

 This work is licensed under a
This work is licensed under a Abstract
Summary
Pituitary apoplexy (PA) is a medical emergency with complex diagnosis and management. In this study, we describe a case of PA in a 63-year-old male treated with oral anticoagulant therapy for atrial fibrillation. In the patient, PA manifested itself with asthenia and severe headache not responsive to common analgesics. Despite the finding of a pituitary mass through CT, and in anticipation of the endocrinological evaluation and pituitary MRI, the patient’s clinical condition worsened with an escalation of headache and asthenia associated with deterioration of the visual field and impairment of consciousness level. The emergency assessments revealed an adrenal failure, whereas MRI showed a haemorrhagic pituitary macroadenoma with compression of the optic chiasm. Intravenous fluids repletion and high-dose hydrocortisone were started with a rapid improvement of the patient’s health and visual field abnormalities. Hydrocortisone was gradually reduced to a replacement dose. During the follow-up, panhypopituitarism was documented, and replacement therapies with l-thyroxine and testosterone were introduced. Three months later, a pituitary MRI showed a 50% reduction in the pituitary adenoma volume.
Learning points
Pituitary apoplexy (PA) is a medical emergency that can result in haemodynamic instability and abnormalities in the level of consciousness.
The management of PA requires a multidisciplinary team that includes endocrinologists, ophthalmologists, neuro-radiologists, and neuro-surgeons.
Pituitary MRI with gadolinium is the diagnostic gold standard for PA.
PA therapy aims to improve general conditions and treat compression symptoms, especially visual field abnormalities.
Adrenocorticotrophic hormone deficiency is a common and severe complication of PA. Thus, all patients with PA must be promptly treated with injective synthetic glucocorticoids (e.g. hydrocortisone 100 mg) and i.v. saline.
PA must be taken into consideration in case of sudden headache in patients with a pituitary macroadenoma, especially if other risk factors are recognized.
Patient Demographics: Adult, Male, White, Italy
Clinical Overview: Pituitary, Pituitary
Related Disciplines: Cardiology, Neurology, Ophthalmology, Radiology/Rheumatology
Keywords: Error in diagnosis/pitfalls and caveats, June, 2022
Background
Pituitary apoplexy (PA) is a clinical syndrome caused by a haemorrhage or an infarction of the pituitary gland, which generally occurs in the context of a pituitary adenoma (1). Recent epidemiological studies indicate that PA has a prevalence of 6.2 cases per 100 000 subjects and an incidence of 2 episodes per million per year (2). The major predisposing factors for PA are the type and dimension of the pituitary adenoma (non-functioning or prolactin-secreting macroadenomas presenting a higher risk) and concomitant anticoagulant therapy. The presence of a pituitary neoplasm may facilitate apoplexy due to the features of its vascularization. Indeed, adenomas usually have arterial vascularization, which is independent of the hypothalamus–pituitary portal system and is characterized by abnormal angiogenesis. This feature and the increased energy requirement of tumoural cells may predispose pituitary adenomas to ischemic or haemorrhagic events, thus leading to apoplexy. In addition, this type of vascularization implies a lower capacity to adapt to metabolic and haemodynamic changes; therefore, conditions inducing changes in blood pressure, such as surgery or pregnancy, or those that cause an increase in the metabolic demand, such as the dynamic pituitary tests, are also associated with a higher risk of PA (1, 3).
PA is classically characterized by a triad of symptoms such as headache, visual disturbances, and altered consciousness. Headache appears to be the most frequent one observed in over 80% of cases, whereas visual disturbances are present in over 50% of cases. Visual disturbances can be (a) campimetry defects caused by optic chiasm compression; (b) alterations in oculomotion due to functional impairment of the III, IV, or VI cranial nerve, and (c) visual acuity deficiency related to compressive phenomena of the optical pathways (rare).
Pituitary damage often results in pituitary hormone deficiency: the most frequent being the adrenocorticotrophic hormone deficiency which occurs in 50–80% of patients. While between 40 and 75% of patients have gonadotropin deficiency, 30–70% suffer from central hypothyroidism. PA is also a cause of prolactin deficiency (10–40% of patients), while the prevalence of growth hormone deficiency has been rarely investigated. Neurohypophysis involvement is overall rare and central diabetes insipidus occurs in about 5% of patients.
In the course of PA, varying degrees of altered state of consciousness can be observed, especially in those patients with acute adrenal failure, ranging from lethargy to coma (1).
