native viroid structure, which may act to facilitate initiation. The GC-boxes may have a similar function to the GC-rich hairpin II in the (–)-strand intermediate, as described previously. The results are compared with the corresponding features of avocado sunblotch viroid, which belongs to a different family of viroids and exhibits different transcription initiation properties.
INTRODUCTION
Viroids are pathogenic RNAs of plants distinguished from viruses by the absence of a protein coat, the absence of protein coding capacity and by their small size. They are circular, single-stranded RNA molecules consisting of a few hundred nucleotides, the smallest having ∼240 and the largest ∼600 nt. Most native viroids adopt an unbranched, rod-like secondary structure with a high degree of intramolecular base pairing (1,2). It is generally assumed that viroid replication and pathogenesis depend completely on the enzyme systems of the host (for reviews see 3–6). The genetic information of the viroid is the RNA sequence and structure, the ability to undergo structural transitions and the capability to interact with host factors.
The viroids of the Pospiviroidae family, including potato spindle tuber viroid (PSTVd, 359 nt), replicate via the so-called asymmetrical rolling circle mechanism (7; for reviews see 4,8), whereas other viroids, like the Avsunviroidae, follow a symmetrical rolling circle model (9,10). The circular PSTVd [cPSTVd, by definition (+)-strand] is transcribed into an oligomeric (–)-strand. The (–)-strand acts as a template for synthesis of an oligomeric (+)-strand. Both transcription steps are catalyzed by a host enzyme, DNA-dependent RNA polymerase II (pol II) (11,12). The oligomeric (+)-strand is cleaved to unit length molecules and ligated to the mature viroid circle by host-encoded RNase(s) and ligase(s) (13,14). Self-cleavage could be ruled out for Pospiviroidae (15) but was found for Avsunviroidae (9,10). For PSTVd, a specific cleavage–ligation site and corresponding structural motifs involved in the processing reaction were elucidated and from that a processing model could be developed (14,16). Well-defined structural elements were also found as motifs critical for transcription. From site-directed mutagenesis of PSTVd it could be concluded that the formation of a thermodynamically metastable structure including a GC-rich hairpin (the so-called hairpin II, HP II) is critical for infectivity (17). Further analysis showed that HP II is a functional element of the (–)-strand replication intermediate (18). In addition, a GC-box has also been described in the circular viroid molecules of the Pospiviroidae family and discussed as a potential promoter element (19). The exact functional relevance of these elements has not yet been elucidated, as the exact start sites for transcription and therefore the spatial arrangement of start sites and specific hairpin structures is unknown.
In this work we wanted to determine the start sites for transcription of the cPSTVd molecule. One should note that the existence of specific start sites cannot be assumed a priori. Early studies have shown that isolated pol II binds to the ends of the rod-like viroid and thus might start transcription close to the ends, but probably not at well-defined sites (20). At a minimum, the finding of specific 5′-ends could be indicative not only of initiation but also of nearby structures and/or sequence elements that act as promotors. In order to identify the start positions, one has to analyze the 5′-ends of (–)-strand PSTVd RNAs. In principle, there exist two alternatives for such an analysis. One would be to isolate and analyze natural (–)-strand transcripts directly. Unfortunately, due to nuclease activities in the cell and, probably, the even more disruptive preparation procedure, it does not appear possible to isolate undegraded transcripts of PSTVd (21). Another approach is in vitro transcription of cPSTVd templates and subsequent analysis of the (–)-strand RNA 5′-ends. Up to now, several in vitro systems for the transcription of viroid RNA have been published (22–26). Since viroid-infected cells were used for these in vitro transcription extracts, it was not possible to differentiate between de novo synthesis of viroid RNA and ‘run-on’ elongation of endogenous linear molecules.
Recently, Navarro and Flores (27) characterized initiation sites of both polarities in the avocado sunblotch viroid (ASBVd). They analyzed the replication intermediates extracted from infected cells by an in vitro capping assay. Linear (+)- and (–)-strands begin with a UAAAA sequence that maps to similar A+U-rich terminal loops. However, ASBVd is quite different from PSTVd in the sense that it is located in the chloroplast and is replicated by a nuclear encoded polymerase, thus the results on the starts sites cannot be transfered a priori. Furthermore, the intermediates of ASBVd accumulate to higher concentrations, so that the method of identifying start sites by analyzing the natural intermediates appears much more favorable for ASBVd as compared to PSTVd.
In order to analyze de novo transcription of exogenous cPSTVd templates without the interference of endogenous viroid molecules, we established an in vitro transcription system based on a nuclear extract from non-infected potato cell cultures. In this system, two start sites with nearly identical sequences of the first 7 nt transcribed could be identified; they are located 15 and 16 nt, respectively, downstream from G:C-rich elements in the secondary structure of cPSTVd. The results are compared to the data on ASBVd mentioned above.
MATERIALS AND METHODS
Buffers and enzymes
Enzymes were obtained from Boehringer Mannheim, Promega, Gibco BRL and Pharmacia and were used according to the suppliers’ instructions.
Samples of cPSTVd and in vitro transcripts
Purified cPSTVd (intermediate strain) was prepared from infected Lycopersicon esculentum plants as described previously (28,29). Higher purity of the cPSTVd samples was achieved by an additional gel electrophoresis step and elution from the gel (30).
Monomeric (–)-strand PSTVd RNAs were transcribed in vitro from plasmids pRH714, pRH716 (21) and pSH1 (31). These plasmids carry a PSTVd (intermediate) sequence that was cloned with different restriction enzymes. Thus, the following 5′→3′ orientations of the transcripts were achieved: pRH 714 (282–359/1–281); pRH 716 (146–1/359–147); pSH1 (337–1/359–338).
Synthetic DNA oligonucleotides and PCR products
In total, 18 primers were used for reverse transcription, PCR amplification and primer extension of PSTVd RNAs.
