

Abstract
In eukaryotic cells, the genome is three dimensionally (3D) organized with DNA interaction dynamics and topology changes that regulate gene expression and drive cell fate. Upon antigen stimulation, naive B cells are activated and form germinal centers (GC) for the generation of memory B cells and plasma cells. Thereby, terminal B-cell differentiation and associated humoral immune response require massive but rigorous 3D DNA reorganization. Here, we review the dynamics of genome reorganization during GC formation and the impact of its alterations on lymphomagenesis from the nucleosome structure to the higher order chromosome organization. We particularly discuss the identified architects of 3D DNA in GC B cells and the role of their mutations in B-cell lymphomas.
Introduction
In eukaryotic cells, DNA is packaged into chromatin through increasing levels of complex structures ranging from the nucleosome to higher order chromosome organization. The nucleosome is the primary structure, composed of 147 nucleotides of DNA wrapped around the core histone octamer, and forms the chromatosome when associated with linker histone H1. Higher order chromosome organization includes specific topological features including segregation in highly compact versus open compartments, topologically associated domains (TADs) and chromatin loops that bridge gene promoters and enhancers [1,2]. Modulation of genome architecture is a major driver of biological processes such as cell fate and cell differentiation, in large part by regulating gene expression, and requires chromatin remodelers and architectural proteins. Chromatin remodelers such as the BAF complex are multiprotein complexes that regulate DNA accessibility by removing or restructuring nucleosomes. Architectural proteins include components of the chromatosome but also cohesin complexes and CCCTC-binding factors that structure chromatin loops and TADs [3].
During the humoral response, B cells form germinal centers (GC) in the secondary lymphoid organs. The GC is composed of a dark zone where B cells rapidly proliferate and undergo somatic hypermutation and a light zone where B cells compete for interaction with T follicular helper cells, which enables them to undergo further differentiation [4]. Phenotypic transitions occurring during the GC reaction require B cells to undergo major reorganization of their three-dimensional (3D) genome architecture to coordinate specific transcription programs [5,6].
Most B-cell lymphomas are of GC B-cell origin, including diffuse large B-cell lymphoma (DLBCL) and follicular lymphoma. Epigenetic dysfunction is a hallmark of GC-derived lymphomas as shown by recurrent mutations of chromatin regulatory proteins and histones [4]. Herein, we highlight recent insights pertaining to DNA architectural regulation of chromatin during GC formation and describe how perturbations spanning from nucleosome structure to high-order chromatin organization contribute to GC lymphomagenesis.
Dynamics of genome architecture during germinal center formation and lymphomagenesis
Epigenetic and 3D genomic remodeling during germinal centers formation
The major phenotypic changes occurring in GC B cells during the humoral immune response involve broad shifts in transcriptional programs, driven by a variety of transcriptional activators and repressors. The genome can be roughly segregated into a highly compacted and inaccessible compartment called ‘compartment B’ where genes are transcriptionally silent, and ‘compartment A’ chromatin that is more accessible and hence available to be activated. Using genome-wide chromosomal conformation capture (Hi-C), Bunting et al. and Vilarassa-Blasi et al. reported that the transition from naive B cells to GC B cells is accompanied by a global decompaction of the genome in mice and humans, respectively [7,8•]. This phenomenon is reversible, with memory B cells showing genome architecture comparable to naive B cells [8]. Decompaction of chromatin in GC B cells allows transcription factors to bind to their cognate DNA elements and recruit chromatin modifiers complexes to introduce activating histone marks. Among these, the H3K4me1 mark is important to license DNA elements to act as gene enhancers, and H3K27ac mark contributes to enhancer–promoter interactions (EPI) and fully activate transcription. Vilarassa-Blasi et al. further identified an intermediate (I) compartment, with compaction states between compartments A and B and enrichment for the H3K27me3 mark, perhaps representing repressed genes with potential to transition to compartment A for activation or compartment B for silencing [8].
