

Abstract
N‐glycans are molecularly diverse sugars borne by over 70% of proteins transiting the secretory pathway and have been implicated in protein folding, stability, and localization. Mutations in genes important for N‐glycosylation result in congenital disorders of glycosylation that are often associated with intellectual disability. Here, we show that structurally distinct N‐glycans regulate an extracellular protein complex involved in the patterning of somatosensory dendrites in Caenorhabditis elegans. Specifically, aman‐2/Golgi alpha‐mannosidase II, a conserved key enzyme in the biosynthesis of specific N‐glycans, regulates the activity of the Menorin adhesion complex without obviously affecting the protein stability and localization of its components. AMAN‐2 functions cell‐autonomously to allow for decoration of the neuronal transmembrane receptor DMA‐1/LRR‐TM with the correct set of high‐mannose/hybrid/paucimannose N‐glycans. Moreover, distinct types of N‐glycans on specific N‐glycosylation sites regulate DMA‐1/LRR‐TM receptor function, which, together with three other extracellular proteins, forms the Menorin adhesion complex. In summary, specific N‐glycan structures regulate dendrite patterning by coordinating the activity of an extracellular adhesion complex, suggesting that the molecular diversity of N‐glycans can contribute to developmental specificity in the nervous system.
Keywords: adhesion, alpha mannosidase II, dendrite, glycosylations, N‐glycans
Subject Categories: Cell Adhesion, Polarity & Cytoskeleton; Neuroscience; Post-translational Modifications & Proteolysis
Structurally distinct N‐glycans regulate the function of the Menorin adhesion complex during patterning of somatosensory dendrites in C. elegans.
Introduction
Development of a nervous system in metazoans requires the coordinated interactions of extracellular molecules to ensure correct neuronal morphogenesis and to establish connectivity (Jan & Jan, 2010; Dong et al, 2015; Lefebvre, 2021). Most of these extracellular proteins are glycoconjugates, that is, carry different types of glycans attached to the protein backbone. Glycans are molecularly the most diverse molecules in nature, in part because they are not genetically encoded. Yet their structures are not random and are therefore conceptually attractive to broaden the molecular diversity and specificity of extracellular proteins and their interactions during development. For example, glycosaminoglycans, a class of glycans, have been suggested to modulate protein–protein interactions and provide information during neural development by way of their structural diversity (Holt & Dickson, 2005; Bülow & Hobert, 2006; Poulain & Yost, 2015; Masu, 2016; Saied‐Santiago & Bülow, 2018).
N‐glycans are another structurally diverse group of glycans that fall into four classes: high‐mannose, hybrid, complex, and paucimannose‐type N‐glycans, and are invariantly attached via an asparagine to a protein backbone (Stanley et al, 2015). Importantly, 70% of all proteins transiting the endoplasmic reticulum are post‐translationally N‐glycosylated (Apweiler et al, 1999). Therefore, the structural diversity of N‐glycans could also significantly expand the repertoire and specificity of protein interactions or functions in the extracellular space. N‐glycans in general have been shown to be important for protein folding, stability, localization, and protein–protein interactions (Stanley et al, 2015). Moreover, mutations in genes involved in N‐glycosylation in humans result in congenital disorders of glycosylation (CDG), which are multi‐syndromic and often include neurological symptoms, including intellectual disability (Freeze, 2006; Jaeken & Peanne, 2017; Chang et al, 2018; Ng & Freeze, 2018). Studies in vertebrates and invertebrates have shown that mutants compromising N‐glycan biosynthesis or N‐glycan attachment result in defects in cell surface localization of cell adhesion molecules and axon guidance cues (Sekine et al, 2013; Medina‐Cano et al, 2018; Mire et al, 2018). Moreover, N‐glycosylation per se has been shown to be important for dendrite development in dissociated rat neurons (Hanus et al, 2016), and the addition of polysialic acid chains to N‐glycans can change the binding properties of cell adhesion molecules (reviewed in Schnaar et al, 2014). However, the question of whether and how specific classes and structures of N‐glycans modulate extracellular pathways or complexes during nervous system development in vivo remains understudied.
Here we use the dendrites of the somatosensory PVD neuron (Posterior cell body, Ventral cord process D), which display complex and stereotyped branching patterns in the nematode Caenorhabditis elegans (Fig 1A; Oren‐Suissa et al, 2010; Smith et al, 2010; Albeg et al, 2011) to investigate the role of different classes of N‐glycans during development. The intricately branched PVD neurons possess polymodal functions of proprioception, nociception, and mechanosensation (Chatzigeorgiou et al, 2010; Albeg et al, 2011; Cohen et al, 2014; Tao et al, 2019). We found that aman‐2/Golgi alpha‐mannosidase II, a conserved enzyme important for the synthesis of complex and paucimannose‐type N‐glycans, is required for PVD dendrite morphogenesis. Specifically, aman‐2/Golgi alpha‐mannosidase II allows for the proper decoration of the leucine‐rich transmembrane receptor DMA‐1/LRR‐TM in PVD with the correct set of high‐mannose/hybrid/paucimannose N‐glycans on specific N‐glycosylation sites. Rather than controlling trafficking or surface localization of DMA‐1/LRR‐TM, we provide evidence that the proper N‐glycosylation of DMA‐1/LRR‐TM in PVD is essential for the function of DMA‐1/LRR‐TM as part of the Menorin pathway during PVD patterning. This pathway comprises two conserved cell adhesion molecules, SAX‐7/L1CAM and MNR‐1/Menorin, that function from the epidermis, and a secreted chemokine LECT‐2/Chondromodulin II from muscle that, together with DMA‐1/LRR‐TM, form a high affinity cell adhesion complex (Fig 1B; Inberg et al, 2019; Sundararajan et al, 2019). Together, our experiments suggest that distinct classes of N‐glycans, rather than N‐glycosylation per se, serve specific functions in dendrite branching and contribute to developmental specificity during neuronal morphogenesis.
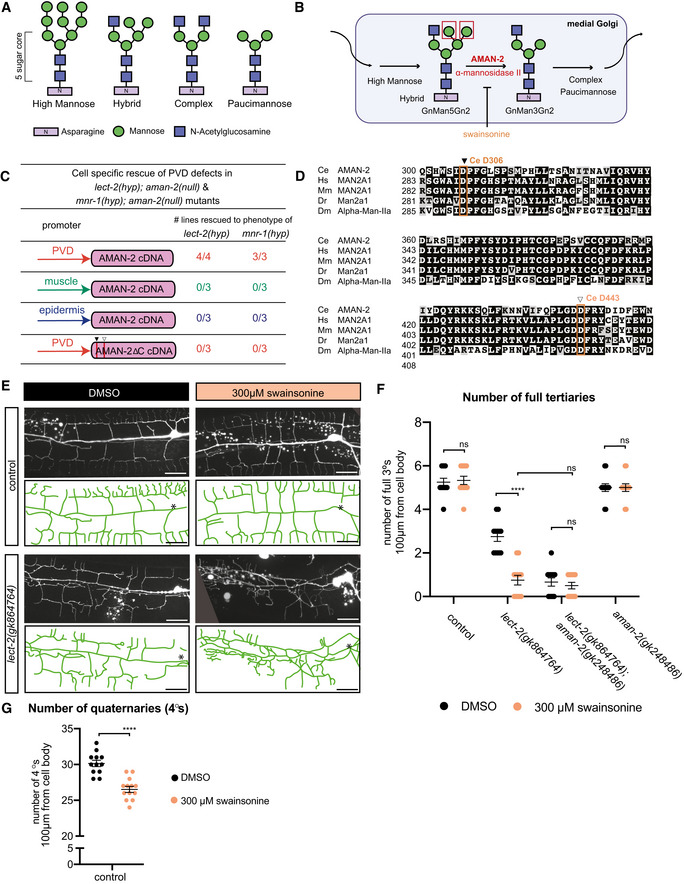
Figure 1. AMAN‐2/Golgi alpha‐mannosidase II is required for PVD dendrite patterning.
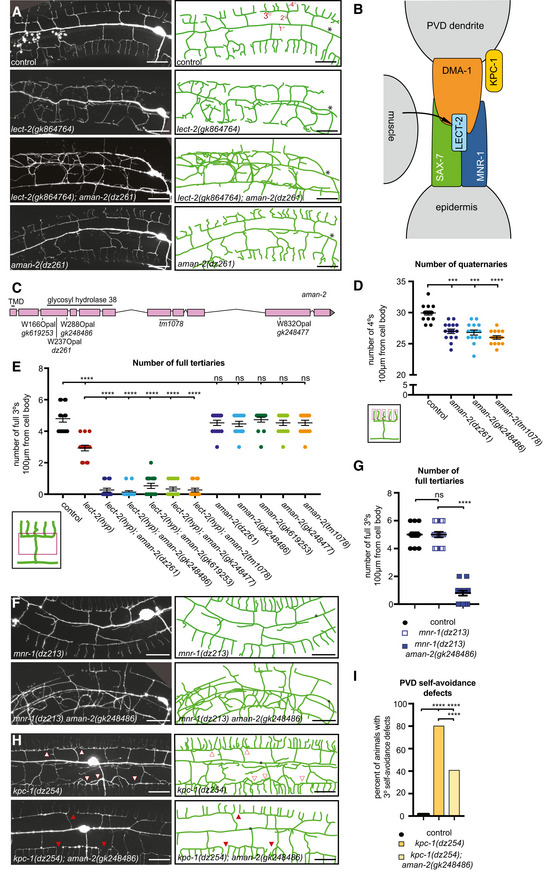
- Fluorescent images (left panels) and tracings (right panels) of PVD neurons of the indicated genotypes. PVD is visualized by the wdIs52 transgene. Primary (1°), secondary (2°), tertiary (3°), and quaternary (4°) dendrites are indicated, and the cell body is marked with an asterisk. Anterior is to the left and dorsal is up in all panels. Scale bars represent 20 μm.
- Schematic of the Menorin complex, including DMA‐1/LRR‐TM, SAX‐7/L1CAM, MNR‐1/Menorin, and LECT‐2/Chondromodulin II, as well as the negative regulator KPC‐1/Furin.
- Genomic environs of aman‐2. An N‐terminal transmembrane domain (TMD) is indicated, encoding a type II transmembrane protein. All alpha‐mannosidase II proteins contain a glycosyl hydrolase 38 domain. Four nonsense alleles and one deletion allele (tm1078) of aman‐2 are denoted.
- Quantification of the number of quaternary branches (indicated in schematic) in different aman‐2 alleles and in wild‐type control animals. All loss of function alleles of aman‐2 display a significant decrease in PVD quaternary branch number. Data are represented as mean ± SEM. Statistical significance was calculated using the Kruskal–Wallis test and is indicated (****P ≤ 0.0001). n = 15 for all genotypes and are biological replicates.
- Quantification of the number of full tertiaries (marked by secondary and tertiary branches as shown in schematic) in the genotypes indicated. Data are represented as mean ± SEM. Statistical significance was calculated using the Kruskal–Wallis test and is indicated (****P ≤ 0.0001, ns = not significant). n = 15 for all genotypes and are biological replicates.
- Fluorescent images and tracings of PVD in mnr‐1(dz213) hypomorphic animals alone and in combination with an aman‐2(gk248486) null mutant. The dz213 introduces a L135F missense mutation in the DUF2181 domain of MNR‐1/Menorin. PVD is visualized by the dzIs53 transgene. The cell body is marked with an asterisk. Scale bars represent 20 μm.
- Quantification of the number of full tertiaries in genotypes indicated and traced in (F). The loss of aman‐2 severely enhances the mnr‐1(dz213) phenotype. Control indicates wild‐type control animals as shown in (A). Data are represented as mean ± SEM. Statistical significance was calculated using the Mann–Whitney test and is indicated (****P ≤ 0.0001, ns = not significant). n > 23 for all genotypes and are biological replicates.
- Fluorescent images and tracings of PVD in kpc‐1(dz254) hypomorph animals alone and in combination with an aman‐2(gk248486) null mutant. PVD is visualized by the dzIs53 transgene. White arrows indicate self‐avoidance defects in tertiary branches. Red arrows show gaps between tertiary branches (no self‐avoidance defects). The cell body is marked with an asterisk. Scale bars represent 20 μm.
- Quantification of the percent of self‐avoidance defects in genotypes indicated and traced in (G). Control indicates wild‐type control animals as shown in (A). The loss of aman‐2 suppresses defects in the kpc‐1(dz254) phenotype. Data are represented as mean. Statistical significance was calculated using the Z‐test and is indicated (****P ≤ 0.0001). n > 15 for all genotypes and are biological replicates.
Source data are available online for this figure.