The following primers were used for reverse transcription of (–)-strand PSTVd RNA [5′→3′, numbers indicating the corresponding nucleotides in the (+)-strand cPSTVd molecule]: AF6, 268GGA AAC AAC TGA AGC TCC CGA GAA C292 (56°C); AF9; 181TCA CCC TTC CTT TCT TCG GGT GTC CTT C209 (55°C); AF15, 100AAC CTG GAG CGA ACT GGC AAA121 (53°C); AF16, 324GAG GGT GTT TAG CCC TTG GAA344 (53°C); AF17, 61GAA GGC GGC TCG GA74 (52°C); AF21, 1CGG AAC TAA ACT CGT GGT TC20 (53°C); TB2, 20CCT GTG GTT CAC ACC TGA CCT CC42 (56°C); XC1, 124GGA CGG TGG GGA GTG C139 (54°C); QFV6, 209TCG CGC CCG CAG GAC CAC226 (56°C). The temperature for reverse transcription with each primer is given in parentheses (calculated according to ref. 32)
These oligonucleotides were used also as first PCR primers, with the following second PCR primers: AF4, 139GCA CTC CCC ACC GTC C124; AF7, 113GTT CGC TCC AGG TTT CCC CGG GGA T89; AF10, 259GTA GCC GAA GCG ACA GCG CAA AGG236; TB1, 171TTT CGG CGG GAA TTA CTC CTG TCG G147; XC2, 354CCA ACT GCG GTT CCA AGG GC335; XC3, 295GCG GTT CTC GGG AGC TTC AGT TGT TTC C268; RGV1, 87CCT GAA GCG CTC CTC CGA G69; RGV2, 41GAG GTC AGG TGT GAA CCA GAG21; RGV5,208GGA AGG ACA CCC AAG AAA GGA AGG GTG AAA A178.
Nine segments were used for PCR amplification. The segments, the PCR primers, the segment lengths and the PCR annealing temperatures are: A, TB2/AF7, 94 nt, 65°C; B, AF17/AF4, 79 nt, 55°C; C, AF15/TB1, 72 nt, 53°C; D, XC1/RGV5, 85 nt, 64°C; E, AF9/AF10, 79 nt, 67°C; F, QFV6/XC3, 87 nt, 73°C; G, AF6/XC2, 87 nt, 70°C; H, AF16/RGV2, 77 nt, 64°C; I, AF21/RGV1, 87 nt, 57°C.
Reverse transcription of (–)-strand PSTVd RNAs
PSTVd in vitro transcripts of (–)-strand polarity, cPSTVd or an aliquot of a nuclear transcription extract were incubated with 20 pmol RT primer in 6 µl of 1× First Strand Buffer (Gibco BRL) for 1 min at 94°C (denaturation) and then transferred to ice for 5 min (fast renaturation). The sample was incubated with 14 µl of RT reaction mix (final volume 20 µl with 10 mM DTT and 500 µM each dNTP in 1× First Strand Buffer). After 10 min incubation at the specific reverse transcription temperature of the primer (cf. above), 80 U reverse transcriptase (Superscript II; Gibco BRL) were added and the mixture incubated at that temperature for 30 min. The reaction was stopped by boiling for 5 min and transfer to ice. Before PCR amplification, the RNA template was degraded by alkaline hydrolysis at 90°C for 1 h in the presence of 105 mM NaOH, then neutralized with 1 M HCl.
PCR amplification of PSTVd cDNAs
An aliquot of 7.5 µl of the sample from reverse transcription was adjusted to 20 mM Tris–HCl, pH 8.9, 50 mM KCl, 0.1% Triton X-100, 20 pmol each PCR primer, 1.5 mM MgCl2, 1 mM dATP, dGTP and dTTP, 0.25 mM dCTP, 3 µCi [α-32P]dCTP and 1.25 U Taq DNA polymerase (Promega) in a final volume of 50 µl. After 4 min denaturation at 94°C, 25 cycles of 94°C for 60 s, annealing at the temperature specified for the different RT–PCR segments for 60 s and 72°C for 60 s, with a final elongation at 72°C for 10 min were performed. After PCR amplification, an aliquot was analyzed by wattage-controlled denaturing PAGE (0.5 M TBE, 8 M urea, 55–60°C; 16). The gels were exposed to X-ray film (Xomat AR; Kodak).
Primer extension analysis of (–)-strand RNA 5′-ends
The RNA was annealed with 5 × 106 c.p.m. labeled primer AF21 or XC1 in 20 µl buffer (50 mM Tris–HCl pH 8.3, 100 mM NaCl) at 85°C for 1 min and slowly (∼2–3 h) renatured to room temperature. The samples were ethanol precipitated, redissolved in 1× TE and the RNA transcribed into cDNA at 53°C (primer AF21) or 54°C (primer XC1) for 1 h in 20 µl buffer [1× First Strand Buffer, 10 mM DTT, 500 µM each dNTP, 40 U RNasin (recombinant RNase inhibitor; Promega) and 200 U reverse transcriptase (Superscript II; Gibco BRL)]. The samples were precipitated with ethanol, redissolved in urea loading solution, heat denatured and analyzed on 8% polyacrylamide denaturing sequencing gels (S2 chamber; Gibco BRL) and exposed to X-ray film (Xomat AR). As a sequence standard, a sequencing reaction of plasmid pRH716 with primer AF21 or pSH1 (compare cPSTVd samples and in vitro transcripts) with primer XC1 was performed with Sequenase II (US Biochemical) following the supplier’s instructions.
Preparation of nuclei and nuclear extracts
Highly purified nuclei from non-infected Solanum tuberosum HH258 suspension cultures (33,34) were prepared according to a protocol similar to that published previously (16). In brief, the cells were converted into protoplasts by incubation with cellulase Onozuka R-10 and Macerozym R-10 (Serva, Heidelberg, Germany), the protoplast were mechanically disrupted and nuclei purified with Ficoll and Percoll gradient centrifugations. As a modification, 10 mM NaF was added to all buffers to inhibit endogenous phosphatases that might decrease the transcriptional activity of the nuclei (35). The purified nuclei were extracted immediately, using a protocol based on a procedure published by Tsagris et al. (13) with two modifications: 10 mM NaF was added to all buffers and the ionic strength of the extraction buffer was increasead from 0.42 to 0.6 M NaCl in order to increase the amount of extracted protein. Approximately 1–2 × 108 nuclei in 20 aliquots of 100 µl each could be obtained as a nuclear extract with a protein concentration of typically 2–2.5 mg/ml, keeping its transcriptional activity for several months at –70°C.
