

SUMMARY
Selective autophagy is a quality control system that specifically eliminates damaged or superfluous organelles, thereby maintaining cellular health. In this process, a double membrane structure called an autophagosome captures target organelles or proteins (i.e. “cargo”) and delivers the cargo to the lysosome for degradation. During the last decade, the attachment of the small protein modifier ubiquitin to cargo has emerged as a common mechanism for initiating organelle or protein capture by the autophagy machinery. In this process, a suite of ubiquitin-binding cargo receptors function to initiate autophagosome assembly in situ on the target cargo, thereby providing selectivity in cargo capture. Here we review recent efforts to understand the biochemical mechanisms and principles by which cargo are marked with ubiquitin and how ubiquitin-binding cargo receptors use conserved structural modules to recruit the autophagosome initiation machinery, with a particular focus on mitochondria and intracellular bacteria as cargo. These emerging mechanisms provide answers to long-standing questions in the field concerning how selectivity in cargo degradation is achieved to maintain cellular health.
INTRODUCTION
Biochemical processes within eukaryotic cells are organized within organelles, whose biogenesis and maintenance requires extensive quality control resources. Indeed, cellular specification relies on diverse organizational attributes for organelles that support specific cellular functions. As such, numerous mechanisms have evolved to both eliminate superfluous organelles and to detect and remove damaged or incompletely assembled organelles (Pohl and Dikic, 2019).
Arguably the best understood system for organelle quality control is selective autophagy. In this process, a double-membrane structure called a phagophore is nucleated on a target organelle (referred to throughout as “cargo”) and through a series of biochemical reactions grows to fully encapsulate the target organelle, pathogens or in some cases large protein complexes (e.g. protein aggregates) (Moehlman and Youle, 2020; Stolz et al., 2014; Wilkinson, 2019). This process is thought to be distinct from so-called “bulk” or non-selective autophagy, occurring under conditions of nutrient deprivation, which captures proteins or certain organelles in the absence of an organelle-specific signal (Pohl and Dikic, 2019). However, many of the same core machineries are used in both selective and non-selective processes. Much of the effort in the selective autophagy field has been directed at understanding three major questions: 1) What mechanisms are used to signal a damaged or supernumerary organelle?, 2) Once signals are activated, how is the organelle specified for autophagosome assembly?, and 3) What biochemical mechanisms physically link the marked organelle with the assembling autophagosome? Together, the over-arching question of concern is how is selectivity achieved in cargo capture such that only specific targets, and not nearby neighbors, are eliminated?
A major part of the answer to these questions has come from the discovery of distinct “eat-me” signals, which mark individual cargo for elimination. While numerous signals have been identified, ubiquitin (Ub)-dependent selective autophagy stands out, in part because it is used across diverse cargo and cellular contexts (Boyle and Randow, 2013; Dikic and Elazar, 2018; Pohl and Dikic, 2019; Tripathi-Giesgen et al., 2021). In this context, Ub is conjugated to macromolecules associated with the cargo, and serves as a platform for recruitment of the autophagy machinery via selective cargo receptors. Diverse Ub conjugating machinery respond to distinct damage signals to promote cargo ubiquitylation, often in a spatially and temporally controlled manner (Evans and Holzbaur, 2020; Harper et al., 2018; Narendra et al., 2008; Tripathi-Giesgen et al., 2021). In some cases, ubiquitylation is opposed by deubiquitylating enzymes that can help set thresholds for engagement of the quality control system (Bingol et al., 2014; Marcassa et al., 2018). At the same time, several forms of selective organelle turnover through autophagy appear to occur in the absence of a requirement for the Ub system, including some forms of endoplasmic reticulum autophagy and BNIP3/BNIP3L-dependent mitophagy, as reviewed elsewhere (Pohl and Dikic, 2019; Wilkinson, 2019).
Recent work in the field has led to significant advances in our understanding of how Ub chains on autophagic cargo are used to recruit a family of Ub-binding cargo receptor proteins and how individual cargo receptors form modular interactions with components of the autophagosome assembly machinery. Despite similar themes in cargo recognition, these studies suggest that unique combinations of receptors are used for specific types of cargo, with evidence of both selectivity and redundancy in promoting autophagic flux of cargo. Principally two types of cargo–damaged mitochondria and invading cytosolic bacteria–have served as model systems for discovery of the underlying regulatory mechanisms and provide a broad canvas for depicting mechanisms and interactions that drive cargo recognition (Harper et al., 2018; Onishi et al., 2021; Tripathi-Giesgen et al., 2021). Here, we first describe the pathways and mechanisms that are used to link Ub with two types of cargo: mitochondria and intracellular bacteria. Taking finding from these systems together with data from biochemical reconstruction experiments, we describe the modular nature of cargo receptors and underlying biochemical steps by which Ub binding is translated into autophagosome assembly. Finally, we relate emerging data from these pathways with parallel systems that have generally been considered to be Ub-independent, thereby providing a convergent model for both Ub-dependent and independent forms of selective organellophagy.
MULTI-STEP MECHANISM FOR Parkin ACTIVATION DURING MITOPHAGY
Mitochondria are subject to turnover by autophagy in diverse cell types and physiological conditions through both Ub-dependent and independent pathways (Montava-Garriga and Ganley, 2020; Ng et al., 2021; Onishi et al., 2021; Pickrell and Youle, 2015). Ub-dependent mitophagy requires the activity of the protein kinase PINK1 and the E3 Ub ligase Parkin (encoded by the PRKN gene), both of which are mutated in early-onset familial forms of Parkinson’s disease (Pickrell and Youle, 2015). In this pathway, a multi-step, feed-forward mechanism involving the mitochondrial localization of PINK1 and Parkin leads to the phosphorylation of Ub and Parkin itself by PINK1 (Kane et al., 2014; Kazlauskaite et al., 2014; Kazlauskaite et al., 2015; Koyano et al., 2014; Ordureau et al., 2014) and culminates in the ubiquitylation of dozens of mitochondrial proteins selectively on damaged mitochondrial to create a Ub “coat” (Sarraf et al., 2013), which in turn supports recruitment of autophagic machinery (Heo et al., 2015; Lazarou et al., 2015; Wong and Holzbaur, 2014) (Figure 1A).
Figure 1. Regulated cargo ubiquitylation to promotes autophagic clearance of damaged mitochondria and intracellular bacteria.
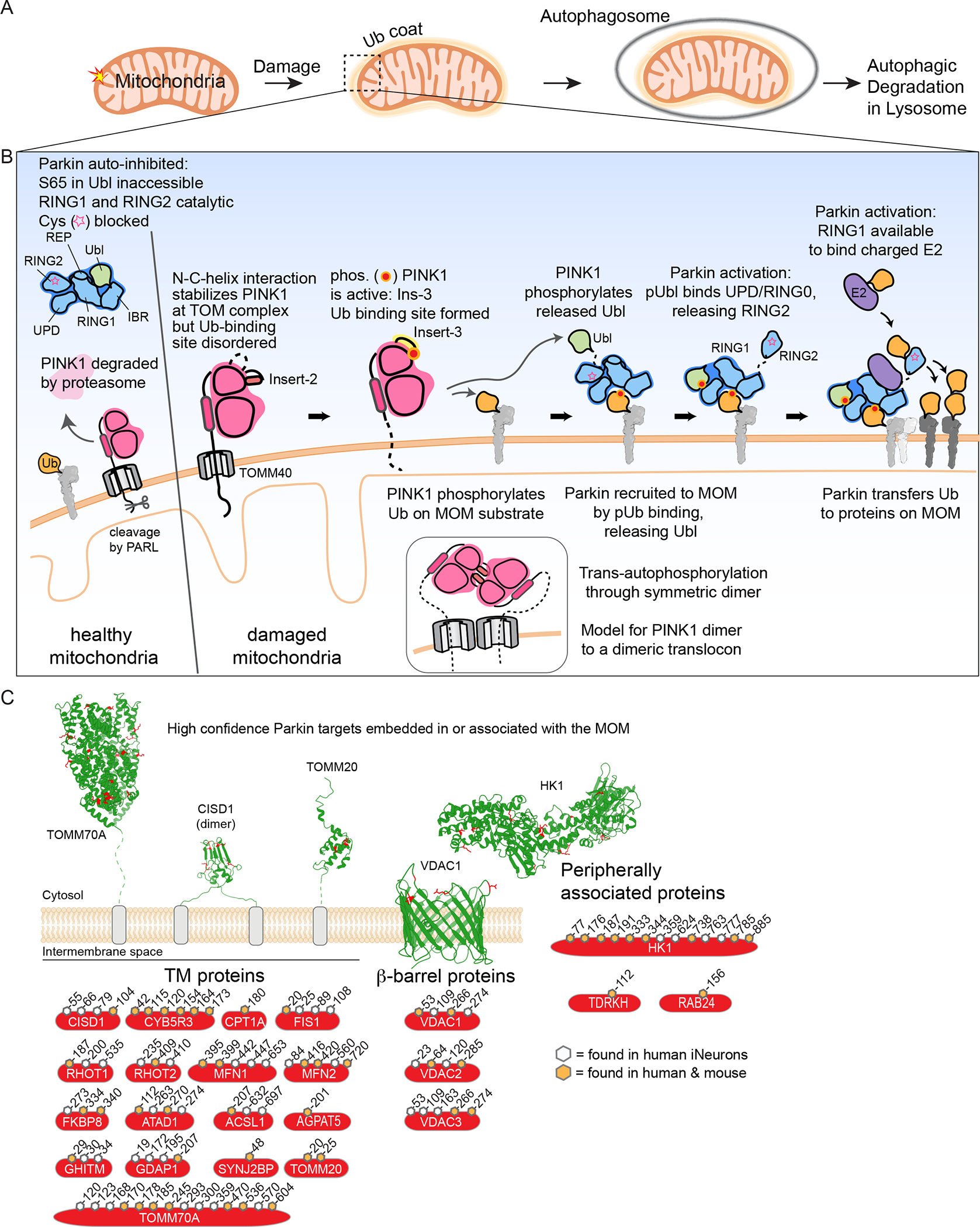
(A) Schematic of Ub-dependent mitophagy. Damaged mitochondria elicits a signal resulting in creation of a Ub “coat” surrounding the mitochondria. This Ub coat functions to promote formation of an autophagosome, which then can fuse with the lysosome to promote mitochondrial clearance.
(B) A feed-forward mechanism for Parkin and PINK1-dependent mitophagy. In cells with healthy mitochondria, cytosolic Parkin is in an auto-inhibited form and PINK1 is rapidly degraded. In response to mitochondrial damage, PINK1 is stabilized on the MOM in association with the translocon and trans-phosphorylates to generate a binding site for both Ub and the Parkin Ubl. MOM-tethered PINK1 can phosphorylate pre-existing Ub in proximity to the outer membrane, which can then bind Parkin to release its Ubl, making it available for phosphorylation by PINK1. Phosphorylated and activated Parkin can ubiquitylate numerous proteins on the MOM, thereby promoting downstream steps in mitophagy. Insets schematically depict dimeric PINK1 as an intermediate in trans-autophosphorylation, possibly templated by association with a dimeric translocon.