Results and Discussion
The N‐glycosylation enzyme AMAN‐2/Golgi alpha‐mannosidase II is required for PVD dendrite patterning
To identify additional factors that regulate the Menorin pathway, we performed an unbiased genetic screen for factors that modify a partial loss of function allele of the chemokine lect‐2/Chondromodulin II (Diaz‐Balzac et al, 2016; Zou et al, 2016). We isolated dz261 as a strong enhancer of the partial loss of function lect‐2 mutation in addition to other alleles in known genes of the Menorin pathway (Figs 1A and D, and EV1A–C). Using a combination of mapping, sequencing, and transformation rescue, we identified dz261 as an allele of aman‐2/Golgi alpha‐mannosidase II (Figs 1C and EV1A–C), which encodes a central enzyme in the N‐glycosylation biosynthetic pathway not previously implicated in dendrite development. The allele dz261 is likely a complete loss of function mutation as it introduces an early stop codon in the key enzymatic domain of the protein (Figs 1C and EV1A–C). Quantifications of PVD branching patterns in aman‐2(dz261) null mutants, as well as four additional nonsense/deletion alleles of aman‐2, demonstrate that this Golgi alpha‐mannosidase II is required for the formation of quaternary branches, but not for the formation of secondary or tertiary branches of PVD dendrites (Figs 1D and E, and EV1D). These observations are reminiscent of partial loss of function alleles of dma‐1/LRR‐TM (Tang et al, 2019) and suggest that aman‐2 may be necessary for full functionality of the Menorin pathway. Of note, we observed no obvious morphological phenotypes or defects in viability in animals lacking AMAN‐2 (Fig EV1E).
Figure EV1. Results of modifier screen and identification of AMAN‐2/Golgi Alpha‐mannosidase II.
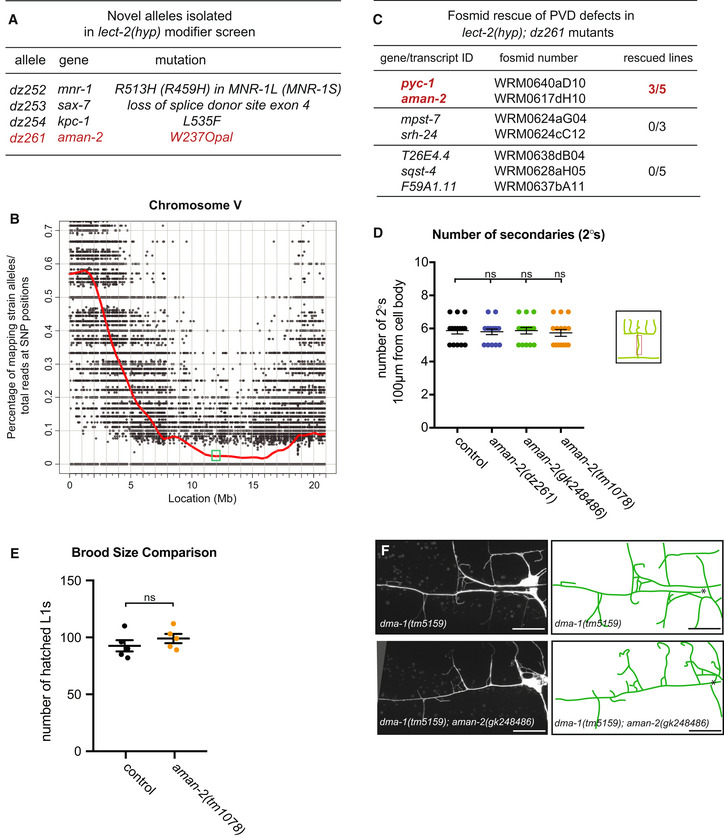
- Table of alleles isolated in the lect‐2(gk864764) hypomorph modifier screen.
- Graph of single nucleotide polymorphism (SNP) results after whole genome sequencing of the dz261 allele (Doitsidou et al, 2016). Axes are denoted above. The green box shows the genomic position of aman‐2.
- Table showing the candidate genes tested as a result of the SNP data in (B). Pools of fosmids covering the regions of indicated transcripts were injected into the lect‐2(hyp); dz261 double mutant. Only the pool containing a fosmid including aman‐2 showed rescue. pyc‐1 was eliminated because a null allele (gk689405) failed to enhance the lect‐2(hyp). Numbers indicate number of biological replicates that showed rescue. 25 animals were scored for each line.
- Quantification of the number of secondary branches (indicated in schematic) in the genotypes indicated. Data are represented as mean ± SEM. Statistical significance was calculated using the Kruskal–Wallis test, ns: no significance. n = 15 for all genotypes and are biological replicates.
- Viability of aman‐2 mutant animals assessed via brood size comparison in the number of eggs that hatched and produced L1 larvae in wild‐type control and aman‐2(tm1078) animals. Data are represented as mean ± SEM. Statistical significance was calculated using the Mann–Whitney test, ns: no significance. n = 5 for all genotypes and are biological replicates.
- Fluorescent images of the dma‐1 null allele, dma‐1(tm5159), alone (top) and in a double null mutant aman‐2(gk248486) background (bottom). Asterisk indicates location of the cell body. Scale bars represent 20 μm.
Source data are available online for this figure.
AMAN‐2/Golgi alpha‐mannosidase II positively regulates the Menorin pathway
To directly test the genetic relationship between aman‐2/Golgi alpha‐mannosidase II and the Menorin pathway, we performed double mutant analyses. We found that the loss of aman‐2/Golgi alpha‐mannosidase II strongly enhances the severity of PVD branching defects in partial loss‐of‐function mutants of lect‐2/Chondromodulin II (gk864764) and mnr‐1/Menorin (dz213, Ramirez‐Suarez & Bülow, unpublished) (Fig 1A and E–G), both of which are essential components of the conserved Menorin cell adhesion complex, and act as positive regulators of PVD development (Fig 1E–G; Dong et al, 2013; Salzberg et al, 2013; Diaz‐Balzac et al, 2016; Zou et al, 2016). In contrast, the loss of aman‐2/Golgi alpha‐mannosidase II suppressed the self‐avoidance defects of tertiary dendrites in a partial loss of function mutation of kpc‐1/Furin (dz254) (Figs 1H and I, and EV1C), a known negative regulator of the Menorin pathway (Schroeder et al, 2013; Salzberg et al, 2014; Dong et al, 2016). Lastly, we found that a dma‐1; aman‐2 double null mutant did not display a more severe dendrite phenotype than the dma‐1 mutant alone (Fig EV1F), suggesting that both genes function in a common pathway. We conclude that aman‐2/Golgi alpha‐mannosidase II normally functions to positively regulate the Menorin pathway to ensure correct PVD dendrite patterning.
AMAN‐2/Golgi alpha‐mannosidase II does not serve obvious functions in regulating transport or abundance of the DMA‐1/LRR‐TM
Mutations in N‐glycosylation are often associated with protein folding defects and trafficking blocks due to misfolding (Stanley et al, 2015) and can, for example, result in lower abundance of cell surface proteins such as cell adhesion proteins in the nervous system (Medina‐Cano et al, 2018). Because protein misfolding is more likely to occur at elevated temperatures (Gasser et al, 2008; Vabulas et al, 2010), we tested whether PVD branching defects in aman‐2/Golgi alpha‐mannosidase II mutants get progressively more severe with increasing temperatures. We found no significant increase in dendrite branching defects at 25°C compared to 15°C in aman‐2(gk248486) mutant animals, in contrast to hypomorphic lect‐2(gk864764) mutant animals (Fig EV2A and B). Previous work showed that mutations causing a secretory block as a result of a defective unfolded protein response trap a DMA‐1::GFP reporter in the cell body of PVD (Wei et al, 2015; Salzberg et al, 2017). We therefore questioned whether the loss of aman‐2/Golgi alpha‐mannosidase II can lead to defects in protein folding and trafficking, and a possible secretory block. We analyzed the amount and number of puncta of the DMA‐1::GFP reporter in the soma, as well as in dendrite branches, and found that DMA‐1::GFP fluorescence in both the soma and primary dendrites, and the number of DMA‐1::GFP puncta in tertiary dendrites, remained unaffected in aman‐2(gk248486) mutant animals (Fig EV2C–E). Moreover, localization or abundance of LECT‐2/Chondromodulin II and SAX‐7/L1CAM were also not obviously affected by loss of aman‐2/Golgi alpha‐mannosidase II (Fig EV2F and G). Taken together, these findings suggest that AMAN‐2/Golgi alpha‐mannosidase II may not primarily function to ensure protein folding, stability, or transport of factors of the Menorin pathway, but may rather regulate more specific aspects of the Menorin pathway during PVD patterning.
Figure EV2. The requirement of AMAN‐2 in PVD may be specific rather than global.
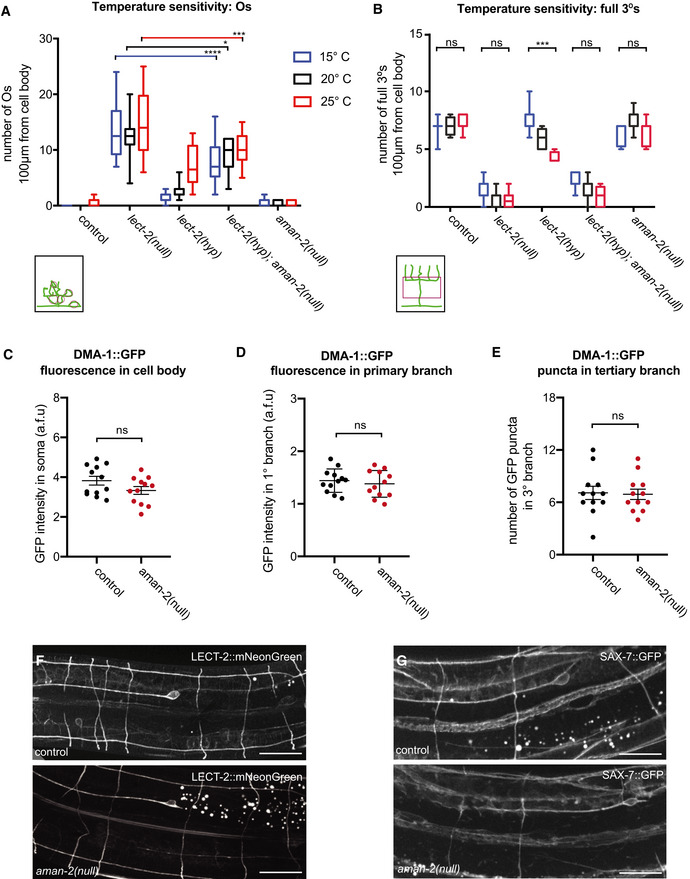
- Quantification of “Os” (overlapping branches as shown in schematic) of denoted genotypes at different temperatures. Data are presented as box and whisker plots, with the median and 25th and 75th percentile indicated. Whiskers show minimum and maximum. Statistical comparisons were performed using two‐way ANOVA tests, *P ≤ 0.05, ***P ≤ 0.001, ****P ≤ 0.0001, ns: not significant. n > 11 for all groups.
- Quantification of full tertiaries of denoted genotypes at different temperatures. Data are presented as box and whisker plots, with the median and 25th and 75th percentile indicated. Whiskers show minimum and maximum. Statistical comparisons were performed using two‐way ANOVA tests, ***P ≤ 0.001, ns: not significant. n > 11 for all groups.
- Quantification of DMA‐1::GFP fluorescence in control and aman‐2(gk248486) animals. GFP intensity is quantified in arbitrary fluorescent units by dividing the fluorescent area of the soma by the background. Data are represented as mean ± SEM. Statistical comparisons were performed using the Mann–Whitney test, ns: not significant. n = 12 for all genotypes and are biological replicates.
- Quantification of DMA‐1::GFP fluorescence in control and aman‐2(gk248486) animals. GFP intensity is quantified in arbitrary fluorescent units by measuring the fluorescence along the primary dendrite, up to 60 µm anterior from the cell body. Data are represented as mean ± SEM. Statistical comparisons were performed using the Mann–Whitney test, ns: not significant. n = 12 for all genotypes and are biological replicates.
- Quantification of DMA‐1::GFP fluorescent puncta in control and aman‐2(gk248486) animals. Puncta in the tertiary branches 60 µm anterior to the cell body were counted. Data are represented as mean ± SEM. Statistical comparisons were performed using the Mann–Whitney test, ns: not significant. n = 12 for all genotypes and are biological replicates.
- Localization of LECT‐2::mNeonGreen (dz249 endogenous reporter) in wild‐type and aman‐2(dz261) null mutant animals. No obvious differences in general neuronal or hypodermal staining were observed. Vesicular gut autofluorescence is visible as white circular staining. Scale bars represent 20 μm. 15 animals were assessed per genotype.
- Localization of SAX‐7::GFP (ddIs290 fosmid reporter) in wild type and aman‐2(dz261) null mutant animals. No obvious differences in general neuronal or hypodermal staining were observed. Vesicular gut autofluorescence is visible as white circular staining. Scale bars represent 20 μm. 15 animals were assessed per genotype.
Source data are available online for this figure.