Transcription of cPSTVd templates with the nuclear extract
Standard transcription reactions of cPSTVd templates were based on a protocol of Nagel-Steger (36). The reaction mix contained 5 µl of nuclear extract thawed on ice, 100 ng cPSTVd template, 40 U RNasin (recombinant RNase inhibitor) and 20 µg BSA (Molecular Biology Grade; Boehringer Mannheim) in 30 µl buffer [50 mM Tris–HCl, pH 7.9, 60 mM (NH4)2SO4, 5 mM MgCl2]. A concentration of 10–6 M α-amanitin was added to a control assay as an inhibitor of pol II. The transcription mix was preincubated at 28°C for 10 min for formation of polymerase preinitiation complexes (35,37) with simultanous digestion of the endogenous DNA template (20 U RNase-free DNase I). Afterwards, NTPs were added to a final concentration of 600 µM and transcription was performed at 28°C for 30 min. The reaction was stopped by addition of 100 µl of stop solution (20 mM EDTA, 200 mM NaCl, 1% SDS; 35) and samples were phenol/chloroform extracted and precipitated with ethanol in the presence of 200 µg/ml glycogen. The products were redissolved in 1× TE and analyzed by RT–PCR and primer extension.
Calculation of RT–PCR primers and sequence homologies
DNA oligonucleotides were chosen according to calculations using a program developed by Nagel-Steger (36) and the GCG program Bestfit. Calculations of sequence homologies were performed with the GCG program Bestfit.
RESULTS
In vitro transcription system and RT–PCR analysis
The (–)-strand synthesis from cPSTVd as template was studied in a nuclear extract prepared from cultured cells of S.tuberosum, which is a host for viroids. The cell culture used for these experiments was free of viroid infection. In the absence of endogenous templates, gel eluted cPSTVd (intermediate strain) was added to the nuclear extract. A particular RT–PCR system was developed to detect even small amounts of de novo synthesized (–)-strand PSTVd transcripts with maximum sensitivity. The RT–PCR amplification had to be highly (–)-strand specific, because the cPSTVd template was still present in the RT reaction and the PSTVd sequence itself is highly self-complementary.
Figure 1A shows the results of a typical RT–PCR analysis of an in vitro transcription assay using segment H (see Table 1 for details) of 77 nt length, which covers the left terminal part of the cPSTVd rod-like secondary structure. With optimized RT–PCR conditions, at least a 5000-fold excess of cPSTVd over the (–)-strand PSTVd transcript as positive control was tolerated; cPSTVd does not yield a band [compare slots cPSTVd and (–)-IVT]. It was possible to detect de novo synthesized (–)-strand PSTVd RNA of at least 77 nt length (slot NE). As a negative control, a transcription assay without endogenous template was performed (slot φ). (–)-Strand synthesis is pol II dependent, because the pol II inhibitor α-amanitin reduces the signal at least 5-fold (slot NE + αA). Comparison of the amounts of products from PCR is only semi-quantitative; one should note, however, that amplification did not proceed to saturation, as is obvious from comparing the bands in slots (–)-IVT and NE. Nevertheless, a faint band for the sample with addition of α-amanitin is still visible (slot NE + αA). This is not due to insufficient (–)-strand specificity, because in the control experiment with a 5000-fold excess of cPSTVd the band was not visible (slot cPSTVd). Further analysis showed that this faint band results from reverse transcriptase catalyzed cDNA synthesis of self-primed linear (+)-strand molecules during the RT reaction (data not shown). A small amount of linear (+)-strand molecules is unavoidable when working with circular PSTVd. Because of the self-priming phenomenon, the RT–PCR conditions for each of the segments selected for PCR had to be optimized to increase the differences in the band intensities of the PCR products resulting from de novo synthesized (–)-strand RNA and resulting from self-primed cDNA synthesis, i.e. NE versus NE + αA. The best differentiation was possible using high RT temperatures (up to 56°C) and only 25 PCR cycles, without reaching the PCR plateau. Whether de novo synthesized products have the appropriate 5′-end or are elongation products from nicked (+)-strand RNA cannot be differentiated at this point using RT–PCR analysis. This discrimination will be described below.
Figure 1.

Analysis of cPSTVd transcription in nuclear extract with an RT–PCR segment. cPSTVd was added as exogenous template to the in vitro transcription assay using a nuclear extract (NE) of cultured healthy S.tuberosum cells. After transcription, (–)-strand-specific RT and subsequent PCR with segment H (77 nt, cf. Table 1) were performed; PCR products were radioactively labeled by incorporation of [32P]dCTP and analyzed on a denaturing 7% polyacrylamide gel. (A) Transcription was carried out under non-optimized conditions. NE, in vitro transcription assay with 0.3 ng cPSTVd as template; NE + αA, NE with 10–6 M α-amanitin added; φ negative control, transcription assay without exogenous template. In order to analyze the (–)-strand specificity of the RT–PCR analysis, the following controls were performed: (–)-IVT, 0.1 pg (–)-strand PSTVd in vitro transcript as RT template; cPSTVd:, 0.5 ng cPSTVd as RT template. (B) Transcription was analyzed after the endogenous DNA template in the nuclear extract was degraded by DNase I; the extract was preincubated with 20 U DNase I before starting the trancription assay by adding dNTPs (lanes marked with DNase I). Other conditions and lanes as in (A).