(C) High-confidence membrane-bound Parkin substrates, classified by membrane interaction type: single or double transmembrane segments, β-barrel proteins, and peripherally associated proteins. Examples displayed with identified ubiquitylated Lys residues shown in red and the cytoplasmic face of the MOM facing up. The high confidence ubiquitylation sites shown are based on biochemical studies in human and mouse cells in the context of endogenous Parkin in either human induced neurons (Ordureau et al., 2020) or primary mouse neurons (Antico et al., 2021). Sites indicated are high confidence sites in human proteins and orange-filled hexagons indicate sites that are also observed to be ubiquitylation by Parkin in mouse primary neurons. PDB files used: TOMM70A (2GW1), CISD1 (3EW0), TOMM20 (1OM2), VDAC1 (2JK4), and HK1 (1CZA).
PINK1 serves as a damage sensor, and in healthy mitochondria is rapidly imported through the translocon of the outer membrane (TOM complex) and degraded through a well-defined mechanism (Sekine and Youle, 2018) (Figure 1B). Various types of mitochondrial damage that affect mitochondrial inner membrane potential or clog the transport system promote PINK1 stabilization on the mitochondrial outer membrane (MOM) in association with the TOM complex (Lazarou et al., 2012), where PINK1 undergoes trans-autophosphorylation (Gan et al., 2021; Okatsu et al., 2012; Rasool et al., 2018; Rasool et al., 2021) (Figure 1B). Recent structural analysis of insect PINK1 revealed that autoactivation occurs through a symmetric dimer mediated by one of three insertions (Insert-2) in the classic bi-lobal kinase fold in PINK1 orthologs (Gan et al., 2021; Rasool et al., 2021) (Figure 1B). PINK1 autophosphorylation stabilizes and positions the Insert-3 region, allowing PINK1 to bind its substrates–Ub and the Parkin Ubl domain–and phosphorylate them at both Ser65 (Gan et al., 2021; Rasool et al., 2022). The PINK1 dimer places the autophosphorylated residue Ser228 in the active site of the other kinase providing a mechanism for trans-autophosphorylation, and mutations in the residues mediating this dimer interaction such as in Ins-2 prevent formation of phosphoSer65 Ub (pUb) in cells upon depolarization and prevent phosphorylation at Ser228 (Gan et al., 2021; Rasool et al., 2021) Further evidence of this dimer formation during PINK1 activation is evidenced by oxidative crosslinking of a Cys residue at the dimer interface and this reversible oxidation of the PINK1 may allow reactive oxygen species to regulate PINK1 activity, as mutation of this Cys residue slows Parkin recruitment to depolarized mitochondria and prevents pUb formation (Gan et al., 2021).
Recent work has also revealed an intramolecular interaction between the C-terminus of PINK1 and a long N-terminal helix, the functional role of which had remained unexplored despite containing residues mutated in genetic forms of Parkinson’s disease (Gan et al., 2021; Kakade et al., 2022; Rasool et al., 2021) (Figure 1B). Mutations that disrupt this helix-packing interaction prevent stabilization of PINK1 on the MOM and PINK1 activity upon mitochondrial damage. Extrapolation from the orientation of the PINK1 N-terminus and the genetic requirements for both TOM complex subunit TOMM7 and an acidic patch in PINK1’s N-terminus for PINK1 stabilization (Heo et al., 2019; Sekine et al., 2019) suggests a possible mechanism where the N-terminal MTS-containing portions of dimeric PINK1 may each bind to a dimeric TOM translocon assembly via symmetrical TOMM7 subunits (Gan et al., 2021; Kakade et al., 2022; Rasool et al., 2021) (Figure 1B).
This newly identified intramolecular helical interaction indicates that PINK1 is held close to the MOM and likely only engages substrates that are membrane proximal (Kakade et al., 2022) (Figure 1B). Numerous studies have found that Ub previously conjugated to proteins associated with the MOM is phosphorylated by PINK1 to generate pUb, which is a rate limiting step for Parkin activation [reviewed in (McWilliams and Muqit, 2017; Harper et al., 2018)]. However, the precise Ub molecules that are preferred PINK1 targets remains a matter of debate, despite a clearer view of the existing ubiquitylation sites on mitochondria revealed in the past few years. Given PINK1’s association with the TOM complex, plausible targets include the Ub attached to K61 of TOMM20 subunit of the TOM complex, a modification which is seen at low stoichiometry in healthy mitochondria in iNeurons and HeLa cells (Bingol et al., 2014; Ordureau et al., 2020; Ordureau et al., 2018). In support of the importance of this site, overexpression of TOMM20K56R/K61R, preventing ubiquitylation, blocks pathway activation (Bingol et al., 2014). Alternatively, basally ubiquitylated K274 in the highly abundant MOM β-barrel protein VDAC1 is also within proximity with the membrane and could be targeted by PINK1 (Ordureau et al., 2018), but a central role for VDAC1 in this context would be inconsistent with the effects of TOMM20K56R/K61R mutant expression. While MFN2 has been suggested to be a preferred PINK1 target (Vranas et al., 2021), cells lacking MFN1/2 maintain functional Parkin-PINK1 mitophagy pathway (Narendra et al., 2008), indicating that other proximal targets are sufficient for feed-forward activation.
pUb serves as a high-affinity receptor (Kd~20 nM) to recruit Parkin to the MOM and allosterically relieves auto-inhibition characteristic of the cytosolic form of Parkin (Gladkova et al., 2018; Ordureau et al., 2014; Sauve et al., 2015; Wauer et al., 2015) (Figure 1B). Multiple interactions contribute to Parkin auto-inhibition: (1) the active-site cysteine in RING2 is occluded by the unique Parkin domain (UPD, also called RING0), (2) RING1, bound by the “repressor” (REP) domain and the N-terminal Ub-like (Ubl) domain, is unable to interact with an E2, and finally (3) the two sites that must be near each other to transfer Ub from the E2 to the active-site Cys residue in RING2 are held far apart from each other. pUb binding drives release of the Parkin Ubl domain from RING1 (Sauve et al., 2018; Wauer et al., 2015). Upon release from RING1, the Parkin Ubl is phosphorylated at S65 by PINK1. Phospho-Ubl binds tightly to the UPD in competition with RING2, allowing for the release of the E2 conjugating enzyme-binding RING2 from its inhibitory contact with the UPD (Gladkova et al., 2018; Harper et al., 2018; Sauve et al., 2018) (Figure 1B). This series of events leads to a 4400-fold increase in Parkin’s Ub transfer activity (Ordureau et al., 2014), and Parkin’s tight association with pUb at the MOM when activated promotes ubiquitylation of proteins at the MOM, marking the mitochondria as damaged via Ub modification. Mutations in Parkin that block binding to pUb abolish both Parkin activation and downstream signaling (Kazlauskaite et al., 2015; Ordureau et al., 2020; Wauer et al., 2015), pointing to its critical role in this process.
Parkin TARGETS AND UBIQUITIN CHAIN ASSEMBLY
Parkin recruitment and activation promotes broad ubiquitylation of several dozen MOM proteins largely on cytosolic-exposed lysine residues, as determined in HeLa cells overexpressed Parkin or in systems employing endogenous Parkin [induced neurons (iNeurons) and primary neurons] (Antico et al., 2021; Bingol et al., 2014; Ordureau et al., 2020; Ordureau et al., 2018; Phu et al., 2020; Sarraf et al., 2013). Ubiquitylation occurs in multiple types of structural elements in proteins of varying abundance on the MOM, suggesting the absence of primary site specificity (Sarraf et al., 2013) (Figure 1C). Many target sites display a large dynamic range of ubiquitylation with stoichiometries of ubiquitylation in iNeurons ranging from 0.2–0.5 for very abundant substrates such as VDACs to <0.1 for much lower abundance substrates such as MFN2 (Ordureau et al., 2018).
While the identity of Parkin targets is quite advanced, our understanding of the Ub proteoforms that are attached to individual substrates and how this is linked with downstream autophagic signaling is more poorly understood. In HeLa cells overexpressing Parkin, mitochondrial depolarization is associated with synthesis of K6, K11, K48, and K63-linked Ub chains, consistent with Parkin’s ability to assemble these linkage types in vitro (Ordureau et al., 2014). By far the most abundant modification in this context is mono-ubiquitylation, with less than half of new Ub synthesis occurring in chains, and the vast majority of pUb occurring either on mono-Ub or on the distal Ub in a chain (Swatek et al., 2019). However, in iNeurons and primary cortical neurons with endogenous Parkin, linkage assembly seems to be more specific to K63 chains (Antico et al., 2021; Ordureau et al., 2020), but as in HeLa cells, the vast majority of pUb is found on mono-Ub or the distal Ub in a chain (Ordureau et al., 2020). Interestingly, HeLa cells expressing only UbK63R display delayed mitophagic flux without apparent impact on primary ubiquitylation of several substrates, consistent with an important role for K63 chains in mitophagy (Ordureau et al., 2015). The reason for the discrepancy in chain linkage types seen in neurons versus HeLa cells is unclear, but the absence of K6 chain assembly could reflect USP30 deubiquitylase in the neuronal setting, as this mitochondrially localized deubiquitylating enzyme is thought to have a preference for K6-linked Ub chains (Gersch et al., 2017). Although linear Ub chains are not detected during Parkin-dependent mitophagy, artificial tethering of linear di-, tetra-, and hexa-Ub chains to the MOM is sufficient to promote mitophagy independent of Parkin and PINK1 (Yamano et al., 2020), as elaborated further below.
These studies suggest that after PINK1-mediated phosphorylation of pre-existing Ub on the MOM, Parkin recruitment and activation promotes additional conjugation of primarily mono-Ub and short unbranched chains on many diverse substrates, which provides additional Ub as substrates for PINK1, thereby extending the feedforward phospho-ubiquitylation cascade (Figure 1A,B). A central design principle seems to be a membrane-proximal regulatory cascade, as both the positive (PINK1) (Rasool et al., 2021) and negative (USP30) (Ordureau et al., 2020) regulatory apparatus have limited access to targets beyond their membrane tethers. This potentially sets up an affinity-driven clustering mechanism for subsequent recognition of presumably short K63-linked Ub chains by cargo receptors, and also ensures that Parkin bound to proximal pUb will be within reach of PINK1 to promote Ubl phosphorylation.
DISTINCT UBIQUITYLATION SYSTEMS IN OTHER FORMS OF ORGANELLOPHAGY
Several other organelles employ Ub signaling to initiate autophagic degradation, using analogous yet molecularly divergent pathways (Pohl and Dikic, 2019). In xenophagy, rupture of vacuoles harboring bacteria result in the rapid recruitment of Galectins providing an initial eat-me signal – unleashing bacterial and host-membrane ubiquitylation (Perrin et al., 2004) and subsequent autophagic degradation (Boyle and Randow, 2013) (Figure 2). Unlike mitochondria, however, ubiquitylation of bacteria involves multiple, diverse Ub ligases and chain linkage types to promote not only autophagosome assembly but also activation of innate immune pathways, as elaborated below. Progress in this area has been extensive and the machinery and mechanisms involved have recently been reviewed in detail (Tripathi-Giesgen et al., 2021).
