

Abstract
Microinjection of spermatozoa or spermatids into oocytes is a major choice for infertility treatment. However, the use of premeiotic spermatocytes has never been considered because of its technical problems. Here, we show that the efficiency of spermatocyte injection in mice can be improved greatly by reducing the size of the recipient oocytes. Live imaging showed that the underlying mechanism involves reduced premature separation of the spermatocyte’s meiotic chromosomes, which produced much greater (19% vs. 1%) birth rates in smaller oocytes. Application of this technique to spermatocyte arrest caused by STX2 deficiency, an azoospermia factor also found in humans, resulted in the production of live offspring. Thus, the microinjection of primary spermatocytes into oocytes may be a potential treatment for overcoming a form of nonobstructive azoospermia caused by meiotic failure.
Keywords: azoospermia, fertilization, meiosis, oocyte, spermatocyte
Subject Categories: Cell Cycle, Development
Mouse primary spermatocytes can complete meiosis following injection into meiotic oocytes, but with severe chromosome segregation errors. Reduction of the cytoplasm of the oocytes prevents these chromosomal errors and enables efficient production of spermatocyte‐derived offspring.
Introduction
Fertilization is the process whereby female and male gametes (oocytes and spermatozoa) unite to form a zygote. From the standpoint of their genomes, the oocyte and spermatozoon are equivalent, but their history and cell type are quite different. Oocytes acquire their large cytoplasm (the ooplasm) during oogenesis to store all the components necessary for embryogenesis, including organelles, proteins, metabolites, mRNAs, and other molecules. By contrast, the contribution of spermatozoa to zygote formation and embryonic development is largely limited to deposition of the paternal genome and oocyte activation. Consequently, a simple injection of a spermatozoon or even the sperm head (nucleus) into a mature oocyte results in normal fertilization, leading to embryo development and the birth of offspring (Palermo et al, 1992; Ogura et al, 2005). Fertilization of oocytes does not even require mature sperm nuclei because injection of nuclei from immature spermatozoa (spermatids) is sufficient for normal fertilization and embryo development to term (Ogura et al, 2005). Indeed, in one clinical study, 90 babies were born following round spermatid injection without any significant adverse effects (Tanaka et al, 2018).
Therefore, a large ooplasm helps determine the embryo’s developmental potential. However, it is known that this feature does not always provide benefits for development. We and others have shown that a large ooplasm is linked to error‐prone chromosomal segregation by analyzing high‐resolution images of meiotic chromosomes in oocytes with artificially increased or decreased ooplasmic volume (Kyogoku & Kitajima, 2017; Lane & Jones, 2017). Thus, the evolution of a particular ooplasmic mass in a species might have arisen as a delicate trade‐off between meiotic fidelity and post‐fertilization developmental competence. In our analysis, it was clear that a large ooplasm was detrimental, because the meiotic chromosomes showed frequent abnormal behavior, which could have led to aneuploidy and embryonic death. Conversely, one can postulate that a smaller ooplasm might be more beneficial than a larger one in terms of chromosomal behavior by ensuring the genetic integrity of the resultant embryos. However, there is no evidence for this postulation because most oocytes of original size undergo meiotic divisions normally and can develop into offspring after fertilization in vivo or in vitro.
As mentioned above, normal diploid embryos can be obtained using spermatids because they are already haploid, as are mature spermatozoa. However, the use of primary spermatocytes for fertilization is considered to be ineffective because they are premeiotic germ cells. Theoretically, the chromosomes of primary spermatocytes might be able to contribute to the construction of diploid embryos after two meiotic divisions within oocytes. Indeed, we and another group have reported the birth of mice following spermatocyte injection into oocytes, but the success rates were low at 1–3% per embryo transferred (Kimura et al, 1998; Ogura et al, 1998). This was mostly caused by the death of embryos shortly after implantation. When we observed the reconstructed oocytes at metaphase II (MII), there was a high incidence of chromosomal aberrations (Ogura et al, 1998; Miki et al, 2006). Since then, there have been no technical improvements in spermatocyte injection. However, the use of primary spermatocytes for conception should be explored, given that many cases of nonobstructive azoospermia in humans are associated with the spermatogenic arrest at the primary spermatocyte stage (Enguita‐Marruedo et al, 2019).
Based on these findings, we expected that reducing the ooplasmic volume might improve the chromosomal integrity of spermatocyte‐injected oocytes and increase the survival rate of the resultant embryos. In this study, by employing high‐resolution live‐imaging techniques, we analyzed the segregation patterns of the maternal and paternal chromosomes within spermatocyte‐injected oocytes with or without reduction of the ooplasm. Furthermore, we examined whether such reduction could improve the birth rate following spermatocyte injection and whether this technology could be applied to azoospermic mice having a mutation causing spermatocyte arrest.
Results
Reduction of the recipient ooplasmic volume increases the rate of normal diploidy in spermatocyte‐injected oocytes
Fertilization with primary spermatocytes was achieved by injecting a spermatocyte nucleus into immature oocytes at the germinal vesicle (GV) stage followed by arrest at the metaphase of meiosis I (MI) induced by cytochalasin D treatment (Fig 1A). Here, the maternal and paternal (spermatocyte‐derived) chromosomes were synchronized, forming a single chromosomal mass. In preliminary experiments, we confirmed that the spermatocytes we selected had tetrad chromosomes that could undergo condensation within MII oocytes, indicating that they were at the midpachytene stage or later (Cobb et al, 1999; Fig 1B). After removal of cytochalasin D, they underwent meiotic division with protrusion of the first polar body to reach the MII stage (Fig 1A). This reconstructed MII “zygote” could be activated artificially to resume meiosis and form a one‐cell embryo having one zygotic nucleus (Fig 1A). To test the developmental ability of reconstructed embryos, we transferred these MII chromosomes to freshly prepared enucleated MII oocytes (Ogura et al, 1998).
Figure 1. Fertilization with primary spermatocytes.
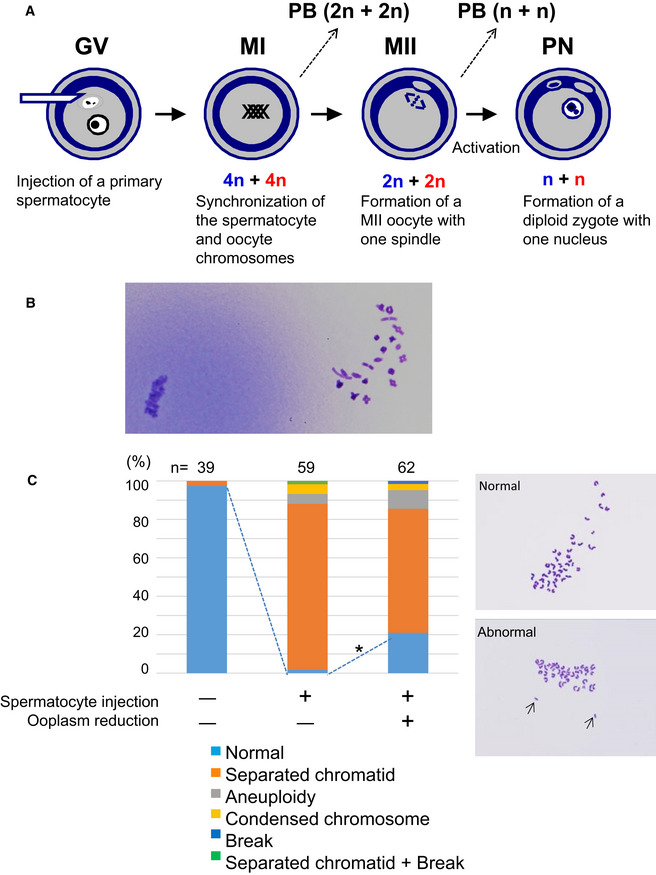
- The scheme of construction of a diploid fertilized oocyte using a primary spermatocyte and a GV‐stage oocyte. The chromosomes of the spermatocyte and the oocyte are intermingled at MI to form a single chromosomal mass.
- Spermatocyte chromosomes that underwent condensation (right) within a MII oocyte (MII chromosome mass, left). In this study, spermatocytes picked up for injection had tetrad chromosomes ready for condensation.
- Chromosomal analysis of MII oocytes that had been injected with primary spermatocytes. In the spermatocyte‐injected groups, normality was improved by reducing the ooplasm mass (*P < 0.005 by Fisher’s exact probability test). Arrows in the right figure indicate prematurely separated chromatids. The numbers of oocytes observed are indicated on the top of the bars. For the exact numbers in each case, see also Appendix Table S1. PB, polar body; GV, germinal vesicle; MI, meiosis I; PN, pronuclear stage.