Table 1. Detection of de novo synthesized (–)-strand PSTVd RNA with different RT–PCR segments.
| RT–PCR segment | Position | Length (bp) | (–)-Strand detection |
|---|---|---|---|
| Numbering is according to the position on cPSTVd, i.e. the segment on the (–)-strand is given in the 3′→5′ direction. | |||
| A | 20–113 | 94 | – |
| B | 61–139 | 79 | – |
| C | 100–171 | 72 | + |
| D | 124–208 | 85 | + |
| E | 181–259 | 79 | + |
| F | 209–295 | 87 | + |
| G | 268–354 | 87 | + |
| H | 324–41 | 77 | + |
| I | 1–87 | 87 | + |
| CD | 100–208 | 109 | + |
| EI | 181–87 | 266 | + |
| DI | 124–87 | 323 | – |
The detection of de novo-synthesized (–)-strands allowed us to improve the transcription activity of the nuclear extract. As shown in Figure 1B, DNase I digestion of the endogenous genomic DNA in the nuclear extracts increased the amount of synthesized (–)-strand PSTVd RNA: one explanation could be an increased number of unbound pol II molecules (compare slot NE DNase I with NE). This example shows that optimal RT–PCR conditions could be found under which signal in the presence of α-amanitin is completely eliminated. Although this elimination could not be established for every segment amplified from de novo synthesized RNA in this report, a strong reduction was always achieved.
Search for specific start sites with overlapping RT–PCR segments
A system of overlapping RT–PCR segments was used to estimate the length and start position of newly synthesized (–)-strand RNAs. Figure 2A shows the results with two short segments (A and I, 94 and 87 nt, respectively; see Table 1 and Fig. 3 for their positions). Whereas segment I clearly demonstrates template-dependent synthesis of (–)-PSTVd RNA because of the significant PCR signal reduction upon α-amanitin addition (compare slots NE and NE + αA), it was not possible to detect an α-amanitin-dependent reduction with segment A (compare slots NE and NE + αA). The very faint but still visible bands represent false positive signals due to a self-priming reaction of the cPSTVd template as described above. Thus, we have to conclude that a start site of (–)-strand transcription from cPSTVd is located between the right sites of segment I (nt 87) and of segment A (nt 113), so that the sequence corresponding to the primer for RT–PCR of segment I is inside the transcript and that of segment A outside.
Figure 2.

Analysis of cPSTVd transcription with a series of RT–PCR segments. The in vitro transcription assays, (–)-strand-specific RT and PCR analysis with a series of amplified segments were carried out as described for Figure 1B. The positions of the segments are depicted in Figure 3 and listed in Table 1. (A) Segments I (87 nt) and A (94 nt); (B) segments EI (266 nt) and DI (323 nt).
Figure 3.

Positions of RT–PCR segments and of primers for primer extension analysis. RT–PCR segments depicted in black indicate de novo synthesized (–)-strand PSTVd RNA, which were detected experimentally (segments I and EI, cf. Fig. 2); segments in grey indicate segments that could not be detected experimentally (segments A and DI, cf. Fig. 2). As indicated with a black dashed line, the potential position of the 5′-end of the synthesized (–)-strand RNA is located between the last amplified 5′-nucleotide of segment I or DI (nt 87) and the last 5′-nucleotide of segment A (nt 113), which could not be amplified. The minimum length of the synthesized (–)-strand RNA is 266 nt, i.e. the length of segment EI, whereas a segment of the length of DI could not be detected. Primers used for the primer extension analysis to evaluate the exact start nucleotides (primers AF21 and XC1) are denoted by arrows.
Altogether, nine short and three longer segments were tested by RT–PCR for their presence in (–)-PSTVd transcripts. The results for four segments (I, A, EI and DI) are depicted in Figure 2. Their positions in relation to the cPSTVd molecule are shown in Figure 3. All segments, together with the corresponding detection of (–)-strands, are listed in Table 1. Figure 2B shows the results for the two long segments (DI and EI, 323 and 266 nt, respectively). The 266 nt long de novo synthesized (–)-strand PSTVd RNA corresponding to segment EI could be detected (compare slots NE and NE + αA), whereas the RNA corresponding to DI was not detectable. One should note that these ‘long’ segments did not exhibit false positive signals like the shorter ones, most probably because a sufficiently long cDNA cannot be produced by self-priming of the template.
As mentioned above, the combination of all segments demonstrates the existence of specific or at least predominant start position(s) and a minimum length of the newly synthesized (–)-strand RNA(s). One start site (RNA 1) is located between nt 87 and 113 with a minimum length of 266 nt, because segment EI was still detectable. Small segments (E, F, G, H and I) amplify part of RNA 1. Similarly, one can conclude from the results on segments CD (positive) and DI (negative) that another start site has to exist positioned between nt 87 and 208 and that the length of the corresponding RNA 2 is at least 108 nt. One should emphasize that evaluation of the results in Table 1 clearly indicated the existence of two different start sites, but that a more detailed interpretation is not required because the exact start positions could be determined as described below.
Determination of exact start sites by primer extension
In order to determine the exact start sites, a primer extension analysis was performed. It should be emphasized that primer extension analysis was only possible after the reverse transcription step of the RT–PCR analysis had been optimized. Figure 4A shows primer extension analysis of the 5′-end of the (–)-strand of RNA 1. The sample from the transcription assay exhibits a main band at position A111 (according to the numbering of cPSTVd); addition of α-amanitin completely abolished this signal (compare slot NE with NE + αA). This band was not caused by a sequence-specific stop of reverse transcriptase, because primer extension of an in vitro (–)-strand PSTVd transcript as a control did not exhibit a band at position A111 [slot (–)]. The minor bands corresponding to shorter primer extended transcripts are either due to early termination or might represent minor start sites adjacent to the predominant start site A111. Addition of non-templated nucleotides was not observed. Thus, a specific or at least predominant start site of pol II-dependent transcription of the cPSTVd molecule was identified at nucleotide A111. This result is in agreement with the RT–PCR analysis of the 5′-end of (–)-strand RNA 1, which predicted a start site between cPSTVd nucleotides 87 and 113.
Figure 4.