Chromatin accessibility has been mapped in human B cells undergoing the GC reaction using ATAC-seq. These data show a massive gain of chromatin accessibility during naive to GC B-cell transition particularly at OCT2 cognate binding sequences [9••]. A transcriptional co-factor called OCAB (or OBF1) is required for GC formation and is necessary for OCT2 transcriptional activation functions. Newly, in-depth mechanistic studies reveal how these proteins drive transition of naive B cells to GC B cells [9–11]. Strikingly, GC B-cell specific enhancer accessibility is predetermined by OCT2 ‘pre-positioning’ at these sites in naive B cells, even though these elements are not yet active. GC-specific expression of OCAB activates these latent OCT2 bound enhancers, leading to gain of accessibility, formation of superenhancers and gain of GC EPIs [9]. This role of OCAB is linked to its binding to OCT2, which enables more stable binding of this factor even to non-optimal OCT2 DNA elements. Notably, many of the key GC-specific superenhancers are formed through the actions of OCT2-OCAB complexes, which are required for them to make long-range EPIs. OCAB loss of function in GC-like cells leads to loss of GC EPIs and hence transcriptional programs including downregulation of the critical GC transcriptional repressor BCL6, and induction of GC exit programming including upregulation of IRF4 [9,11]. BCL6 is essential for GC formation, and its high level of expression in GC B cells is driven by a GC-specific locus control region (LCR) located more than 100 kb upstream [7]. Recent studies show that the BCL6 LCR forms around an OCT2 pre-positioned binding site in an OCAB-dependent manner, leading to a gain of accessibility that opens additional OCT2-OCAB binding sites (BS). Full LCR activation and interaction with the BCL6 promoter requires formation of a ternary complex between OCT2-OCAB and the critical GC transcription factor MEF2B, as well as the mediator complex [12]. These ternary complexes only form at three of the many enhancer elements that compose the BCL6-LCR, each of which is required to maintain BCL6 expression and viability of GC-derived lymphoma cells. In contrast, the remaining enhancers were not essential for these processes (Figure 1).
Figure 1.
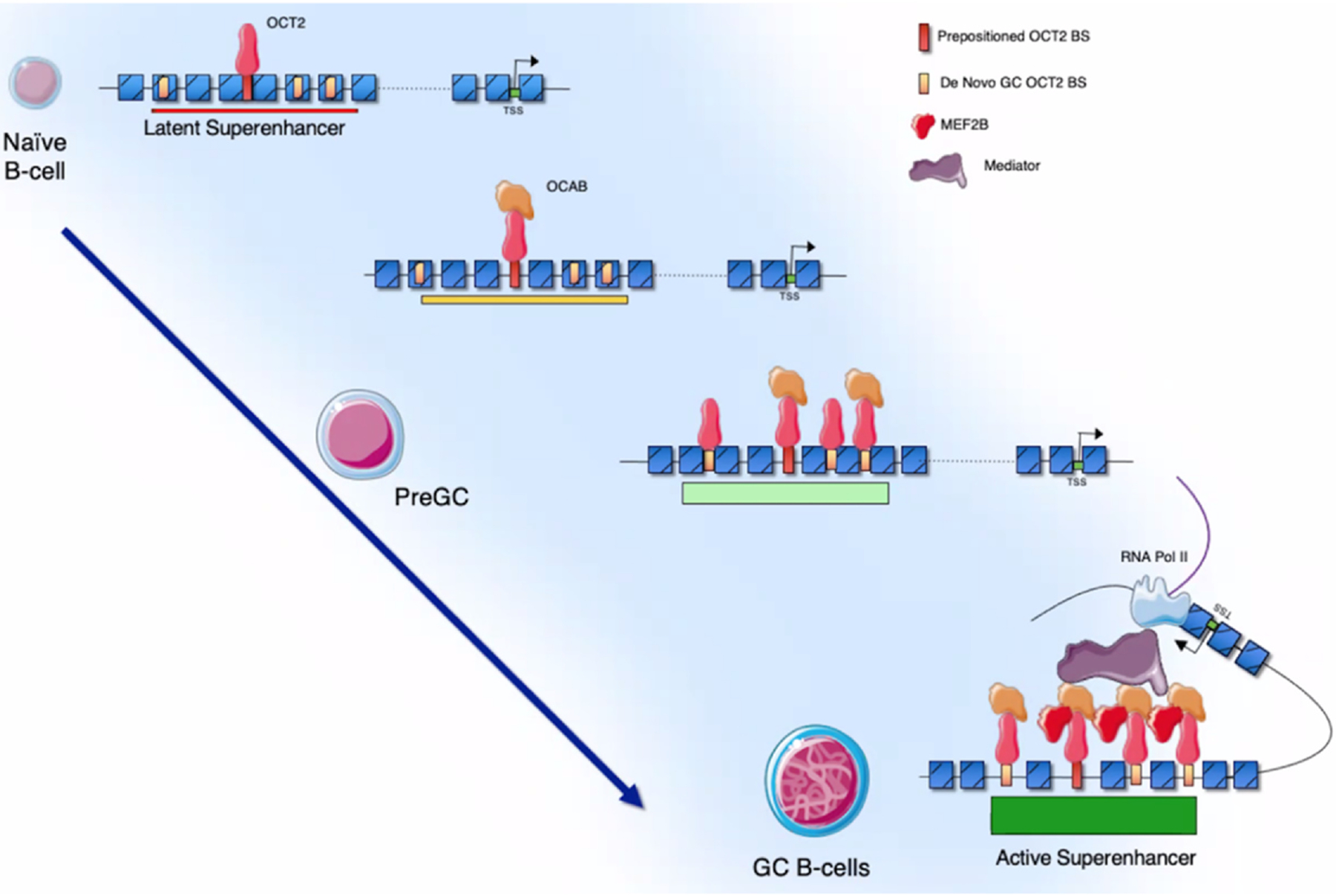