Figure 2. Pathway for ubiquitylation of bacterial vacuole and bacterial surface during xenophagy.
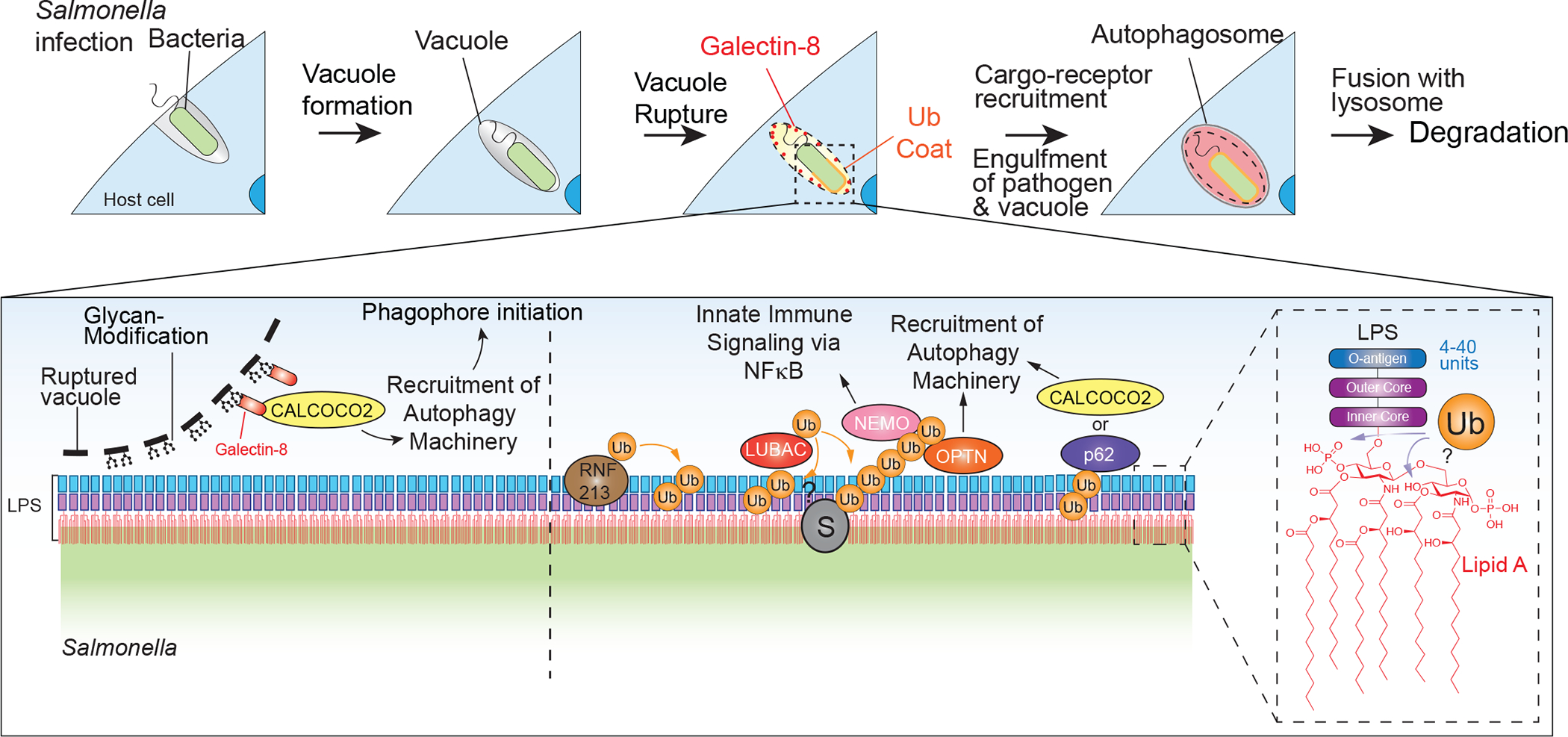
Bacteria enter mammalian cells within a vacuole, and upon vacuole rupture, Galectins are recruited to glycan-modified proteins on the inner leaflet of the vacuole membrane. Galectins such as LGALS8 can recruit cargo receptors such as CALCOCO2, which in turn associates with autophagy machinery to initiate in situ phagophore initiation. Vacuole rupture also allows access of RNF213, which initiates a ubiquitylation cascade by catalyzing ubiquitylation of LPS on the bacterial surface. This ubiquitylation recruits LUBAC, which then can assemble M1 Ub chains to promote recruitment of OPTN and NEMO to promote innate immunity and autophagy activation. CALCOCO2 and SQSTM1 are also recruited to ubiquitylated bacteria to further support activation of autophagy.
Salmonella, arguably the best understood xenophagy model-system, is targeted by cellular autophagy systems upon its escape from its vacuolar compartment using a three-pronged mechanism. First, vacuole rupture is associated with rapid recruitment of Galectin-3, 8 and 9 that bind exposed lumenal glycans, but only depletion of Galectin-8 affects bacterial restriction (Boyle and Randow, 2013; Thurston et al., 2009; Thurston et al., 2012). Early work suggested that Galectin-8 can directly bind the cargo receptor CALCOCO2 as part of the initial response to bacterial infection, and while Galectin-8 recruitment is correlated with detection of Ub co-incident with Galectin, Ub abundance co-incident with bacteria continues to increase after the initial Galectin-8 signal has decreased (Thurston et al., 2009; Thurston et al., 2012).
The molecular basis of increased ubiquitylation occurs through the action of RNF213 Ub ligase, which ubiquitylates the bacterium directly via a non-canonical linkage to the abundant bacterial outer membrane component lipopolysaccharide (LPS) upon vacuole rupture (Otten et al., 2021) (Figure 2). A recent cryoEM structure of RNF213 illustrates that in addition to a C-terminal multidomain Ub ligase module harboring a canonical RING domain, it contains six dynein-like AAA+ domains (two that are catalytically active ATPases and four that are inactive) (Ahel et al., 2020). Surprisingly, LPS ubiquitylation requires ATP binding and hydrolysis by the dynein-like domain but does not require the canonical RING domain (Otten et al., 2021). Instead, LPS lipidation is dependent on a CHC3H motif located in the large C-terminal multidomain, although this motif is not resolved in the RNF213 cryoEM structure (Otten et al., 2021; Ahel et al., 2020).
RNF213-dependent LPS ubiquitylation is a prerequisite for the third step, the recruitment of a second Ub ligase–the linear (M1) Ub assembly complex (LUBAC) (Noad et al., 2017; van Wijk et al., 2017)–that builds M1 chains on either bacterial surface proteins or Ub molecules previously conjugated by RNF213 (Otten et al., 2021). Thus, RNF213 can be viewed as a sensor of the bacterial surface that is directly hard-wired to initiate a ubiquitylation cascade on highly abundant and specific LPS molecules on the bacterial surface, providing hundreds of signal amplification sites by LUBAC as measured by super-resolution microscopy (Noad et al., 2017; van Wijk et al., 2017). M1 Ub chains activate innate immune signaling via the adaptor protein NEMO (Noad et al., 2017; van Wijk et al., 2017), as well as inducing recruitment of the autophagy machinery (Figure 2). Several other Ub ligases, including Parkin, LSRAM1, and ARIH1, have been reported to function in various aspects of xenophagy (Huett et al., 2012; Polajnar et al., 2017; Tripathi-Giesgen et al., 2021), but precisely how these systems interface with the two central E3s – RNF213 and LUBAC – remains poorly understood. The net result is the initiation of autophagosome formation on both the vacuolar membrane and the bacteria, ensuring that both components are eliminated via lysosomal degradation (Boyle and Randow, 2013; Tripathi-Giesgen et al., 2021) (Figure 2). As with Parkin-dependent mitochondrial elimination, Ub chain assembly is rate-limiting for bacterial elimination (Otten et al., 2021), pointing to the critical role of Ub-binding cargo receptors for efficient xenophagy. The identification of RNF213 as an enzyme capable of transferring Ub to a non-proteinaceous substrate suggests the possibility that other E3s may display non-canonical Ub transfer activity. Indeed, recent work suggests that LUBAC can also transfer Ub to sugars (Kelsall et al., 2021), raising the possibility that it ubiquitylates non-proteinaceous substrates during xenophagy. These results also raise questions as to whether mutations in RNF213 found in Moyamoya disease affect its non-canonical Ub transfer activity, although in the case of LPS, this does not seem to be the case (Otten et al., 2021).
LINKING UBIQUITYLATED CARGO TO CORE AUTOPHAGY MACHINERY
The marking of cargo with Ub is a rate-limiting step in the initiation of an autophagy cascade resulting in the encapsulation of the organelle in a double-membrane autophagosome. But how is this Ub signal decoded to initiate autophagosome formation? Our understanding of molecular mechanisms for autophagosome assembly have emerged largely from analysis of macroautophagy. Four major complexes are instrumental in macroautophagy (Chang et al., 2021b; Lamb et al., 2013): 1) the ULK kinase complex (composed of FIP200-ATG101-ATG13 and either the catalytic protein kinase ULK1 or ULK2), 2) the PI3 kinase complexes (composed of kinase VPS34 in complex with VPS15, ATG14, and BECLIN) that produces PI3P at the site of an incipient phagophore, 3) WD40-domain containing WIPI proteins in complex with ATG2 or ATG16 that nucleate lipid transport and recruitment of lipidation machinery at PI3P-enriched sites, and 4) the Ub-like ATG8 protein lipidation complex (composed of an ATG12-ATG5 complex that binds ATG16) and acts to promote ATG7 (E1)/ATG3 (E2)-dependent conjugation of ATG8 proteins to phosphatidylserine located in the phagophore. In mammals, there are six ATG8 proteins – MAP1LC3A/B/C and GABARAP/L1/L2 that play partially redundant roles in autophagosome maturation (Nguyen et al., 2016; Vaites et al., 2018).
A group of proteins classified as cargo receptors – defined as containing domains linking damage signals on autophagic cargo with the core autophagy machinery – link the macroautophagy pathway to specific cargo. These cargo receptors were initially identified as recruiting autophagy machinery via ATG8 proteins through LC3-interacting regions (LIR) within cargo receptors (Lamb et al., 2013; Stolz et al., 2014) (Figure 3A). In this model, the cargo receptor directly binds ATG8 previously deposited onto phagophore membranes through the action of the ULK1, VPS34, WIPI, and ATG7 cascade in response to an independent signal. However, mechanisms of autophagosome initiation during selective autophagy are unexplained by this model, and this model also does not explain how phagophore formation would independently be initiated concomitantly with activation of cargo ubiquitylation. Additionally, this ATG8-interaction-templated selective autophagy model is incongruent with observations that autophagosomes still form around cargo during Parkin-dependent mitophagy in cells lacking ATG8 conjugation activity (Nguyen et al., 2016; Vaites et al., 2018). Recent work from several laboratories now suggests an alternative model whereby organelle-associated Ub-binding cargo receptors initially recruit the ULK-FIP200 complex to support autophagosome assembly in situ, with ATG8 binding to cargo receptor occurring in a subsequent step (Figure 3B). This new perspective in autophagosome initiation helps explain how selectivity in autophagosome assembly around the target organelle is achieved while also highlighting the central role of specific cargo receptors (Chang et al., 2021a).