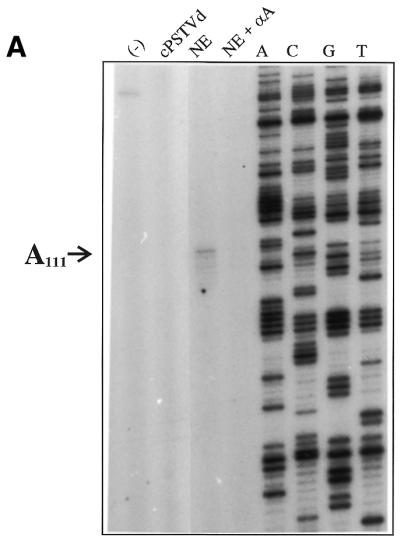

Determination of the exact start sites of cPSTVd transcription by primer extension. The whole sample of a primer extension assay (cf. Materials and Methods) was analyzed on an 8% polyacrylamide sequencing gel. Slot NE, in vitro transcription with cPSTVd template (applied amount ∼5 ng cPSTVd); NE + αA, NE with 10–6 M α-amanitin added. The following controls for (–)-strand specificity of the primer extension reaction were used: (–), 100 pg (–)-strand PSTVd in vitro transcript [IVT pRH 716 with 5′-G146 as the start site in (A) and IVT pSH1 with 5′-C227 in (B)]; cPSTVd, 10 ng cPSTVd. (A) The 5′-end of RNA 1 was analyzed using primer AF21 (cf. Fig. 3); the same primer was used for the sequencing ladder of pSH1. (B) The 5′-end of RNA 2 was analyzed using primer XC1 (cf. Fig. 3); the same primer was used for the sequencing ladder of pSH1.
Figure 4B shows primer extension of the 5′-end of (–)-strand RNA 2. One band at position A325 was found; controls were carried out corresponding to those with (–)-strand RNA 1. A very faint band was observed five to six bases upstream of A325, with only minor reduction by α-amanitin (visible only in the original, not in the reproduction). This band might be from less specific transcription and was not considered further. The second start site at nucleotide A325 is also in agreement with the RT–PCR analysis, which predicted a second (–)-strand RNA start site between cPSTVd nucleotides 208 and 359/1 and 87. Further primer extensions using additional primers did not show additional 5′-ends of the newly synthesized RNAs (data not shown).
Exclusion of nuclease degradation as a cause of the appearance of ‘artifical start sites’
A priori it could not be excluded that the nuclear extract used for transcription might contain some nuclease activity, which could lead to nicks in the viroid RNA. These nicks might occur at selective sites as a consequence of the sequence and secondary structure of the viroid. One might imagine two possibilities of selective nicks simulating specific start sites: (i) nicks in circular PSTVd could generate a promotor-independent start site for pol II on the 3′-site of the nick, which is known for nicks in double-stranded DNA templates for pol II transcription (38,39); (ii) nicks in the (–)-strand RNA could simulate a transcription product start site when the start site is determined by primer extension.
In order to exclude both possibilities, cPSTVd and (–)-strand PSTVd transcript were incubated in nuclear extract under the conditions of the transcription reaction and were then analyzed for selective nicks by primer extension. In addition to cPSTVd, an in vitro (+)-strand transcript was used in the test to obtain a known 5′-end for both polarities in the primer extension analysis. The 5′-ends of the in vitro transcripts and the primers for primer extension analysis were selected to optimize detection in the region of the start sites A111 and A325 (listed in Table 2).
Table 2. In vitro transcripts (IVT) as templates, their 5′-ends and the primers used for each combination of the primer extension analysis.
| Template | 5′-End | Primer | |
|---|---|---|---|
| The numbers indicate the corresponding nucleotides in the (+)-strand cPSTVd molecule given in the 3′→5′ direction. | |||
| A111 | (+)pFR2-IVT | G85 | 5′-280GGAAGGACACCCGAAGAAAGGAAGGGTGAA178-3′ |
| (–)pRH716-IVT | G146 | 5′-1CGGAACTAAACTCGTGGTTC20-3′ | |
| A325 | (+)pRH713-IVT | C282 | 5′-41GAGGTCAGGTGTGAACCAGAG21-3′ |
| (–)pSH1-IVT | C337 | 5′-237CTTTGCGCTGTCGCTTCGGC256-3′ | |
Figure 5 shows two examples of primer extension analysis, one for site A111 in the (–)-strand and one for site A325 in the (+)-strand. Analysis of the other two combinations showed very similar results (data not shown). No significant bands are visible at positions A111 and A325. Apart from the dominant band for the 5′-end of the in vitro transcript, only minor bands originating from non-specific primer extension stops are visible. Even if these stops originated from nicks in the (–)-strand transcript, none of them were observed at position A111. A more intense band in the (+)-strand, which is still more than an order of magnitude less than that of the 5′-end, is close to A325, but is actually at G324. The band at G324 is most probably from a stop induced by the secondary structure of the template, and these stops may not be restricted only to G324. A transcription start site, however, was visible only at A325 (cf. Fig. 4B) and it appears highly improbable that it was due to a structure-induced stop. When the products from incubation in buffer are compared with those from incubation in nuclear extracts (slot +NE), additional bands were not observed, thus nuclease activity leading to selective nicks in PSTVd RNA could not be observed in the nuclear extract used for the analysis of specific transcription start sites.
Figure 5.

Search for selective nicks due to RNase activity in the nuclear extract. A (–)-strand in vitro transcript [(–)-IVT or (–)-pRH716; Table 2] (left) and cPSTVd and a (+)-strand in vitro transcript [(+)-IVT or (+)-pRH713; Table 2] (right) were incubated in buffer (left slot) and in nuclear extract (+NE) and 10 ng of each was analyzed by primer extension. The 5′-ends of the in vitro transcription and the primer sequences of the primer extension are listed in Table 2. Electrophoresis was carried out on an 8% polyacrylamide sequencing gel. The positions and the lengths of the extension products and the positions A111, G324 and A325 are denoted by arrows. It should be noted that in the case of (–) polarity, the sequencing ladder reads the complementary sequence of cPSTVd, i.e. U325 at position A325 and C324 at position G324.