Recipient oocytes with half the normal ooplasmic volume were prepared by aspiration using a large glass pipette (Movie EV1). We confirmed previously that oocyte manipulation by itself did not affect the fidelity of chromosome segregation by sham operations, including aspirating half the cytoplasm and putting it back again (Kyogoku & Kitajima, 2017). Using these halved oocytes, we first analyzed the chromosomal integrity of spermatocyte‐injected oocytes. In control oocytes without spermatocyte injection (i.e., oocyte chromosomes only), the proportion of oocytes with normal chromosomes was 97% at MII (Fig 1C and Appendix Table S1). In spermatocyte‐injected oocytes with intact ooplasm, the proportion of normal chromosomes was decreased significantly to 2% (1/59, P < 0.0001; Fig 1C and Appendix Table S1). The most frequent abnormality (86%, 51/58) was the presence of prematurely separated sister chromatids (Fig 1C and Appendix Table S1). By contrast, when spermatocytes were injected into half‐sized oocytes, the proportion of MII oocytes with normal chromosomes improved significantly to 13/62 (21%; P < 0.005, vs. 2% in the intact cytoplasm group) because of the decreased number of prematurely separated chromatids (Fig 1C and Appendix Table S1). Thus, in spermatocyte‐injected oocytes, while chromosomal normality was largely lost during meiosis I within the ooplasm of the original size, these chromosomal aberrations could be significantly prevented by reduction of the ooplasmic volume.
Reduction of the recipient ooplasm corrects the behavior of spermatocyte‐derived chromosomes during meiosis
Next, we sought to study how chromosomal behavior was influenced by the ooplasmic volume and which of the two parental (maternal or paternal) chromosomes was more vulnerable to the stress of biparental meiosis. The high‐resolution three‐dimensional (3D) live‐imaging system reported in our previous studies was employed for analyzing the chromosomal behavior during meiosis I (Kitajima et al, 2011; Kyogoku & Kitajima, 2017). To this end, it was essential to discriminate the origins of the chromosomes via fluorescence microscopy. Interestingly, the paternal (spermatocyte‐derived) chromosomes could be distinguished from the maternal chromosomes by the relatively lower fluorescent intensities of the histone H2B–mCherry marker (Fig 2A). Our 3D visualization of individual chromosomal positions showed that biparental meiosis exhibited more frequent misalignment of paternal chromosomes at late MI, compared with the maternal chromosomes (Fig 2B and C, and Movie EV2). Halving ooplasmic volume significantly reduced the number of misaligned paternal chromosomes (Fig 2B and C, and Movie EV3), an effect that we expected based on our previous observations (Kyogoku & Kitajima, 2017). Thus, paternal chromosomes are susceptible to errors in ooplasm‐hosted biparental meiosis, which can be tuned by reducing the ooplasmic volume.
Figure 2. Live imaging of biparental meiosis.
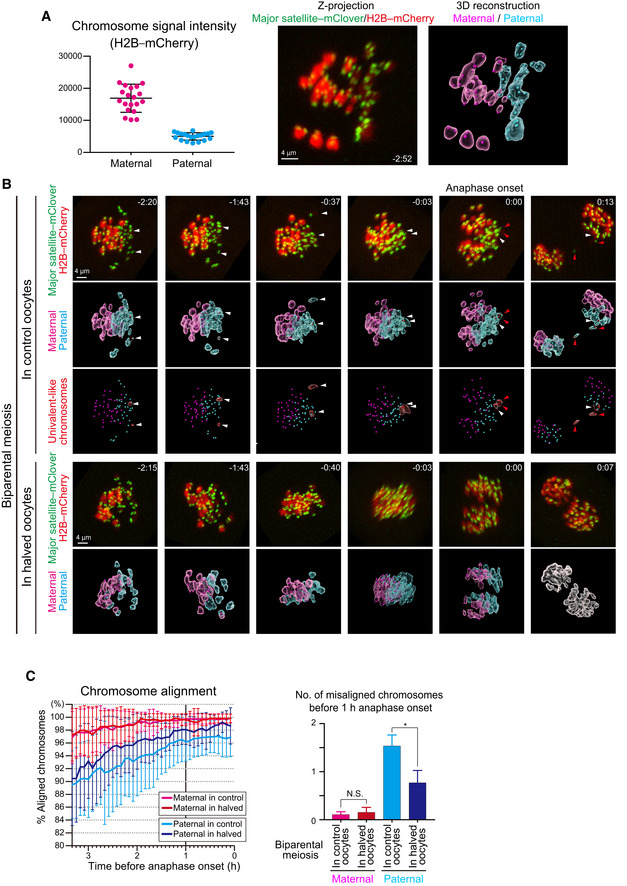
- Identification of parental origin of the chromosomes was distinguishable based on H2B–mCherry fluorescent intensities (paternal chromosomes exhibit lower intensities; n = 20 oocytes). Bars and error bars denote means ± SD. The z‐projection image shows major satellite–mClover (centromeres, green) and H2B–mCherry (chromosomes, red). Time from anaphase onset is shown in h:min. Scale bar = 4 μm. The 3D‐reconstructed image shows maternal (magenta) and paternal (cyan) chromosomes. Spots indicate centromeres.
- Chromosome tracking in 3D. The reconstructed images are viewed from the side of the metaphase plate. Signals are interpolated in the Z axis for visualization. White and red arrowheads, as well as red surfaces, indicate univalent‐like chromosomes that underwent unbalanced predivision (premature segregation of sister chromatids). Scale bar = 4 μm.
- Halving the recipient ooplasmic mass rescued chromosome alignment. The numbers of misaligned chromosomes and their parental origin were determined in 3D (n = 39 and 17 oocytes). Error bars show the standard deviation. Student’s t‐test was used to compare means. *P < 0.05. N.S., not significant.
We then analyzed how biparental meiosis in normal‐sized ooplasms results in chromosomal abnormality. Our technique of complete centromere tracking using 3D imaging (Kitajima et al, 2011; Sakakibara et al, 2015) enabled us to demonstrate that 89% of biparental meiotic divisions showed errors in chromosomal segregation at anaphase I (Figs 2B and 3A, and Movie EV2). Almost all of the errors were of spermatocyte origin (Fig 3A). Categorization of anaphase trajectories showed that predominant error patterns were balanced and unbalanced predivisions (premature segregation of sister chromatids at MI; Fig 3B), consistent with our observation of separated chromatids in MII spreads (Fig 1B and Appendix Table S1). Meanwhile, chromosome breakages and nondisjunction were relatively minor (Fig 3B).
Figure 3. Halving the recipient ooplasm prevents premature separation of paternal chromosomes in biparental meiosis.
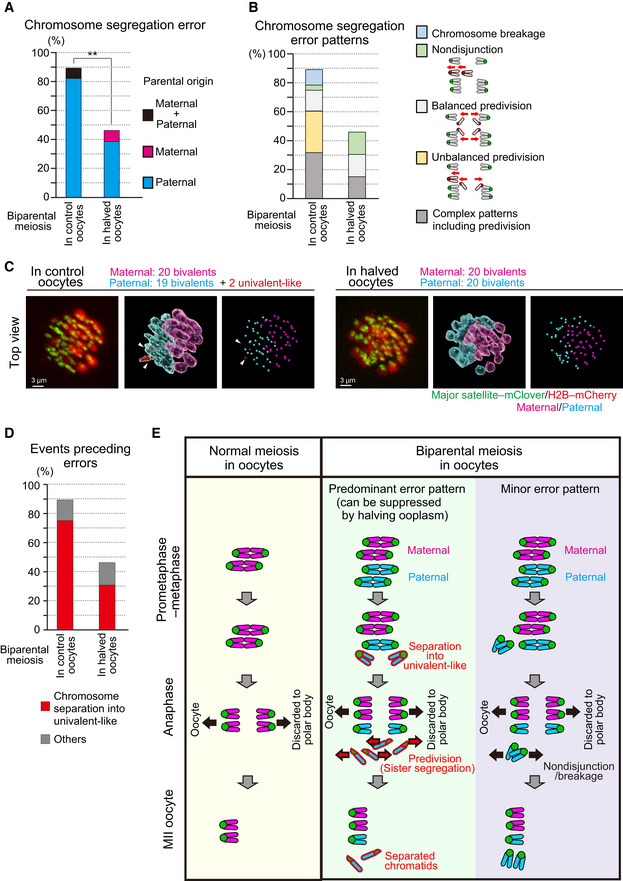
- Halving the recipient ooplasm mass reduced chromosome segregation errors. Errors were determined by tracking all chromosomes at anaphase (n = 39 and 17 oocytes; See also Fig 2B). The parental origin of errors is shown. Ooplasmic halving significantly reduced the rate of errors (**P < 0.01, Chi‐square test).
- Predivision was predominant in biparental meiosis. Chromosome segregation error patterns were categorized based on anaphase trajectories: nondisjunction (0:4 segregation), balanced predivision (2:2 sister chromatid segregation), unbalanced predivision (1:3 segregation including sister chromatid segregation), and complex patterns including predivision (multiple errors including sister chromatid segregation). Chromosome breakages (chromosomes lacking centromeres) were also observed. Data from 39 and 17 oocytes, respectively.