Figure 3. Model for in situ autophagosome nucleation.

(A) Early models for selective autophagy posited that LIR motifs within cargo receptors associate with ATG8 proteins on pre-existing phagophores, generated by the action of ULK1, VPS34, lipidation, and WIPI complexes.
(B) In newly emerging models for autophagosome nucleation, cargo receptors directly associate with the FIP200 component of the ULK1 complex, including through a FIR/LIR-claw domain interaction, to initiate autophagosome assembly adjacent to the surface of the target cargo. Receptor-ULK1 complexes can be recruited either via galectins in association with glycans or via ubiquitylated cargo. As ATG8 becomes conjugated to the growing phagophore, ATG8 proteins can subsequently bind to LIR motifs in cargo receptors to ensure cargo capture and autophagosome maturation in proximity to the target surface.
ORGANIZATION AND FUNCTIONAL SELECTIVITY OF Ub-BINDING CARGO RECEPTORS
Five Ub-binding cargo receptors have been characterized extensively: OPTN (also called optineurin), TAX1BP1, CALCOCO2 (also called NDP52), NBR1, and SQSTM1 (also called p62) (Figure 4A). Each of these cargo receptors contains structurally distinct C-terminal Ub-interacting domains (UBZ1, UBA, ZnF, or UBAN), which vary somewhat in their Ub binding specificity, as well as domains through which these receptors can homo- or hetero-oligomerize (Figure 4A). Although the analysis of Ub specificity in early studies was limited somewhat by the availability of various types of Ub chain linkages, CALCOCO2 was found to bind to M1, K63 and K48-linked chains (Ordureau et al., 2015; Thurston et al., 2016; Xie et al., 2015), and in some experiments showed a preference for K63 over K48-linked chains (Ordureau et al., 2015; Xie et al., 2015). In contrast, OPTN’s UBAN domain binds linear di-Ub and K63-linked di-Ub with a preference for linear chains but does not detectably associate with K48-linked di-Ub (Li et al., 2018). For OPTN, phosphorylation within the UBAN domain at Ser473 preferentially increases the affinity for K63-linked Ub, modulating the selectivity for Ub chain linkage type (Heo et al., 2015; Herhaus et al., 2019; Richter et al., 2016).
Figure 4. Cargo receptors nucleate autophagosome formation in situ.
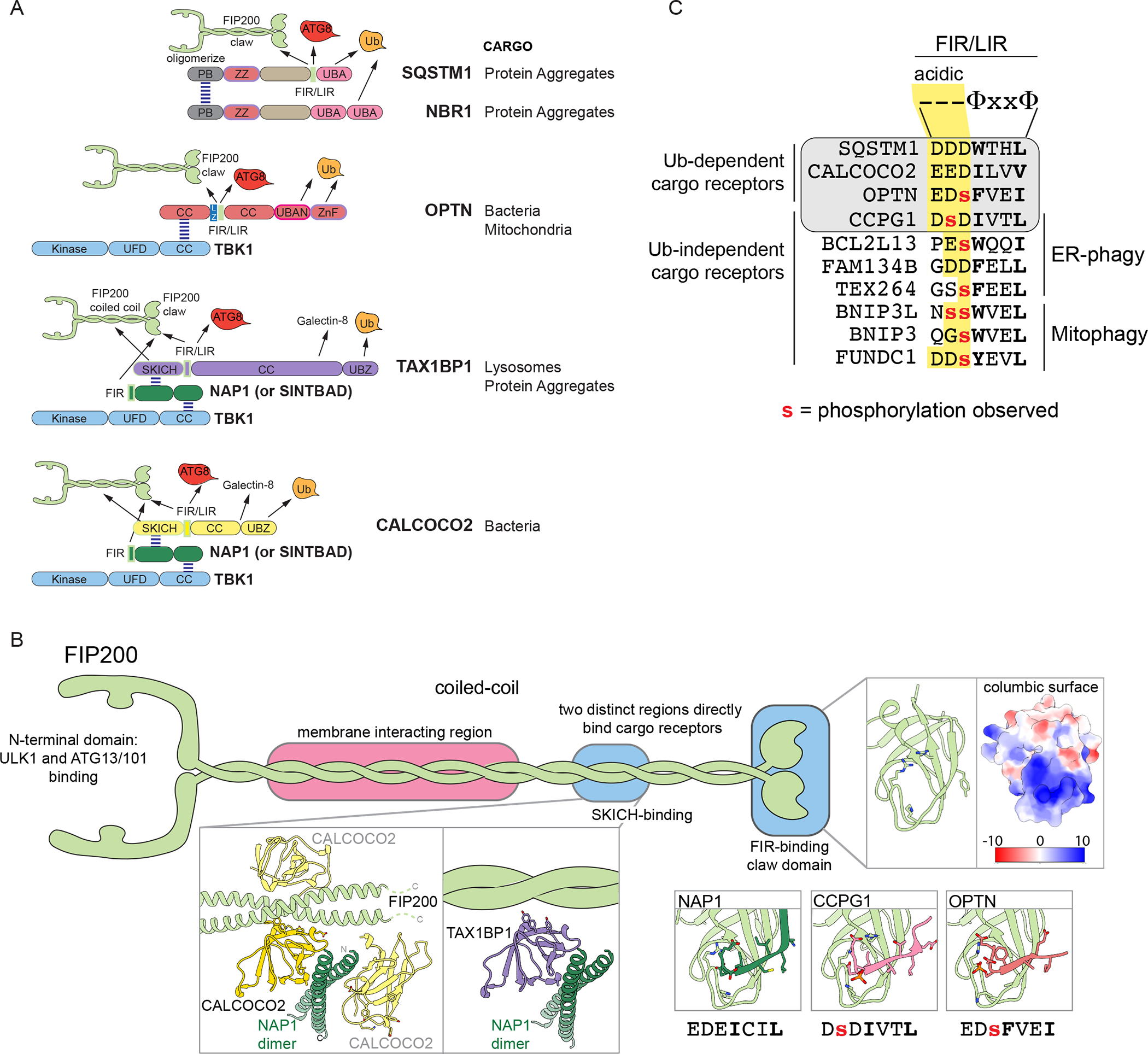
(A) Structural organization of major Ub-binding cargo receptors highlighting each receptor’s ability to self-associate, bind cargo through Ub binding domains, and recruit key components of the autophagy machinery: ULK1 complex through FIP200 and TBK1. CC, coiled-coil; UBA, Ub-associated domain; LZ, leucine zipper; UFD, Ub fold domain; ZnF, Zinc finger; UBAN, Ub binding domain in ABIN and NEMA; PB, Phox and BEM1-related domain; ZZ, ZZ-type zinc finger.
(B) Structural details of FIP200 interaction with autophagy cargo receptors. Dimeric FIP200 contains an N-terminal region sharing structural homology with the UFD of TBK1 scaffolding the other components of the ULK1 complex and is connected through a long coiled-coil to the C-terminal claw domain (PDB: 6DCE (Turco et al., 2019)). A C-terminal region of the coiled coil of FIP200 forms a trimeric complex with NAP1 and SKICH-domain-containing receptors CALCOCO2 (yellow) or TAX1BP1 (purple). Inset, structures of CALCOCO2 SKICH domain (dark yellow and light yellow for independent CALCOCO2 SKICH domains bound to dimeric FIP200 or NAP1) with FIP200 (light green) (PDB: 7EAA, (Fu et al., 2021)) and with NAP1 (dark green) (PDB: 5Z7L, (Fu et al., 2018)) overlaid to demonstrate possible orientations of the trimeric CALCOCO2 SKICH complex (light yellow). Inset, Negatively charged residues FIR sequences from NAP1 (PDB:7EA2 (Fu et al., 2018), CCPG1 (PDB:7D0E (Zhou et al., 2021)), and OPTN (PDB: 7CZM (Zhou et al., 2021)) bind in the same register on the claw domain of FIP200 and hydrophobic residues contribute to a β-sheet interaction with the claw.
(C) Consensus and selected sequences of FIR/LIR motifs in autophagy receptors. Lower case red residues have been detected to be phosphorylated in one or more cellular settings. Hydrophobic residues, Φ. Sequences highlighted with gray background have been shown to bind to the claw, while others are predicted FIR/LIR motifs in other Ub-independent cargo receptors, including those involved in mitophagy and ER-phagy.
Cargo receptors display evidence of both selectivity for cargo and functional redundancy (Figure 4A), consistent with their ability to bind each other and their diversity in Ub-binding domains. However, differences in Ub-binding specificity do not fully explain the selectivity or necessity of distinct cargo receptors in different types of selective autophagy. In the majority of cells examined to date, OPTN and to a lesser extent CALCOCO2 are required in Parkin-dependent mitochondrial turnover, and both proteins are rapidly recruited to mitochondria upon MOM ubiquitylation in a manner that requires their Ub-binding domains (Heo et al., 2015; Lazarou et al., 2015; Richter et al., 2016; Wong and Holzbaur, 2014). The finding that K63-linked Ub chains support mitophagy is consistent with the ability of OPTN to bind this chain type (Heo et al., 2015; Ordureau et al., 2015; Richter et al., 2016). Moreover, the ability of artificially recruited M1 Ub chains to promote Parkin-independent mitophagy (Yamano et al., 2020) likely reflects binding to M1 Ub chains by OPTN’s UBAN domain. In contrast, while SQSTM1 and TAX1BP1 are also recruited to damaged mitochondria with kinetics that are comparable with OPTN, they are not necessary for Parkin-dependent mitophagic flux (Heo et al., 2015; Lazarou et al., 2015). In cells lacking OPTN, CALCOCO2, and TAX1BP1, expression of OPTN alone is sufficient to support mitophagic flux and this requires TBK1 kinase activity, described below. Moreover, CALCOCO2, SQSTM1, and NBR1 are much less effective than OPTN in promoting mitophagy in the context of artificially tethered M1 chains, consistent with their more limited ability to bind linear Ub chains (Yamano et al., 2020).