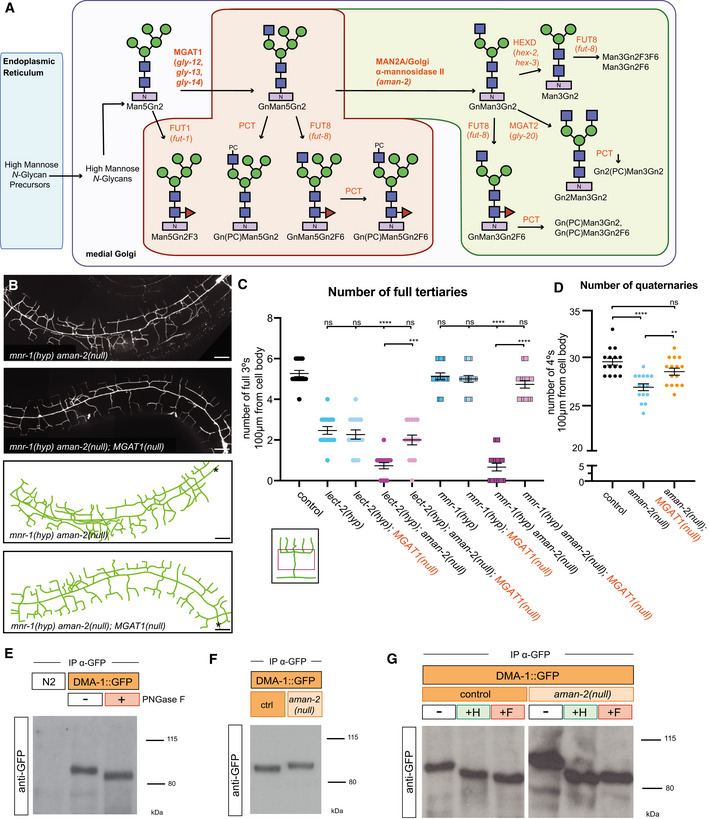
Enzymatic activity of AMAN‐2/Golgi alpha‐mannosidase II is required cell‐autonomously in PVD to form higher order branches
The octasaccharide GnMan5Gn2 in a specific linkage configuration is the unique precursor to hybrid, complex, and paucimannose N‐glycans (Fig 2A and B; Moremen, 2002; Paschinger et al, 2019). AMAN‐2 is a Golgi alpha‐mannosidase II, which is conserved from yeast to humans and cleaves two specific mannose residues from GnMan5Gn2, thereby generating the substrate for the formation of complex and paucimannose N‐glycans (Fig 2A and B; Moremen, 2002; Paschinger et al, 2019). To determine where AMAN‐2/Golgi alpha‐mannosidase II functions and whether enzymatic activity is required for its role in PVD dendrite morphogenesis, we investigated transgenic expression of a wild‐type AMAN‐2 cDNA under the control of heterologous promoters in PVD, muscle, or epidermis for their ability to rescue aman‐2 mutant defects. Expression in PVD, but not muscle or epidermis, rescued the enhanced phenotypes in PVD dendrite branching of lect‐2(gk864764); aman‐2(gk248486) and mnr‐1(dz213) aman‐2(gk248486) double mutants (Fig 2C). These observations suggest that AMAN‐2/Golgi alpha‐mannosidase II functions cell‐autonomously in PVD to pattern dendritic arbors.
Figure 2. AMAN‐2/Golgi alpha‐mannosidase II requires enzymatic activity in PVD to form higher order branches.
- Types of N‐linked glycans. The shared pentasaccharide core consists of two N‐acetylglucosamines (blue squares) and 3 mannoses (green circles) attached onto an Asparagine residue with an N‐X‐S/T consensus site. Glycan types vary by identity of additional sugars onto the pentasaccharide core.
- Drawing showing the enzymatic activity of AMAN‐2 in the medial Golgi. AMAN‐2 cleaves two specific α1,3 and α1,6 mannose linked residues boxed in red, allowing for the formation of complex and paucimannose type N‐glycans. Arrows denote other enzymes. Swainsonine specifically inhibits enzymatic activity of alpha‐mannosidase II. (Man = mannose, Gn = N‐acetylglucosamine).
- Table showing cell‐specific rescue experiments of PVD defects. AMAN‐2 cDNAs are expressed under the control of PVD, muscle, and epidermal specific promoters in the indicated double mutant backgrounds. Rescue is defined by restoration of the enhanced PVD phenotype back to that of the single hypomorphic mutants alone in aman‐2(gk248486) null mutant backgrounds enhanced by lect‐2(gk846764) or mnr‐1(dz213). 25 transgenic animals and their non‐transgenic siblings were scored for each line. Numbers indicate number of biological replicates that showed rescue.
- Multiple sequence alignment of human MAN2A1 (Hs; acc# NP_002363.2), mouse MAN2A1 (Mm; acc# NP_032575.2), zebrafish Man2a1 (Dr; acc# NP_001103497.2), fruit fly Alpha‐Man‐IIa (Dm; acc# NP_650494.2) and C. elegans AMAN‐2 (Ce; acc# NP_505995.2) created by COBALT (constrained based multiple sequence alignment tool). Conserved catalytic sites D306 (black arrow) and D443 (white arrow) are boxed in orange.
- Fluorescent images and tracings of wild‐type control (top) and lect‐2(gk846764) hypomorphic animals (bottom) fed on plates with 300 µm swainsonine vs. a DMSO control. PVD is visualized by the wyIs581 transgene. The cell body is denoted with an asterisk. Anterior is to the left and dorsal is up in all panels. Vesicular gut autofluorescence is visible as white circular staining. Scale bars represent 20 μm.
- Quantification of the number of full tertiaries in denoted genetic backgrounds (aman‐2 null is gk248486). Black data points indicate DMSO and orange data points show swainsonine treated animals. Data are represented as mean ± SEM. Statistical significance was calculated using the Mann–Whitney test and is indicated (****P ≤ 0.0001, ns = not significant). n = 12 for all genotypes and are biological replicates.
- Quantification of the number of quaternary dendrites in wild‐type control animals fed on plates with and without 300 µm swainsonine. Animals treated with swainsonine show a significant decrease in quaternary branch number, akin to the data in Fig 1C. Data are represented as mean ± SEM. Statistical significance was calculated using the Mann–Whitney test and is indicated (****P ≤ 0.0001). n = 12 for each experiment and are biological replicates.
Source data are available online for this figure.
Since AMAN‐2/Golgi alpha‐mannosidase II canonically functions as an enzyme (Moremen, 2002; Shah et al, 2008), we next asked whether catalytic activity is required for its role in PVD dendrite branching. We approached this both genetically and pharmacologically. Prior studies showed that two highly conserved aspartates are part of the conserved catalytic site in AMAN‐2/Golgi alpha‐mannosidases II (D306 and D443) and act sequentially to cleave off two mannose residues (Fig 2D; Shah et al, 2008). We found that an AMAN‐2 cDNA with both aspartates mutated, and hence likely catalytically dead, failed to rescue the defects in lect‐2(gk864764); aman‐2(gk248486) and mnr‐1(dz213) aman‐2(gk248486) double mutants (Fig 2C). To address the possibility that mutating the catalytic residues compromised the stability or structure of AMAN‐2, we took advantage of swainsonine, a compound that specifically inhibits Golgi alpha‐mannosidase II (Lu et al, 2014). We found that exposing animals to swainsonine resulted in PVD defects that were indistinguishable from the effects of a null mutation in aman‐2, either in combination with a partial loss of function allele of lect‐2, or in wild‐type animals (Fig 2E–G). In other words, the pharmacological inhibition of AMAN‐2/Golgi alpha‐mannosidase II activity resulted in the same phenotypic consequences as genetically inactivating or removing the enzyme. Collectively, these findings lead us to conclude that the catalytic activity of AMAN‐2/Golgi alpha‐mannosidase II is essential to support dendrite patterning in PVD. This further implies that N‐glycosylation of a molecule expressed in PVD is crucial for normal dendrite arborization.
The presence of abnormal N‐glycans in aman‐2/Golgi alpha‐mannosidase II mutants results in defective PVD arborization
In eukaryotes, N‐glycosylation is initiated in the endoplasmic reticulum (ER) with the synthesis of a 14‐saccharide glycan on the phosphorylated polyisoprenol lipid dolichol‐P‐P (Stanley et al, 2015). Subsequently, the saccharide is transferred by a multiprotein complex termed oligosaccharyltransferase (OST) from dolichol‐P‐P to the aspartate within a N‐X‐S/T motif in nascent proteins as they are translocated into the ER (Stanley et al, 2015). As N‐glycosylated proteins transit the Golgi, the glycans undergo a series of enzymatic modifications that add and remove specific sugar residues, leading to a wide array of possible N‐glycan structures (Fig 3A; Stanley et al, 2015). For example, in one of the earlier steps, the enzyme MGAT1 adds a N‐acetylglucosamine residue to Man5Gn2 to form GnMan5Gn2 (Fig 3A). Genetically removing MGAT1 in mice results in the complete loss of complex and hybrid N‐glycans and early embryonic death, demonstrating that these N‐glycans are essential for mammalian development (Ioffe & Stanley, 1994; Metzler et al, 1994). GnMan5Gn2 is the substrate for AMAN‐2/MAN2A, which sequentially removes two mannose residues to form GnMan3Gn2 (Fig 3A) (Stanley et al, 2015). These reactions are followed by either removal or addition of additional sugars, or modification by a host of other conserved enzymes that lead to either complex or paucimannose N‐glycans (Fig 3A). To determine which specific N‐glycans are missing in aman‐2 mutants and are therefore required for normal branching of PVD dendrites, we systematically tested whether mutations in any of the genes downstream of aman‐2 (including hex‐2/hexosaminidase, hex‐3/hexosaminidase, fut‐8/FUT8 Fucosyltransferase, and gly‐20/MGAT II) would also enhance the lect‐2 partial loss of function allele. We found that removing the genes encoding these enzymes alone, or in combination, did not enhance the lect‐2 partial loss of function allele (Fig EV3A). Mutating MGAT1/N‐acetylglucosaminyltransferase‐I (in worms encoded by three paralogous genes gly‐12, gly‐13, gly‐14) (Chen et al, 1999, 2003), which acts immediately before AMAN‐2/Golgi alpha‐mannosidase II, also showed no effects (Fig EV3B). Collectively, these findings suggest that no lack of specific N‐glycans downstream of MGAT1, or of AMAN‐2, alone are responsible for the observed defects in PVD dendrites.
Figure 3. The presence of abnormal N‐glycans in mutants of AMAN‐2/Golgi alpha‐mannosidase II results in defects in PVD arborization.
- Schematic of the conserved N‐glycosylation pathway in C. elegans. The blue box represents the Endoplasmic Reticulum, while the purple box represents the medial Golgi. Glycan residues are consistent with Fig 2A, with the addition of red triangles denoting fucose residues. Arrows and orange text represent enzymes. The green area marks wild‐type N‐glycan chains whereas the red area represents abnormal N‐glycan chains that arise in the absence of AMAN‐2. The glycans in the green area are not formed in the absence of AMAN‐2. (Man = mannose, Gn = N‐acetylglucosamine, F = fucose, PC = phosphorylcholine, MGAT1 = N‐acetylglucosaminyltransferase I, FUT = fucosyltransferase, HEXD = hexosaminidase).
- Fluorescent composite images (top) and tracings (bottom) of mnr‐1(dz213) in an aman‐2(gk248486) and an aman‐2(gk248486); MGAT‐1(null) background. An MGAT null mutant lacks the three C. elegans paralogs: gly‐12, gly‐13, and gly‐14. PVD is visualized by the wyIs581 transgene. The cell body is denoted with an asterisk. Anterior is to the left and dorsal is up in all panels. Scale bars represent 20 μm.
- Quantification of full tertiaries of denoted genotypes. Data are represented as mean ± SEM. Statistical comparisons were performed using the Kruskal–Wallis test. Statistical significance is indicated (***P ≤ 0.001, ****P ≤ 0.0001, ns = not significant). n = 15 for all genotypes and are biological replicates.
- Quantification of the number of quaternary branches in wild‐type control, aman‐2(gk248486) null, and MGAT1(null) animals. Loss of MGAT1 suppresses the decrease in quaternary branch number in the aman‐2(null) background. Data are represented as mean ± SEM. Statistical significance was calculated using the Kruskal–Wallis test and is indicated (**P ≤ 0.01, ****P ≤ 0.0001). n = 15 for all genotypes and are biological replicates.
- Western blot against GFP in C. elegans lysate expressing no transgenes (N2) and expressing DMA‐1::GFP (qyIs369), after precipitating with anti‐GFP antibody. The red boxed plus sign indicates that the lysate is treated with the PNGase F glycosidase. The downward size shift reveals that N‐glycan structures are present on DMA‐1. Ladder is marked in kilodaltons (kDa). The GFP tag contains no N‐glycosylation sites. The experiment was repeated four times with biological replicates.
- Western blot against GFP in C. elegans lysate DMA‐1::GFP (qyIs369), after precipitating with anti‐GFP antibody. Control indicates an otherwise wild‐type background as opposed to an aman‐2(gk248486) null background. The upward size shift in the mutant reveals that the loss of aman‐2 alters the identity of N‐glycan structures on DMA‐1. The experiment was repeated four times with biological replicates.