Graphical representation of GC identity establishment by OCT2-OCAB-MEF2B. OCT2 is ‘pre-positioned’ at latent GC enhancers and superenhancers in naive B cells, and de novo GC OCT2 BS are not accessible at this stage. Expression of OCAB in B cells transitioning to the GC reaction results in its binding to OCT2, with subsequent increase in chromatin accessibility. This in turn opens additional canonical and non-canonical OCT2 BS (de novo GC OCT2 BS) for further binding and chromatin opening by these complexes and the formation of GC-specific superenhancers. The further expression of MEF2B in GC B cells results in the formation of OCT2-OCAB-MEF2B ternary complexes, specifically at the key superenhancer subunits most essential for EPIs and GC B cells, which is further supported by OCAB recruitment of the mediator complex. Loss of OCAB reverses this process and leads to loss of accessibility at GC enhancers and loss of expression of GC-specific genes.
Chromatin remodeling and looping complexes control germinal center formation and are often disrupted in lymphomas
Chromatin accessibility involves nucleosome remodeling mediated by the ATP-dependent chromatin remodeling BAF complexes among others. There are several types of BAF complexes composed of up to 15 subunits which can eject or reposition nucleosomes at enhancers and promoters [13]. BAF subunits are frequently mutated in GC lymphomas. In particular, DLBCLs feature recurrent mutations in the BAF subunits genes such as ARID1A (7%), ARID1B (7%), SMARCA4 (6%) or BCL7A (5%) [4]. SMARCA4, encoding the BRG1 protein, is known to be important for the development of the B-cell lineage [14•]. Conditional deletion of Smarca4 in GC B cells abrogates GC formation in mice, where it makes critical contributions to enhancer chromatin accessibility required for the GC program [15]. In addition, in human DLBCL cell lines, a mutant of the SMARCA4 interactor BCL7A which lacks the N-terminal SMARCA4 interaction domain, reduces expression of checkpoint genes that control B-cell proliferation, such as CDKN1A (p21), highlighting a tumor suppressor role of BCL7A [16]. These studies point to critical roles for BAF complexes in GC B-cell proliferation and potential tumor suppressor functions during lymphomagenesis [15,16] (Figure 2).
Figure 2.