- Univalent‐like chromosomes. Images were 3D‐reconstructed as in Fig 2B and viewed from the top of the metaphase plate. Red surfaces with white arrowheads indicate univalent‐like chromosomes. Scale bar = 3 μm.
- Halving the ooplasm volume suppressed the premature separation of paternal chromosomes. Oocytes were categorized based on whether the chromosomes exhibited premature separation into univalent‐like structures prior to segregation errors. Data from 39 and 17 oocytes, respectively.
- Summary of biparental meiosis. In normal‐sized oocytes, biparental meiosis frequently exhibits premature separation of paternal chromosomes into univalent‐like structures. These chromosomes undergo predivision (premature segregation of sister chromatids), and thus result in separated chromatids in MII oocytes. Chromosome nondisjunction and breakage were relatively minor. Halving the ooplasmic volume reduced premature separation of paternal chromosomes.
Predivisions are error patterns observed following the premature separation of bivalent chromosomes into univalents during the prometaphase and metaphase in naturally aged oocytes (Sakakibara et al, 2015). Therefore, we carefully analyzed the prometaphase–metaphase trajectories of the chromosomes that underwent segregation errors in biparental meiosis. This analysis revealed that most of the errors were preceded by premature separation of bivalent chromosomes into univalent‐like structures (Fig 2B and 3C, and Movies EV2 and EV3). Importantly, decreasing (halving) the recipient ooplasm mass significantly suppressed the premature bivalent separation of chromosomes (75% in controls vs. 31% in halved oocytes; Fig 3D) and chromosome segregation errors (89% in control vs. 46% in halved oocytes; Fig 3A, B and D). Thus, the chromosomal aberrations found in spermatocyte‐injected oocytes were largely attributable to the premature separation of spermatocyte‐derived chromosomes, and about half of such aberrations could be prevented by reducing the size of the recipient ooplasm (Fig 3E).
Behaviors of meiosis‐regulating proteins in spermatocyte‐injected oocytes
Predivisions can be promoted by defects in MI‐specific centromere and kinetochore functions that include centromeric cohesion protection and sister kinetochore mono‐orientation. We found that spermatocyte‐derived chromosomes were associated with SGO2, a protein required for centromeric cohesion protection at their centromeres (Lee et al, 2008; Llano et al, 2008), similar to what is seen in maternal chromosomes (Fig 4A). Likewise, PLK1, a protein required for both centromeric protection and sister kinetochore mono‐orientation (Kim et al, 2015), was localized at the kinetochores of spermatocyte‐derived chromosomes (Fig 4B). Thus, the predivision of spermatocyte‐derived chromosomes is unlikely to be explained by defects in the recruitment of these proteins.
Figure 4. Behavior of meiosis‐regulating proteins in spermatocyte‐injected oocytes.
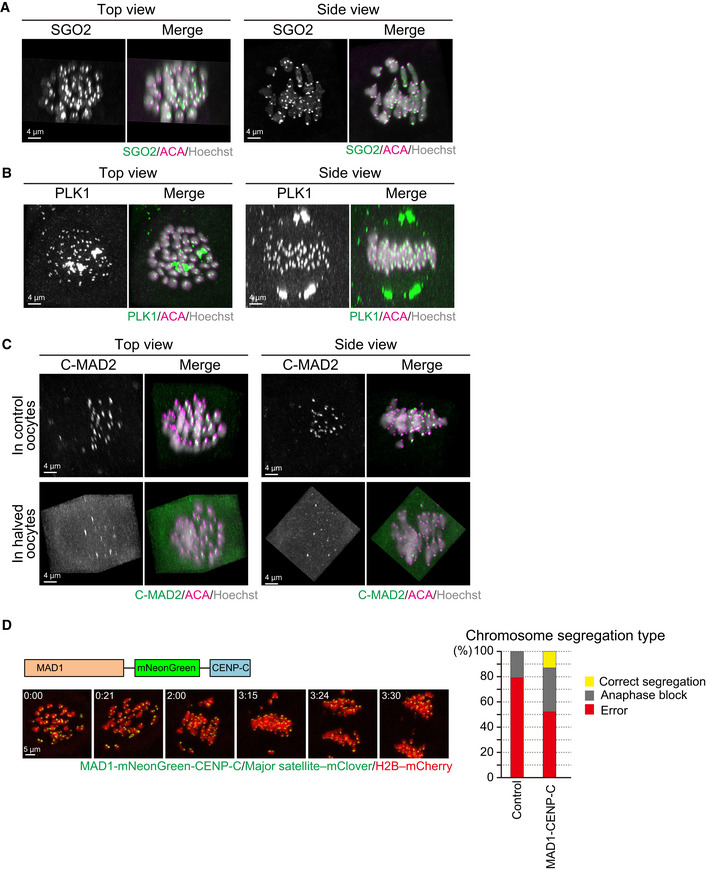
- SGO2 localizes at the centromeres of spermatocyte‐derived and maternal chromosomes. Oocytes were fixed at metaphase I and stained for SGO2 (green), ACA (centromeres, magenta), and Hoechst33342 (DNA, blue). Images were reconstructed in 3D and viewed from the top and side of the metaphase plate. Note that signals are interpolated in z.
- PLK1 localizes at the kinetochores of spermatocyte‐derived and maternal chromosomes. PLK1 (green) localization was investigated as in (A).
- MAD2 kinetochore localization is defective on spermatocyte‐derived chromosomes. The localization of the closed (active) form of MAD2 (C‐MAD2, green) was investigated as in (A). Note that the kinetochore enrichment of MAD2 was less on the chromosomes in half of the metaphase plate, which likely corresponded to spermatocyte‐derived chromosomes, in control and halved oocytes.
- Effect of forced SAC activation. Oocytes expressing MAD1‐mNeonGreen‐CENP‐C (green), together with major satellite–mClover (green) and H2B–mCherry (red), were imaged. Time after the start of imaging (h:mm) is shown. Oocytes were categorized based on anaphase figures (n = 24, 23 oocytes).
Another possible explanation is that spermatocyte‐derived chromosomes are defective in activating the spindle assembly checkpoint (SAC) in the ooplasm. Consistent with this idea, we found that spermatocyte‐derived chromosomes appeared to fail the kinetochore recruitment of the SAC activator MAD2 (Fig 4C). However, even halving ooplasmic volume did not recover MAD2 localization on spermatocyte‐derived chromosomes (Fig 4C). Moreover, unlike halving ooplasmic volume, the forced activation of the SAC by tethering MAD1 at spermatocyte kinetochores with the MAD1‐CENP‐C fusion construct (Kyogoku & Kitajima, 2017) did not efficiently rescue chromosome segregation errors in biparental meiosis (Fig 4D). Thus, it is likely that halving ooplasmic volume rescued chromosome segregation errors in biparental meiosis largely due to effects other than modifying the SAC activity.
These experimental results imply that the amount of chromosome mass, rather than a spermatocyte‐specific chromosomal property, might be responsible for the chromosomal aberrations during biparental meiosis. To test this possibility, we transferred MI karyoplasts into recipient MI oocytes instead of spermatocyte nuclei. As expected, following the first meiosis in vitro, the reconstructed oocytes formed a large MII chromosome mass and a large first polar body (Appendix Fig S1A). However, 64% (9/14) of these MII oocytes contained chromosomal abnormalities (Appendix Fig S1B), which suggests that the frequent segregation errors encountered during biparental meiosis can be primarily attributable to the doubled MI chromosomal mass, although the presence of the spermatocyte chromosomes might aggravate these errors.
Reduction in the recipient ooplasm improved the birth rates following spermatocyte injection
Next, we examined whether reduction of the ooplasm volume could improve the developmental ability of spermatocyte‐derived embryos. When we reconstructed embryos using normal‐sized oocytes and transferred them into recipient females, only 1% (1/96) developed into offspring (Fig 5A and Appendix Table S2), consistent with our previous reports (Ogura et al, 1998; Miki et al, 2006). By contrast, when we used halved oocytes, 19% (17/90) of the reconstructed embryos developed into live offspring, achieving a nearly 20‐fold improvement (P < 0.0001, Fig 5A and Appendix Table S2). The pups born by this improved method had body and placental weights within the normal ranges (Appendix Fig S2). We allowed three male pups to grow into adults and confirmed that they were all fertile by mating them with normal female mice.
Figure 5. Birth of spermatocyte‐derived offspring following embryo transfer.