Data from literature uncover that most PA patients were initially submitted to a brain CT scan to rule out the occurrence of a cerebrovascular event. Unfortunately, CT has a sensitivity of 80% for the recognition of intrasellar masses and only 20–30% for the identification of bleeding components in their context, so PA may initially be overlooked. Therefore, if a PA is suspected, a pituitary MRI is required due to its excellent accuracy in the characterization of pituitary lesions and vascular events (4).
Once the diagnosis of PA is established, treatment aims to resolve pituitary mass compression symptoms and replace hormonal defects. The therapeutic cornerstone of PA is administering high dose of i.v. glucocorticoids which are effective in alleviating compressive symptoms and replacing a likely adrenal insufficiency. This medical approach must be provided to all patients with PA, independently of the availability of hormonal blood sample results. Pituitary neurosurgery is a further option that must be considered in the case of neurological symptoms that do not recede or even worsen after glucocorticoid therapy has started (5).
Case presentation
In August 2019, a 63-year-old man entered the local emergency room complaining of intense headache and asthenia started 1 week before but got worse over the last 24 h. He was suffering from arterial hypertension and atrial fibrillation treated with lercanidipine and rivaroxaban, respectively. To rule out the suspicion of a cerebrovascular event, the patient underwent a non-contrast CT scan which revealed a sellar lesion of about 15 mm characterized by intralesional hyperdensity. The patient was scheduled for an endocrinological evaluation and a pituitary MRI.
After 5 days, his conditions broadly worsened, and the headache was accompanied by diplopia and alteration of the state of consciousness; he appeared soporous, with a Glasgow coma scale of 13/15. There were no lateralizing motor deficits and osteo-tendon reflexes and sensitivity remained normal. The patient underwent an endocrinological evaluation which highlighted normal characteristics of the skin, normal mucous membranes, normal external genitalia, normal distribution of the pilitium, and irregular heart rhythm with arterial pressure of 105/70 mmHg.
Investigation
MRI of the sella turcica with contrast media administration revealed an expansive intra and supra-sellar lesion of 20 × 18 × 18 mm characterized by meta-haemoglobin content. The lesion had a latero- and supra-sellar extension causing compression of both cavernous sinuses and the optic chiasm. These radiological findings were suggestive of a haemorrhagic pituitary macroadenoma (Fig. 1).
Figure 1.
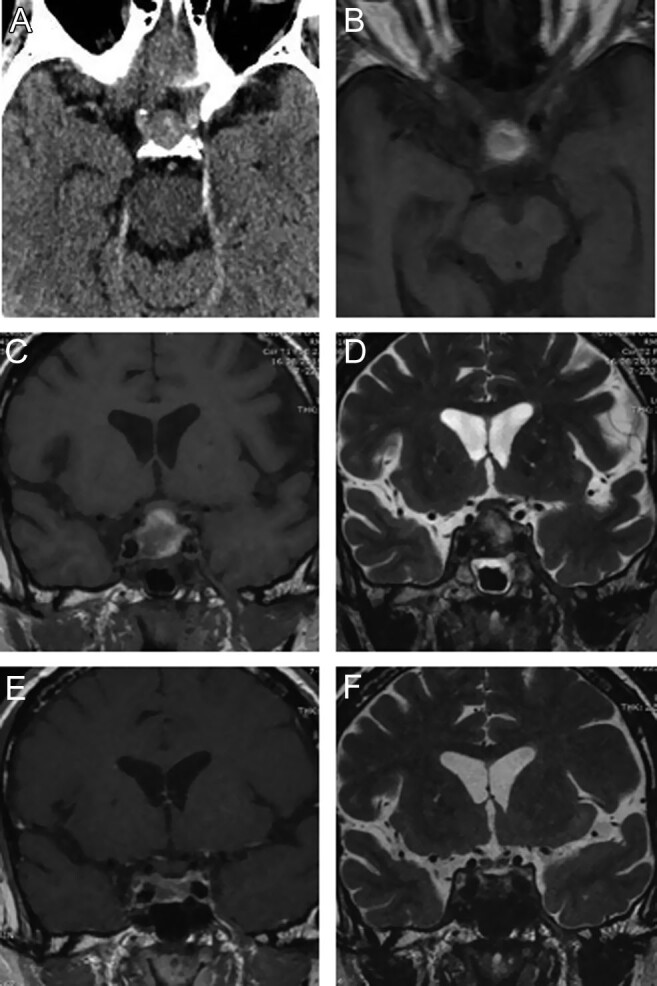
Brain CT scan at the admission to emergency room reveals an endosellar and suprasellar adenomatous mass with intralesional hyperdensity (A). Axial MR T1-weighted images (B) show signal hyperintensity of the suprasellar component confirming bleeding of adenoma. Coronal T1 (C) and T2 (D)-weighted images of the sellar region clearly depict the adenoma morphology with impingement of the optic chiasm. Note the inhomogeneous signal for the presence of hematic and necrotic components. The same images (E and F) obtained 2 months later confirm the shrinkage of the adenoma with residual hematic catabolites for apoplexy.