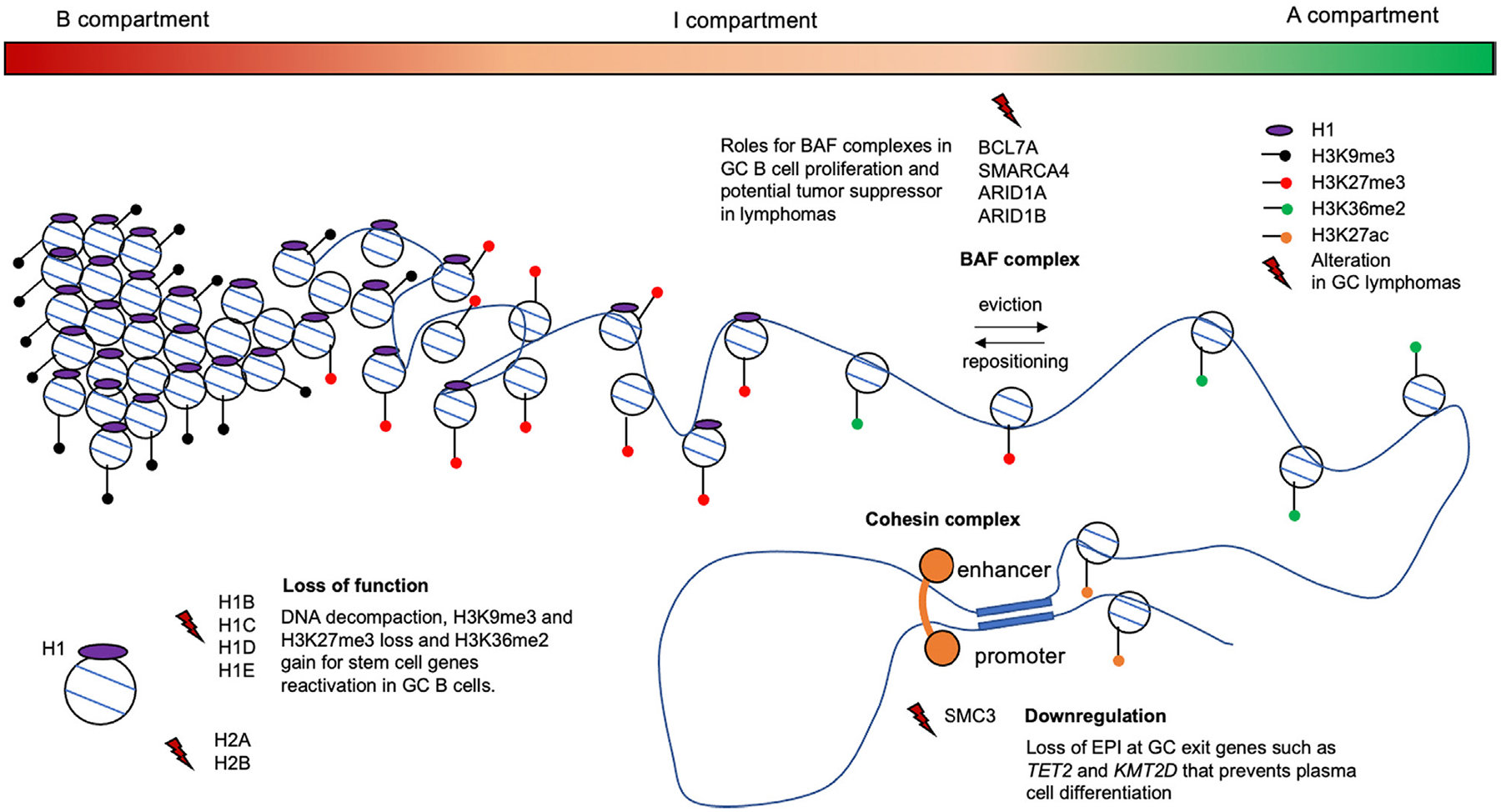
Graphical representation of chromatin architecture regulatory protein alterations in GC-derived lymphomas. The genome ranges from highly compacted to decompacted through three continuous compartments (B to I to A). These are associated with histone marks that either silence (H3K9me3 and H3K27me3) or facilitate activation (H3K36me2) of gene expression. The gradient of H1 density is associated with the range of chromatin compaction and hence epigenetic and transcriptional activation potential. H1 loss of function mutations lead to decompaction and activation of embryonic stem cell genes. Cohesin complexes create chromatin loops such as EPIs. SMC3 haploinsufficiency impairs GC B-cell differentiation by reducing such EPIs at GC exit genes and tumor suppressors thus contributing to lymphomagenesis. BAF subunits are involved in nucleosome ejection and repositioning and are frequently mutated in GC lymphomas. Their precise role in lymphomagenesis still needs to be clarified, but recent studies suggest that BAF components have a tumor suppressor role [30]. Histone H1 proteins are represented in purple, H3 post-modifications by dots: H3K9me3 (black), H3K27me3 (red), H3K36me2 (green), H3K27ac (orange) and lightning indicate alterations of chromatin regulatory proteins and histones.