- Western blot against GFP in C. elegans lysate DMA‐1::GFP (qyIs369), after precipitating with anti‐GFP antibody. Control indicates an otherwise wild‐type background as opposed to an aman‐2(gk248486) null background. The red boxed +F indicates that the lysate is treated with the PNGase F glycosidase, while the green boxed +H corresponds to the Endo H glycosidase, which cleaves high‐mannose and hybrid type N‐glycans. For complementary experiments using the Endo D glycosidase, which cleaves paucimannose type N‐glycans, see Fig EV4B. Size shifts indicate that some hybrid/high‐mannose structures are present on DMA‐1 (left), and that the aman‐2 mutant results in only hybrid/high‐mannose structures on DMA‐1 (right). The experiment was repeated four times with biological replicates.
Source data are available online for this figure.
Figure EV3. Quantification of branching in N‐glycosylation pathway mutants.
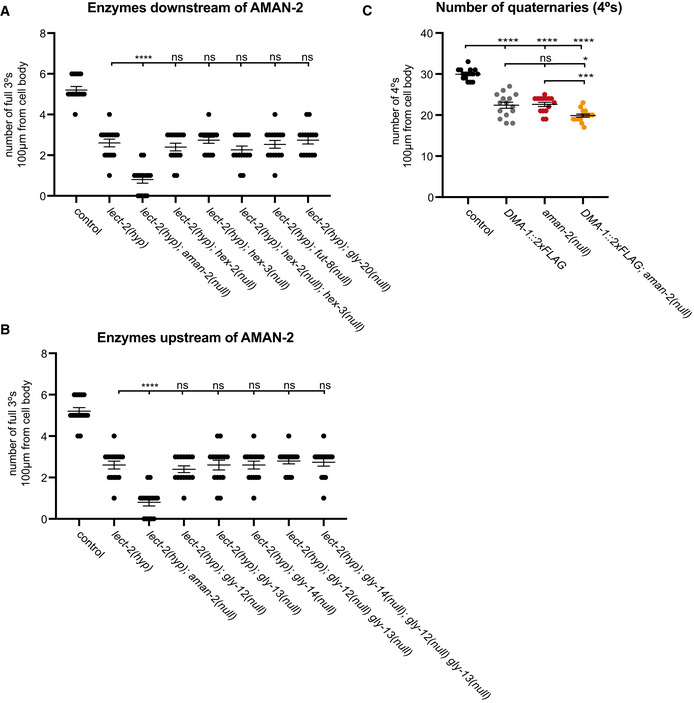
- Quantification of the number of full tertiaries in wild‐type controls, lect‐2(gk864764) hypomorphs, and lect‐2(gk864764) in combination with mutants of MGAT1 orthologs. See strain list for alleles. Data are represented as mean ± SEM. Statistical significance was calculated using the Kruskal–Wallis test, ****P ≤ 0.0001, ns: not significant. n = 15 for all genotypes and are biological replicates.
- Quantification of the number of full tertiaries in wild‐type controls, lect‐2(gk864764) hypomorphs, and lect‐2(gk864764) in combination with mutants of enzymes acting downstream of aman‐2. See strain list for alleles. Data are represented as mean ± SEM. Statistical significance was calculated using the Kruskal–Wallis test, ****P ≤ 0.0001, ns: not significant). n = 15 for all genotypes and are biological replicates.
- Quantification of the number of quaternary branches in wild‐type control animals, DMA‐1::2XFLAG (denoted as DMA‐1 wild type in Fig 4), and in combination with aman‐2(gk248486). In the DMA‐1::2XFLAG (wy1041) background, there is a baseline decrease in the number of branches, possibly due to the insertion of the tag (Dong et al, 2016). While equivalent to the phenotype of aman‐2(gk248486), combining the two backgrounds results in a further decrease in quaternary branches, providing an additional example of how hypomorphic alleles in the Menorin pathway can be enhanced by the loss of aman‐2. Data are represented as mean ± SEM. Statistical significance was calculated using the Kruskal–Wallis test, *P ≤ 0.05, ***P ≤ 0.001, ****P ≤ 0.0001. n = 15 for all genotypes and are biological replicates.
Source data are available online for this figure.
Previous structural studies of N‐glycans in aman‐2(tm1078) null mutant animals (Paschinger et al, 2006) established that the loss of aman‐2/Golgi alpha‐mannosidase II in C. elegans caused (i) a loss of the normal products of AMAN‐2, including complex N‐glycans (Fig 3A, shaded in green) and (ii) a buildup of GnMan5Gn2, the substrate of AMAN‐2 (Fig 3A; Paschinger et al, 2006). This GnMan5Gn2 intermediate was found to serve as substrate for other enzymes further downstream (including FUT‐8/Fut8 Fucosyltransferase and PCT/Phosphorylcholine‐transferase) leading to the appearance of abnormal N‐glycans, not normally present in wild‐type animals (Fig 3A, shaded in red) (Paschinger et al, 2006). To determine whether the defects in PVD branching were caused by the absence of wild‐type N‐glycans, or the presence of abnormal GnMan5Gn2 N‐glycans, we mutated both MGAT1 and AMAN‐2 in the partial loss of function backgrounds of lect‐2 and mnr‐1. The prediction was that if the enhancement of the partial loss of function alleles of lect‐2 or mnr‐1 by the loss of aman‐2 is caused by abnormal N‐glycans, then removal of MGAT1, the preceding enzyme would suppress that enhancement. Indeed, the loss of MGAT1 did suppress the enhanced phenotypes in lect‐2; aman‐2 and mnr‐1; aman‐2 double mutants (Fig 3B and C). Moreover, the decrease in quaternary PVD branches in AMAN‐2 mutants is also suppressed by the loss of MGAT1 (Fig 3D). These data indicate that one or more structurally abnormal N‐glycan(s) with a terminal N‐acetylglucosamine introduced by MGAT1 are responsible for the observed defects in PVD patterning.
DMA‐1/LRR‐TM N‐glycosylation is changed in aman‐2/Golgi alpha‐mannosidase II mutants
Since we demonstrated that (i) aman‐2/Golgi alpha‐mannosidase II genetically interacts with the Menorin pathway, and (ii) AMAN‐2/Golgi alpha‐mannosidase II activity is required cell‐autonomously in PVD to regulate branching, we hypothesized that AMAN‐2/Golgi alpha‐mannosidase II directly regulates N‐glycans on at least one component of the Menorin complex in PVD. Treatment of whole worm lysates with the bacterial PNGase F glycosidase, which cleaves all N‐glycans from Asn, resulted in distinct downward shifts in molecular weight of both DMA‐1 and KPC‐1, indicating that N‐glycans were present on both proteins in vivo and had been removed (Figs 3E and EV4C). Thus, both PVD‐expressed proteins, DMA/LRR‐TM and KPC‐1/Furin, are N‐glycosylated, consistent with a previous report for DMA‐1/LRR‐TM (Feng et al, 2020). Another cell‐autonomous factor and possible candidate, HPO‐30/Claudin (Smith et al, 2013), contains no predicted N‐glycan consensus motifs.
Figure EV4. Members of the Menorin pathway are N‐glycosylated.
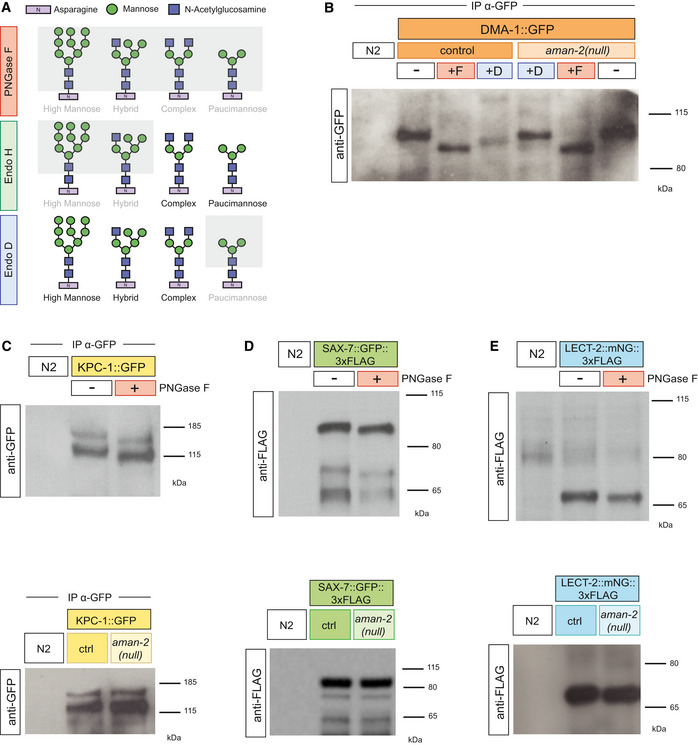
- Western blot against GFP in C. elegans lysate DMA‐1::GFP (qyIs369), after precipitating with anti‐GFP antibody. Control indicates an otherwise wild‐type background as opposed to an aman‐2(gk248486) background. The red boxed +F indicates that the lysate is treated with the PNGase F glycosidase, while the green boxed +D corresponds to the Endo D glycosidase, which cleaves paucimannose type N‐glycans. Size shifts indicate that some paucimannose structures are present on DMA‐1 (left), and that the aman‐2 mutant results in the loss of paucimannose structures on DMA‐1 (right). Ladder is marked in kilodaltons (kDa). The experiment was repeated four times with biological replicates.
- Western blot against GFP in C. elegans lysate expressing no transgenes (N2) and expressing KPC‐1::GFP (dzEx1865), after precipitating with anti‐GFP antibody. The molecular weights of wild‐type lysate are 130 and 150 kDa. The red boxed plus sign indicates that the lysate is treated with the PNGase F glycosidase. The downward size shift reveals that N‐glycan structures are present on KPC‐1. In the bottom blot, control indicates an otherwise wild‐type background as opposed to an aman‐2(gk248486) background. No visible size shift is observed, and the experiment was repeated three times with unique samples.
- Western blot against FLAG in C. elegans lysate expressing no transgenes (N2) and expressing SAX‐7::GFP::3XFLAG (ddIs290). Robust expression precludes the need for immunoprecipitation. The molecular weights of wild‐type lysate are 100, 70, and 65 kDa. The red boxed plus sign indicates that the lysate is treated with the PNGase F glycosidase. The downward size shift reveals that N‐glycan structures are present on SAX‐7. In the bottom blot, control indicates an otherwise wild‐type background as opposed to an aman‐2(gk248486) background. No visible size shift is observed, and the experiment was repeated three times with unique samples. The FLAG epitope contains no N‐glycosylation sites.
- Western blot against FLAG in C. elegans lysate expressing no transgenes (N2) and expressing endogenous LECT‐2::mNeonGreen::3XFLAG (dz249). Robust expression precludes the need for immunoprecipitation. The molecular weight of wild‐type lysate is 70 kDa. The red boxed plus sign indicates that the lysate is treated with the PNGase F glycosidase. The small downward size shift reveals that N‐glycan structures are present on LECT‐2. In the bottom blot, control indicates an otherwise wild‐type background as opposed to an aman‐2(gk248486) background. No visible size shift is observed, and the experiment was repeated three times with unique samples.
We next determined whether the loss of AMAN‐2 resulted in altered N‐glycosylation of DMA‐1/LRR‐TM. Interestingly, the absence of aman‐2/Golgi alpha‐mannosidase II resulted in a clear increase in DMA/LRR‐TM molecular weight, whereas the size of KPC‐1/Furin remained unaffected (Figs 3F and EV4C). The upward shift in the size of DMA‐1/LRR‐TM following the loss of aman‐2 is consistent with our genetic data establishing that the presence of abnormal GnMan5Gn2 N‐glycans gives rise to the PVD mutant phenotype. To determine what types of N‐glycans are attached to DMA‐1 in wild‐type and aman‐2 mutant backgrounds, we treated lysates with additional endoglycosidases: Endo H, which cleaves hybrid/high‐mannose N‐glycans, and Endo D, which cleaves only paucimannose N‐glycans. Based on the size of the shifts observed, we conclude that in wild‐type animals, DMA‐1/LRR‐TM possesses primarily hybrid/high‐mannose N‐glycans with a smaller amount of paucimannose N‐glycans. In contrast, in the absence of AMAN‐2, all N‐glycans on DMA‐1/LRR‐TM were converted to hybrid/high‐mannose type (likely GnMan5Gn2‐derived N‐glycans) with no or little detectable paucimannose N‐glycans (Figs 3G and EV4A and B). Additionally, proteomic studies identified C. elegans SAX‐7/L1CAM and LECT‐2/Chondromodulin II as glycoproteins (Kaji et al, 2007), which we confirmed by a shift in molecular weight upon digestion of all N‐glycans by PNGase F (Fig EV4D and E). However, in the absence of aman‐2/Golgi alpha‐mannosidase II, neither SAX‐7/L1CAM and LECT‐2/Chondromodulin II displayed obvious changes in molecular weight, suggesting that they may not carry abnormal N‐glycans in an aman‐2 mutant background. Collectively, our data show that among the N‐glycosylated proteins of the Menorin complex, only DMA‐1/LRR‐TM was significantly affected by the loss of aman‐2/Golgi alpha‐mannosidase II and carried altered N‐glycans in aman‐2 null animals.