DISCUSSION
Transcription of circular PSTVd template into (–)-strand RNA could be performed in vitro in a nuclear extract prepared from a non-infected S.tuberosum cell culture. Start sites for synthesis of (–)-strand RNA are not randomly distributed over the PSTVd circle, but are positioned at two sites in distant parts of PSTVd. This result was obtained by two independent approaches, first by analysis with shorter PCR segments, so-called amplicons, of (–)-strand transcripts and second by identifying the 5′-ends by primer extension. Recently Navarro and Flores (27) analyzed transcription of ASBVd for both polarities: it also starts specifically, but at a single site for each polarity. Compared with PSTVd, ASBVd belongs to a different class of viroids. It is transcribed by a different cellular enzyme, undergoes self-cleavage and is located in a different cellular compartment. Thus, the results on ASBVd (27) and PSTVd (this work) can be regarded as complementary and could give a fairly complete picture of viroid replication.
Transcription of circular PSTVd in a nuclear extract
Preparation of the nuclear extract was carried out in two steps, i.e. isolation of nuclei from protoplasts as described earlier (16) and extraction of the nuclear proteins. The extraction protocol was systematically optimized to gain maximal transcriptional activity and the resulting protocol differed in several features from that used for viroid processing (16) and also from that of the transcription extract published earlier (13). Besides the optimum NaCl concentration for extraction of protein, the addition of NaF to all buffers to inhibit endogenous phosphatases was found to be essential (35). As mentioned above, in vivo analysis as carried out for ASBVd (27) would be much more difficult for PSTVd. Thus, we regard our cell-free system as the closest approach possible to the in vivo situation.
The transcriptional activity was sufficient to analyze the start sites of the newly synthesized (–)-strand RNA and products somewhat longer than 264 nt (RT–PCR segment EI, Table 1) could be detected. However, even after optimization of the extract, (–)-strand intermediates longer than unit length, as present in infected cells, could not be produced in measurable amounts. One has to keep in mind that viroid replication by DNA-dependent RNA pol II is per se artifical in the plant and proceeds with low activity, and with even lower activity in the cell-free extract. Combining the transcripts from both start sites the transcripts covered the whole circular viroid, i.e. the transcripts were not restricted to only part of the viroid. The restricted lengths of the transcripts is due to restricted transcriptional activity of the nuclear extract rather than degradative activity, because particular care was taken to avoid artifacts due to RNase activity in the nuclear extract. As shown in the incubation experiments (cf. Fig. 5) for cPSTVd and for in vitro transcripts in the absence of rNTPs, degradation at selected sites in the viroid structure could not be detected and a close inspection of the start sites, A111 and A325, did not show any significant bands at these sites. Thus, open ends in circular PSTVd, i.e. the (+)-strand template as well as selected sites of nicking in the (–)-strand RNA product, could not be detected. Transcription stops in the secondary structure of viroid RNA could be observed in control experiments, but were more than an order of magnitude less effective as compared with 5′-ends and were not located at the start sites. This led us to exclude the possibility that the start sites could be artifacts from structure-induced transcription stops or from RNase nicking activity in the nuclear extract. Since the start sites were identified as 5′-ends by primer extension, the newly synthesized (–)-strand RNA cannot be mixed up with elongation products from nicked cPSTVd. One should note, however, that we cannot exclude additional start sites of minor activity, if these were not visible by our approach.
Both start sites have similar sequences and secondary structure contexts
Since two distinct sites were found, we looked for common features of the start sites with respect to sequence and secondary structure. In both cases the start nucleotide, i.e. the first nucleotide to be transcribed, is an adenine (A111 and A325; cf. Fig. 6) and the sequences of the six 5′-located nucleotides that are transcribed next are very similar, differing only by a single purine exchange (A107 versus G321; cf. Fig. 6). A corresponding mutation G321→A321 leads to identity and was found to be infectious (40). A pyrimidine exchange C323→U323 also led to an infectious mutant (40). We have to admit, however, that it is not known whether both initiation sites are used in these mutants. Interestingly, sequence specificity was also found for the first nucleotides transcribed from the ASBVd template. Rather than two start sites of the same polarity, one start site of (+) and another of (–) polarity were compared; both replication intermediates started transcription with 5′-UAAA-3′ (27). Specific 5′-neighboring sequences have not been reported for plant genes (41). It is not known whether the synthesized (–)-strand PSTVd RNA contains the 5′-capping structure typical of mRNA synthesized by pol II. Since viroids do not undergo translation they would not need a 5′-cap. The specific sequence could even prevent capping or induce some specific, as yet unknown modification of the 5′-end of viroid (–)-strand RNA.
Figure 6.

Positions of the transcription start sites A111 and A325 in the secondary structure of cPSTVd. Two segments containing the initiation sites and the GC-boxes are enlarged. The direction of transcriptional synthesis by pol II is indicated by arrows. The parts of the PSTVd molecule forming hairpins I, II and III (cf. Introduction for details) are denoted.
The two initiation regions have another structural element in common: a GC-box is located upstream of the start nucleotide (cf. Fig. 6). The sequences of the two GC-boxes are different, but their distances from the start nucleotides (16 nt for A111 and 15 nt for A325) are very similar. Thus, we assume that the correct distance is more critical for correct initiation than the exact sequence of the GC-box. A GC-rich double helical segment is also essential for transcription of (–)-strand PSTVd. The so-called HP II structure is, however, not part of the thermodynamically most stable, i.e. rod-like, structure, but is formed by sequential folding during the transcription process. Its relevance to transcription of the (–)-strand was concluded originally from site-directed mutagenesis studies (17,18). Thus, PSTVd replication seems to involve GC-box elements for both polarities. Although the present results were obtained for PSTVd, one might expect their general relevance for all viroids of the PSTVd group, since the sequences of the start sites and the GC-box elements are highly conserved in that group (G.Steger and D.Riesner, unpublished results).
Most eukaryotic genes that are transcribed by pol II have a TATA box as a promotor element; exceptions are some housekeeping genes. The PSTVd molecule apparently does not have an element that could be the RNA equivalent of the TATA box. GC-boxes have been described as binding sites for transcription factor SP1, which is necessary for transcription of housekeeping genes (42). GC elements have also been found as parts of promotors in plants, even at a comparable distance from the start site (43–46). The probability of GC-box promotors without TATA boxes has been discussed by Roeder (47). The transcription start sites in ASBVd do not correlate with the presence and location of GC-boxes but are close to or in a unique secondary structure element, i.e. the right terminal loop (27). It is not surprising that viroids using different polymerases have adapted by using different start sites and a different context of sequence and secondary structure elements.