The cohesin complex is required for the formation of TADs as well as most EPIs. Cohesin complexes form rings through which they can create interactions between distal elements. This process is critically dependent on the ATPase subunit SMC3 [17]. Cohesin-mediated loop exclusion is required for antibody class switch of GC B cells during the humoral immune response [18•]. Whereas homozygous deletion of Smc3 abrogates GC formation, its haploinsufficiency results in hyperplasia and lymphomagenesis in mice [19•,20]. This was linked to loss of EPI among critical GC exit genes known to have tumor suppressor functions including Kmt2d and Tet2. Smc3 haploinsufficient cells were hence impaired in their ability to undergo the terminal phases of plasma cell differentiation [19]. One interpretation of this result is that a reduction in the abundance of cohesin complexes reduces the chance that newly forming transcriptional complexes that mediate cell lineage transition would be able to form de novo stable, long-range enhancer-based chromatin loops with their respective promoter elements. Consistent with this notion, Smc3 haploinsufficiency in hematopoietic stem cells also impairs lineage commitment to all cell fates, except monocytes which are the evolutionary default for these cells [21]. Smc3 haploinsufficiency also increases DNA damage in GC B cells and accumulation of mutations in murine lymphomas [20]. Although cohesin mutations are rare in lymphoma, many patients feature low expression of these genes, which was shown to be an independent predictor of inferior clinical outcome [19,20] (Figure 2).
Chromatosome dysfunction as a driver of germinal center-derived lymphoma
Core histones H2A/H2B and linker histones H1 are frequently mutated in germinal center-lymphomas
The chromatosome is formed by a nucleosome in a complex with a linker histone H1. GC-derived lymphomas are the only tumors that feature high incidence of H1 mutations, specifically affecting replication-associated HIST1H1B (H1–5) (8%), HIST1H1C (H1–2) (12%), HIST1H1D (H1–3) (8%) and HIST1H1E (H1–4) (18%) genes. Although less frequent, mutations of core histones H2A and H2B are also present in lymphomas affecting HIST1H2AC (H2AC6) (6%), HIST1H2AM (H2AC17) (7%) and HIST1H2BK (H2BC12) (5%) genes, whereas H3 and H4 mutations are rare. Interestingly, H1 and H2 mutations are often co-occurrent in DLBCL [22–26]. Histone H1 missense mutations are widely spread across the globular domain, important for DNA and nucleosome binding, and the C-terminal tail, playing roles for higher affinity binding to the nucleosome and chromatin compaction [27]. Of note, H1 mutations are more common at certain residues (A164 and A123 in HIST1H1E and A65 and A101 in HIST1H1C), perhaps reflecting their deleterious impact on globular domain folding or contacts with DNA and other proteins. H2A and H2B mutations are also widespread with overrepresented mutations at A11, A127 and K96 for H2A and R87, S124 and S37 for H2B. These specific H2A/B mutations are not enriched in other cancer types and may be especially relevant to DLBCL (Figure 2).
Linker histone H1 drives lymphomagenesis through disruption of chromatin compartmentalization
H1 proteins are known to cause chromatin compaction and gene silencing. Lower H1 density is associated with transcriptionally active chromatin and H1 eviction can facilitate gene activation [1]. For example, during early B-cell differentiation, H1 eviction is mediated by NAP1-p300 at specific immune activation genes such as CD40 [28]. Gene expression and HPLC data from mouse tissues show that the H1c, H1d and H1e isoforms represent 90% of total H1 in mature B cells [29]. The most recurrent mutations in patients with lymphoma are on the HIST1H1C (H1–2) and HIST1H1E (H1–4) genes and are often co-occurrent [30••]. Mechanistically, H1 globular domain mutations impaired their association with chromatin, whereas C-terminal mutants bind normally but disrupt chromatin compaction [30]. Loss of H1c and H1e induced GC hyperplasia in mice and was associated with prominent shifting of chromatin from the B to A compartments indicating decompaction of specific regions of the genome. Strikingly, combining Hi-C and ChIP-seq, the data suggest a gradient of histone modifications according to genomic compaction state, whereby H1 loss of function leads to reduction of H3K9me3 from compartment B, reduction of H3K27me3 from regions of the genome that shift to compartment A and gain of H3K36me2 peaking at the most decompacted compartment A regions. As a consequence, many early stem cell genes that are normally sequestered in compacted chromatin in mature B cells become reactivated, due to compartment shift and gain of chromatin accessibility allowing transcription factors to bind to formerly inaccessible elements. Aberrant expression of these stem programs in H1 deficient GC B cells is associated with aberrant self-renewal of GC centrocytes, development of highly malignant lymphomas in mice, and inferior clinical outcomes in humans [30]. These data suggest that H1 mutations can confer stem-like activity in these lymphomas that arise from fully differentiated B cells (Figure 2). Interestingly, stem cell enhancers reactivated by H1 loss of function are enriched in OCT2 BS, perhaps indicating that OCT2-OCAB complexes could ‘hijack’ these enhancers to aberrantly drive expression of the associated genes. There remain many unanswered questions about H1 in lymphomas, such as those regarding specific functions of different subtypes and missense mutations and the role of H1 post-translational modifications [27,31–34].