AMAN‐2/Golgi alpha‐mannosidase II modulates N‐glycans on DMA‐1/LRR‐TM to regulate PVD morphogenesis
The DMA‐1/LRR‐TM receptor contains four predicted N‐glycosylation motifs, all of which reside in leucine rich repeats (Fig 4A). To establish whether the N‐glycosylation of DMA‐1/LRR‐TM is essential for its role in PVD dendrite branching, we mutated all four sites, alone and in combinations. Using CRISPR/Cas9‐based genome editing, we converted the asparagine residues of the four predicted N‐glycan attachment sites to glutamine to maintain chemical similarity, but eliminate the possibility of N‐glycosylation. We cannot formally exclude that these mutations affect stability, transport or folding of the mutant DMA‐1/LRR proteins, but consider this less likely, because these point mutants displayed different behaviors in different genetic backgrounds (see below). We found that only when abolishing predicted N‐glycan attachment site 4 (N386), alone or in combination with other sites, was PVD quaternary branching compromised (Fig 4B). These results reveal that N‐glycosylation of DMA‐1/LRR‐TM is required for its role in PVD patterning of quaternary branches and highlight the importance of N386 within a membrane proximal LRR repeat. We then assessed whether abolishing N‐glycan attachment sites in an aman‐2/Golgi alpha‐mannosidase II loss of function background had any effects on dendrite patterning. Analysis of different site‐specific mutations revealed that some N‐glycosylation sites on DMA‐1/LLR‐TM enhance the severity of PVD dendrite branching defects in an aman‐2 mutant background (Fig 4C–E). The mutant phenotype resulting from a presumptive loss of all N‐glycans on DMA‐1 (S1234) is enhanced in an aman‐2 mutant background, suggesting that N‐glycosylated proteins other than DMA‐1/LRR‐TM may serve additional functions during PVD morphogenesis, or that abnormal N‐glycans on cryptic N‐glycosylation sites further compromise function (Fig 4C). The results in an aman‐2 mutant background also suggest that having some type of N‐glycan on site 4 of DMA‐1/LRR‐TM, even if abnormal, is better than having no N‐glycan at all (cf. S4 and S123); second having such abnormal N‐glycans on sites 1‐3 further compromises DMA‐1 function during PVD development (cf. S4 in control vs aman‐2 mutant background, Fig 4B–E). In line with the latter, we show that the loss of MGAT1 and AMAN‐2 when site 4 of DMA‐1 is mutant (S4), results in a suppression of the mutant quaternary phenotype (Fig 4C). This suggests that the presence of the abnormal N‐glycans on sites 1‐3, rather than 4, may be the major driver for the developmental defects in PVD dendrites. Of note, glycoproteomic studies in mammalian brains showed that different attachment sites in a given protein are not equal and can bear distinct N‐glycans (Riley et al, 2019). While prior studies established the importance of N‐glycosylation during neuronal development, our studies establish an important role for specific classes and structures of N‐glycans in mediating neuronal development, and specifically dendrite patterning. They suggest that the DMA‐1/LRR‐TM receptor in PVD must be decorated with specific hybrid/high‐mannose or paucimannose N‐glycans as a result of AMAN‐2 activity and that these specific N‐glycans are important for Menorin complex function in PVD dendrite morphogenesis. A possible explanation is that the N‐glycans on specific N‐glycosylation sites in DMA‐1/LRR‐TM function permissively to maintain high affinity binding of DMA‐1/LRR‐TM to other members of the Menorin complex through specific N‐glycans (Fig 4F). This interaction could be compromised by the formation of abnormal glycans in the absence of AMAN‐2, leading to destabilization of the complex. Alternatively, the affinity of the proteins in the complex could be increased by different N‐glycans, or modified in their function by other means. Regardless, our genetic and biochemical data, together with analytical data of N‐glycans in aman‐2/Golgi alpha‐mannosidase II mutants (Paschinger et al, 2006), underscore the importance of AMAN‐2/Golgi alpha‐mannosidase II as a linchpin for the creation of complex and paucimannose N‐glycans, and avoidance of abnormal hybrid and high‐mannose‐type N‐glycans. Since metabolite availability can influence N‐glycan synthesis and flux (reviewed in Dennis et al, 2009), these findings also raise the possibility that environmental factors can intersect with intrinsic genetic programs to regulate the function of extracellular adhesion complexes during neural development by modulating N‐glycosylation. While AMAN‐2 is most highly expressed in PVD neurons (CeNGEN; Taylor et al, 2021), it may be of interest to determine systematically whether it plays a role in the development of other neurons in which it is expressed.
Figure 4. AMAN‐2/Golgi alpha‐mannosidase II modulates N‐glycans on DMA‐1/LRR‐TM to regulate PVD morphogenesis.
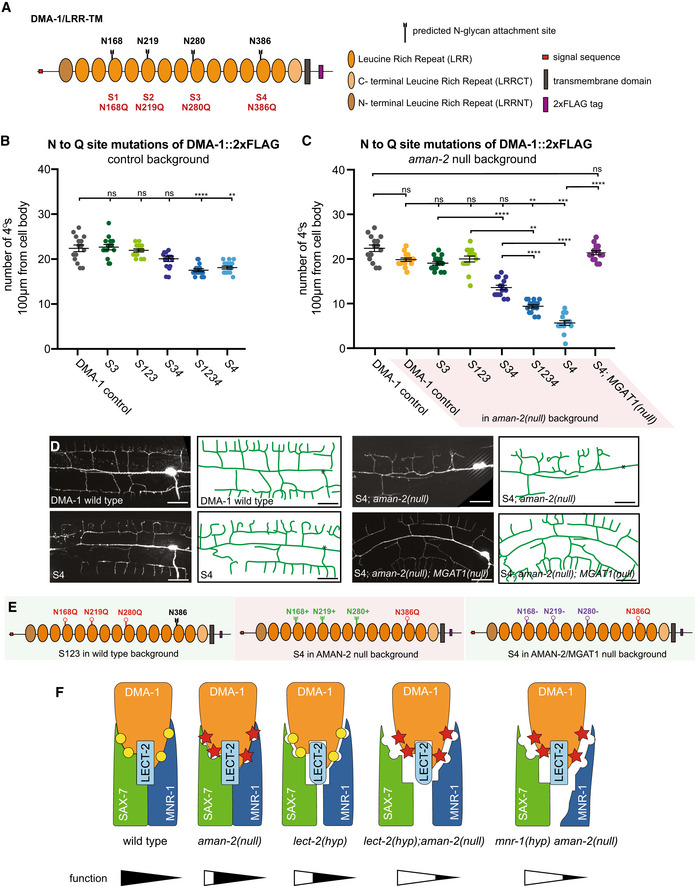
- Schematic of DMA‐1/LRR‐TM with mutated N‐glycan attachment sites S1‐S4 indicated.
- Quantification of the number of quaternary branches in DMA‐1::2xFLAG control animals and animals with combinations of DMA‐1::2xFLAG N‐glycan attachment sites mutated. Note that DMA‐1::2xFLAG control animals display a slightly reduced number of quaternary dendrites compared to wild‐type animals (Fig EV3C). Data are represented as mean ± SEM. Statistical significance was calculated using the Kruskal–Wallis test, and is indicated (**P ≤ 0.01, ****P ≤ 0.0001, ns = not significant). n = 15 for all genotypes and are biological replicates.
- Quantification of the number of quaternary branches in DMA‐1::2xFLAG control animals alone and in combination with an aman‐2(gk248486) null mutant (shaded in red). Control data is identical as in (B) and shown for comparison only. Data are represented as mean ± SEM. Statistical significance was calculated using the Kruskal–Wallis test and Mann–Whitney between individual comparisons, and is indicated (**P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001, ns = not significant). n = 15 for all genotypes and are biological replicates.
- Fluorescent images (left) and tracings (center) of PVD in animals of denoted backgrounds (aman‐2(null) is gk248486). S4 corresponds to the endogenous alteration of N‐glycan attachment site N386 of DMA‐1. PVD is visualized by the dzIs117 transgene. The cell body is marked with an asterisk. Scale bars represent 20 μm.
- The schematics on the right detail the molecular context of the indicated backgrounds. Green indicates a relatively normal PVD structure and red a heavily defective arborization. Black attachments indicate wild‐type, red indicate blocked sites, green indicates abnormal N‐glycans chains, and purple indicates presumably incomplete but present N‐glycan chains.
- Proposed model of how the function of the Menorin complex could be modulated by complex stability. Note that other mechanisms are possible (see discussion). Yellow circles represent normal N‐glycan chains, while red stars represent abnormal N‐glycans in aman‐2 mutant animals. Putative level of functionality is indicated below.
Source data are available online for this figure.
Previous studies demonstrated that N‐glycosylation is important for folding and surface localization of cell adhesion molecules and axon guidance factors in vertebrates and invertebrates, such as L1CAMs and ephrins, respectively (Sekine et al, 2013; Medina‐Cano et al, 2018; Mire et al, 2018). On the other hand, structural studies and in vitro experiments suggested that N‐glycans can regulate protein–protein interactions of cell adhesion molecules (Fogel et al, 2010; Labasque et al, 2014; Li et al, 2016). Our findings demonstrate in vivo, that not only N‐glycans per se, but that specific classes of N‐glycan structures are important to modulate cell‐cell signaling, and possibly, receptor‐ligand binding, complex formation, or function. This is reminiscent of the role of O‐fucose glycans on the Notch receptor extracellular domain, which affect its signaling and ligand interactions (Moloney et al, 2000). Given that over 70% of proteins transiting the secretory pathway are N‐glycosylated (Apweiler et al, 1999), these findings raise the possibility that specific N‐glycan structures are important determinants to regulate the interactions of extracellular complexes during nervous system development more broadly, and could contribute to the specificity that is required in the nervous system of both vertebrates and invertebrates. In this context, it is interesting to note that over 70 congenital disorders of glycosylation affecting genes in the N‐glycosylation pathway have been described (Freeze, 2006; Ng & Freeze, 2018), of which many are associated with intellectual disability or other neurological symptoms (Jaeken & Peanne, 2017; Chang et al, 2018). While no mutations in Golgi alpha‐mannosidase II in humans have been described to date, it is conceivable that such mutations exist, and may result in neurological phenotypes. Our studies provide the conceptual framework for the investigation of developmental defects of the nervous system in mutants of genes involved in N‐glycosylation and this growing class of congenital disorders.