Ectopic recruitment of PINK1 to the MOM has been shown to promote low levels of OPTN-dependent mitophagic flux independently of Parkin. While this suggests that Ub phosphorylation directly serves as a recognition element for OPTN (Lazarou et al., 2015), other evidence argues against a direct mechanism. First, pUb within K63-linked Ub chains is strongly inhibitory to OPTN binding in vitro (Heo et al., 2015; Richter et al., 2016). Second, a 3-fold increase in the stoichiometry of pUb on damaged mitochondria as a result of PINK1 overexpression dramatically decreases recruitment of both OPTN and CALCOCO2 to the MOM without affecting primary site ubiquitylation of MOM proteins by Parkin (Ordureau et al., 2018). Third, as mentioned above, recruitment of M1-linked di-Ub chains to the MOM promotes OPTN-dependent mitophagy in cells lacking PINK1 (Yamano et al., 2020), the only known Ser65-Ub kinase, formally ruling out an obligate direct role for Ser65 phosphorylation in OPTN recruitment in support of mitophagy. Fourth, the structures of the conserved UBAN domains of OPTN or NEMO with M1 or K63 di-Ub appear incompatible with direct interaction of pS65 with the UBAN coiled-coil (Herhaus et al., 2019; Yoshikawa et al., 2009). These results argue against a direct positive role for Ub phosphorylation in binding of cargo receptors to Ub chains on the MOM, and are more consistent with the primary role of Ub phosphorylation being Parkin recruitment and activation (Yamano et al., 2020). Moreover, the ability of artificially recruited linear di-Ub to fully support OPTN-dependent mitophagic flux (Yamano et al., 2020) is also consistent with the idea that short chains built by Parkin can promote receptor recruitment (Ordureau et al., 2020; Swatek et al., 2019).
Other types of ubiquitylated cargo display distinct requirements for cargo receptors. Damaged lysosomes accumulate K63-linked and K48-linked Ub chains in response to membrane rupture, but removal of K48-linked chains by a p97-YOD1 deubiquitylase-dependent pathway is required for flux of damaged lysosomes via autophagy (Papadopoulos et al., 2017). Thus, it would appear that K63-linked Ub chains are critical to lysophagy. But in contrast to mitophagy, lysophagy specifically requires the cargo receptor TAX1BP1, despite also recruiting OPTN, SQSTM1 and CALCOCO2 with similar kinetics (Eapen et al., 2021). Similar to CALCOCO2 (Thurston et al., 2012), TAX1BP1 also binds Galectin-8 (Bell et al., 2021; Huttlin et al., 2021), allowing direct recruitment to cargo via interaction of Galectin-8 recruited to glycans from within the lysosomal lumen exposed by damage. Nevertheless, TAX1BP1-dependent lysophagy is still dependent on lysosomal ubiquitylation (Eapen et al., 2021), suggesting that recruitment via Galectin-8 is insufficient for flux. Despite similar functional properties with TAX1BP1, overexpression of CALCOCO2 in cells lacking TAX1BP1, OPTN, and CALCOCO2 does not rescue lysophagic flux. In contrast, OPTN overexpression rescues lysophagic flux in cells lacking TAX1BP1, OPTN, and CALCOCO2 even though OPTN is not required for lysophagic flux when other cargo receptors are present (Eapen et al., 2021). Still more complexity exists with xenophagy, where individual elimination of CALCOCO2, OPTN, and SQSTM1 reduce gram-negative bacterial clearance to varying extents (Ravenhill et al., 2019; Thurston et al., 2009; Verlhac et al., 2015; Wild et al., 2011; Zheng et al., 2009), but co-depletion of CALCOCO2 and OPTN does not result in an additive phenotype (Wild et al., 2011). One limitation of these xenophagy studies is that they have primarily been done using RNAi, and systematic analysis using gene deletions could clarify the quantitative contributions of individual receptors. Nevertheless, it is possible that specific receptors are independently required for distinct steps that promote autophagosome formation. There may be two distinct types of cargo that must be recognized by distinct cargo receptors in xenophagy: the bacterium itself as well as the damaged vacuole each with their own ubiquitylation responses (Boyle and Randow, 2013) (Figure 2). Finally, rather than their differences in Ub and cargo recognition, differences in how cargo receptors recruit autophagy machinery may underlie the observed behaviors in selective autophagy processes.
CARGO RECEPTORS DIRECTLY RECRUIT AUTOPHAGY MACHINERY
Answers to the question of how autophagosomes are selectively assembled on specific cargo have emerged from a series of experiments that reveal direct recruitment of the ULK1-FIP200 kinase complex to cargo receptors. Together with other core autophagy machinery, this recruitment initiates autophagosome formation in situ on the target organelle. The first indication of direct recruitment of early autophagy machinery by cargo receptors was described in the context of Ub-independent ER-phagy via the CCPG1 receptor, where a previously identified LIR in CCPG1 was found to bind to a C-terminal region of FIP200 (Smith et al., 2018). This LIR motif was coined “FIR” for FIP200-interacting region and was shown to be required for CCPG1-dependent ER-phagy (Smith et al., 2018). This finding also revealed that homologous complexes in budding yeast autophagy likely function similarly. The yeast ULK1 kinase complex homolog Atg1-Atg11 is recruited to the cytoplasmic-to-vacuole targeting substrate pApe1 in complex with autophagy cargo receptor Atg19, resulting in activation of Atg1 kinase activity and induction of downstream autophagy steps (Kamber et al., 2015). Atg11 contains a domain that is similar to the C-terminus of FIP200, while Atg19 contains a LIR-like sequence and phosphorylation sites. As described below, a unifying model is emerging wherein cargo receptors can use overlapping sequences–FIR/LIR motifs–to sequential bind FIP200-ULK1 and ATG8 proteins thereby templating autophagosome assembly on cargo in situ.
Several structural themes have emerged that link cargo receptors with the FIP200-ULK1 complex to initiate autophagosome assembly in situ, although the precise interactions differ among receptors. At least two distinct sites on FIP200 interact with cargo receptors. The first involves a region within the homodimeric coiled-coil portion of FIP200, which binds to SKICH domains in TAX1BP1 and CALCOCO2 (Shi et al., 2020). These SKICH-domains can simultaneously bind both the related adaptor proteins NAP1 or SINTBAD and the coiled-coil region of FIP200, forming a trimeric complex (Ravenhill et al., 2019; Thurston et al., 2009; Vargas et al., 2019) (Figure 4A and B). In addition to stabilizing these cargo receptors’ interaction with FIP200, NAP1/SINTBAD binding supports formation of a larger complex containing the TBK1 protein kinase (Fu et al., 2018; Ravenhill et al., 2019; Shi et al., 2020), thereby linking autophagy receptors to a TBK1-dependent signaling function that enhances multiple forms of selective autophagy (Figure 4B). SKICH binding to the coiled-coil of FIP200 may also promote autophagy by allosteric activation of FIP200 membrane binding (Shi et al., 2020). This effect was proposed to facilitate recruitment of ATG9 vesicles to support in situ autophagosome formation (Shi et al., 2020), but additional studies are needed to elucidate the underlying mechanisms and to test the requirement for this region of FIP200 in autophagic flux.
A second cargo adaptor binding site in the C-terminal globular domain of FIP200–referred to as the “claw” domain–has basic and hydrophobic bindings surfaces that interacts with FIR motifs flanked by acidic residues (Turco et al., 2019; Zhou et al., 2021) (Figure 4B). Both CALCOCO2 and NAP1 contain FIR/LIR motifs that interact with the FIP200 claw (Zhou et al., 2021). Thus, the CALCOCO2-NAP1 complex has the potential to make bivalent interactions with the both C-terminal coiled-coil of FIP200 through SKICH interactions and claw domain of FIP200 through FIR interactions (Figure 4B). Beyond CALCOCO2’s FIR/LIR motif, analogous motifs in SQSTM1 and OPTN interact with the claw domain (Turco et al., 2019; Zhou et al., 2021) (Figure 4B). While acidic residues are present in a subset of FIR motifs, in others such as OPTN, the motif appear to be switchable with changing affinity for the claw domain based on the phosphorylation state of Ser or Thr residues N-terminal to the hydrophobic motif (Zhou et al., 2021). Phosphorylation of Ser177 in OPTN increases the affinity for the FIP200 claw by ~20-fold (Zhou et al., 2021). In SQSTM1, the FIR/LIR sequence is preceded by multiple acidic residues and therefore could be compatible with binding to the basic pocket in the claw domain without requiring phosphorylation, however SQSTM1 binding to the claw is enhanced by phosphomimetic substitutions C-terminal to the FIR/LIR (Turco et al., 2019), suggesting the possibility of multiple binding modes or additional interaction surfaces.
The functional importance of recruitment of FIP200 by cargo receptors for selective autophagy is demonstrated by the finding that artificial recruitment of CALCOCO2 to mitochondria in the absence of PINK1 induces mitophagy and requires the SKICH domain to do so (Vargas et al., 2019). This also demonstrates that the sole requirement for Parkin and PINK1 for mitophagy is the assembly of Ub chains as a signal promoting recruitment of the cargo receptor via its Ub-binding domain. Analogous requirements for CALCOCO2 in xenophagy have also been identified (Ravenhill et al., 2019). The SKICH domain of TAX1BP1 promotes ATG7-independent autophagic flux of NBR1, and employs its central coiled-coil domain to associate with NBR1, suggesting the possibility of receptor-receptor interactions (Ohnstad et al., 2020). Likewise for TAX1BP1-dependent lysophagy, loss of its SKICH domain or a single mutation disrupting the formation of the SKICH domain:NAP1:FIP200 complex, also abolishes lysophagic flux (Eapen et al., 2021). Moreover, in vitro reconstitution of MAP1LC3B lipidation on giant unilamellar vesicles – performed in the presence of the major autophagy complexes (ATG12-ATG5-ATG16, ATG7, ATG3, WIPIs, and VPS34 complex) and linear tetra-Ub–is stimulated by addition of CALCOCO2 or TAX1BP1 and more potently stimulated by co-addition of the ULK1-FIP200 complex (Chang et al., 2021b). Interestingly, reconstitution of MAP1LC3B lipidation is independent of ULK1 kinase activity, possibly reflecting a scaffolding role for the ULK1 complex in promoting Ub and receptor-dependent MAP1LC3B lipidation. Alternatively, the other machinery (e.g. purified VPS34 complex) may already be modified at activating phosphorylation sites, thereby removing the necessity of ULK1 kinase activity in this setting. Importantly, while CALCOCO2 and TAX1BP1 can weakly promote recruitment of the ULK1-FIP200 in the presence of core factors, it is stimulated by addition of the VPS34 complex (Chang et al., 2021b).
Taken together, these studies indicate that recruitment of the ULK1 kinase complex to target cargo via a FIP200-receptor interaction allows nucleation of phagophore formation within the vicinity of the cargo (Figure 3B). Such nucleation likely requires coupling to other major complexes in autophagy such as the VPS34 kinase complex, and ATG2 to support channeling of membrane building blocks for autophagosome assembly. In this model, ATG8 association with LIR motifs in cargo receptors would still be a relevant step as the phagophore grows and encapsulates the cargo, but occurs subsequent to ULK1 complex recruitment via claw-FIR interactions (Chang et al., 2021a) (Figure 3B). Indeed, it has been proposed that the affinity of ATG8 proteins for LIR motifs in cargo receptors coupled with the high density of ATG8 proteins on phagophores may effectively displace the claw domain of FIP200 from cargo receptors, thereby allowing for assembly of a phagophore membrane directly on the target cargo (Fu et al., 2021; Turco et al., 2019; Zhou et al., 2021) (Figure 3B).