Core nucleosome mutations in lymphomas: a mechanism to understand
The contribution of H2A and H2B mutations in GC-lymphomas remains unknown. However, studies in solid tumors suggest ways through which these mutations could also play an important role in lymphomas [35]. Indeed, screening of 160 distinct histones harboring different mutations for several biochemical assays identified that H2A and H2B mutants, mostly in the globular domain of these histones, alter both nucleosome stability and nucleosome sliding abilities. Overexpression of H2B mutants (H2B-E71, E76, and E113) dysregulated many pathways implicated in tumorigenesis [36]. Notably, H2B mutations at the E76 residue impair nucleosome stability, induce aberrant chromatin accessibility at gene promoters and confer aberrant growth patterns without affecting histone 3 post-translational modification marks [37•]. Another group showed that H2B-G53D mutations disrupt H2B interaction with DNA, leading to transcriptional activation of cancer-related genes and aberrant functional properties [38,39].
Conclusions and perspectives
Recent efforts have led to the characterization of the mutational landscape of GC lymphomas and described 3D DNA dynamics during GC formation. An emerging area of interest is how mutations in transcription factors and chromatin modifiers can affect 3D architecture. For example, recent studies demonstrated an aberrant gain of H3K27 acetylation at oncogene enhancers and aberrant compaction of chromatin occurring with a gain of function of PRC2 [40–42]. This knowledge will form the basis to reveal mechanistically how specific mutations impact the function of DNA architectural proteins, their biological contribution to epigenetic and transcriptional regulation during GC formation and lymphomagenesis, and whether these architectural effects might confer novel therapeutic vulnerabilities.
Acknowledgements
For this work, A.P., E.C., and A.M. are funded through (National Institutes of Health/National Cancer Institute) NIH/NCI R01 CA231561. A.P. is awarded the ACCR-Incyte Immuno-oncology Research Fellowship 2021. A.M. and E.C. are also funded by (Leukemia Lymphoma Society) LLS SCOR 7012. A.M. is funded by NIH/NCI R35 CA220499, LLS TRP 6572, the Follicular Lymphoma Consortium, and the Samuel Waxman Cancer Research Foundation. E.C. is also funded by NIH/NCI R01 CA250074 and NIH/NCI R01 CA260691.
Conflict of interest statement
The authors declare no direct competing financial or non-financial interests. However, A.M. has research funding from Janssen Pharmaceuticals, Sanofi, Epizyme and Daiichi Sankyo; has consulted for Epizyme and Constellation and is on the advisory board for KDAC Pharma.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest.
- 1.Fyodorov DV, Zhou B-R, Skoultchi AI, Bai Y: Emerging roles of linker histones in regulating chromatin structure and function. Nat Rev Mol Cell Biol 2018, 19:192–206, 10.1038/nrm.2017.94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McCord RP, Kaplan N, Giorgetti L: Chromosome conformation capture and beyond: toward an integrative view of chromosome structure and function. Mol Cell 2020, 77:688–708, 10.1016/j.molcel.2019.12.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rowley MJ, Corces VG: Organizational principles of 3D genome architecture. Nat Rev Genet 2018, 19:789–800, 10.1038/s41576-018-0060-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mlynarczyk C, Fontán L, Melnick A: Germinal center-derived lymphomas: the darkest side of humoral immunity. Immunol Rev 2019, 288:214–239, 10.1111/imr.12755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scourzic L, Salataj E, Apostolou E: Deciphering the complexity of 3D chromatin organization driving lymphopoiesis and lymphoid malignancies. Front Immunol 2021, 12:669881, 10.3389/fimmu.2021.669881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Azagra A, Marina-Zárate E, Ramiro AR, Javierre BM, Parra M: From loops to looks: transcription factors and chromatin organization shaping terminal B cell differentiation. Trends Immunol 2020, 41:46–60, 10.1016/j.it.2019.11.006 [DOI] [PubMed] [Google Scholar]
- 7.Bunting KL, et al. : Multi-tiered reorganization of the genome during B cell affinity maturation anchored by a germinal center-specific locus control region. Immunity 2016, 45:497–512, 10.1016/j.immuni.2016.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vilarrasa-Blasi R, et al. : Dynamics of genome architecture and chromatin function during human B cell differentiation and neoplastic transformation. Nat Commun 2021, 12:651, 10.1038/s41467-020-20849-y. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This study describes 3D DNA architecture dynamics during human terminal B-cell differentiation and defines the newly identified intermediate chromatin compartment.