Blood chemistry tests showed mild hyponatremia (blood sodium level = 134 mEq/L), normal potassium levels (4.05 mEq/L), and increased creatinine values (2.08 mg/dL). Hormonal tests showed panhypopituitarism (Table 1).
Table 1.
Hormonal tests performed at the time of PA diagnosis, after 1 and after 10 months.
| Test | At the time of diagnosis | After 1 montha | After 10 monthsb | Reference range |
|---|---|---|---|---|
| FT4 | 0.78 | 0.92 | 1.36 | 0.7–1.7 ng/dL |
| TSH | 0.049 | 0.174 | 0.5 | 0.4–4 IU/mL |
| Cortisol | 1.5 | 0.9 | 15.3 | 6.7–22.8 μg/dL |
| ACTH | <5 | <5 | NA | <50 ng/L |
| LH | 0.2 | 0.4 | NA | 1.4–12.7 IU/L |
| FSH | 0.7 | 0.9 | NA | 1.3–19.5 IU/L |
| Total testosterone | <0.1 | <0.1 | 6.5 | 1.75–7.8 μg/L |
| PRL | <0.25 | NA | <0.25 | 2–13 μg/L |
| IGF-1 | 72.9 | NA | 38 | 42–192 μg/L |
aExaminations were repeated during l-thyroxine (62.5 μg/day) and hydrocortisone (20 mg/day) therapy; bExaminations were repeated during l-thyroxine (75 μg/day), hydrocortisone (20 mg/day), and testosterone (30 mg/day) therapy.
FT, free thyroxine; NA, not available; PRL, prolactin.
Neurosurgical and ophthalmological evaluations were performed to assess the need for neurosurgical debulking. The eye examination revealed constant diplopia in the primary position and in the lateral gaze and oedema of the papilla, while visual acuity evaluation and computerized visual field appeared normal. The team decided to closely monitor neurological signs, especially those suggesting intracranial hypertension.
Treatment
Therapy with hydrocortisone 100 mg intravenously every 6 h and fluid resuscitation with 0.9% saline solution, 1 L in the first hour and 4 L in the following 24 hours, was started.
In the following hours, a rapid improvement of the general and neurological condition took place and headache receded. In the following days, a gradual resolution of papilledema was also documented. After 3 days, hydrocortisone dose was tapered from 100 to 50 mg intravenously every 6 h, and l-thyroxine 50 μg/day was started.
In consideration of the improvement of neurological symptoms and resolution of papilledema, neurosurgery was not necessary at this point. After 7 days of high-dose hydrocortisone, the patient appeared in good general condition and asymptomatic, except for the persistence of diplopia, but only in the lateral gaze; he was therefore discharged with hydrocortisone therapy 20 mg/day and l-thyroxine 62.5 μg/day.
Outcome and follow-up
At the outpatient visit, conducted 30 days after discharge, the patient was in good general condition, complaining of sexual disturbances (erectile deficit and reduced libido) as the only symptoms. The physical examination revealed irregular heart rhythm, target blood pressure values (130/75 mmHg), and persistence of diplopia in the gaze to the right. No signs of hypogonadism were evident. Hormonal tests (carried out after fasting and before hormone therapy) confirmed panhypopituitarism (Table 1). The dosage of l-thyroxine was increased to 75 μg/day. Testosterone therapy was introduced, 2% gel formulation, 30 mg/day and, for hypocortisolism, to favour patient compliance and therapeutic efficacy, switching from hydrocortisone to modified-release hydrocortisone (plenadren, 20 mg/day) was recommended. MRI of the sella turcica with contrast medium carried out 3 months after the diagnosis of PA showed a 50% reduction in the size of the macroadenoma that now was confined within the sella turcica. Furthermore, the optic chiasm was no longer compressed compared to the previous examination. The patient returned to our attention 10 months after the apoplectic event in good general conditions and with biochemical evidence of the adequacy of hormone replacement therapy (Table 1).
Discussion
In this study, we present the case of PA occurring in a man treated with anticoagulant therapy which manifested with rapidly worsening neurological symptoms, adrenal crisis, and panhypopituitarism. Medical therapy with high-dose glucocorticoids proven successful in treating symptoms, thus avoiding neurosurgical intervention.