ROLE OF TBK1 IN CARGO RECEPTOR FUNCTION
Multiple links between the TBK1 kinase and Ub-binding cargo receptors have been found in the context of autophagy dependent degradation of mitochondria, lysosomes, and bacteria, and all three pathways are promoted by TBK1 kinase activity. Several cargo receptors associate with TBK1 and/or are phosphorylated by TBK1 (Eapen et al., 2021; Harding et al., 2021; Heo et al., 2015; Lazarou et al., 2015; Morton et al., 2008; Richter et al., 2016; Thurston et al., 2009; Wild et al., 2011). TBK1 associates with the SKICH domains of TAX1BP1 and CALCOCO2 via NAP1/SINTBAD adaptor proteins (Fu et al., 2021; Thurston et al., 2009; Vargas et al., 2019). While OPTN lacks a SKICH domain, it directly binds TBK1 through an N-terminal coiled-coil region with sequence similarity to the region of NAP1 and SINTBAD that bind TBK1 (Li et al., 2016; Morton et al., 2008) (Figure 3A). In the context of mitochondrial damage, TBK1 activation depends on OPTN’s Ub-binding domain, indicating coupling of activation with recruitment to damaged mitochondria (Heo et al., 2015; Richter et al., 2016; Vargas et al., 2019). Upon activation, TBK1 phosphorylates the FIR/LIR motif in OPTN and in or near the UBAN domain (S473, S513). These modifications promote association of OPTN with K63-linked ubiquitin chains (Heo et al., 2015; Richter et al., 2016) and increase the affinity of the OPTN FIR/LIR motif (Wild et al., 2011) and FIP200 claw domain (Zhou et al., 2021), suggesting a mechanism for coupling TBK1 activation with OPTN binding to the FIP200-ULK1 complex. Additional data based on artificial recruitment of TBK1 in the context of CALCOCO2-dependent mitophagy is also consistent with trans-autoactivation, but here TBK1 primarily stabilizes the association of CALCOCO2 with FIP200 (Vargas et al., 2019). In in vitro reconstitution of lipidation, OPTN containing phosphomimetic substitutions promotes MAP1LC3 lipidation in the presence of the VPS34 complex but lipidation was not enhanced by addition of the ULK1-FIP200 complex (Chang et al., 2021b). Moreover, phospho-mimetic OPTN cannot associate with the ULK1 complex in vitro, while both CALCOCO2 and TAX1BP1 can recruit the ULK1-FIP200 complex (Chang et al., 2021b). These biochemical results are in contrast to the cellular context, where expression of OPTN in cells lacking both OPTN and CALCOCO2 rescues ULK1 recruitment to damaged mitochondria (Lazarou et al., 2015). At face value, this suggests the possibility of missing components in the minimal reconstitution system that could support OPTN-dependent ULK1-FIP200 recruitment, as seen in cells. Beyond ULK1-FIP200 recruitment to cargo, OPTN has also been demonstrated to form phase separated structures containing ATG9A-positive vesicles (Yamano et al., 2020). This co-association requires the leucine zipper region of OPTN adjacent to the FIR/LIR region (Figure 4A). Although the underlying mechanism and regulation still needs to be explored, this work suggests an independent role for OPTN in recruitment of autophagic machinery that is distinct from other cargo receptors.
Beyond cargo adaptors, TBK1 has additional substrates that may contribute to autophagic flux. These include GABARAPL2 and MAP1LC3C, whose phosphorylation by TBK1 blocks hydrolysis of their lipidated forms by the ATG4 hydrolases (Herhaus et al., 2020), and RAB7, which is transiently phosphorylated on Ser72 by TBK1 during mitophagy (Heo et al., 2018). RABGEFs are recruited to damaged mitochondria and co-recruit RAB5 and RAB7, which can then be acted on by RABGAPs to promote delivery of ATG9-containing vesicles to the site of autophagosome assembly (Yamano et al., 2014; Yamano et al., 2018). The precise role of RAB7 Ser72 phosphorylation is unknown, although phosphorylation is thought to interrupt the normal membrane cycling via the GDP dissociation inhibitor (GDI) proteins.
FIR-CLAW PARADIGM IN UB-INDEPENDENT FORMS OF AUTOPHAGY
The LIR motif is found broadly in both core autophagy proteins, especially those involved in conjugation or processing of ATG8 proteins, as well as in a variety of cargo receptors (Stolz et al., 2014). Indeed, mutation of LIR motifs within candidate selective autophagy receptors with concomitant loss of cargo flux is often considered diagnostic of such a function (Stolz et al., 2014). Several Ub-independent cargo receptors, including CCPG1 discussed above, typically have membrane imbedded or associated domains as well as LIR-containing regions exposed to the cytosol. CCPG1 also contains acidic residues N-terminal to its FIR/LIR motif and moreover, has a Ser residue N-terminal to the FIR/LIR whose phosphorylation greatly stimulates binding to the claw domain in vitro (Zhou et al., 2021) (Figure 4B). Thus, it is plausible that many previously identified LIR motifs function as FIR motifs to initiate selective autophagy. This includes BNIP3 and BNIP3L, factors required for Parkin-independent forms of mitophagy in multiple cellular settings (Ng et al., 2021). The C-terminus of BNIP3 is embedded in the MOM and its cytoplasm-exposed N-terminus contains a LIR motif preceded by two Ser residues. Phosphorylation of these Ser residues by ULK1 can promote mitophagy (Poole et al., 2021). Recent work has shown that BNIP3L promotes differentiation-dependent mitophagy during neurogenesis from stem cells (Ordureau et al., 2021). Yet stem cells harboring S36A/L39A LIR mutant in the BNIP3L gene still retain mitophagic flux, albeit at a reduced level (Ordureau et al., 2021). Thus, if BNIP3L’s FIR/LIR motif functions by interaction with the claw domain of FIP200, phosphorylation may not be absolutely required. Interestingly, numerous other cargo adaptors have candidate FIR/LIR motifs with acidic or potentially phosphorylatable N-terminal sequences, and there is evidence of phosphorylation of several of these based on phosphoproteomics (Figure 4C). Thus, a key question for several Ub-independent cargo receptors across diverse organellar cargo concerns the identity of signaling pathways that promote association of the FIR/LIR with FIP200 to initiate autophagy. In this regard, CAMK2 phosphorylation of the ER-phagy receptor FAM134B promotes flux, but modification occurs at sequences far from the FIR/LIR (Jiang et al., 2020), and how CAMK2 activity is integrated with FIR/LIR function remains unclear.
NEXT PHASES IN UNDERSTANDING SELECTIVE AUTOPHAGY
Recent work in the field has led to remarkable advances in our understanding of autophagosome biogenesis, which parallels advances in signals and mechanisms that couple cargo to the autophagosome assembly machinery. This includes recent reconstitution of autophagosome nucleation via ATG9 vesicle seeds (Sawa-Makarska et al., 2020), as well as the description of ATG2’s contribution to lipid transfer during autophagosomal growth (Chang et al., 2021a). The emerging view is of a highly complex array of proteins and processes present at the cargo-phagophore “synapse” that dictate both cargo recognition and phagophore expansion. This has been visualized in the case of mitophagy through proximity biotinylation of cargo receptors, which reveals molecular scale proximity of receptors to both each other as well as the autophagosome machinery itself (Heo et al., 2019). A central theme to emerge is multivalent interactions between various components, some of which are comparatively weak on their own, which creates a robust system for cargo capture. It is also likely that avidity through dense clustering of initiating membrane proximal Ub signals or cargo receptors themselves contributes to robustness of the autophagy system (Chang et al., 2021a). The development of more advanced reconstitution systems that more closely represent cellular cargo, and the application of similar techniques to a wider set of different types of selective autophagy, may reveal shared and divergent mechanisms explaining how such networks of multivalent interactions direct formation of an autophagosome on distinct cargo. In addition, studies are needed to more fully elucidate how the state of Ub proteoforms on cargo dictate and limit receptor recruitment. Advances in cellular imaging using tomography will likely provide spatial visualization of the autophagy process at high resolution, with a long-term goal of visualizing autophagy molecular machines in action. Finally, while the connections between factors in selective autophagy are becoming clearer, and mutations in OPTN, SQSTM1, and TBK1 are linked to amyotrophic lateral sclerosis and frontal temporal dementia (Harper et al., 2018), the critical cargoes whose elimination is defective in disease, as well as how this relates to analogous pathways in diseases such as Parkinson’s disease, remain elusive.
ACKNOWLEDGMENTS
J.W.H. is supported by grants from the NIH (AG011085, NS083524, NS110395, U24 HG006673), Aligning Science Across Parkinson’s (ASAP), Chan-Zuckerberg Neurodegeneration Challenge Network Collaborative Grant, and the Bluefield Project. The Michael J. Fox Foundation administers the grant ASAP-000282 on behalf of ASAP and itself. For the purpose of open access, the author has applied a CC-BY public copyright license to the Author Accepted Manuscript (AAM) version arising from this submission. We thank Lukas Henneberg for assistance with Figure 1.