- 9.Doane AS, et al. : OCT2 pre-positioning facilitates cell fate transition and chromatin architecture changes in humoral immunity. Nat Immunol 2021, 22:1327–1340, 10.1038/s41590-021-01025-w. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This study highlights the role of OCT2 prepositioning at specific chromatin sites in naive B cells to become specific enhancers in GC B cells with OCAB overexpression that drives establishment of GC B-cell integrity.
- 10.Betzler AC, Fiedler K, Hoffmann TK, Fehling HJ, Wirth T, Brunner C: BOB.1/OBF.1 is required during B-cell ontogeny for B-cell differentiation and germinal center function. Eur J Immunol 2022, 52:404–417. [DOI] [PubMed] [Google Scholar]
- 11.Song S, et al. : OBF1 and Oct factors control the germinal center transcriptional program. Blood 2021, 137:2920–2934, 10.1182/blood.2020010175 [DOI] [PubMed] [Google Scholar]
- 12.Chu C-S, et al. : Unique immune cell coactivators specify locus control region function and cell stage. Mol Cell 2020, 80:845–861, 10.1016/j.molcel.2020.10.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mittal P, Roberts CWM: The SWI/SNF complex in cancer — biology, biomarkers and therapy. Nat Rev Clin Oncol 2020, 17:435–448, 10.1038/s41571-020-0357-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Y, Zolotarev N, Yang C-Y, Rambold A, Mittler G, Grosschedl R: A prion-like domain in transcription factor EBF1 promotes phase separation and enables B cell programming of progenitor chromatin. Immunity 2020, 53:1151–1167, 10.1016/j.immuni.2020.10.009. [DOI] [PubMed] [Google Scholar]; • This study documents the recruitment of BRG1 by EBF1 to promote phase separation and chromatin accessibility for establishment of B-cell lineage.
- 15.Schmiedel D, Hezroni H, Hamburg A, Shulman Z: Brg1 supports B cell proliferation and germinal center formation through enhancer activation. Front Immunol 2021, 12:3482, 10.3389/fimmu.2021.705848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baliñas-Gavira C, et al. : Frequent mutations in the amino-terminal domain of BCL7A impair its tumor suppressor role in DLBCL. Leukemia 2020, 34:2722–2735, 10.1038/s41375-020-0919-5 [DOI] [PubMed] [Google Scholar]
- 17.Davidson IF, Peters J-M: Genome folding through loop extrusion by SMC complexes. Nat Rev Mol Cell Biol 2021, 22:445–464, 10.1038/s41580-021-00349-7 [DOI] [PubMed] [Google Scholar]
- 18.Zhang X, Zhang Y, Ba Z, Kyritsis N, Casellas R, Alt FW: Fundamental roles of chromatin loop extrusion in antibody class switching. Nature 2019, 575:385–389, 10.1038/s41586-019-1723-0. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This study highlights the role of chromatin loop extrusion in antibody class switching through accumulation of cohesin at switch regions in class switch recombination center.
- 19.Rivas MA, et al. : Smc3 dosage regulates B cell transit through germinal centers and restricts their malignant transformation. Nat Immunol 2021, 22:240–253, 10.1038/s41590-020-00827-8. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This study documents a dose-dependent function of cohesin complex in GC B cells and its impact in lymphomagenesis.
