

Abstract
Interpretability of machine learning (ML) models represents the extent to which a model’s decision-making process can be understood by model developers and/or end users. Transcriptomics-based cancer prognosis models, for example, while achieving good accuracy, are usually hard to interpret, due to the high-dimensional feature space and the complexity of models. As interpretability is critical for the transparency and fairness of ML models, several algorithms have been proposed to improve the interpretability of arbitrary classifiers. However, evaluation of these algorithms often requires substantial domain knowledge. Here, we propose a breast cancer metastasis prediction model using a very small number of biologically interpretable features, and a simple yet novel model interpretation approach that can provide personalized interpretations. In addition, we contributed, to the best of our knowledge, the first method to quantitatively compare different interpretation algorithms. Experimental results show that our model not only achieved competitive prediction accuracy, but also higher inter-classifier interpretation consistency than state-of-the-art interpretation methods. Importantly, our interpretation results can improve the generalizability of the prediction models. Overall, this work provides several novel ideas to construct and evaluate interpretable ML models that can be valuable to both the cancer machine learning community and related application domains.
Keywords: Cancer metastasis, Interpretable machine learning, Feature engineering, Performance evaluation
1. INTRODUCTION
Machine Learning (ML) models have been widely used for decision making in different fields such as disease diagnosis and image classification. With proper training, ML models are often capable of capturing complex linear or non-linear relationships among features, with predictive performance comparable or superior to domain experts. However, as shown in several examples, ML models tend to have biases that can lead to, for example, gender / socioeconomic unfairness, and other undesirable behaviors, which are often too subtle to be detected, especially for users without a deeper understanding of the ML algorithms or training/predicting processes [1], [2]. In addition, in many applications, the prediction is not the end point; rather, a user may be interested to understand the cause of a prediction, and design intervention strategies to improve the outcome in the future (e.g., disease status). When a ML model is complex, it may be difficult for a human to understand the decision process. Therefore, it is important for a classification model to provide an interpretation, or explanation, of each prediction, so that a human can understand the prediction process and/or the basis of the decision, which helps to detect possible biases and artifacts from the ML model, and to better utilize the knowledge learned from the prediction model to achieve desired outcomes in the future [3].
The extent for a ML model to have such human-friendly interpretations is referred to as the interpretability of the model. Model interpretability can come from two sources: (1) intrinsic model interpretability and (2) post hoc model interpretability. Intrinsic model interpretability is obtained from ML models that readily provides the underlying relationships within the data. These include, for instance, sparse linear models and decision trees with a limited number of nodes, where a user can understand the model parameters in a relatively straightforward way or is able to simulate the entire decision process. On the other hand, post hoc interpretability comes into play when an ML model has been trained and its decision process is difficult to understand or simulate due to either the complexity of the decision function or the large number of features. In these cases, interpretations can be provided by a separate interpretation model on top of the classification models. For example, several interpretation methods have been proposed in recent years to provide a global or local explanation of neural networks [4], [5], [6], [7], [8]. More recently, model agnostic post hoc interpretation methods, which can provide interpretation to a wider range of classification models, have become more popular (for example, [9], [10], [11], [12], [13]). Post hoc interpretations is further categorized into individual-level and dataset-level interpretations. Individual-level interpretation is useful when a practitioner wants to understand the prediction of a particular instance by the model [9], [10], [12], [13]. A practitioner is mostly interested in identifying which features and/or feature interactions led to that particular prediction. In contrast, dataset-level interpretation provides a more general view of the relationships learned by an ML model on an entire dataset [11]. By analyzing the interpretation obtained from the entire dataset, a practitioner can potentially identify if the ML model has any bias or which features are most important and validate through his/her domain expertise [14].