PA should be considered, in all respects, an endocrinological emergency. The acute clinical setting mimics that of other more common cerebrovascular events and is usually evaluated by a CT scan in the emergency room. Although cerebral MRI has greater sensitivity and specificity to identify PA, in our case, the CT scan documented the presence of a pituitary mass presenting with suspected bleeding. When MRI was performed 7 days after the CT scan, it showed extracellular methaemoglobin that indicated that the haemorrhagic event was in a subacute phase and therefore to be traced back to the date of the onset of symptoms. It is likely that an overdue endocrinological investigation led to the deterioration of the patient's condition in the following days certainly due to acute adrenal insufficiency onset and worsening of intracranial hypertension. It is worth noting that the patient had two of the most common risk factors for adenoma bleeding, such as a large-size adenoma and the concomitant anticoagulant therapy. The latter, in particular, has been already observed in patients treated with new oral anticoagulant drugs (6).
According to the Pituitary Apoplexy Guidelines Development Group of the University of Oxford, since acute secondary adrenal insufficiency is seen in approximately two-thirds of patients with PA, immediate medical management of patients with PA should include careful assessment of fluid and electrolyte balance, supportive measures to ensure haemodynamic stability, and replacement of corticosteroids. In particular, indications to start empirical steroid therapy in patients with PA are haemodynamic instability, altered consciousness level, reduced visual acuity, and severe visual field defects. In fact, glucocorticoid therapy, in addition to having effects on haemodynamic instability, is effective for anti-inflammatory and anti-oedema effects on neurological symptoms overall.
Regarding the therapeutic choice, patients with PA who are without any neuro-ophthalmic signs or presenting mildly stable signs can be considered for conservative management with careful monitoring. On the other hand, patients with severe neuro-ophthalmic signs such as severely reduced visual acuity, severe and persistent or deteriorating visual field defects, or deteriorating level of consciousness should be considered for surgical management (5).
It is worth noting that only five retrospective comparative studies have analyzed differences in pituitary and visual outcome between surgical and medical therapy (7, 8, 9, 10, 11). Altogether, they showed a better visual outcome with conservative therapy and no differences in pituitary outcome. However, patients in the conservative group generally had less severe ocular defects than those in the surgical group (Table 2). Therefore, further studies with patients homogeneously distributed in the two treatment groups are needed in order to more clearly define therapeutic indications.
Table 2.
Patients’ outcomes in the five retrospective studies that compared medical and surgical therapy.
| Author | Ayuk et al. (6) | Gruber et al. (7) | Sibal et al. (8) | Leyer et al. (9) | Bujavansa et al. (10) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Type of therapy | Medical | Surgery | Medical | Surgery | Medical | Surgery | Medical | Surgery | Medical | Surgery |
| Number of patients | 18 | 15 | 20 | 10 | 15 | 24 | 25 | 19 | 22 | 33 |
| Age | NA | NA | 54 | 46 | 45.7 | 50.7 | 58 | 50 | NA | NA |
| Males/females | NA | NA | 16/4 | 7/3 | 9/9 | 19/8 | 10/15 | 7/12 | NA | NA |
| Reduced visual acuity at presentation (%) | NA | NA | 11 (55) | 7 (70) | 4 (26) | 14 (58) | 8 (32) | 16 (84) | NA | NA |
| Resolution of visual acuity after therapy (%) | NA | NA | 5 (45) | 4 (57) | 3 (75) | 8 (57) | 6 (75) | 7 (44) | NA | NA |
| Visual field defects at presentation (%) | 6 (33) | 7 (46) | 4 (20) | 6 (60) | 4 (26) | 16 (64) | 5 (20) | 14 (74) | 10 (45) | 13 (39) |
| Resolution of visual field defects after therapy (%) | 6 (100) | 4 (57) | 2 (50) | 2 (33) | 3 (75) | 7 (43) | 4 (80) | 8 (57) | 6 (60) | 4 (31) |
| Oculomotion defects at presentation (%) | 7 (39) | 8 (53) | 12 (60) | 3 (37) | 8 (52) | 14 (58) | 12 (48) | 10 (53) | 15 (68) | 18 (54) |
| Resolution of oculomotion defects after therapy (%) | 7 (100) | 5 (63) | 10 (83) | 2 (66) | 6 (75) | 9 (64) | 11 (92) | 6 (60) | 15 (100) | 15 (83) |
| Hypopituitarism at presentation (%) | 13 (87) | 15(83) | 15 (75) | 9 (90) | 13 (72) | 21 (87) | 20 (80) | 15 (79) | NA | NA |
| Normal pituitary function after therapy (%) | NA | NA | 1 (5) | 2 (20) | 2 (11) | 5 (19) | 9 (37) | 3 (16) | 2 (9) | 3(9) |
NA, not available.