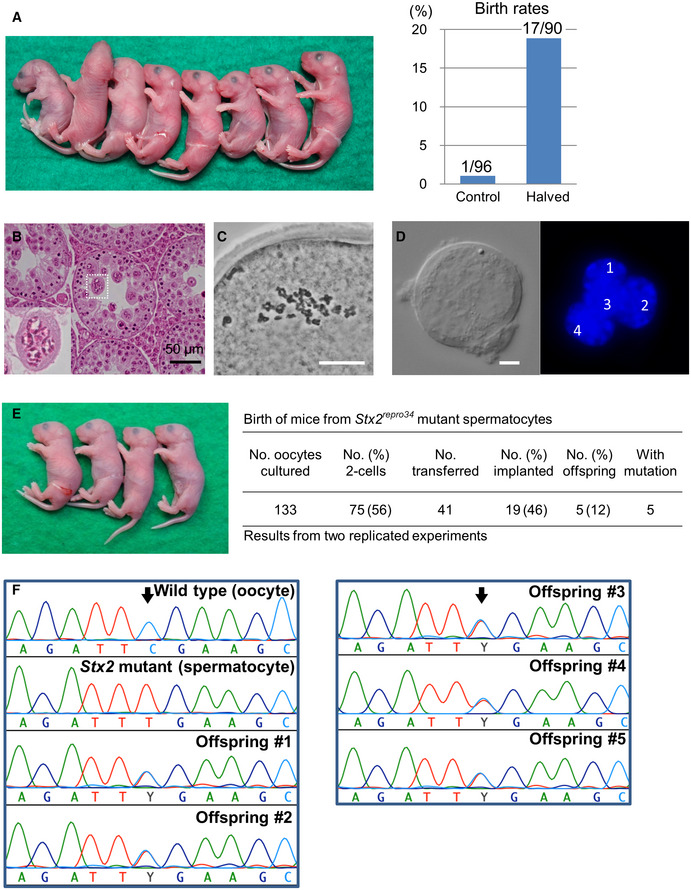
- Mouse pups born following spermatocyte injection (left) and the birth rates following embryo transfer (right). The numbers in the graph indicate: (number of pups born)/(number of embryos transferred). For detailed results, see also Appendix Table S2.
- Histology of the testis of a Stx2repro34 mouse. Arrowheads indicate multinucleated cells containing spermatocyte‐like nuclei. There are no spermatids or spermatozoa. Bar = 50 μm.
- An MII oocyte injected with a putative spermatocyte nucleus from a multinucleated cell in a Stx2repro34 mouse testis, showing the typical paired meiotic chromosomes. Bar = 20 μm.
- A multinucleated cell isolated from a Stx2repro34 mouse testis, showing four nuclei. Differential interference contrast (left) and Hoechst‐stained (right) images. Bar = 10 μm.
- Left: mouse pups born following microinjection with putative primary spermatocyte nuclei isolated from multinucleated cells; Right: birth rate of pups following Stx2repro34 spermatocyte microinjection.
- Genomic sequencing confirming the origin of pups from Stx2repro34 spermatocytes. Arrows indicate the expected point mutation of Stx2repro34 . Y indicates a hybrid status with T and C bases.
Spermatocyte injection rescued azoospermia caused by meiotic arrest
Finally, we applied this improved spermatocyte injection method to mouse strains with azoospermia caused by spermatogenic arrest at the primary spermatocyte stage. If the chromosomes of spermatocytes are functionally and structurally intact, we surmised that their normal meiotic divisions might be induced by the meiotic machinery of recipient oocytes. We performed our studies on Stx2repro34 mice (hereafter, repro34 mice) that carry a mutation in the Stx2 (syntaxin 2) gene induced by N‐ethyl‐N‐nitrosourea (ENU) mutagenesis (Fujiwara et al, 2013). Its human homologue, STX2, has been identified as a causal factor of nonobstructive azoospermia (Nakamura et al, 2018). Both mouse Stx2 and human STX2 mutations are characterized by the formation of large syncytial spermatocytes because of their inability to maintain intercellular bridges (Fujiwara et al, 2013; Nakamura et al, 2018; Fig 5B). We confirmed that the nuclei within these syncytial cells of repro34 mice were derived from spermatocytes by examining their prophase I chromosomes following injection into MII oocytes (Fig 5C). We reconstructed embryos using the nuclei collected from these syncytial spermatocytes (Fig 5D and Movie EV4). After 41 embryos were transferred into recipient females, five pups (four female and one male) were born (Fig 5E). All of these pups carried the point mutation in the Stx2 gene (Fig 5F). We also applied this technique to spermatocytes from Exoc1 (exocyst complex component 1)‐deficient mice that also show syncytial spermatocytes (Osawa et al, 2021; Fig EV1A). Three pups (two female and one male) carrying the mutation were born at term (Fig EV1B and C). All the eight pups derived from Stx2‐ or Exoc1‐deficient spermatocytes looked normal in appearance and their body and placental weights were within normal ranges, except for the bodyweight of pups from Stx2‐deficient spermatocytes (Appendix Fig S2). They grew into normal‐looking adults and were proven to be fertile. We also attempted to rescue azoospermic male mice deficient in D1Pas1 (Inoue et al, 2016). D1Pas1 is a mouse autosomal DEAD‐box RNA helicase expressed predominantly in the testis. Its deficiency causes spermatocyte arrest at or before the midpachytene stage by unknown causes (Inoue et al, 2016). We transferred 43 reconstructed two‐cell embryos into recipient female mice, but no implantation sites were found in the recipient uteri (Appendix Table S3). It is probable that only primary spermatocytes that develop beyond the midpachytene stage can undergo normal meiosis within oocytes.
Figure EV1. Birth of mice following injection of oocytes with primary spermatocytes collected from germline‐specific Exoc1‐knockout mice.
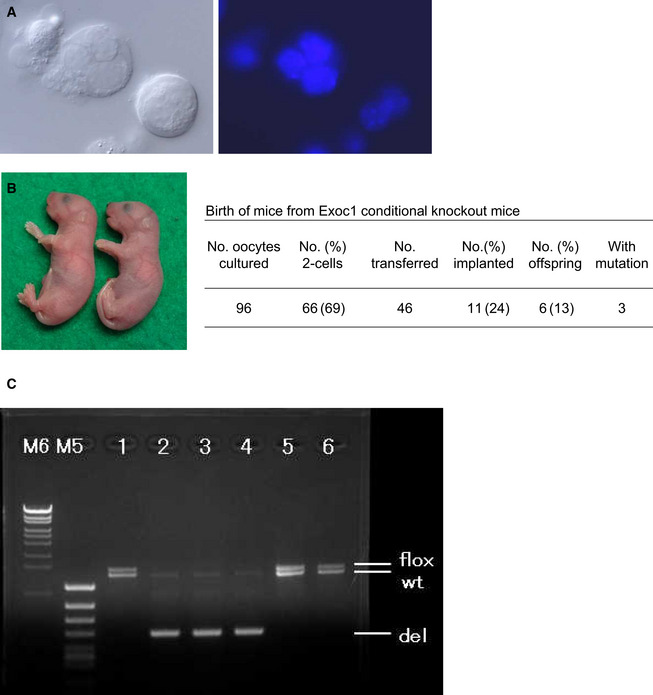
- Multinucleated cells (syncytial spermatocytes) obtained from an Exoc1‐knockout male mouse. Each multinucleated cell contained 2–4 spermatocyte nuclei. Differential interference contrast microscopy (left) and Hoechst‐staining (right) images. Bar = 10 μm.
- Mice born from Exoc1‐knockout spermatocytes.
- Polymerase chain reaction analysis in mice born from Exoc1‐knockout spermatocytes. Mice #2, #3, and #4 carried the Exoc1‐knockout allele (del) while mice #1, #5, and #6 did not. Three pups that did not carry the Exoc1 mutation were most likely derived from spermatogonia that escaped the Cre‐induced Exoc1 deletion. The expected amplicon sizes are as follows: flox allele, 1,426 bp; wild type allele, 1,291 bp; deletion allele, 490 bp.
Chromosomal analysis of offspring born following spermatocyte injection
As described above, all 11 spermatocyte‐derived pups (three wild‐type‐derived and eight knockout‐derived) nursed by the foster mothers grew into fertile adults. We then analyzed their chromosomal constitution in detail by multicolor fluorescence in situ hybridization (FISH). Among the three male mice derived from wild‐type spermatocytes, two had a normal karyotype, but one had XYY sex chromosomes (Fig 6). This XYY male mouse was not fully fertile and could impregnate only one out of 10 female mice. This observation is consistent with a report that XYY male mice are not always completely infertile (Obata et al, 2008). Note that we cannot exclude the possibility of XY/XYY mosaicism in this mouse. Among the five mice (four female and one male) derived from Stx2‐deficient spermatocytes, two female mice and one male mouse were normal, but one female had an XO chromosome and another had a shortened X chromosome (Fig EV2). Among the three female mice derived from Exoc1‐deficient spermatocytes, one had an XO chromosomal configuration. No abnormalities were found in the autosomes of the mice examined.
Figure 6. Chromosomal multicolor FISH analysis of the offspring derived from wild‐type spermatocytes.
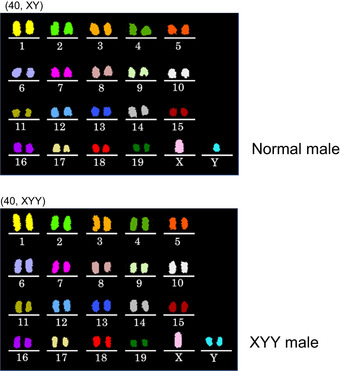
Of the three males analyzed, one had XYY sex chromosomes. No abnormalities were found in autosomes in these mice.