Materials and Methods
Reagents and Tools table
| Reagent/Resource | Source | Identifier/Catalog # |
|---|---|---|
| Bacterial and Virus strains | ||
| Escherichia coli: OP50 | Caenorhabditis Genetics Center | OP50 |
| Experimental Models: Organisms/Strains | ||
| C. elegans: wild‐type isolate | Caenorhabditis Genetics Center | N2 |
| C. elegans: wdIs52 II | Kind gift of David Miller | NC1687 |
| C. elegans: wyIs378 X | Kind gift of Kang Shen | wyIs378 |
| C. elegans: wyIs581 IV | Kind gift of Kang Shen | wyIs581 |
| C. elegans: dzIs117 II | This paper | dzIs117 |
| C. elegans: lect‐2(dz249[lect‐2::mNeonGreen::3XFLAG]) II | Diaz‐Balzac et al (2016) | EB2724 |
| C. elegans: lect‐2(dz249[lect‐2::mNeonGreen::3XFLAG]) II; him‐5(ok1896) V | Diaz‐Balzac et al (2016) | EB3894 |
| C. elegans: dzIs53 ddIs290 (SAX‐7::GFP::3XFLAG) II; him‐5(ok1896) V | Diaz‐Balzac et al (2016) | EB2808 |
| C. elegans: lect‐2(dz249[lect‐2::mNeonGreen::3XFLAG]) II; aman‐2(dz261) V | This paper | EB3185 |
| C. elegans: dzIs53 ddIs290 (SAX‐7::GFP::3XFLAG) II; aman‐2(dz261) V | This paper | EB3182 |
| C. elegans: qyIs369 X | Kind gift of Kang Shen | qyIs369 |
| C. elegans: wyIs581 IV; qyIs369 X | This paper | EB3534 |
| C. elegans: wyIs581 IV; qyIs369 X; aman‐2(gk248486) V | This paper | EB3634 |
| C. elegans: dzIs53 II; him‐5(ok1896) V | This paper | EB2824 |
| C. elegans: lect‐2(gk864764) II; wyIs378 X | This paper | EB2795 |
| C. elegans: wdIs52 lect‐2(gk864764) II | This paper | EB2805 |
| C. elegans: kpc‐1(dz254) I; wdIs52 lect‐2(gk864764) II | This paper | EB2908 |
| C. elegans: wdIs52 lect‐2(gk864764) II; mnr‐1(dz252) V | This paper | EB2906 |
| C. elegans: wdIs52 lect‐2(gk864764) II; sax‐7(dz253) IV | This paper | EB2907 |
| C. elegans: wdIs52 lect‐2(gk864764) II; aman‐2(dz261) V | This paper | EB2915 |
| C. elegans: wyIs581 IV; him‐5(ok1896) V | This paper | EB2926 |
| C. elegans: kpc‐1(dz254) I; dzIs53 II | This paper | EB2983 |
| C. elegans: kpc‐1(dz254) I; dzIs53 II; him‐5(ok1896) V | This paper | EB3282 |
| C. elegans: kpc‐1(dz254) I; dzIs53 II; aman‐2(gk248486) V | This paper | EB3299 |
| C. elegans: lect‐2(gk864764) II; wyIs581 IV; him‐5(ok1896) V | This paper | EB3061 |
| C. elegans: dzIs53 II; mnr‐1(dz213) V | This paper | EB3281 |
| C. elegans: wyIs581 IV; mnr‐1(dz213) aman‐2(gk248486) V | This paper | EB3403 |
| C. elegans: aman‐2(gk248486) V | Caenorhabditis Genetics Center | VC20294 |
| C. elegans: aman‐2(gk248477) V | Caenorhabditis Genetics Center | VC20422 |
| C. elegans: aman‐2(gk619253) V | Caenorhabditis Genetics Center | VC40398 |
| C. elegans: aman‐2(tm1078) V | Kind gift of the Shohei Mitani | tm1078 |
| C. elegans: lect‐2(gk864764) II; wyIs581 IV | This paper | EB3228 |
| C. elegans: lect‐2(gk864764) II; wyIs581 IV; aman‐2(gk248486) V | This paper | EB3260 |
| C. elegans: wyIs581 IV; aman‐2(gk248486) V | This paper | EB3261 |
| C. elegans: lect‐2(gk864764) II; wyIs581 IV; aman‐2(gk248477) V | This paper | EB3293 |
| C. elegans: lect‐2(gk864764) II; wyIs581 IV; aman‐2(gk619253) V | This paper | EB3294 |
| C. elegans: lect‐2(gk864764) II; wyIs581 IV; aman‐2(tm1078) V | This paper | EB4422 |
| C. elegans: wyIs581 IV; aman‐2(gk248477) V | This paper | EB3405 |
| C. elegans: wyIs581 IV; aman‐2(gk619253) V | This paper | EB3406 |
| C. elegans: wyIs581 IV; aman‐2(tm1078) V | This paper | EB4183 |
| C. elegans: dzIs117 II; him‐5(ok1896) V | This paper | EB3353 |
| C. elegans: pyc‐1(gk689405) V | Caenorhabditis Genetics Center | VC40551 |
| C. elegans: lect‐2(gk864764) II; wyIs581 IV; pyc‐1(gk689405) V | This paper | EB3298 |
| C. elegans: gly‐12(id47) X | Caenorhabditis Genetics Center | AS270 |
| C. elegans: dpy‐6(e14) gly‐13(ok712) X | Caenorhabditis Genetics Center | AS338 |
| C. elegans: gly‐14(id48) III | Caenorhabditis Genetics Center | AS271 |
| C. elegans: gly‐14(id48) III; gly‐12(id47) gly‐13(ok712) X | Kind gift of Iain Wilson | AS341 |
| C. elegans: gly‐20(ok826) V | Caenorhabditis Genetics Center | RB943 |
| C. elegans: fut‐8(ok2558) V | Caenorhabditis Genetics Center | RB1945 |
| C. elegans: hex‐2(ok2985) V | Caenorhabditis Genetics Center | RB2205 |
| C. elegans: hex‐3(gk944651) III | Caenorhabditis Genetics Center | VC40929 |
| C. elegans: lect‐2(gk864764) II; wyIs581 IV; gly‐12(id47) X | This paper | EB3407 |
| C. elegans: lect‐2(gk864764) II; wyIs581 IV; dpy‐6(e14) gly‐13(ok712) X | This paper | EB3408 |
| C. elegans: wyIs581 IV; gly‐20(ok826) V | This paper | EB3410 |
| C. elegans: wyIs581 IV; fut‐8(ok2558) V | This paper | EB3411 |
| C. elegans: lect‐2(gk864764) II; otIs173 III; wyIs581 IV; hex‐2(ok2985) V | This paper | EB3653 |
| C. elegans: lect‐2(gk864764) II; hex‐3(gk944651) III; wyIs581 IV; him‐5(ok1896) V | This paper | EB3905 |
| C. elegans: lect‐2(gk864764) II; hex‐3(gk944651) III; wyIs581 IV; hex‐2(ok2985) V | This paper | EB3906 |
| C. elegans: ddIs290 lect‐2(gk864764) II; gly‐14(id48) III; wyIs581 IV | This paper | EB3638 |
| C. elegans: ddIs290 lect‐2(gk864764) II; wyIs581 IV; gly‐12(id47) gly‐13(ok712) X | This paper | EB3637 |
| C. elegans: ddIs290 lect‐2(gk864764) II; gly‐14(id48) III; wyIs581 IV; gly‐12(id47) gly‐13(ok712) X | This paper | EB3636 |
| C. elegans: dzIs53 II; gly‐14(id48) III; wyIs581 IV; mnr‐1(dz213) V; gly‐12(id47) gly‐13(ok712) X | This paper | EB4376 |
| C. elegans: dzIs53 II; gly‐14(id48) III; wyIs581 IV; mnr‐1(dz213) aman‐2(gk248486) V; gly‐12(id47) gly‐13(ok712) X | This paper | EB4377 |
| C. elegans: dzIs117 II; gly‐14(id48) III; wyIs581 IV; aman‐2(gk248486) V; gly‐12(id47) gly‐13(ok712) X | This paper | EB4420 |
| C. elegans: dzEx1865(Pser‐2prom3::kpc‐1::GFP); dzIs53 I | This paper | EB3533 |
| C. elegans: dzEx1865(Pser‐2prom3::kpc‐1::GFP); dzIs53 II; aman‐2(gk248486) V | This paper | EB3667 |
| C. elegans: dma‐1(wy1041[dma‐1::2XFLAG]) I; dzIs117 II | This paper | EB4239 |
| C. elegans: dma‐1(wy1041[dma‐1::2XFLAG]) I; dzIs117 II; him‐5(ok1896) V | This paper | EB4378 |
| C. elegans: dma‐1(dz294[dma‐1(N280Q)::2xFLAG]) I; dzIs117 II; him‐5(ok1896) V | This paper | EB4219 |
| C. elegans: dma‐1(dz294[dma‐1(N280Q)::2xFLAG]) I; dzIs117 II; aman‐2(gk248486) V | This paper | EB4220 |
| C. elegans: dma‐1(dz295[dma‐1(N168Q, N219Q, N280Q)::2xFLAG]) I; dzIs117 II; him‐5(ok1896) V | This paper | EB4223 |
| C. elegans: dma‐1(dz295[dma‐1(N168Q, N219Q, N280Q)::2xFLAG]) I; dzIs117 II; aman‐2(gk248486) V | This paper | EB4224 |
| C. elegans: dma‐1(dz296[dma‐1(N280Q; N386Q)::2xFLAG]) I; dzIs117 II; him‐5(ok1896) V | This paper | EB4227 |
| C. elegans: dma‐1(dz296[dma‐1(N280Q; N386Q)::2xFLAG]) I; dzIs117 II; aman‐2(gk248486) V | This paper | EB4228 |
| C. elegans: dma‐1(dz297[dma‐1(N168Q, N219Q, N280Q, N386Q)::2xFLAG]) I; dzIs117 II; him‐5(ok1896) V | This paper | EB4231 |
| C. elegans: dma‐1(dz297[dma‐1(N168Q, N219Q, N280Q, N386Q)::2xFLAG]) I; dzIs117 II; aman‐2(gk248486) V | This paper | EB4232 |
| C. elegans: dma‐1(dz298[dma‐1(N386Q)::2xFLAG]) I; dzIs117 II; him‐5(ok1896) V | This paper | EB4235 |
| C. elegans: dma‐1(dz298[dma‐1(N386Q)::2xFLAG]) I; dzIs117 II; aman‐2(gk248486) V | This paper | EB4236 |
| C. elegans: dma‐1(dz298[dma‐1(N386Q)::2xFLAG]) I; dzIs117 II; gly‐14(id48) III; aman‐2(gk248486) V; gly‐12(id47) gly‐13(ok712) X | This paper | EB4421 |
| Oligonucleotides | ||
| Guide RNA for S1: N126 [TAATACGACTCACTATAGGATAGAGACAATGATCGCAGTTTTAGAGCTAGAAATAGCAAG] | This paper | oMR103 |
| Guide RNA for S2: N219 [TAATACGACTCACTATAgTTCCAAGTTGACGGAGTTGGTTTTAGAGCTAGAAATAGCAAG] | This paper | oMR122 |
| Guide RNA for S3: N280 [TAATACGACTCACTATAgTTCGTATTGCAAAATGCACGTTTTAGAGCTAGAAATAGCAAG] | This paper | oMR123 |
| guide RNA for S4: N386 [TAATACGACTCACTATAgAATGTTCAATGAAATCAGGGTTTTAGAGCTAGAAATAGCAAG] | This paper | oMR124 |
| Repair for S1: N126Q [CGAAGTAAATTTGTAGAAAGATCACTGATAGTATTCTGAGAGAGGGACAGGGAGCGGAGAGCTCTAAGATATGTGAATACTCCAGTTGGAAGAAT] | This paper | oMR130 |
| Repair for S2: N219Q [GTTTCTCAATTGGATGAATTATATTTGAATCATTGCCAGCTCTCCTCCATCTACTCCCTCGCACTCGACCGCATCCCTCAGCTGCGCCAGCTGGGAATCGGAGGAAATAATCTCAAAATGGTTCCAAC] | This paper | oMR131 |
| Repair for S3: N280Q [CCGAGCAAATTGTGGGAAAGATCTAATTTTGAGATCTGCGTATTGCAAAATGCACAAGCTGTGATTTCTTGAATGGAATTGTGAGACAAGTC] | This paper | oMR127 |
| Repair for S4: N386Q [TTGTCAGGCAAATAAGTCAATTCATTTCCAGAAATCTGCAGTGAGATGAGGTGATAATAGCGAGACGGCAGCTGTACCGGGATGAATTT] | This paper | oMR132 |
| Genotype S1 forward [GAGTTCTTCGTCTCATCAATTG] | This paper | oMR107 |
| Genotype S1 reverse [GAATAGGATTCCGGTCTAGTCG] | This paper | oMR108 |
| Genotype S2, S3 forward [CTGCGATCATTGTCTCTATCC] | This paper | oMR109 |
| Genotype S2 reverse [CCATCCCAACCATTTTGAGTC] | This paper | oMR129 |
| Genotype S3 reverse [GGTCCATGACTCCTCGAAA] | This paper | oMR111 |
| Genotype S4 forward [GCACATGAACAATCCCAAGG] | This paper | oMR112 |
| To clone Pser2prom3::aman‐2, Pdpy‐7::aman‐2, Pmyo‐3::aman‐2 forward [TTCAGGAGGACCCTTGGAGGGTACcATGGGAAAACGCAATTTCTATATTATCCTA] | This paper | oMR34 |
| To clone Pser2prom3::aman‐2, Pdpy‐7::aman‐2, Pmyo‐3::aman‐2 reverse [GATATTTCCAGTATTCTTGTATCATTTTAAtCCGGAACCCGCCTTTGTCTGATCT] | This paper | oMR35 |
| To clone Pser2prom3::aman‐2 catalytic dead site 1 forward [CATTGGTCAATTGCCCCATTCGGTTTATC] | This paper | oMR64 |
| To clone Pser2prom3::aman‐2 catalytic dead site 1 reverse [GATAAACCGAATGGGGCAATTGACCAATG] | This paper | oMR65 |
| To clone Pser2prom3::aman‐2 catalytic dead site 2 forward [CCACTTGGAGATGCCTTCAGGTACGAC] | This paper | oMR66 |
| To clone Pser2prom3::aman‐2 catalytic dead site 2 reverse [GTCGTACCTGAAGGCATCTCCAAGTGG] | This paper | oMR67 |
| Recombinant DNA | ||
| Plasmid: Pser2prom3::aman‐2 | The aman‐2 cDNA was PCR amplified with KpnI/BspeI cloning sites attached and cloned into Pser2prom3::lect‐2. | N/A |
| Plasmid: Pdpy‐7::aman‐2 | The aman‐2 cDNA from Pser2prom3::aman‐2 was cloned into Pdpy‐7::lect‐2 with KpnI/BspeI. | N/A |
| Plasmid: Pmyo‐3::aman‐2 | The aman‐2cDNA from Pser2prom3::aman‐2 was cloned into Pmyo‐3::lect‐2 with KpnI/ BspeI. | N/A |
| Plasmid: Pser2prom3::aman‐2 catalytic dead | The Pser2prom3::aman‐2 was PCR amplified with the NEB Q5 site directed mutagenesis kit. | N/A |
| Endoglycosidases | ||
| PNGase F (#P0704S) | NEB https://www.neb.com/products/p0704‐pngase‐f#Product%20Information | P0704S |
| Endo H (#P0702L) |
NEB https://www.neb.com/products/p0702‐endo‐h#Product%20Information |
P0702L |
| Endo D (#P0742S) |
NEB https://www.neb.com/products/p0742‐remove‐it‐endo‐d#Product%20Information |
P0742S |
| Antibodies | ||
| Monoclonal primary Anti‐GFP in mouse (Roche 11814460001) |
Roche https://www.sigmaaldrich.com/US/en/product/roche/11814460001 |
11814460001 |
| Monoclonal primary Anti‐FLAG in mouse (Sigma F1804) |
Sigma |
F1804 |
| Polyclonal secondary Anti‐mouse HRP (Millipore AP308P) |
Millipore |
AP308P |
| Western Blot reagents | ||
| 4–12% Bis‐Tris Gradient Gels |
GenScript https://www.genscript.com/molecule/M00653‐SurePAGE_Bis_Tris_10x8_4_12_12_wells.html |
M00653 |
| Protein A/G agarose beads (sc‐2003) |
Santa Cruz |
Sc‐2003 |
| Software and Algorithms | ||
| Fiji | https://fiji.sc/ | N/A |
| GraphPad PRISM 8 | https://www.graphpad.com/scientific‐software/prism/ | N/A |
Methods and Protocols
Caenorhabditis elegans handling
All strains were maintained using standard methods (Brenner, 1974) and experiments were performed at 20°C, except where indicated otherwise. Phenotypic analysis was performed in 1‐day‐old adults, with no more than 4–5 eggs present. Brood size was determined by single picking late L4 adults and allowing them to lay eggs on seeded plates for 48 h. Number of L1 larvae were subsequently counted per plate. For details and a complete list of strains used and generated in this study, see resources table.