REFERENCES
- Ahel J, Lehner A, Vogel A, Schleiffer A, Meinhart A, Haselbach D, and Clausen T (2020). Moyamoya disease factor RNF213 is a giant E3 ligase with a dynein-like core and a distinct ubiquitin-transfer mechanism. Elife 9. e56185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antico O, Ordureau A, Stevens M, Singh F, Nirujogi RS, Gierlinski M, Barini E, Rickwood ML, Prescott A, Toth R, et al. (2021). Global ubiquitylation analysis of mitochondria in primary neurons identifies endogenous Parkin targets following activation of PINK1. Sci Adv 7, eabj0722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell SL, Lopez KL, Cox JS, Patrick KL, and Watson RO (2021). Galectin-8 Senses Phagosomal Damage and Recruits Selective Autophagy Adapter TAX1BP1 To Control Mycobacterium tuberculosis Infection in Macrophages. mBio 12, e0187120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingol B, Tea JS, Phu L, Reichelt M, Bakalarski CE, Song Q, Foreman O, Kirkpatrick DS, and Sheng M (2014). The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature 510, 370–375. [DOI] [PubMed] [Google Scholar]
- Boyle KB, and Randow F (2013). The role of ‘eat-me’ signals and autophagy cargo receptors in innate immunity. Curr Opin Microbiol 16, 339–348. [DOI] [PubMed] [Google Scholar]
- Chang C, Jensen LE, and Hurley JH (2021a). Autophagosome biogenesis comes out of the black box. Nat Cell Biol 23, 450–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C, Shi X, Jensen LE, Yokom AL, Fracchiolla D, Martens S, and Hurley JH (2021b). Reconstitution of cargo-induced LC3 lipidation in mammalian selective autophagy. Sci Adv 7, eabg4922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikic I, and Elazar Z (2018). Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol 19, 349–364. [DOI] [PubMed] [Google Scholar]
- Eapen VV, Swarup S, Hoyer MJ, Paulo JA, and Harper JW (2021). Quantitative proteomics reveals the selectivity of ubiquitin-binding autophagy receptors in the turnover of damaged lysosomes by lysophagy. Elife 10, e72328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans CS, and Holzbaur EL (2020). Degradation of engulfed mitochondria is rate-limiting in Optineurin-mediated mitophagy in neurons. Elife 9, e50260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu T, Liu J, Wang Y, Xie X, Hu S, and Pan L (2018). Mechanistic insights into the interactions of NAP1 with the SKICH domains of NDP52 and TAX1BP1. Proc Natl Acad Sci U S A 115, E11651–E11660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu T, Zhang M, Zhou Z, Wu P, Peng C, Wang Y, Gong X, Li Y, Wang Y, Xu X, et al. (2021). Structural and biochemical advances on the recruitment of the autophagy-initiating ULK and TBK1 complexes by autophagy receptor NDP52. Sci Adv 7, eabi6582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan ZY, Callegari S, Cobbold SA, Cotton TR, Mlodzianoski MJ, Schubert AF, Geoghegan ND, Rogers KL, Leis A, Dewson G, et al. (2021). Activation mechanism of PINK1. Nature, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gersch M, Gladkova C, Schubert AF, Michel MA, Maslen S, and Komander D (2017). Mechanism and regulation of the Lys6-selective deubiquitinase USP30. Nat Struct Mol Biol 24, 920–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladkova C, Maslen SL, Skehel JM, and Komander D (2018). Mechanism of parkin activation by PINK1. Nature 559, 410–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding O, Evans CS, Ye J, Cheung J, Maniatis T, and Holzbaur ELF (2021). ALS- and FTD-associated missense mutations in TBK1 differentially disrupt mitophagy. Proc Natl Acad Sci U S A 118, e2025053118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper JW, Ordureau A, and Heo JM (2018). Building and decoding ubiquitin chains for mitophagy. Nat Rev Mol Cell Biol 19, 93–108. [DOI] [PubMed] [Google Scholar]
- Heo JM, Harper NJ, Paulo JA, Li M, Xu Q, Coughlin M, Elledge SJ, and Harper JW (2019). Integrated proteogenetic analysis reveals the landscape of a mitochondrial-autophagosome synapse during PARK2-dependent mitophagy. Sci Adv 5, eaay4624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo JM, Ordureau A, Paulo JA, Rinehart J, and Harper JW (2015). The PINK1-PARKIN Mitochondrial Ubiquitylation Pathway Drives a Program of OPTN/NDP52 Recruitment and TBK1 Activation to Promote Mitophagy. Mol Cell 60, 7–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo JM, Ordureau A, Swarup S, Paulo JA, Shen K, Sabatini DM, and Harper JW (2018). RAB7A phosphorylation by TBK1 promotes mitophagy via the PINK-PARKIN pathway. Sci Adv 4, eaav0443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herhaus L, Bhaskara RM, Lystad AH, Gestal-Mato U, Covarrubias-Pinto A, Bonn F, Simonsen A, Hummer G, and Dikic I (2020). TBK1-mediated phosphorylation of LC3C and GABARAP-L2 controls autophagosome shedding by ATG4 protease. EMBO Rep 21, e48317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herhaus L, van den Bedem H, Tang S, Maslennikov I, Wakatsuki S, Dikic I, and Rahighi S (2019). Molecular Recognition of M1-Linked Ubiquitin Chains by Native and Phosphorylated UBAN Domains. J Mol Biol 431, 3146–3156. [DOI] [PubMed] [Google Scholar]
- Huett A, Heath RJ, Begun J, Sassi SO, Baxt LA, Vyas JM, Goldberg MB, and Xavier RJ (2012). The LRR and RING domain protein LRSAM1 is an E3 ligase crucial for ubiquitin-dependent autophagy of intracellular Salmonella Typhimurium. Cell Host Microbe 12, 778–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttlin EL, Bruckner RJ, Navarrete-Perea J, Cannon JR, Baltier K, Gebreab F, Gygi MP, Thornock A, Zarraga G, Tam S, et al. (2021). Dual proteome-scale networks reveal cell-specific remodeling of the human interactome. Cell 184, 3022–3040 e3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Wang X, Ding X, Du M, Li B, Weng X, Zhang J, Li L, Tian R, Zhu Q, et al. (2020). FAM134B oligomerization drives endoplasmic reticulum membrane scission for ER-phagy. EMBO J 39, e102608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakade P, Ojha H, Raimi OG, Shaw A, Waddell AD, Ault JR, Burel S, Brockmann K, Kumar A, Ahangar MS, et al. (2022). Mapping of a N-terminal alpha-helix domain required for human PINK1 stabilization, Serine228 autophosphorylation and activation in cells. Open Biol 12, 210264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamber RA, Shoemaker CJ, and Denic V (2015). Receptor-Bound Targets of Selective Autophagy Use a Scaffold Protein to Activate the Atg1 Kinase. Mol Cell 59, 372–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, Banerjee S, and Youle RJ (2014). PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol 205, 143–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazlauskaite A, Kelly V, Johnson C, Baillie C, Hastie CJ, Peggie M, Macartney T, Woodroof HI, Alessi DR, Pedrioli PG, et al. (2014). Phosphorylation of Parkin at Serine65 is essential for activation: elaboration of a Miro1 substrate-based assay of Parkin E3 ligase activity. Open Biol 4,130213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazlauskaite A, Martinez-Torres RJ, Wilkie S, Kumar A, Peltier J, Gonzalez A, Johnson C, Zhang J, Hope AG, Peggie M, et al. (2015). Binding to serine 65-phosphorylated ubiquitin primes Parkin for optimal PINK1-dependent phosphorylation and activation. EMBO Rep 16, 939–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelsall IR, McCrory EH, Xu Y, Scudamore CL, Nanda SK, Mancebo-Gamella P, Wood NT, Knebel A, Matthews SJ, and Cohen P (2021). HOIL-1-catalysed ubiquitylation of unbranched glucosaccharides and its activation by ubiquitin oligomers. BioRxiv 10.1101/2021.09.10.459791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, Kimura Y, Tsuchiya H, Yoshihara H, Hirokawa T, et al. (2014). Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 510, 162–166. [DOI] [PubMed] [Google Scholar]
- Lamb CA, Yoshimori T, and Tooze SA (2013). The autophagosome: origins unknown, biogenesis complex. Nat Rev Mol Cell Biol 14, 759–774. [DOI] [PubMed] [Google Scholar]
- Lazarou M, Jin SM, Kane LA, and Youle RJ (2012). Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev Cell 22, 320–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, and Youle RJ (2015). The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524, 309–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Xie X, Wang Y, Liu J, Cheng X, Guo Y, Gong Y, Hu S, and Pan L (2016). Structural insights into the interaction and disease mechanism of neurodegenerative disease-associated optineurin and TBK1 proteins. Nat Commun 7, 12708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Xu D, Wang Y, Zhou Z, Liu J, Hu S, Gong Y, Yuan J, and Pan L (2018). Structural insights into the ubiquitin recognition by OPTN (optineurin) and its regulation by TBK1-mediated phosphorylation. Autophagy 14, 66–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcassa E, Kallinos A, Jardine J, Rusilowicz-Jones EV, Martinez A, Kuehl S, Islinger M, Clague MJ, and Urbe S (2018). Dual role of USP30 in controlling basal pexophagy and mitophagy. EMBO Rep 19, e45595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWilliams TG, and Muqit MM (2017). PINK1 and Parkin: emerging themes in mitochondrial homeostasis. Curr Opin Cell Biol 45, 83–91. [DOI] [PubMed] [Google Scholar]
- Moehlman AT, and Youle RJ (2020). Mitochondrial Quality Control and Restraining Innate Immunity. Annu Rev Cell Dev Biol 36, 265–289. [DOI] [PubMed] [Google Scholar]
- Montava-Garriga L, and Ganley IG (2020). Outstanding Questions in Mitophagy: What We Do and Do Not Know. J Mol Biol 432, 206–230. [DOI] [PubMed] [Google Scholar]
- Morton S, Hesson L, Peggie M, and Cohen P (2008). Enhanced binding of TBK1 by an optineurin mutant that causes a familial form of primary open angle glaucoma. FEBS Lett 582, 997–1002. [DOI] [PubMed] [Google Scholar]
- Narendra D, Tanaka A, Suen DF, and Youle RJ (2008). Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183, 795–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng MYW, Wai T, and Simonsen A (2021). Quality control of the mitochondrion. Dev Cell 56, 881–905. [DOI] [PubMed] [Google Scholar]