The functional relevance of the finding of two start sites, A111 and A325, is not known. Future experiments will have to determine whether the two sites are used independently, cooperatively or exclude each other. We are currently using the more general approach of site-directed mutagenesis to analyse the functional interdependence and the roles of single nucleotides in the start position, in the initiating sequence and in the GC-box elements.
Another open question concerns unknown start site(s) on the (–)-strand PSTVd intermediate. As already mentioned, at least a GC-box motif (HP II) was found to be critical for (–)-strand to (+)-strand transcription. Transcriptional analysis of (–)-strand transcription is much more difficult to perform, although the nuclear extract used in this work should also be applicable to the other polarity. If, however, the template is the (–)-strand and the transcript the (+)-strand, both strands are linear. Because of the self-complementarity of the sequence, a specific PCR is more difficult as compared to the situation in this study, where the (+)-strand template was circular and only the (–)-strand transcript was linear.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Drs T. Baumstark and P. Klaff for stimulating discussions and Ms H. Gruber for help in preparing the manuscript. This work was supported by grants from the Deutsche Forschungsgemeinschaft and Fonds der Chemischen Industrie.
REFERENCES
- 1.Gross H.J., Domdey,H., Lossow,C., Jank,P., Raba,M., Alberty,H. and Sänger,H.L. (1978) Nucleotide sequence and secondary structure of potato spindle tuber viroid. Nature, 273, 203–208. [DOI] [PubMed] [Google Scholar]
- 2.Riesner D., Henco,K., Rokohl,U., Klotz,G., Kleinschmidt,A.K., Domdey,H., Jank,P., Gross,H.J. and Sänger,H.L. (1979) Structure and structure formation of viroids. J. Mol. Biol., 133, 85–115. [DOI] [PubMed] [Google Scholar]
- 3.Riesner D. and Gross,H.J. (1985) Viroids. Annu. Rev. Biochem., 54, 531–564. [DOI] [PubMed] [Google Scholar]
- 4.Diener T.O. (ed.) (1987) The Viroids. Plenum Publishing Corp., New York, NY.
- 5.Semancik J.S. (ed.) (1987) Viroids and Viroid-like Pathogens. CRC Press, Boca Raton, FL.
- 6.Symons R.H. (ed.) (1990) Viroids and related pathogenic RNAs. Semin. Virol., 1, 75–134. [Google Scholar]
- 7.Branch A.D. and Robertson,H.D. (1984) A replication cycle for viroids and other small infectious RNAs. Science, 223, 450–455. [DOI] [PubMed] [Google Scholar]
- 8.Sänger H.L. (1987) Viroid replication. In Diener,T.O. (ed.), The Viroids. Plenum Publishing Corp., New York, NY.
- 9.Symons R.H. (1992) Small catalytic RNAs. Annu. Rev. Biochem., 61, 641–671. [DOI] [PubMed] [Google Scholar]
- 10.Darós J.A., Marcos,J.F., Hernández,C. and Flores,R. (1994) Replication of avocado sunblotch viroid: evidence for a symmetric pathway with two rolling circles and hammerhead ribozyme processing. Proc. Natl Acad. Sci. USA, 91, 12813–12817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mühlbach H.-P. and Sänger,H.L. (1979) Viroid replication is inhibited by α-amanitin. Nature, 278, 185–188. [DOI] [PubMed] [Google Scholar]
- 12.Schindler I.M. and Mühlbach,H.-P. (1992) Involvement of nuclear DNA-dependent RNA-polymerases in potato spindle tuber viroid replication: a reevaluation. Plant Sci., 84, 221–229. [Google Scholar]
- 13.Tsagris M., Tabler,M., Mühlbach,H.P. and Sänger,H.L. (1987) Linear oligomeric potato spindle tuber viroid (PSTV) RNAs are accurately processed in vitro to the monomeric circular viroid proper when incubated with a nuclear extract from healthy potato cells. EMBO J., 6, 2173–2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baumstark T., Schröder,A.R.W. and Riesner,D. (1997) Switch from cleavage to ligation is driven by a change from a tetraloop to a loop E conformation. EMBO J., 16, 599–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsagris M., Tabler,M. and Sänger,H.L. (1987) Oligomeric potato spindle tuber viroid RNA does not process autocatalytically under conditions where other RNAs do. Virology, 157, 227–231. [DOI] [PubMed] [Google Scholar]
- 16.Baumstark T. and Riesner,D. (1995) Only one of four possible secondary structures of the central conserved region of potato spindle tuber viroid is a substrate for processing in a potato nuclear extract. Nucleic Acids Res., 23, 4246–4254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Loss P., Schmitz,M., Steger,G. and Riesner,D. (1991) Formation of a thermodynamically metastable structure containing hairpin II is critical for infectivity of potato spindle tuber viroid RNA. EMBO J., 10, 719–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qu F., Heinrich,C., Loss,P., Steger,G., Tien,P. and Riesner,D. (1993) Multiple pathways of reversion in viroids for conservation of structural elements. EMBO J., 12, 2129–2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heinrich C. (1991) Basenpaaraustausche mit gerichteter Mutagenese in der konservierten Region der Haarnadelstruktur II des Potato Spindle Tuber Viroid (PSTVd). Diplomarbeit, Heinrich-Heine-Universität, Düsseldorf, Germany.