Figure EV2. Chromosomal multicolor FISH analysis of the offspring derived from Stx2‐deficient spermatocytes.
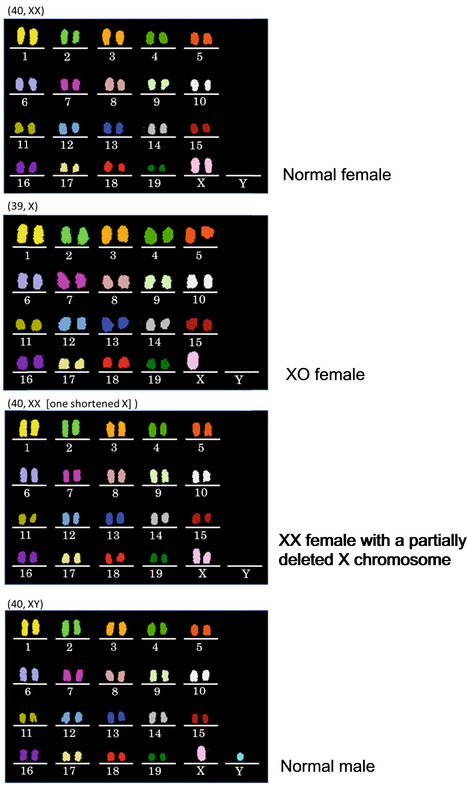
No abnormalities were found in autosomes, but there were two cases of sex chromosomal abnormalities.
Thus, while mice born from primary spermatocytes often suffered from sex chromosomal aberrations, they were largely normal phenotypically and grew to adulthood. Two possibilities could explain these sex chromosome‐biased anomalies: that is, either segregation errors occurred predominantly in sex chromosomes, or they occurred randomly in all autosomes as well as sex chromosomes. Embryos with autosomal abnormalities died before birth. To distinguish between these possibilities, we applied multicolor FISH analysis to MII oocytes derived from spermatocyte injection. As shown in Fig EV3A and B, chromosomal abnormalities including chromatid separations were also found in most autosomes and the X chromosome. This finding indicated that chromosomal abnormalities occurred randomly in all chromosomes and only embryos that had normal autosomes survived to term. This may be the main reason why the spermatocyte‐derived pups exclusively showed sex chromosome abnormalities, if any.
Figure EV3. Chromosomal multicolor FISH analysis of MII oocytes derived from spermatocyte injection.
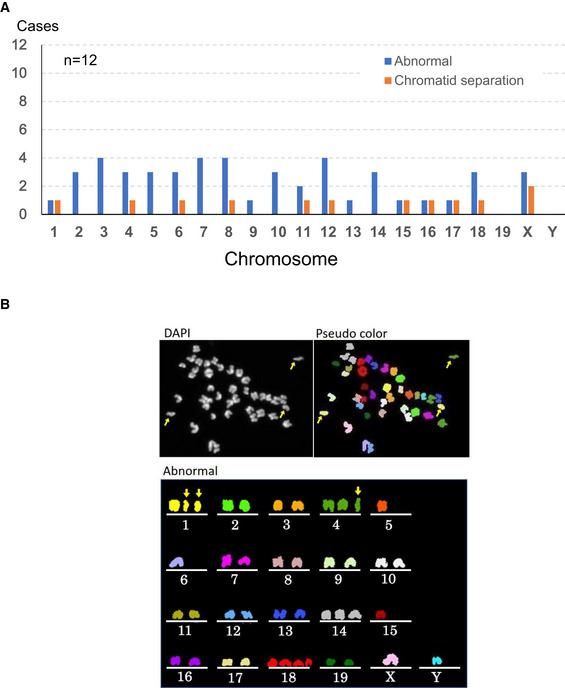
- Chromosomal abnormalities were found in both autosomes and sex chromosomes.
- A representative image of an oocyte with chromosomal aberrations. Arrows indicate prematurely separated chromatids. Besides them, chromosomes 5, 6, 14, 15 and 18 were numerically abnormal.
Discussion
Here, we addressed whether reducing the recipient ooplasm could ameliorate the embryonic death rate caused by the meiotic errors that can occur in mammalian oocytes. To this end, we employed an assisted fertilization system using primary spermatocytes, which need simultaneous biparental meiosis within oocytes: namely, meiosis with doubled chromosomes. Following spermatocyte injection into halved oocytes, the proportion of normal chromosomes at MII increased from 2 to 21% and the birth rate increased from 1 to 19%. These results demonstrate unequivocally that reducing the mass of the ooplasm indeed helps to normalize chromosomal behavior, leading to better survival of embryos to term. It would be interesting to test whether this strategy could also correct the meiotic errors that are frequently found in oocytes from aged female mammals (Mihajlović & FitzHarris, 2018). In humans, these meiotic errors in oocytes are known to increase with advanced age and to reduce conception rates significantly (El Yakoubi & Wassmann, 2017; Mikwar et al, 2020). In these errors, diverse mechanistic defects are involved, such as defects in chromosomal cross‐over formation, cohesin loss, and spindle deformation (Mihajlović & FitzHarris, 2018; Ma et al, 2020). We suspect that reducing the mass of the ooplasm might help rescue or prevent at least some of these defects.
The nearly 20‐fold improvement in the birth rate following spermatocyte injection into halved oocytes was much better than we expected. It is known that meiosis in female and male mammals differs largely with respect to the underlying molecular mechanisms and cell cycle progression patterns, such as acentriolar meiotic spindles and the absence of the interphase between two meiotic divisions in female germ cells. Consistent with this, many strains of gene knockout mice carrying mutations of meiosis‐related factors show either male or female infertility, not both (Jamsai & O’Bryan, 2011; Biswas et al, 2021). Our findings imply that the meiotic chromosomes of female and male germ cells have structural commonalities that allow mechanistic interchangeability between them. Nevertheless, our live imaging demonstrated that premature bivalent separation occurred predominantly in spermatocyte‐derived chromosomes, preceding chromosome segregation errors. The spermatocyte chromosomes might be intrinsically more error‐prone than oocyte chromosomes. Alternatively, the biochemical environment of the oocyte cytoplasm or spindle forces might have selectively promoted the separation of the spermatocyte chromosomes. The lack of the SAC activator MAD2 and delayed alignment of the spermatocyte chromosomes (Figs 2B and C, and 4C) may reflect spermatocyte‐chromosome‐specific difficulties in biparental meiosis. The doubled chromosome mass likely represents a general difficulty in biparental meiosis because doubling the chromosomal mass with another MI karyoplast also caused chromosomal aberrations (Appendix Fig S1). In sum, chromosome segregation errors in biparental meiosis are likely attributed to multiple factors. Among them, premature separation of spermatocyte chromosomes can be prevented by halving ooplasmic volume, which greatly increases the likelihood of successful biparental meiosis.
Chromosomal analysis by multicolor FISH revealed that four of the 11 spermatocyte‐derived offspring carried chromosomal abnormalities that were restricted to the sex chromosomes. However, within MII‐stage oocytes following spermatocyte injection, there were also substantial numbers of abnormalities in autosomes (Fig EV3). These findings indicate that the sex‐chromosome‐biased chromosomal aberration may be explained by the high embryonic lethality of autosomal aneuploidy, which might have selected embryos with normal autosomes for survival to term.
This study has practical implications for treating spermatogenic arrest caused by the meiotic arrest. Given the complexity of meiosis, many genes are involved in its regulation, as revealed by mouse gene knockout models (Jamsai & O’Bryan, 2011). Defects in some of these genes might cause the failure of meiosis and spermatogenic arrest at the primary spermatocyte stage. The results of this study will help identify the types of meiotic arrest that can be rescued or prevented by the spermatocyte injection technique we developed here. Such information would provide invaluable clues for human clinical research aiming to develop treatments for meiosis‐related male infertility. In addition, we propose another important implication of this study: at present, although complete in vitro gametogenesis is possible for female germ cells (Hikabe et al, 2016; Yoshino et al, 2021), male germ cells still require in vivo‐derived tissue pieces for the completion of meiosis (Ishikura et al, 2021). If male primordial germ cell‐like cells derived from induced pluripotent stem cells (Hayashi et al, 2011) could be cultured up to the pachytene spermatocyte stage in vitro, their injection into immature oocytes might produce offspring by skipping in vivo male gametogenesis completely. This could be an alternative strategy to enable conception in cases of male patients with germ cell loss. These scenarios could open up new methods for treating human male infertility, although there are a number of ethical and technical issues—for example, the high incidence of chromosome abnormalities—that must be resolved before these strategies could be used by clinics offering assisted reproductive technology.