In recent years, ML models have been used for the prediction of cancer outcomes using whole-genome gene expression or other omics data (for example, [15], [16], [17], [18]). Although these models provided good prediction accuracy, they usually lack interpretability, and are likely biased or overfitted, due to the high dimensional feature space. To resolve this deficiency, in this study, we propose an effective method to construct an interpretable classification model for the prediction of breast cancer metastasis, which is one of the leading causes of cancer-related deaths [19], [20]. Our method starts with a robust parameter-free graph clustering algorithm to identify highly compact biologically significant gene clusters as features, which make simple interpretation possible. We then propose a novel post hoc interpretation algorithm, Probability Changed-based Interpretation (PCI), to obtain both individual-level and dataset-level interpretations for any classification model. Considering the lack of existing standards in evaluating interpretation methods, we designed a simple metric to evaluate the quality of the interpretation based on the recently proposed PDR evaluation framework [14]. Experimental results on a large breast cancer patient cohort showed that the unsupervised learning approach resulted in a significantly reduced number of features, while achieving similar or better accuracy than gene-based features. Importantly, PCI outperformed several existing interpretation methods with respect to the robustness measure of the interpretation results, and identified biologically relevant pathways on both individual level and dataset level. Finally, we show that the interpretations obtained by PCI as well as other interpretation methods can be utilized to design better classifier for improved prediction performance. While our method is only demonstrated in breast cancer metastasis prediction, the design is fairly general and can be easily adapted to many other applications in medical field and beyond.
2. MATERIALS AND METHODS
2.1. Engineering biologically relevant and interpretable features
To construct interpretable classification models for metastasis prediction using whole-genome gene expression data, we aimed to significantly reduce the number of features without sacrificing the classification performance. As genes form coordinated functional groups / pathways, classification models are usually more robust on pathway level than on individual gene level. While there are many known pathways in existing databases, it has been shown that using known pathways as features does not necessarily lead to more accurate predictive models [21]. In addition, the number of features remains large, due to extensive overlap between different pathways, which makes interpretation still a difficult task. Therefore, we employed an in-house network-based clustering algorithm to identify densely connected subnetworks, also known as communities, to represent high-quality gene clusters [22]. The algorithm constructs a symmetric k-nearest neighbor gene co-expression network, where two genes are connected if they are the top k nearest neighbors of each other, measured by the Pearson correlation coefficient of their expression profiles across all patients. The algorithm has the advantage of determining the optimal k in network construction by optimizing the modularity difference between actual and randomized data, and subsequently can estimate the number of clusters by itself [22]. It is also important to note that many genes were left unclustered due to the symmetric requirement of the network construction. To further reduce the number of features, genes not in any cluster or in small clusters with less than 10 genes were discarded. The remaining gene clusters were used as features, and the feature value of a gene cluster was defined as the average expression of the genes belonging to that cluster.
2.2. Probability Change-based Interpretation (PCI)
To capture the relationship between feature values and class labels, we perturb the feature values in the training or testing data, one feature and one instance at a time, and use the change in the prediction result as a measure of the importance of the feature towards the prediction. The idea can be applied to any modern classification algorithm that is capable of providing numerical predictions, such as the probability for each class label, and therefore belong to the class of post hoc interpretation methods. The change can be measured both on the entire dataset level and on individual instance level, representing dataset-level and individual-level interpretations, respectively. See section 2.2.4 for the key differences between our method and related methods.
2.2.1. Data Perturbation
Let Xm×n be a gene expression matrix with m instances and n features, and y the label of each instance. Let Γ be a classification algorithm, and ΓX,y (or ΓX for simplicity) be the classification function trained on X and y. Given an instance x as input, the classification function must provide as output a class predicted probability (CPP), which is the probability of x being a particular class obtained from the prediction model. For simplicity, only binary classification is described here; however, generalization to multi-class is possible. Let t1×n be a test instance not in X and ti the i-th element of t. Then, Pn×n is the perturbation matrix whose elements pij are given by:
| (1) |
In order words, the i-th row of P, pi, contains test instance t with the i-th feature set to ν, a value that effectively nullifies the feature’s contribution towards prediction. In our study, gene expression values are log ratios, and therefore ν is set to 0. When absolute values of gene expression are used as features, ν can be set as the average value of the feature from all instances.