PA requires a high grade of suspicion. In particular, headache, progressive alterations of state of consciousness, and radiological signs of adenoma bleeding (especially in the case of anticoagulant therapy) were all suggestive signs of PA. Prompt diagnosis and the initiation of medical therapy are necessary in order to avoid surgery and to prevent the worsening of clinical conditions or fatal events.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This work did not receive any specific grant from any funding agency in the public, commercial, or not-for-profit sector.
Patient consent
Written informed consent for publication of their clinical details and/or clinical images was obtained from the patient/parent/guardian/relative of the patient.
Author contribution statement
N V, C U, and I L conceived the study and wrote the paper. G C, G M, and A A did table and figures. L M, F B, C M, and M C revised and corrected the paper.
References
- 1.Briet C, Salenave S, Bonneville JF, Laws ER, Chanson P. Pituitary apoplexy. Endocrine Reviews 201536622–645. ( 10.1210/er.2015-1042) [DOI] [PubMed] [Google Scholar]
- 2.Fernandez A, Karavitaki N, Wass JA. Prevalence of pituitary adenomas: a community-based, cross-sectional study in Banbury (Oxfordshire, UK). Clinical Endocrinology 201072377–382. ( 10.1111/j.1365-2265.2009.03667.x) [DOI] [PubMed] [Google Scholar]
- 3.Oguz SH, Soylemezoglu F, Dagdelen S, Erbas T. A case of atypical macroprolactinoma presenting with pituitary apoplexy during pregnancy and review of the literature. Gynecological Endocrinology 202036109–116. ( 10.1080/09513590.2019.1650339) [DOI] [PubMed] [Google Scholar]
- 4.Chanson P, Lepeintre JF, Ducreux D. Management of pituitary apoplexy. Expert Opinion on Pharmacotherapy 200451287–1298. ( 10.1517/14656566.5.6.1287) [DOI] [PubMed] [Google Scholar]
- 5.Rajasekaran S, Vanderpump M, Baldeweg S, Drake W, Reddy N, Lanyon M, Markey A, Plant G, Powell M, Sinha S, et al. UK guidelines for the management of pituitary apoplexy. Clinical Endocrinology 2011749–20. ( 10.1111/j.1365-2265.2010.03913.x) [DOI] [PubMed] [Google Scholar]
- 6.Doglietto F, Costi E, Villaret AB, Mardighian D, Fontanella MM, Giustina A. New oral anticoagulants and pituitary apoplexy. Pituitary 201619232–234. ( 10.1007/s11102-014-0616-3) [DOI] [PubMed] [Google Scholar]
- 7.Ayuk J, McGregor EJ, Mitchell RD, Gittoes NJ. Acute management of pituitary apoplexy–surgery or conservative management? Clinical Endocrinology 200461747–752. ( 10.1111/j.1365-2265.2004.02162.x) [DOI] [PubMed] [Google Scholar]
- 8.Sibal L, Ball SG, Connolly V, James RA, Kane P, Kelly WF, Kendall-Taylor P, Mathias D, Perros P, Quinton R, et al. Pituitary apoplexy: a review of clinical presentation, management and outcome in 45 cases. Pituitary 20047157–163. ( 10.1007/s11102-005-1050-3) [DOI] [PubMed] [Google Scholar]
- 9.Gruber A, Clayton J, Kumar S, Robertson I, Howlett TA, Mansell P. Pituitary apoplexy: retrospective review of 30 patients – is surgical intervention always necessary? British Journal of Neurosurgery 200620379–385. ( 10.1080/02688690601046678) [DOI] [PubMed] [Google Scholar]
- 10.Leyer C, Castinetti F, Morange I, Gueydan M, Oliver C, Conte-Devolx B, Dufour H, Brue T. A conservative management is preferable in milder forms of pituitary tumor apoplexy. Journal of Endocrinological Investigation 201134502–509. ( 10.3275/7241) [DOI] [PubMed] [Google Scholar]
- 11.Bujawansa S, Thondam SK, Steele C, Cuthbertson DJ, Gilkes CE, Noonan C, Bleaney CW, Macfarlane IA, Javadpour M, Daousi C. Presentation, management and outcome in acute pituitary apoplexy: a large single-centre experience from the United Kingdom. Clinical Endocrinology 201480419–424. ( 10.1111/cen.12307) [DOI] [PubMed] [Google Scholar]