Materials and Methods
Mice
All animal experiments were approved by the Institutional Animal Care and Use Committees at RIKEN Tsukuba (T2020‐Jitsu004 and T2021‐Jitsu004) and Kobe (A2011‐05‐18) Branches and were performed in accordance with the ARRIVE guidelines. B6D2F1 and C57BL/6NCrSlc mice were purchased from Japan SLC. ICR mice were purchased from CLEA, Japan. Repro 34 mice carrying the Stxrepro34 mutation were introduced from Okayama University. The repro34 mutation was induced in a C57BL/6J male and subsequently, a congenic line with the C3HeB/FeJ background was created (Akiyama et al, 2008). Germline‐specific Exoc1 conditional knockout mice were generated by breeding Exoc1tm1c(EUCOMM)Hmgu mice (Skarnes et al, 2011) with Nanos3‐Cre driver mice (kindly gifted by Dr Y. Saga, RIKEN BRC RBRC02568), which express Cre in spermatogonia (Suzuki et al, 2008; Osawa et al, 2021). D1Pas1 knockout mice were provided by RIKEN BioResource Center (RBRC05432). All mice were maintained under specific‐pathogen‐free conditions, provided with water and commercial laboratory mouse chow ad libitum, and housed under controlled lighting conditions (daily light period, 07:00 to 21:00).
Genotyping of mice
In experiments using primary spermatocytes from mutant mice, genotyping of offspring was performed by polymerase chain reaction (PCR) using the following specific primer sets for genomic DNA. For the Stxrepro34 mutation, Stx2 4_F (GTGGGATCACGAGTCACTCACT) and Stx2 4_R (GAGTGAGTTCCACGACAGCCA) were used (Fujiwara et al, 2013). The PCR products were sequenced to detect the point mutation. For Exoc1 conditional knockout, Exoc1‐cKO genotyping 3‐1 (AGTCTTCCTTCCCTGGGTTG) and Exoc1‐cKO genotyping n3‐3 (CCGGATTGATGGTAGTGGTC) were used (modified from Osawa et al, 2021).
Collection of oocytes
Female B6D2F1 mice (9–12 weeks old) were injected with 7.5 IU of equine chorionic gonadotropin (eCG, ASKA Pharmaceutical, Tokyo, Japan). Forty‐four to forty‐eight hours after injection, fully grown oocytes at the GV stage were collected from large antral follicles and released into an M2 medium supplemented with 150 μg/ml dibutyryl cyclic (dbc) AMP (Merck KGaA). After being freed from cumulus cells by pipetting, oocytes were cultured for at least 1 h in MEM) Merck KGaA) supplemented with 50 μg/ml gentamicin, 0.22 mM Na‐pyruvate, 1 μg/ml epidermal growth factor (EGF), 150 μg/ml dbc AMP, and 4 mg/ml bovine serum albumin (BSA), (mMEM; Fulka & Langerova, 2014) at 37°C in an atmosphere of 5% CO2 in humidified air, until micromanipulation.
Collection of primary spermatocytes
Spermatogenic cells were collected from the testes of male B6D2F1, C57BL/6N, and ICR mice (12–16 weeks old) by a mechanical method as reported in a previous study (Ogura & Yanagimachi, 1993). Briefly, the testes were placed in an erythrocyte‐lysing buffer (155 mM NH4Cl, 10 mM KHCO3, 2 mM EDTA; pH 7.2). After the tunica albuginea had been removed, the testes were transferred into a cold (4°C) Dulbecco’s phosphate‐buffered saline (PBS) supplemented with 5.6 mM glucose, 5.4 mM sodium lactate, and 3 mg/ml BSA (GL‐PBS; Ogura et al, 1996). The seminiferous tubules were cut into small pieces using a pair of fine scissors and pipetted gently to allow spermatogenic cells to be released into the medium. The cell suspension was filtered through a 38‐μm nylon mesh and washed twice by centrifugation (200 g for 4 min). After gentle washing, the cells were resuspended in GL‐PBS and stored at 4°C until microinjection.
Micromanipulation
To make half‐sized oocytes, oocytes at the GV stage were transferred to an M2 medium (Merck Millipore) containing 7.5 μg/ml cytochalasin D (Merck KGaA) and 60 mM NaCl for 10 min at 37°C. All manipulations were performed under an inverted microscope with a Piezo‐driven micromanipulator (PrimeTech). The zona pellucida was opened by piezo drilling and one‐third to half of the ooplasmic volume was aspirated with an injection pipette (inner diameter 25 μm) at 37°C (Movie EV1). After manipulation, oocytes were cultured in mMEM containing 7.5 μg/ml cytochalasin D and 40 mM NaCl at 37°C in an atmosphere of 5% CO2 in the air. About 1–1.5 h later, primary spermatocytes (pachytene to diplotene stages) were injected into oocytes that were induced to arrest at the MI stage by cytochalasin D. Oocytes were cultured in mMEM containing 7.5 μg/ml cytochalasin D and 40 mM NaCl for 2 h at 37°C in an atmosphere of 5% CO2 in humidified air. After washing in mMEM, the oocytes were cultured for 14–17 h until they reached the MII stage. The karyoplasts containing chromosomes were removed and were then fused with fresh enucleated oocytes using Sendai virus (HVJ; Ishihara Sangyo Co., Ltd.) in a Hepes‐buffered CZB medium (Chatot et al, 1990) containing 7.5 μg/ml cytochalasin D. After manipulation, the oocytes were cultured in CZB medium containing 7.5 μg/ml cytochalasin D for 1 h at 37°C in an atmosphere of 5% CO2 in humidified air until complete fusion occurred. Reconstructed oocytes were activated by culturing them in a Ca2+‐free CZB medium containing 8 mM SrCl2 for 20 min. After washing, the oocytes were cultured in a CZB medium for 24 h under 5% CO2 in humidified air at 37°C.
Embryo transfer
Embryos that reached the 2‐cell stage by 24 h were transferred into the oviducts of Day 1 pseudopregnant ICR strain female mice (9–12 weeks old). On Day 19.5, recipient females were euthanized and their uteri were examined for live fetuses. In some experiments, live fetuses were nursed by lactating foster ICR strain mothers. After weaning, they were checked for fertility by mating with ICR mice of the opposite sex.
Chromosome preparation of oocytes
The MII oocytes were treated with 0.5% actinase E (Kaken Pharmaceutical Co.) for 5 min at room temperature to loosen the zona pellucida and then treated with a hypotonic solution (1:1 mixture of 1.2% sodium citrate and 60% fetal bovine serum, FBS; Merck KGaA) for 10 min at room temperature. Chromosome slides were prepared using a gradual‐fixation/air‐drying method (Mikamo & Kamiguchi, 1983). Briefly, oocytes were treated with Fixative I (methanol:acetic acid:distilled water = 5:1:4) for 6–8 min and put onto a glass slide with a small amount of Fixative I. Then, the oocytes were treated with Fixative II (methanol:acetic acid = 3:1) for 2 min, followed by treatment with Fixative III (methanol:acetic acid:distilled water = 3:3:1) for 1 min. The slides were air‐dried under conditions of 50–60% humidity at 22–24°C. For conventional chromosome analysis, the slides were stained with 2% Giemsa (Merck KGaA) for 8 min. C‐band staining was used to distinguish between structural chromosome aberrations and aneuploidy (Tateno & Kamiguchi, 2007).
Chromosome analysis by multicolor FISH
Spleens were removed under sterile conditions from mice produced by spermatocyte injection. Lymphocytes were isolated from the spleen and incubated in a tissue culture tube at a cell concentration of 1 × 106/ml in RPMI1640 (Nacalai Tesque) containing lipopolysaccharide (10 μg/ml, Merck KGaA), concanavalin A (3 μg/ml, Nacalai Tesque), 2‐mercaptoethanol (50 μM, Nacalai Tesque), and FBS (6%) at 37°C under 5% CO2 in humidified air for 48 h. Colcemid (KaryoMAX, Gibco) at a concentration of 0.02 μg/ml was added to the cell suspension for the last 2 h of culture to arrest the cell cycle at metaphase. The cells were centrifuged at 420 g for 5 min and resuspended in 3 ml of a hypotonic solution (0.075 M KCl). Twenty minutes later, 2 ml of Carnoy’s fixative (methanol:acetic acid = 3:1) was added to the cell suspension. Cells were centrifuged at 420 g for 5 min and resuspended in 5 ml of fresh Carnoy’s fixative. Centrifugations and fixations were repeated three times. Chromosome preparations were made using a Hanabi metaphase spreader (ADSTEC). For multicolor FISH analysis, the chromosome slides were hybridized with 21XMouse (MetaSystems) according to the manufacturer’s protocol. For denaturation of chromosomal DNA, the slides were incubated in 2× saline sodium buffer (SSC) at 70°C for 30 min and then treated with 0.07 M NaOH at room temperature for 1 min. The denatured slides were washed in 0.1× SSC and 2× SSC at 4°C for 1 min each and dehydrated with a series of 70, 95, and 100% ethanol. Multicolor FISH probes were denatured at 75°C for 5 min and applied to the chromosome slides. After hybridization at 37°C for 48 h in a humidified chamber, the chromosome slides were treated with 0.4× SSC at 72°C for 2 min, washed in 2× SSC with 0.05% Tween20 (Merck KGaA) at room temperature for 30 s, and rinsed in distilled water. For counterstaining, the slides were covered by a coverslip with DAPI/Antifade (MetaSystems). The chromosome slides were observed using fluorescent microscopy. Fluorescence images were captured using a high‐sensitive digital camera (α7s, SONY). The images were imported into the ChromaWizard software (Auer et al, 2018) to assign fluorescence colors to each chromosome. Based on these fluorescence colors, the chromosome numbering was determined. Ten metaphase cells per mouse were analyzed for karyotyping.