2.2.2. Individual-level interpretation
Individual-level interpretation captures the impact of each feature on the prediction result of an individual instance. The individual-level interpretation for the prediction of t by ΓX is a vector r1×n, whose elements are defined as follows:
| (2) |
Here, r1×n is the individual-level interpretation based on function ΓX, where the contribution of a feature towards the final prediction for that test instance is quantified by ri and the sign indicates the positive or negative contribution of a feature. We also define unsigned interpretation, , whose elements are defined by:
| (3) |
2.2.3. Dataset-level interpretation
Dataset-level interpretation is a measurement of feature importance score (FIS) on an entire dataset. We first obtain individual-level interpretation of each of the instances belonging to the entire dataset by leave-one-out cross validation. Final FIS for the entire dataset is a vector s1×n, whose element si is calculated by averaging the unsigned individual-level interpretation of each instance of the dataset:
| (4) |
where xj is the j-th instance from X, and is the classification function learned from the training data with xj removed from X.
2.2.4. Related methods
In essence, the idea of PCI is similar to several existing interpretation algorithms, namely, Individual Conditional Expectation (ICE) [12], Accumulated Local Effects (ALE) [13], Permutation Feature Importance (PermImp) [11], and Local Surrogate (LIME) [9]. However, the key difference lies on how data is perturbed, and how the change is measured and/or presented as an interpretation. For example, both ICE and ALE implement a series of perturbations in a grid search fashion, and present the changes as a set of plots showing the relationship between feature values and the prediction results, for each feature and each patient. The outcome is more suitable to be read by ML experts / model developers for diagnosis of the underlying prediction model rather than by an end user of the model such as domain experts or patients for particular prediction results. On the other hand, PermImp attempts to summarize the change of prediction results across a range of feature values and all instances. As a result, it will not provide any interpretation for the prediction of an individual instance after the model is deployed. In contrast, our method provides both dataset-level and individual-level interpretations, which can be useful for both ML experts / model developers and end users. Finally, both LIME and PermImp rely on random perturbation of feature values, which can result in unrealistic input instances and consequently inaccurate interpretations. In comparison, our method provides a more controlled feature perturbation that is tied with the meaning of the features which is more realistic and can be understood by domain experts and users. For example, in the case of gene expression-based disease diagnosis, we choose to replace the original feature values (expressed as log ratios) to zero, which is essentially nullifying the impact of the feature on the disease outcome. Our method will be compared with both LIME and PermImp in the experimental results section to demonstrate the effectiveness of this strategy.
2.3. Evaluation of interpretation methods under the PDR (Predictive, Descriptive and Relevancy) framework
While several post hoc interpretation methods have been proposed, it is often difficult to evaluate the usefulness of such methods in real applications. The evaluations of existing interpretation methods often involve human subjects and are based on carefully crafted datasets with “ground truth” interpretations, which do not necessarily generalize to other datasets. In fact, the exact definition of interpretablity is often obscure and domain dependent [23]. To introduce a common ground for the development and evaluation of interpretation methods, several studies attempted to define the main requirements of interpretable models, which can be applied to evaluate both inherently interpretable ML models as well as interpretations provided by interpretation methods [14], [24], [25]. The recent proposed PDR framework [14] suggests that an interpretable model (and its interpretations) needs to satisfy three requirements: predictive accuracy, descriptive accuracy and relevancy, which we will briefly describe below. It is important to note that, while the PDR framework defines how the model should be evaluated, it does not provide specific measurements for evaluation, especially for descriptive accuracy and relevancy. In this work, we attempt to provide several simple measures that, taken together, provide a quantitative and relatively domain-independent approach to evaluate interpretation methods.