- 20.Rivas MA, et al. : Cohesin core complex gene dosage contributes to germinal center derived lymphoma phenotypes and outcomes. Front Immunol 2021, 12:3686, 10.3389/fimmu.2021.688493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Viny AD, et al. : Dose-dependent role of the cohesin complex in normal and malignant hematopoiesis. J Exp Med 2015, 212:1819–1832, 10.1084/jem.20151317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schmitz R, et al. : Genetics and pathogenesis of diffuse large B-cell lymphoma. N Engl J Med 2018, 378:1396–1407, 10.1056/NEJMoa1801445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chapuy B, et al. : Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat Med 2018, 24:679–690, 10.1038/s41591-018-0016-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lacy SE, et al. : Targeted sequencing in DLBCL, molecular subtypes, and outcomes: a haematological malignancy research network report. Blood 2020, 135:1759–1771, 10.1182/blood.2019003535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ennishi D, et al. : Double-hit gene expression signature defines a distinct subgroup of germinal center B-cell-like diffuse large B-cell lymphoma. J Clin Oncol 2019, 37:190–201, 10.1200/JCO.18.01583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reddy A, et al. : Genetic and functional drivers of diffuse large B cell lymphoma. Cell 2017, 171:481–494, 10.1016/j.cell.2017.09.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou B-R, et al. : Distinct structures and dynamics of chromatosomes with different human linker histone isoforms. Mol Cell 2021, 81:166–182, 10.1016/j.molcel.2020.10.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shimada M, et al. : Gene-specific H1 eviction through a transcriptional activator→p300→NAP1→H1 pathway. Mol Cell 2019, 74:268–283, 10.1016/j.molcel.2019.02.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Willcockson MA, et al. : H1 histones control the epigenetic landscape by local chromatin compaction. Nature 2021, 589:293–298, 10.1038/s41586-020-3032-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yusufova N, et al. : Histone H1 loss drives lymphoma by disrupting 3D chromatin architecture. Nature 2021, 589:299–305, 10.1038/s41586-020-3017-y. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This study documents H1 mutations as loss of function in lymphomas and demonstrates that H1 loss leads to embryonic stem cell gene reactivation through disruption of DNA architecture in GC B cells.
- 31.Osunsade A, et al. : A robust method for the purification and characterization of recombinant human histone H1 variants. Biochemistry 2019, 58:171–176, 10.1021/acs.biochem.8b01060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yoneda M, Yasui K, Nakagawa T, Hattori N, Ito T: Nucleosome assembly protein 1 (NAP-1) is a regulator of histone H1 acetylation. J Biochem (6) 2021, 170:763–773, 10.1093/jb/mvab098 [DOI] [PubMed] [Google Scholar]
- 33.Saha A, Seward CH, Stubbs L, Mizzen CA: Site-specific phosphorylation of histone H1.4 is associated with transcription activation. Int J Mol Sci 2020, 21:E8861, 10.3390/ijms21228861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Höllmüller E, et al. : Site-specific ubiquitylation acts as a regulator of linker histone H1. Nat Commun 2021, 12:3497, 10.1038/s41467-021-23636-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nacev BA, et al. : The expanding landscape of “oncohistone” mutations in human cancers. Nature 2019, 567:473–478, 10.1038/s41586-019-1038-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bagert JD, et al. : Oncohistone mutations enhance chromatin remodeling and alter cell fates. Nat Chem Biol 2021, 17:403–411, 10.1038/s41589-021-00738-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bennett RL, et al. : A mutation in histone H2B represents a new class of oncogenic driver. Cancer Discov 2019, 9:1438–1451, 10.1158/2159-8290.CD-19-0393. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This study refers H2B mutations in cancers and characterizes H2B-E76 mutations as involved in nucleosome destabilization, chromatin accessibility perturbation and gene expression alteration to trigger onco-genesis.
- 38.Wan YCE, et al. : Cancer-associated histone mutation H2BG53D disrupts DNA-histone octamer interaction and promotes oncogenic phenotypes. Signal Transduct Target Ther 2020, 5:27, 10.1038/s41392-020-0131-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wan YCE, et al. : The H2BG53D oncohistone directly upregulates ANXA3 transcription and enhances cell migration in pancreatic ductal adenocarcinoma. Signal Transduct Target Ther 2020, 5:106, 10.1038/s41392-020-00219-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sungalee S, et al. : Histone acetylation dynamics modulates chromatin conformation and allele-specific interactions at oncogenic loci. Nat Genet 2021, 53:650–662, 10.1038/s41588-021-00842-x [DOI] [PubMed] [Google Scholar]
- 41.Donaldson-Collier MC, et al. : EZH2 oncogenic mutations drive epigenetic, transcriptional, and structural changes within chromatin domains. Nat Genet 2019, 51:517–528, 10.1038/s41588-018-0338-y [DOI] [PubMed] [Google Scholar]
- 42.Béguelin W, et al. : Mutant EZH2 induces a pre-malignant lymphoma niche by reprogramming the immune response. Cancer Cell 2020, 37:655–673, 10.1016/j.ccell.2020.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]