Multicolor FISH analysis of MII oocytes was performed as described above, except that the chromosome slides were treated with NaOH, 0.07 M at 4°C (instead of at room temperature) for 1 min.
Chromosome analysis by Giemsa banding (G‐banding)
When multicolor FISH analysis revealed a possible chromosome deletion, additional G‐band staining was performed to identify the lost part of the chromosomes. The chromosome slides were treated with 0.025% trypsin (FUJIFILM Wako Pure Chemical Corporation) for 2 min at room temperature, washed in PBS, and stained with 4% Giemsa for 8 min. Deletion sites were determined according to the band pattern nomenclature of mouse chromosomes (Nesbitt & Francke, 1973).
Whole‐mount immunostaining
Immunofluorescence staining was conducted as described previously (Kyogoku & Kitajima, 2017). Metaphase I oocytes were collected 4–5 h after spermatocyte injection. They were fixed in 2% paraformaldehyde in PBS–polyvinyl alcohol (PVA; pH 7.4) for 30 min. After samples were blocked and permeabilized in PBS–PVA containing 1 mg/ml BSA (PBS–PVA–BSA) and 0.1% Triton X‐100 (Nacalai Tesque Inc.), the oocytes were incubated with the appropriate primary antibodies at 4°C overnight, washed several times in PBS–PVA–BSA and incubated with secondary antibodies for 90 min at room temperature. DNA was counterstained with 40 µg/ml of Hoechst 33342. Finally, the oocytes were washed and transferred to BSA–PVA for imaging with a Zeiss LSM780 confocal microscope. The following primary antibodies were used: Mouse anti‐C‐MAD2 (1:200, sc‐65492; Santa Cruz), human anti‐centromere antibodies (ACA; 1:200, 15‐234; Antibodies Incorporated), rabbit anti‐SGO2 (1:500; Lee et al, 2008), and mouse anti‐PLK1 (1:500. ab17057; Abcam). The secondary antibodies were Alexa Fluor 555 goat anti‐human IgG (H + L) (A21433), Alexa Fluor 488 goat anti‐mouse IgG (H + L) (A11029), and Alexa Fluor 488 goat anti‐rabbit IgG (H + L) (A11034; 1:400; Molecular Probes).
Live cell imaging
After linearization of the template plasmids, mRNA was synthesized using the mMESSAGE mMACHINE KIT (Ambion). The synthesized RNAs were stored at −80°C until use. The in vitro‐transcribed mRNAs (1.2 pl of 650 ng/μl major satellite–mClover (Miyanari et al, 2013), 0.6 pl of 350 ng/μl MAD1‐mNeonGreen‐CENP‐C (Kyogoku & Kitajima, 2017), and 0.6 pl of 350 ng/μl H2B–mCherry) were microinjected into oocytes. These were cultured for 1 h and then subjected to micromanipulation. Live cell imaging was performed as described (Kitajima et al, 2011; Sakakibara et al, 2015), with some modifications. Briefly, a Zeiss LSM710 or LSM880 confocal microscope equipped with a 40× C‐Apochromat 1.2NA water immersion objective lens (Carl Zeiss) was controlled by a multi‐position autofocus macro (Politi et al, 2018). For centromere tracking (Fig 3), 19 confocal z‐sections (every 1.5 μm) of 512 × 512 pixel x/y images covering a total volume of 35.4 × 35.4 × 28.5 μm were acquired at 200‐s intervals for at least 10 h after spermatocyte injection into oocytes expressing major satellite–mClover and H2B–mCherry. Centromere tracking was performed as described (Kitajima et al, 2011; Sakakibara et al, 2015). The parental origin of chromosomes was identified by the intensities of the chromosomes and the centromeres (lower fluorescent intensity for the spermatocyte chromosomes; Fig 2A).
Statistical analysis
The rates of chromosomal abnormalities and embryo development were evaluated using Fisher’s exact probability test. The body and placental weights of pups were evaluated using Student’s t‐test. Other statistical tests are indicated in the figure legends, as appropriate.
Author contributions
Narumi Ogonuki: Conceptualization; Investigation; Methodology; Writing—original draft. Hirohisa Kyogoku: Data curation; Software; Formal analysis; Investigation; Methodology. Toshiaki Hino: Formal analysis; Investigation; Methodology. Yuki Osawa: Resources; Methodology. Yasuhiro Fujiwara: Resources; Data curation; Investigation. Kimiko Inoue: Formal analysis; Methodology. Tetsuo Kunieda: Resources; Investigation; Methodology. Seiya Mizuno: Resources; Formal analysis; Investigation; Methodology. Hiroyuki Tateno: Data curation; Formal analysis. Fumihiro Sugiyama: Resources; Data curation; Methodology; Project administration. Tomoya S Kitajima: Conceptualization; Data curation; Formal analysis; Supervision; Funding acquisition; Investigation; Methodology; Writing—original draft; Project administration. Atsuo Ogura: Conceptualization; Data curation; Formal analysis; Supervision; Funding acquisition; Investigation; Visualization; Methodology; Writing—original draft; Project administration; Writing—review and editing.
In addition to the CRediT author contributions listed above, the contributions in detail are:
NO and AO conceived the project. The project was developed jointly by AO, TK, SM, HT, TSK, and FS. Experiments were carried out by NO, TH, HK, YO, YF, and KI. The paper was written by NO, TH, HK, TSK, and AO.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Acknowledgements
This study was supported by KAKENHI Grant Numbers 23500507 (N.O.), JP19H05758 (A.O. and T.H.), 20H05376 (H.K.), and 18H05549 (T.S.K.). Nanos3‐Cre driver mice (RBRC02568) and D1Pas1 knockout mice (RBRC05432) were provided by RIKEN BRC through the National BioResource Project of the MEXT/AMED, Japan.
EMBO reports (2022) 23: e54992.
See also: N Bouftas & Katja Wassmann (July 2022)
Contributor Information
Tomoya S Kitajima, Email: tomoya.kitajima@riken.jp.
Atsuo Ogura, Email: ogura@rtc.riken.go.jp.
Data availability
No data were deposited in a public database.