Details of genetic screen and mutant alleles
The lect‐2(gk864764) hypomorphic strain was treated with EMS (Kutscher & Shaham, 2014) and the progeny of clonal F1 animals scored for enhancement, suppression, or modification of the PVD branching phenotype. This screen resulted in the isolation of four mutant alleles (Fig EV1A). Guided by the phenotypes of dz252, dz253, and dz254, we used complementation tests and Sanger sequencing to identify the molecular lesions. For dz261, we used a combination of whole genome sequencing and single nucleotide polymorphism mapping (Minevich et al, 2012) to narrow down the region to a 5 Mb interval (11MB‐16Mb) on chromosome V (Fig EV1B). This region contained 7 polymorphisms with predicted functional consequences. We injected seven fosmids covering those genes in pools and found that only the pool which contained a fosmid covering aman‐2 resulted in rescue (Fig EV1B). In addition, we obtained three nonsense alleles of aman‐2 (gk248486, gk248477, gk619253) from the Million Mutation Project (Thompson et al, 2013) and one deletion allele (tm1078, kind gift from the Mitani lab). The dz261 mutation was further confirmed by Sanger sequencing of the original isolate, identifying a W237Opal nonsense mutation in the aman‐2 locus.
Molecular biology and transgenesis
To assemble tissue specific expression constructs used for heterologous rescue experiments, the aman‐2 cDNA clone yk11g705 (kind gift of Yoji Kohara) was cloned under control of the following promoters: PVD ser2prom‐3 (Tsalik et al, 2003), hypodermal dpy‐7p (Gilleard et al, 1997), body wall muscle myo‐3p (Okkema et al, 1993). These constructs were injected into N2 animals at 5 ng/µl, together with myo‐2p::mCherry as an injection marker at 50 ng/µl and BlueScript as DNA filler up to a final DNA concentration of 100 ng/µl. Males from transgenic lines were then crossed into lect‐2(gk864764) II; wyIs581 IV; aman‐2(gk248477 )V and wyIs581 IV; mnr‐1(dz213) aman‐2(gk248486) V. Point mutants in the aman‐2 cDNA were introduced by site‐specific mutagenesis (NEB Q5 Site‐Directed Mutagenesis). All plasmids contained the unc‐54 3’UTR.
Pharmacology
Experiments in which the activity of AMAN‐2 was blocked pharmacologically were performed with the compound swainsonine (1 mg swainsonine #16860 vials, Fisher Scientific catalog #NC1670046). Dosage experiments were performed from 50–500 µM of swainsonine dissolved in agar of NGM plates, with 300 µM being sufficient to elicit phenotypes in PVD. After drying agar plates for 24 h, 200 µl of OP50 E. coli was seeded onto each plate, and 5 young adult worms were left to self‐fertilize and lay eggs. The F1 generations were analyzed, imaged, and quantified. DMSO was used as a control in plates not treated with swainsonine.
Western blot analysis and immunoprecipitation
Whole C. elegans lysates were generally prepared in RIPA buffer and sonicated in a Biorupter benchtop waterbath sonicator for 15 min. If applicable, lysates were treated with 1 unit of endoglycosidases PNGase F, Endo H, or Endo D, respectively, at temperatures as indicated in NEB protocols (linked in the Reagents and Tools table). Overnight immunoprecipitation of lysates with anti‐GFP antibody prior to SDS–PAGE was performed for proteins with low expression levels (DMA‐1::GFP and KPC‐1::GFP) as follows. Five full plates of DMA‐1::GFP and KPC‐1::GFP tagged worms were washed in RIPA buffer pH7.0 and lysed for 15 min in a Biorupter water bath as previously described for whole worm protein extraction (Li & Zinovyeva, 2020). Post lysis, 20 uL of Protein A/G Plus Agarose beads (Santa Cruz sc‐2003) and 1 μl of anti‐GFP antibody (Roche 11814460001) were used to pull down DMA‐1::GFP and KPC‐1::GFP overnight at 4°C. For SAX‐7::GFP::FLAG and LECT‐2::mNG::FLAG, ten and twenty gravid adult animals, respectively, were sufficient to see robust expression on Western Blot without the necessity for precipitation. These samples were boiled and loaded directly into the gels. Gradient gels (4–12% GenScript) were used in all experiments. For all anti‐FLAG blots, a concentration of 1:800 anti‐Flag (Sigma F1804) and 1:5,000 anti‐mouse HRP (Millipore AP308P) were used. For all anti‐GFP blots, a concentration of 1:500 anti‐GFP (Roche 11814460001) and 1:5,000 anti‐mouse HRP (Millipore AP308P) was used.
CRISPR/Cas9 mediated gene editing
CRISPR‐Cas9 constructs were designed and the dpy‐10 co‐crispr protocol was followed as previously described (Dickinson & Goldstein, 2016) to introduce single point mutations of predicted N‐glycan attachment Asparagine residues. Predicted sites were identified using NetNGlyc 1.0 Server (Gupta & Brunak, 2002). A battery of guideRNAs were designed to direct Cas9 cuts near sites of interest, and homologous repair template oligomers were designed to mutate Asparagine residues to Glutamine. Strains with combinations of mutated sites were generated by sequential injections and/or multiple simultaneous edits. Note, that all edits were made in strains with the C‐terminus of DMA‐1 already tagged with a 2XFLAG immunotag before the PDZ binding domain (Dong et al, 2016). Strains EB4219 through EB4238 in the Reagents and Tools table were obtained using these methods.
Imaging
Fluorescent images were captured in live C. elegans using a Plan‐Apochromat 40×/1.4 or 63x/1.4 objective on a Zeiss Axioimager Z1 Apotome. Worms were immobilized using 1 mM Levamisole and Z stacks were collected. Maximum intensity projections were used for further analysis and tracing of dendrites. For quantification of branching, 1‐day‐old adults were mounted onto slides and immobilized with 1mM Levamisole. In both cases of capturing images and counting live on the microscope, the number of secondary and tertiary branches, “Os,” (self‐avoidance defects), and/or quaternary branches within 100 µm of the primary branch anterior to the cell body were quantified.
Quantification and statistical analysis
For quantifications of PVD branching, the experimenter was blinded to genotypes of point mutants in dma‐1. Blinding for other genotypes was not possible, because genotypes could be inferred from phenotype. Statistical comparisons were conducted on Prism 8 GraphPad Software using Mann–Whitney, Kruskal–Wallis with Dunn’s Multiple Comparison ad‐hoc, Z‐, or two‐sided ANOVA with Dunnett’s Multiple Comparison ad‐hoc tests as appropriate. Statistical significance is indicated as ns, not significant; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001 and ****P ≤ 0.0001.
Author contributions
Maisha Rahman: Conceptualization; Data curation; Formal analysis; Funding acquisition; Investigation; Visualization; Methodology; Writing—original draft; Writing—review and editing. Nelson J Ramirez‐Suarez: Resources; Funding acquisition; Writing—review and editing. Carlos A Diaz‐Balzac: Resources; Funding acquisition; Writing—review and editing. Hannes E Bülow: Conceptualization; Formal analysis; Supervision; Funding acquisition; Validation; Visualization; Project administration; Writing—review and editing.
In addition to the CRediT author contributions listed above, the contributions in detail are:
Conceptualization Ideas: MR, HEB; Validation: MR; Formal Analysis: MR, HEB; Investigation: MR; Resources: NJR‐S, CAD‐B; Writing – Original Draft: MR; Writing – Review & Editing: MR, NJR‐S, CAD‐B, HEB; Visualization Preparation: MR; Project Administration: HEB; Funding Acquisition: MR, NJR‐S, CAD‐B, HEB.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Acknowledgements
We thank Scott Garforth, Sarah Garrett, Peri Kurshan, Yehuda Salzberg, Pamela Stanley, Robert Townley, and members of the Bülow laboratory for comments on the manuscript or helpful discussions during the course of this work. We thank David Miller, Shohei Mitani, Kang Shen, and Iain Wilson for reagents, and Yuji Kohara for the yk11g705 cDNA clone. We are grateful to Meera Trivedi for sharing the dzIs117 strain prior to publication. Some strains were provided by the Caenorhabditis Genome Center (funded by the NIH Office of Research Infrastructure Programs P40 OD010440). This work was supported by grants from the National Institute of Health (NIH): R01NS096672 and R21NS111145 to HEB; F31NS100370 to MR; T32GM007288 and F31HD066967 to CADB; P30HD071593 to Albert Einstein College of Medicine. We acknowledge support to MR by the Department of Neuroscience. NJRS was the recipient of a Colciencias‐Fulbright Fellowship and HEB of an Irma T. Hirschl/Monique Weill‐Caulier research fellowship.
EMBO reports (2022) 23: e54163.
Data availability
This study includes no data deposited in external repositories.