- 20.Goodman T.C., Nagel,L., Rappold,W., Klotz,G. and Riesner,D. (1984) Viroid replication: equilibrium association constant and comparative activity measurements for the viroid–polymerase interaction. Nucleic Acids Res., 12, 6231–6246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hecker R., Wang,Z., Steger,G. and Riesner,D. (1988) Analysis of RNA structures by temperature gradient gel electrophoresis: viroid replication and processing. Gene, 72, 59–74. [DOI] [PubMed] [Google Scholar]
- 22.Flores R. and Semancik,J.S. (1982) Properties of a cell-free system for the synthesis of citrus exocortis viroid. Proc. Natl Acad. Sci. USA, 79, 6285–6288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Semancik J.S. and Harper,K.L. (1984) Optimal conditions for cell-free synthesis of citrus exocortis viroid and the question of specificity of RNA polymerase activity. Proc. Natl Acad. Sci. USA, 81, 4429–4433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Flores R. (1989) Synthesis of RNAs specific to citrus exocortis viroid by a fraction rich in nuclei from infected Gynura aurantiaca: examination of the nature of the products and solubilization of the polymerase-template complex. J. Gen. Virol., 70, 2695–2706. [Google Scholar]
- 25.Rivera-Bustamante R.F. and Semancik,J.S. (1989) Properties of a viroid-replicating complex solubilized from nuclei. J. Gen. Virol., 70, 2707–2716. [Google Scholar]
- 26.Marcos J.F. and Flores,R. (1992) Characterization of RNAs specific to avocado sunblotch viroid synthesized in vitro by a cell-free system from infected avocado leaves. Virology, 186, 481–488. [DOI] [PubMed] [Google Scholar]
- 27.Navarro J. and Flores,F. (2000) Characterization of the initiation site of both polarity strands of a viroid RNA reveals a motif conserved in sequence and structure. EMBO J., 19, 2662–2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Colpan M., Schumacher,J., Brüggemann,W., Sänger,H.L. and Riesner,D. (1983) Large scale purification of viroid RNA using Cs2SO4 gradient centrifugation and high performance liquid chromatography. Anal. Biochem., 131, 257–265. [DOI] [PubMed] [Google Scholar]
- 29.Riesner D., Klaff,P., Steger,G. and Hecker,R. (1987) Viroids: subcellular location and structure of replicative intermediates. Ann. N. Y. Acad. Sci., 503, 212–237. [DOI] [PubMed] [Google Scholar]
- 30.Krupp G. (1988) RNA synthesis: strategies for the use of bacteriophage RNA polymerase. Gene, 72, 75–89. [DOI] [PubMed] [Google Scholar]
- 31.Repsilber D., Wiese,S., Rachen,M., Schröder,A.R., Riesner,D. and Steger,G. (1999) Formation of metastable RNA structures by sequential folding during transcription: time-resolved structural analysis of potato spindle tuber viroid (–)-stranded RNA by temperature-gradient gel electrophoresis. RNA, 5, 574–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Steger G. (1994) Thermal denaturation of double-stranded nucleic acids: prediction of temperatures critical for gradient gel electrophoresis and polymerase chain reaction. Nucleic Acids Res., 22, 2760–2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Behnke M. (1975) Regeneration in Gewebekulturen einiger dihaploider Solanum tuberosum Klone. Z. Pflanzenzuecht., 75, 262–265. [Google Scholar]
- 34.Mühlbach H.-P. and Sänger,H.L. (1981) Continuous replication of potato spindle tuber viroid (PSTV) in permanent cell cultures of potato and tomato. Biosci. Rep., 1, 79–87. [DOI] [PubMed] [Google Scholar]
- 35.Fan H. and Sugiura,M. (1995) A plant basal in vitro system supporting accurate transcription of both RNA polymerase II- and III-dependent genes: supplement of green leaf component(s) drives accurate transcription of a light-responsive rbcS gene. EMBO J., 14, 1024–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nagel-Steger L. (1990) Untersuchungen zur Replikation des Potato Spindle Tuber Viroids durch die DNA-abhängige RNA-Polymerase II aus Wirtsgewebe. Thesis, Heinrich-Heine-Universität, Düsseldorf, Germany.
- 37.Schweizer P. and Mösinger,E. (1994) Initiator-dependent transcription in vitro by a wheat germ chromatin extract. Plant Mol. Biol., 25, 115–120. [DOI] [PubMed] [Google Scholar]
- 38.Lewis M.K. and Burgess,R.R. (1980) Transcription of the simian virus-40 DNA by wheat germ polymerase II. J. Biol. Chem., 255, 4928–4936. [PubMed] [Google Scholar]
- 39.Matsui T., Segall,J., Weil,A. and Roeder,R.G. (1980) Multiple factors required for accurate initiation of transcription by purified RNA polymerase II. J. Biol. Chem., 255, 11992–11996. [PubMed] [Google Scholar]
- 40.Owens R.A., Thompson,S. and Steger,G. (1991) Effects of random mutagenesis upon potato spindle tuber viroid replication and symptom expression. Virology, 185, 18–31. [DOI] [PubMed] [Google Scholar]
- 41.Schweizer P. (1994) In vitro transcription of plant nuclear genes. In Nover,L. (ed.), Plant Promotors and Transcription Factors. Springer Verlag, Berlin, Germany.
- 42.Gidoni D., Dynan,W.S. and Tijan,R. (1984) Multiple specific contacts between a mammalian transcription factor and its cognate promoters. Nature, 312, 409–413. [DOI] [PubMed] [Google Scholar]
- 43.Davies J.P. and Grossmann,A.R. (1994) Sequences controlling transcription of Chlamydomonas reinhardtiiβ2-tubulin gene after deflagellation and during the cell cycle. Mol. Cell. Biol., 14, 5165–5174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schindler U. and Cashmore,A.R. (1990) Photoregulated gene expression may involve ubiquitous DNA binding proteins. EMBO J., 9, 3415–3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Neuteboom S.T.C., Stoffels,A., Hullemann,E., Memelink,J., Schilperoort,R.A. and Hoge,J.H.C. (1993) Interaction between the tobacco DNA-binding activity CBF and the cyt-1 promoter element of the Agrobacterium tumefaciens T-DNA gene T-CYT correlates with cyt-1 directed gene expression in multiple tobacco tissue types. Plant J., 4, 525–534. [DOI] [PubMed] [Google Scholar]
- 46.Olive M.R., Reacock,W.J. and Dennis,E.S. (1991) The anaerobis responsive element contains two GC-rich sequences essential for binding a nuclear protein and hypoxic activation of the maize Adh1 promoter. Nucleic Acids Res., 19, 7053–7060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roeder R.G. (1996) The role of general transcription factors in transcription by RNA polymerase II. Trends Biochem. Sci., 21, 327–335. [PubMed] [Google Scholar]