- Nguyen TN, Padman BS, Usher J, Oorschot V, Ramm G, and Lazarou M (2016). Atg8 family LC3/GABARAP proteins are crucial for autophagosome-lysosome fusion but not autophagosome formation during PINK1/Parkin mitophagy and starvation. J Cell Biol 215, 857–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noad J, von der Malsburg A, Pathe C, Michel MA, Komander D, and Randow F (2017). LUBAC-synthesized linear ubiquitin chains restrict cytosol-invading bacteria by activating autophagy and NF-kappaB. Nat Microbiol 2, 17063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnstad AE, Delgado JM, North BJ, Nasa I, Kettenbach AN, Schultz SW, and Shoemaker CJ (2020). Receptor-mediated clustering of FIP200 bypasses the role of LC3 lipidation in autophagy. EMBO J 39, e104948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okatsu K, Oka T, Iguchi M, Imamura K, Kosako H, Tani N, Kimura M, Go E, Koyano F, Funayama M, et al. (2012). PINK1 autophosphorylation upon membrane potential dissipation is essential for Parkin recruitment to damaged mitochondria. Nat Commun 3, 1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onishi M, Yamano K, Sato M, Matsuda N, and Okamoto K (2021). Molecular mechanisms and physiological functions of mitophagy. EMBO J 40, e104705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordureau A, Heo JM, Duda DM, Paulo JA, Olszewski JL, Yanishevski D, Rinehart J, Schulman BA, and Harper JW (2015). Defining roles of PARKIN and ubiquitin phosphorylation by PINK1 in mitochondrial quality control using a ubiquitin replacement strategy. Proc Natl Acad Sci U S A 112, 6637–6642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordureau A, Kraus F, Zhang J, An H, Park S, Ahfeldt T, Paulo JA, and Harper JW (2021). Temporal proteomics during neurogenesis reveals large-scale proteome and organelle remodeling via selective autophagy. Mol Cell, 81, 5082–5098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordureau A, Paulo JA, Zhang J, An H, Swatek KN, Cannon JR, Wan Q, Komander D, and Harper JW (2020). Global Landscape and Dynamics of Parkin and USP30-Dependent Ubiquitylomes in iNeurons during Mitophagic Signaling. Mol Cell 77, 1124–1142 e1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordureau A, Paulo JA, Zhang W, Ahfeldt T, Zhang J, Cohn EF, Hou Z, Heo JM, Rubin LL, Sidhu SS, et al. (2018). Dynamics of PARKIN-Dependent Mitochondrial Ubiquitylation in Induced Neurons and Model Systems Revealed by Digital Snapshot Proteomics. Mol Cell 70, 211–227 e218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordureau A, Sarraf SA, Duda DM, Heo JM, Jedrychowski MP, Sviderskiy VO, Olszewski JL, Koerber JT, Xie T, Beausoleil SA, et al. (2014). Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol Cell 56, 360–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otten EG, Werner E, Crespillo-Casado A, Boyle KB, Dharamdasani V, Pathe C, Santhanam B, and Randow F (2021). Ubiquitylation of lipopolysaccharide by RNF213 during bacterial infection. Nature 594, 111–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos C, Kirchner P, Bug M, Grum D, Koerver L, Schulze N, Poehler R, Dressler A, Fengler S, Arhzaouy K, et al. (2017). VCP/p97 cooperates with YOD1, UBXD1 and PLAA to drive clearance of ruptured lysosomes by autophagy. EMBO J 36, 135–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrin AJ, Jiang X, Birmingham CL, So NS, and Brumell JH (2004). Recognition of bacteria in the cytosol of Mammalian cells by the ubiquitin system. Curr Biol 14, 806–811. [DOI] [PubMed] [Google Scholar]
- Phu L, Rose CM, Tea JS, Wall CE, Verschueren E, Cheung TK, Kirkpatrick DS, and Bingol B (2020). Dynamic Regulation of Mitochondrial Import by the Ubiquitin System. Mol Cell 77, 1107–1123 e1110. [DOI] [PubMed] [Google Scholar]
- Pickrell AM, and Youle RJ (2015). The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 85, 257–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohl C, and Dikic I (2019). Cellular quality control by the ubiquitin-proteasome system and autophagy. Science 366, 818–822. [DOI] [PubMed] [Google Scholar]
- Polajnar M, Dietz MS, Heilemann M, and Behrends C (2017). Expanding the host cell ubiquitylation machinery targeting cytosolic Salmonella. EMBO Rep 18, 1572–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole LP, Bock-Hughes A, Berardi DE, and Macleod KF (2021). ULK1 promotes mitophagy via phosphorylation and stabilization of BNIP3. Sci Rep 11, 20526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasool S, Soya N, Truong L, Croteau N, Lukacs GL, and Trempe JF (2018). PINK1 autophosphorylation is required for ubiquitin recognition. EMBO Rep 19, e44981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasool S, Veyron S, Soya N, Eldeeb MA, Lukacs GL, Fon EA, and Trempe JF (2021). Mechanism of PINK1 activation by autophosphorylation and insights into assembly on the TOM complex. Mol Cell 82, 44–59. [DOI] [PubMed] [Google Scholar]
- Ravenhill BJ, Boyle KB, von Muhlinen N, Ellison CJ, Masson GR, Otten EG, Foeglein A, Williams R, and Randow F (2019). The Cargo Receptor NDP52 Initiates Selective Autophagy by Recruiting the ULK Complex to Cytosol-Invading Bacteria. Mol Cell 74, 320–329 e326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter B, Sliter DA, Herhaus L, Stolz A, Wang C, Beli P, Zaffagnini G, Wild P, Martens S, Wagner SA, et al. (2016). Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc Natl Acad Sci U S A 113, 4039–4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarraf SA, Raman M, Guarani-Pereira V, Sowa ME, Huttlin EL, Gygi SP, and Harper JW (2013). Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature 496, 372–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauve V, Lilov A, Seirafi M, Vranas M, Rasool S, Kozlov G, Sprules T, Wang J, Trempe JF, and Gehring K (2015). A Ubl/ubiquitin switch in the activation of Parkin. EMBO J 34, 2492–2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauve V, Sung G, Soya N, Kozlov G, Blaimschein N, Miotto LS, Trempe JF, Lukacs GL, and Gehring K (2018). Mechanism of parkin activation by phosphorylation. Nat Struct Mol Biol 25, 623–630. [DOI] [PubMed] [Google Scholar]
- Sawa-Makarska J, Baumann V, Coudevylle N, von Bulow S, Nogellova V, Abert C, Schuschnig M, Graef M, Hummer G, and Martens S (2020). Reconstitution of autophagosome nucleation defines Atg9 vesicles as seeds for membrane formation. Science 369, eaaz7714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekine S, Wang C, Sideris DP, Bunker E, Zhang Z, and Youle RJ (2019). Reciprocal Roles of Tom7 and OMA1 during Mitochondrial Import and Activation of PINK1. Mol Cell 73, 1028–1043 e1025. [DOI] [PubMed] [Google Scholar]
- Sekine S, and Youle RJ (2018). PINK1 import regulation; a fine system to convey mitochondrial stress to the cytosol. BMC Biol 16, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Chang C, Yokom AL, Jensen LE, and Hurley JH (2020). The autophagy adaptor NDP52 and the FIP200 coiled-coil allosterically activate ULK1 complex membrane recruitment. Elife 9, e59099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MD, Harley ME, Kemp AJ, Wills J, Lee M, Arends M, von Kriegsheim A, Behrends C, and Wilkinson S (2018). CCPG1 Is a Non-canonical Autophagy Cargo Receptor Essential for ER-Phagy and Pancreatic ER Proteostasis. Dev Cell 44, 217–232 e211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolz A, Ernst A, and Dikic I (2014). Cargo recognition and trafficking in selective autophagy. Nat Cell Biol 16, 495–501. [DOI] [PubMed] [Google Scholar]
- Swatek KN, Usher JL, Kueck AF, Gladkova C, Mevissen TET, Pruneda JN, Skern T, and Komander D (2019). Insights into ubiquitin chain architecture using Ub-clipping. Nature 572, 533–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurston TL, Boyle KB, Allen M, Ravenhill BJ, Karpiyevich M, Bloor S, Kaul A, Noad J, Foeglein A, Matthews SA, et al. (2016). Recruitment of TBK1 to cytosol-invading Salmonella induces WIPI2-dependent antibacterial autophagy. EMBO J 35, 1779–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurston TL, Ryzhakov G, Bloor S, von Muhlinen N, and Randow F (2009). The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat Immunol 10, 1215–1221. [DOI] [PubMed] [Google Scholar]
- Thurston TL, Wandel MP, von Muhlinen N, Foeglein A, and Randow F (2012). Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature 482, 414–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi-Giesgen I, Behrends C, and Alpi AF (2021). The ubiquitin ligation machinery in the defense against bacterial pathogens. EMBO Rep 22, e52864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turco E, Witt M, Abert C, Bock-Bierbaum T, Su MY, Trapannone R, Sztacho M, Danieli A, Shi X, Zaffagnini G, et al. (2019). FIP200 Claw Domain Binding to p62 Promotes Autophagosome Formation at Ubiquitin Condensates. Mol Cell 74, 330–346 e311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaites LP, Paulo JA, Huttlin EL, and Harper JW (2018). Systematic Analysis of Human Cells Lacking ATG8 Proteins Uncovers Roles for GABARAPs and the CCZ1/MON1 Regulator C18orf8/RMC1 in Macroautophagic and Selective Autophagic Flux. Mol Cell Biol 38, e00392, [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Wijk SJL, Fricke F, Herhaus L, Gupta J, Hotte K, Pampaloni F, Grumati P, Kaulich M, Sou YS, Komatsu M, et al. (2017). Linear ubiquitination of cytosolic Salmonella Typhimurium activates NF-kappaB and restricts bacterial proliferation. Nat Microbiol 2, 17066. [DOI] [PubMed] [Google Scholar]
- Vargas JNS, Wang C, Bunker E, Hao L, Maric D, Schiavo G, Randow F, and Youle RJ (2019). Spatiotemporal Control of ULK1 Activation by NDP52 and TBK1 during Selective Autophagy. Mol Cell 74, 347–362 e346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verlhac P, Gregoire IP, Azocar O, Petkova DS, Baguet J, Viret C, and Faure M (2015). Autophagy receptor NDP52 regulates pathogen-containing autophagosome maturation. Cell Host Microbe 17, 515–525. [DOI] [PubMed] [Google Scholar]
- Vranas M, Lu Y, Rasool S, Croteau N, Krett JD, Sauvé V, Gehring K, Fon EA, Durcan TM, and Trempe J-F (2022). Selective localization of Mfn2 near PINK1 enable its preferential ubiquitination by Parkin on mitochondria. Open Biol. 12, 210255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wauer T, Simicek M, Schubert A, and Komander D (2015). Mechanism of phospho-ubiquitin-induced PARKIN activation. Nature 524, 370–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, Richter B, Korac J, Waidmann O, Choudhary C, et al. (2011). Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 333, 228–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson S (2019). ER-phagy: shaping up and destressing the endoplasmic reticulum. FEBS J 286, 2645–2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong YC, and Holzbaur EL (2014). Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc Natl Acad Sci U S A 111, E4439–4448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X, Li F, Wang Y, Wang Y, Lin Z, Cheng X, Liu J, Chen C, and Pan L (2015). Molecular basis of ubiquitin recognition by the autophagy receptor CALCOCO2. Autophagy 11, 1775–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamano K, Fogel AI, Wang C, van der Bliek AM, and Youle RJ (2014). Mitochondrial Rab GAPs govern autophagosome biogenesis during mitophagy. Elife 3, e01612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamano K, Kikuchi R, Kojima W, Hayashida R, Koyano F, Kawawaki J, Shoda T, Demizu Y, Naito M, Tanaka K, et al. (2020). Critical role of mitochondrial ubiquitination and the OPTN-ATG9A axis in mitophagy. J Cell Biol 219, e201912144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamano K, Wang C, Sarraf SA, Munch C, Kikuchi R, Noda NN, Hizukuri Y, Kanemaki MT, Harper W, Tanaka K, et al. (2018). Endosomal Rab cycles regulate Parkin-mediated mitophagy. Elife 7, e31326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikawa A, Sato Y, Yamashita M, Mimura H, Yamagata A, and Fukai S (2009). Crystal structure of the NEMO ubiquitin-binding domain in complex with Lys 63-linked di-ubiquitin. FEBS Lett 583, 3317–3322. [DOI] [PubMed] [Google Scholar]
- Zheng YT, Shahnazari S, Brech A, Lamark T, Johansen T, and Brumell JH (2009). The adaptor protein p62/SQSTM1 targets invading bacteria to the autophagy pathway. J Immunol 183, 5909–5916. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Liu J, Fu T, Wu P, Peng C, Gong X, Wang Y, Zhang M, Li Y, Wang Y, et al. (2021). Phosphorylation regulates the binding of autophagy receptors to FIP200 Claw domain for selective autophagy initiation. Nat Commun 12, 1570. [DOI] [PMC free article] [PubMed] [Google Scholar]