References
- Albeg A, Smith CJ, Chatzigeorgiou M, Feitelson DG, Hall DH, Schafer WR, Miller DM III, Treinin M (2011) C. elegans multi‐dendritic sensory neurons: morphology and function. Mol Cell Neurosci 46: 308–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apweiler R, Hermjakob H, Sharon N (1999) On the frequency of protein glycosylation, as deduced from analysis of the SWISS‐PROT database. Biochim Biophys Acta 1473: 4–8 [DOI] [PubMed] [Google Scholar]
- Brenner S (1974) The genetics of Caenorhabditis elegans . Genetics 77: 71–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bülow HE, Hobert O (2006) The molecular diversity of glycosaminoglycans shapes animal development. Ann Rev Cell Dev Biol 22: 375–407 [DOI] [PubMed] [Google Scholar]
- Chang IJ, He M, Lam CT (2018) Congenital disorders of glycosylation. Ann Transl Med 6: 477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatzigeorgiou M, Yoo S, Watson JD, Lee W‐H, Spencer WC, Kindt KS, Hwang SW, Miller DM III, Treinin M, Driscoll M et al (2010) Specific roles for DEG/ENaC and TRP channels in touch and thermosensation in C. elegans nociceptors. Nat Neurosci 13: 861–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Spence Andrew M, Schachter H (2003) Isolation of null alleles of the Caenorhabditis elegans gly‐12, gly‐13 and gly‐14 genes, all of which encode UDP‐GlcNAc: α‐3‐D‐mannoside β1,2‐N‐acetylglucosaminyltransferase I activity. Biochimie 85: 391–401 [DOI] [PubMed] [Google Scholar]
- Chen S, Zhou S, Sarkar M, Spence AM, Schachter H (1999) Expression of three Caenorhabditis elegans N‐acetylglucosaminyltransferase I genes during development. J Biol Chem 274: 288–297 [DOI] [PubMed] [Google Scholar]
- Cohen E, Chatzigeorgiou M, Husson SJ, Steuer‐Costa W, Gottschalk A, Schafer WR, Treinin M (2014) Caenorhabditis elegans nicotinic acetylcholine receptors are required for nociception. Mol Cell Neurosci 59: 85–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis JW, Nabi IR, Demetriou M (2009) Metabolism, cell surface organization, and disease. Cell 139: 1229–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz‐Balzac CA, Rahman M, Lazaro‐Pena MI, Martin Hernandez LA, Salzberg Y, Aguirre‐Chen C, Kaprielian Z, Bülow HE (2016) Muscle‐ and skin‐derived cues jointly orchestrate patterning of somatosensory dendrites. Curr Biol 26: 2379–2387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson DJ, Goldstein B (2016) CRISPR‐based methods for Caenorhabditis elegans genome engineering. Genetics 202: 885–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doitsidou M, Jarriault S, Poole RJ (2016) Next‐generation sequencing‐based approaches for mutation mapping and identification in Caenorhabditis elegans . Genetics 204: 451–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X, Chiu H, Park YJ, Zou W, Zou Y, Ozkan E, Chang C, Shen K (2016) Precise regulation of the guidance receptor DMA‐1 by KPC‐1/Furin instructs dendritic branching decisions. Elife 5: e11008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X, Liu OW, Howell AS, Shen K (2013) An extracellular adhesion molecule complex patterns dendritic branching and morphogenesis. Cell 155: 296–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X, Shen K, Bülow HE (2015) Intrinsic and extrinsic mechanisms of dendritic morphogenesis. Annu Rev Physiol 77: 271–300 [DOI] [PubMed] [Google Scholar]
- Feng Z, Zhao Y, Li T, Nie W, Yang X, Wang X, Wu J, Liao J, Zou Y (2020) CATP‐8/P5A ATPase regulates ER processing of the DMA‐1 receptor for dendritic branching. Cell Rep 32: 108101 [DOI] [PubMed] [Google Scholar]
- Fogel AI, Li Y, Giza J, Wang Q, Lam TT, Modis Y, Biederer T (2010) N‐glycosylation at the SynCAM (synaptic cell adhesion molecule) immunoglobulin interface modulates synaptic adhesion. J Biol Chem 285: 34864–34874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeze HH (2006) Genetic defects in the human glycome. Nat Rev Genet 7: 537–551 [DOI] [PubMed] [Google Scholar]
- Gasser B, Saloheimo M, Rinas U, Dragosits M, Rodríguez‐Carmona E, Baumann K, Giuliani M, Parrilli E, Branduardi P, Lang C et al (2008) Protein folding and conformational stress in microbial cells producing recombinant proteins: a host comparative overview. Microb Cell Fact 7: 11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilleard JS, Barry JD, Johnstone IL (1997) cis regulatory requirements for hypodermal cell‐specific expression of the Caenorhabditis elegans cuticle collagen gene dpy‐7. Mol Cell Biol 17: 2301–2311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta R, Brunak S (2002) Prediction of glycosylation across the human proteome and the correlation to protein function. Pac Symp Biocomput 2002: 310–322 [PubMed] [Google Scholar]
- Hanus C, Geptin H, Tushev G, Garg S, Alvarez‐Castelao B, Sambandan S, Kochen L, Hafner AS, Langer JD, Schuman EM (2016) Unconventional secretory processing diversifies neuronal ion channel properties. Elife 5: e20609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt CE, Dickson BJ (2005) Sugar codes for axons? Neuron 46: 169–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inberg S, Meledin A, Kravtsov V, Iosilevskii Y, Oren‐Suissa M, Podbilewicz B (2019) Lessons from worm dendritic patterning. Annu Rev Neurosci 42: 365–383 [DOI] [PubMed] [Google Scholar]
- Ioffe E, Stanley P (1994) Mice lacking N‐acetylglucosaminyltransferase I activity die at mid‐gestation, revealing an essential role for complex or hybrid N‐linked carbohydrates. Proc Natl Acad Sci USA 91: 728–732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeken J, Peanne R (2017) What is new in CDG? J Inherit Metab Dis 40: 569–586 [DOI] [PubMed] [Google Scholar]
- Jan YN, Jan LY (2010) Branching out: mechanisms of dendritic arborization. Nat Rev Neurosci 11: 316–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaji H, Kamiie J, Kawakami H, Kido K, Yamauchi Y, Shinkawa T, Taoka M, Takahashi N, Isobe T (2007) Proteomics reveals N‐linked glycoprotein diversity in Caenorhabditis elegans and suggests an atypical translocation mechanism for integral membrane proteins. Mol Cell Proteomics 6: 2100–2109 [DOI] [PubMed] [Google Scholar]
- Kutscher LM, Shaham S (2014) Forward and reverse mutagenesis in C. elegans . WormBook 1–26 10.1895/wormbook.1.167.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labasque M, Hivert B, Nogales‐Gadea G, Querol L, Illa I, Faivre‐Sarrailh C (2014) Specific contactin N‐glycans are implicated in neurofascin binding and autoimmune targeting in peripheral neuropathies. J Biol Chem 289: 7907–7918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre JL (2021) Molecular mechanisms that mediate dendrite morphogenesis. In Current Topics in Developmental Biology, Bashaw GJ (ed), Vol. 142, pp 233–282. Cambridge, MA: Academic Press; [DOI] [PubMed] [Google Scholar]
- Li L, Zinovyeva AY (2020) Protein extract preparation and co‐immunoprecipitation from Caenorhabditis elegans . J Vis Exp 10.3791/61243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li SA, Cheng L, Yu Y, Wang JH, Chen Q (2016) Structural basis of Dscam1 homodimerization: insights into context constraint for protein recognition. Sci Adv 2: e1501118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Wang S‐S, Wang W‐L, Zhang L, Zhao B‐Y (2014) Effect of swainsonine in Oxytropis kansuensis on Golgi α‐mannosidase II expression in the brain tissues of Sprague‐Dawley rats. J Agric Food Chem 62: 7407–7412 [DOI] [PubMed] [Google Scholar]
- Masu M (2016) Proteoglycans and axon guidance: a new relationship between old partners. J Neurochem 139(Suppl 2): 58–75 [DOI] [PubMed] [Google Scholar]
- Medina‐Cano D, Ucuncu E, Nguyen LS, Nicouleau M, Lipecka J, Bizot J‐C, Thiel C, Foulquier F, Lefort N, Faivre‐Sarrailh C et al (2018) High N‐glycan multiplicity is critical for neuronal adhesion and sensitizes the developing cerebellum to N‐glycosylation defect. Elife 7: e38309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzler M, Gertz A, Sarkar M, Schachter H, Schrader JW, Marth JD (1994) Complex asparagine‐linked oligosaccharides are required for morphogenic events during post‐implantation development. EMBO J 13: 2056–2065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minevich G, Park DS, Blankenberg D, Poole RJ, Hobert O (2012) CloudMap: a cloud‐based pipeline for analysis of mutant genome sequences. Genetics 192: 1249–1269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mire E, Hocine M, Bazellieres E, Jungas T, Davy A, Chauvet S, Mann F (2018) Developmental upregulation of Ephrin‐B1 silences Sema3C/Neuropilin‐1 signaling during post‐crossing navigation of corpus callosum axons. Curr Biol 28: 1768–1782 [DOI] [PubMed] [Google Scholar]
- Moloney DJ, Panin VM, Johnston SH, Chen J, Shao LI, Wilson R, Wang Y, Stanley P, Irvine KD, Haltiwanger RS et al (2000) Fringe is a glycosyltransferase that modifies Notch. Nature 406: 369–375 [DOI] [PubMed] [Google Scholar]
- Moremen KW (2002) Golgi α‐mannosidase II deficiency in vertebrate systems: implications for asparagine‐linked oligosaccharide processing in mammals. Biochem Biophys Acta 1573: 225–235 [DOI] [PubMed] [Google Scholar]
- Ng BG, Freeze HH (2018) Perspectives on glycosylation and its congenital disorders. Trends Genet 34: 466–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okkema PG, Harrison SW, Plunger V, Aryana A, Fire A (1993) Sequence requirements for myosin gene expression and regulation in Caenorhabditis elegans . Genetics 135: 385–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oren‐Suissa M, Hall DH, Treinin M, Shemer G, Podbilewicz B (2010) The fusogen EFF‐1 controls sculpting of mechanosensory dendrites. Science 328: 1285–1288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paschinger K, Hackl M, Gutternigg M, Kretschmer‐Lubich D, Stemmer U, Jantsch V, Lochnit G, Wilson IB (2006) A deletion in the golgi alpha‐mannosidase II gene of Caenorhabditis elegans results in unexpected non‐wild‐type N‐glycan structures. J Biol Chem 281: 28265–28277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paschinger K, Yan S, Wilson IBH (2019) N‐glycomic complexity in anatomical simplicity: Caenorhabditis elegans as a non‐model nematode? Front Mol Biosci 6: 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulain FE, Yost HJ (2015) Heparan sulfate proteoglycans: a sugar code for vertebrate development? Development 142: 3456–3467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley NM, Hebert AS, Westphall MS, Coon JJ (2019) Capturing site‐specific heterogeneity with large‐scale N‐glycoproteome analysis. Nat Commun 10: 1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saied‐Santiago K, Bülow HE (2018) Diverse roles for glycosaminoglycans in neural patterning. Dev Dyn 247: 54–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzberg Y, Coleman AJ, Celestrin K, Cohen‐Berkman M, Biederer T, Henis‐Korenblit S, Bülow HE (2017) Reduced Insulin/Insulin‐like growth factor receptor signaling mitigates defective dendrite morphogenesis in mutants of the ER stress sensor IRE‐1. PLoS Genet 13: e1006579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzberg Y, Diaz‐Balzac CA, Ramirez‐Suarez NJ, Attreed M, Tecle E, Desbois M, Kaprielian Z, Bülow HE (2013) Skin‐derived cues control arborization of sensory dendrites in Caenorhabditis elegans . Cell 155: 308–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzberg Y, Ramirez‐Suarez NJ, Bülow HE (2014) The proprotein convertase KPC‐1/furin controls branching and self‐avoidance of sensory dendrites in Caenorhabditis elegans. PLoS Genet 10: e1004657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnaar RL, Gerardy‐Schahn R, Hildebrandt H (2014) Sialic acids in the brain: gangliosides and polysialic acid in nervous system development, stability, disease, and regeneration. Physiol Rev 94: 461–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder NE, Androwski RJ, Rashid A, Lee H, Lee J, Barr MM (2013) Dauer‐specific dendrite arborization in C. elegans is regulated by KPC‐1/Furin. Curr Biol 23: 1527–1535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekine SU, Haraguchi S, Chao K, Kato T, Luo L, Miura M, Chihara T (2013) Meigo governs dendrite targeting specificity by modulating ephrin level and N‐glycosylation. Nat Neurosci 16: 683–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah N, Kuntz DA, Rose DR (2008) Golgi alpha‐mannosidase II cleaves two sugars sequentially in the same catalytic site. Proc Natl Acad Sci USA 105: 9570–9575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C, O’Brien T, Chatzigeorgiou M, Spencer W, Feingold‐Link E, Husson S, Hori S, Mitani S, Gottschalk A, Schafer W et al (2013) Sensory neuron fates are distinguished by a transcriptional switch that regulates dendrite branch stabilization. Neuron 79: 266–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CJ, Watson JD, Spencer WC, O'Brien T, Cha B, Albeg A, Treinin M, Miller DM III (2010) Time‐lapse imaging and cell‐specific expression profiling reveal dynamic branching and molecular determinants of a multi‐dendritic nociceptor in C. elegans. Dev Biol 345: 18–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley P, Taniguchi N, Aebi M (2015) N‐Glycans, In Essentials of Glycobiology, Stanley P, Taniguchi N, Aebi M, Varki A, Cummings RD, Esko JD, Stanley P, Hart GW, Aebi M, Darvill AG, Kinoshita T, Packer NH et al (eds), pp 99–111. New York, NY: Cold Spring Harbor; [Google Scholar]
- Sundararajan L, Stern J, Miller DM 3rd (2019) Mechanisms that regulate morphogenesis of a highly branched neuron in C. elegans . Dev Biol 451: 53–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang LT, Diaz‐Balzac CA, Rahman M, Ramirez‐Suarez NJ, Salzberg Y, Lazaro‐Pena MI, Bülow HE (2019) TIAM‐1/GEF can shape somatosensory dendrites independently of its GEF activity by regulating F‐actin localization. Elife 8: e38949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao L, Porto D, Li Z, Fechner S, Lee SA, Goodman MB, Xu XZS, Lu H, Shen K (2019) Parallel processing of two mechanosensory modalities by a single neuron in C. elegans . Dev Cell 51: 617–631.e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor SR, Santpere G, Weinreb A, Barrett A, Reilly MB, Xu C, Varol E, Oikonomou P, Glenwinkel L, McWhirter R et al (2021) Molecular topography of an entire nervous system. Cell 184: 4329–4347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson O, Edgley M, Strasbourger P, Flibotte S, Ewing B, Adair R, Au V, Chaudhry I, Fernando L, Hutter H et al (2013) The million mutation project: a new approach to genetics in Caenorhabditis elegans . Genome Res 23: 1749–1762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsalik EL, Niacaris T, Wenick AS, Pau K, Avery L, Hobert O (2003) LIM homeobox gene‐dependent expression of biogenic amine receptors in restricted regions of the C. elegans nervous system. Dev Biol 263: 81–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vabulas RM, Raychaudhuri S, Hayer‐Hartl M, Hartl FU (2010) Protein folding in the cytoplasm and the heat shock response. Cold Spring Harb Perspect Biol 2: a004390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei X, Howell AS, Dong X, Taylor CA, Cooper RC, Zhang J, Zou W, Sherwood DR, Shen K (2015) The unfolded protein response is required for dendrite morphogenesis. Elife 4: e06963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou W, Shen A, Dong X, Tugizova M, Xiang YK, Shen K (2016) A multi‐protein receptor‐ligand complex underlies combinatorial dendrite guidance choices in C. elegans . Elife 5: e18345 [DOI] [PMC free article] [PubMed] [Google Scholar]