References
- Akiyama K, Akimaru S, Asano Y, Khalaj M, Kiyosu C, Masoudi AA, Takahashi S, Katayama K, Tsuji T, Noguchi J et al (2008) A new ENU‐induced mutant mouse with defective spermatogenesis caused by a nonsense mutation of the Syntaxin 2/Epimorphin (Stx2/Epim) gene. J Reprod Dev 58: 122–128 [DOI] [PubMed] [Google Scholar]
- Auer N, Hrdina A, Hiremath C, Vcelar S, Baumann M, Borth N, Jadhav V (2018) ChromaWizard: an open source image analysis software for multicolor fluorescence in situ hybridization analysis. Cytometry A 93: 749–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas L, Tyc K, El Yakoubi W, Morgan K, Xing J, Schindler K (2021) Meiosis interrupted: the genetics of female infertility via meiotic failure. Reproduction 161: R13–R35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatot CL, Lewis JL, Torres I, Ziomek CA (1990) Development of 1‐cell embryos from different strains of mice in CZB medium. Biol Reprod 42: 432–440 [DOI] [PubMed] [Google Scholar]
- Cobb J, Cargile B, Handel MA (1999) Acquisition of competence to condense metaphase I chromosomes during spermatogenesis. Dev Biol 205: 49–64 [DOI] [PubMed] [Google Scholar]
- El Yakoubi W, Wassmann K (2017) Meiotic divisions: no place for gender equality. Adv Exp Med Biol 1002: 1–17 [DOI] [PubMed] [Google Scholar]
- Enguita‐Marruedo A, Sleddens‐Linkels E, Ooms M, de Geus V, Wilke M, Blom E, Dohle GR, Looijenga LHJ, van Cappellen W, Baart EB et al (2019) Meiotic arrest occurs most frequently at metaphase and is often incomplete in azoospermic men. Fertil Steril 112: 1059–1070.e1053 [DOI] [PubMed] [Google Scholar]
- Fujiwara Y, Ogonuki N, Inoue K, Ogura A, Handel MA, Noguchi J, Kunieda T (2013) t‐SNARE Syntaxin2 (STX2) is implicated in intracellular transport of sulfoglycolipids during meiotic prophase in mouse spermatogenesis. Biol Reprod 88: 141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulka H, Langerova A (2014) The maternal nucleolus plays a key role in centromere satellite maintenance during the oocyte to embryo transition. Development 141: 1694–1704 [DOI] [PubMed] [Google Scholar]
- Hayashi K, Ohta H, Kurimoto K, Aramaki S, Saitou M (2011) Reconstitution of the mouse germ cell specification pathway in culture by pluripotent stem cells. Cell 146: 519–532 [DOI] [PubMed] [Google Scholar]
- Hikabe O, Hamazaki N, Nagamatsu GO, Obata Y, Hirao Y, Hamada N, Shimamoto SO, Imamura T, Nakashima K, Saitou M et al (2016) Reconstitution in vitro of the entire cycle of the mouse female germ line. Nature 539: 299–303 [DOI] [PubMed] [Google Scholar]
- Inoue H, Ogonuki N, Hirose M, Hatanaka Y, Matoba S, Chuma S, Kobayashi K, Wakana S, Noguchi J, Inoue K et al (2016) Mouse D1Pas1, a DEAD‐box RNA helicase, is required for the completion of first meiotic prophase in male germ cells. Biochem Biophys Res Commun 478: 592–598 [DOI] [PubMed] [Google Scholar]
- Ishikura Y, Ohta H, Sato T, Murase Y, Yabuta Y, Kojima Y, Yamashiro C, Nakamura T, Yamamoto T, Ogawa T et al (2021) In vitro reconstitution of the whole male germ‐cell development from mouse pluripotent stem cells. Cell Stem Cell 28: 2167–2179.e9 [DOI] [PubMed] [Google Scholar]
- Jamsai D, O’Bryan MK (2011) Mouse models in male fertility research. Asian J Androl 13: 139–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Ishiguro K‐I, Nambu A, Akiyoshi B, Yokobayashi S, Kagami A, Ishiguro T, Pendas AM, Takeda N, Sakakibara Y et al (2015) Meikin is a conserved regulator of meiosis‐I‐specific kinetochore function. Nature 517: 466–471 [DOI] [PubMed] [Google Scholar]
- Kimura Y, Tateno H, Handel MA, Yanagimachi R (1998) Factors affecting meiotic and developmental competence of primary spermatocyte nuclei injected into mouse oocytes. Biol Reprod 59: 871–877 [DOI] [PubMed] [Google Scholar]
- Kitajima TS, Ohsugi M, Ellenberg J (2011) Complete kinetochore tracking reveals error‐prone homologous chromosome biorientation in mammalian oocytes. Cell 146: 568–581 [DOI] [PubMed] [Google Scholar]
- Kyogoku H, Kitajima TS (2017) Large cytoplasm is linked to the error‐prone nature of oocytes. Dev Cell 41: 287–298 [DOI] [PubMed] [Google Scholar]
- Lane SIR, Jones KT (2017) Chromosome biorientation and APC activity remain uncoupled in oocytes with reduced volume. J Cell Biol 216: 3949–3957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Kitajima TS, Tanno Y, Yoshida K, Morita T, Miyano T, Miyake M, Watanabe Y (2008) Unified mode of centromeric protection by shugoshin in mammalian oocytes and somatic cells. Nat Cell Biol 10: 42–52 [DOI] [PubMed] [Google Scholar]
- Llano E, Gómez R, Gutiérrez‐Caballero C, Herrán Y, Sánchez‐Martín M, Vázquez‐Quiñones L, Hernández T, de Álava E, Cuadrado A, Barbero JL et al (2008) Shugoshin‐2 is essential for the completion of meiosis but not for mitotic cell division in mice. Genes Dev 22: 2400–2413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma JY, Li S, Chen LN, Schatten H, Ou XH, Sun QY (2020) Why is oocyte aneuploidy increased with maternal aging? J Genet Genomics 47: 659–671 [DOI] [PubMed] [Google Scholar]
- Mihajlović AI, FitzHarris G (2018) Segregating chromosomes in the mammalian oocyte. Curr Biol 28: R895–R907 [DOI] [PubMed] [Google Scholar]
- Mikamo K, Kamiguchi Y (1983) A new assessment system for chromosomal mutagenicity using Chinese hamster oocytes and early zygotes. J Toxicol Sci 8: 316 [Google Scholar]
- Miki H, Ogonuki N, Inoue K, Baba T, Ogura A (2006) Improvement of cumulus‐free oocyte maturation in vitro and its application to microinsemination with primary spermatocytes in mice. J Reprod Dev 52: 239–248 [DOI] [PubMed] [Google Scholar]
- Mikwar M, MacFarlane AJ, Marchetti F (2020) Mechanisms of oocyte aneuploidy associated with advanced maternal age. Mutat Res Rev Mutat Res 785: 108320 [DOI] [PubMed] [Google Scholar]
- Miyanari Y, Ziegler‐Birling C, Torres‐Padilla ME (2013) Live visualization of chromatin dynamics with fluorescent TALEs. Nat Struct Mol Biol 20: 1321–1324 [DOI] [PubMed] [Google Scholar]
- Nakamura S, Kobori Y, Ueda Y, Tanaka Y, Ishikawa H, Yoshida A, Katsumi M, Saito K, Nakamura A, Ogata T et al (2018) STX2 is a causative gene for nonobstructive azoospermia. Hum Mutat 39: 830–833 [DOI] [PubMed] [Google Scholar]
- Nesbitt MN, Francke U (1973) A system of nomenclature for band patterns of mouse chromosomes. Chromosoma 41: 145–158 [DOI] [PubMed] [Google Scholar]
- Obata Y, Villemure M, Kono T, Taketo T (2008) Transmission of Y chromosomes from XY female mice was made possible by the replacement of cytoplasm during oocyte maturation. Proc Natl Acad Sci USA 105: 13918–13923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogura A, Matsuda J, Asano T, Suzuki O, Yanagimachi R (1996) Mouse oocytes injected with cryopreserved round spermatids can develop into normal offspring. J Assist Reprod Genet 13: 431–434 [DOI] [PubMed] [Google Scholar]
- Ogura A, Ogonuki N, Miki H, Inoue K (2005) Microinsemination and nuclear transfer using male germ cells. Int Rev Cytol 246: 189–229 [DOI] [PubMed] [Google Scholar]
- Ogura A, Suzuki O, Tanemura K, Mochida K, Kobayashi Y, Matsuda J (1998) Development of normal mice from metaphase I oocytes fertilized with primary spermatocytes. Proc Nat Acad Sci USA 95: 5611–5615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogura A, Yanagimachi R (1993) Round spermatid nuclei injected into hamster oocytes form pronuclei and participate in syngamy. Biol Reprod 48: 219–225 [DOI] [PubMed] [Google Scholar]
- Osawa Y, Murata K, Usui M, Kuba Y, Le HT, Mikami N, Nakagawa T, Daitoku Y, Kato K, Shawki HH et al (2021) EXOC1 plays an integral role in spermatogonia pseudopod elongation and spermatocyte stable syncytium formation in mice. eLife 10: e59759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palermo G, Joris H, Devroey P, Van Steirteghem AC (1992) Pregnancies after intracytoplasmic injection of single spermatozoon into an oocyte. Lancet 340: 17–18 [DOI] [PubMed] [Google Scholar]
- Politi AZ, Cai Y, Walther N, Hossain MJ, Koch B, Wachsmuth M, Ellenberg J (2018) Quantitative mapping of fluorescently tagged cellular proteins using FCS‐calibrated four‐dimensional imaging. Nat Protoc 13: 1445–1464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakakibara Y, Hashimoto S, Nakaoka Y, Kouznetsova A, Höög C, Kitajima TS (2015) Bivalent separation into univalents precedes age‐related meiosis I errors in oocytes. Nat Commun 6: 7550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skarnes WC, Rosen B, West AP, Koutsourakis M, Bushell W, Iyer V, Mujica AO, Thomas M, Harrow J, Cox T et al (2011) A conditional knockout resource for the genome‐wide study of mouse gene function. Nature 474: 337–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H, Tsuda M, Kiso M, Saga Y (2008) Nanos3 maintains the germ cell lineage in the mouse by suppressing both Bax‐dependent and ‐independent apoptotic pathways. Dev Biol 318: 133–142 [DOI] [PubMed] [Google Scholar]
- Tanaka A, Suzuki K, Nagayoshi M, Tanaka A, Takemoto Y, Watanabe S, Takeda S, Irahara M, Kuji N, Yamagata Z et al (2018) Ninety babies born after round spermatid injection into oocytes: survey of their development from fertilization to 2 years of age. Fertil Steril 110: 443–451 [DOI] [PubMed] [Google Scholar]
- Tateno H, Kamiguchi Y (2007) Evaluation of chromosomal risk following intracytoplasmic sperm injection in the mouse. Biol Reprod 77: 336–342 [DOI] [PubMed] [Google Scholar]
- Yoshino T, Suzuki T, Nagamatsu GO, Yabukami H, Ikegaya M, Kishima M, Kita H, Imamura T, Nakashima K, Nishinakamura R et al (2021) Generation of ovarian follicles from mouse pluripotent stem cells. Science 373: eabe0237 [DOI] [PubMed] [Google Scholar]