2.3.1. Evaluation of predictive accuracy
Predictive accuracy measures how well a classification model (interpretable or not) captures the underlying relationships between the features and the labels [14]. A classification model has to provide acceptable prediction accuracy on unseen data to gain confidence from the end-users. Therefore, any effort attempting to improve the interpretablity of a classification model should not be at the cost of predictive performance. For post hoc interpretation approaches, this is generally not an issue because interpretation can be supplied to any classification model. However, it is important to note that, even with post hoc interpretation method, classification models utilizing less features will be more likely to achieve better interpretability. In our experiment, therefore, we compared the AUC of multiple classification models utilizing different features, including gene features and different versions of gene clusters as features.
2.3.2. Evaluation of descriptive accuracy
Descriptive accuracy defines the extent that an interpretation (implicitly from a classification model or explicitly from a separate interpretation model) helps a practitioner to better understand the relationships captured by an ML model [14]. While the definition of descriptive accuracy is intrinsically domain dependent, and a direct evaluation would involve human subjects, we propose an indirect, quantitative measure that can be easily computed. We believe that genuine interpretations should be relatively independent of the underlying classification algorithms, assuming the algorithms have sufficient predictive accuracy. As most interpretation models provide a single numerical value for each feature as an interpretation (for individual instances or on a dataset level), we hypothesize that the ranking of features from the interpretation of a test instance should be stable with respect to different classification algorithm. Therefore, we propose to measure descriptive accuracy using inter-classifier stability (ICS). The individual instance-level ICS is measured by the average Spearman correlation coefficient (i.e., SP_CC) between the individual-level interpretations from two classification algorithms, Γ1 and Γ2.
| (5) |
In addition, we define the inter-classifier stability using unsigned interpretations, which may be necessary as some interpretation method do not include signs as part of the interpretations such as Random forest. Additionally, finding the top most significant features is useful in many applications where sign is not considered.
| (6) |
Finally, inter-classifier stability can be measured for dataset-level interpretations:
| (7) |
2.3.3. Evaluation of relevancy
Relevancy is described as that an interpretation method should not only provide sufficient transparency but also this transparency should enable domain experts to understand the complex relationships within the data and to diagnose the ML model in identifying any potential biases in the ML models [14]. In our case, evaluation of relevancy depends on whether the interpretation is able to identify important features which are biologically relevant to the progression of the breast cancer metastasis. This is evaluated from several different perspectives. (1) First, feature-level relevancy is evaluated by the statistically significant enrichment of biologically relevant functional terms (e.g., Gene Ontology terms or KEGG pathways) in the gene clusters. (2) Second, to evaluate the relevancy of dataset-level interpretations, features with the highest feature importance scores are inspected for their known involvement in the classification problem at hand. (3) Finally, it is reasonable to assume that, if the individual-level interpretation provided by an interpretation method is truly relevant, the interpretation for that same instance from different underlying classification functions should be similar. Consequently, we hypothesize that, given an interpretation method with reasonable descriptive accuracy (as measured above), instances with lower inter-classifier stability are ones that are hard to be modeled by the current features and therefore may have poor generalizability. Therefore, by removing these instances from the training data can help build models with better prediction performance, especially under cross-dataset settings. Utilizing this idea, we ranked each patient in our training data by its average inter-classifier stability across several pairs of classification algorithms, and removed those with the lowest stability scores and rebuilt a classifier using the remaining “purified” data, and tested the performance of the model using a separate patient cohort that was not used during any stage of the model training.
2.4. Experimental settings
Amsterdam Classification Evaluation Suite (ACES) [26], a combined breast cancer gene expression dataset from 12 different patient cohorts was used for the model development and the majority of the comparative evaluation (cohort details are provided in Table 1). The class label of a patient was determined as non-metastatic if the patient survived without cancer recurrence for at least 5 years after the first occurrence of cancer. ACES is composed of 1616 patients, among which 455 is metastatic. There were 12,750 gene probes in the ACES dataset. To evaluate the prediction accuracy of the gene-cluster, two cross-validation (CV) approaches were employed. In the first approach, 10-fold CV (10-FCV) was repeated on the whole dataset for 10 times resulting in 100 folds. In the second approach, leave-one-study-out cross-validation (LOSO-CV) where one cohort is kept as a test set and the remaining ones are used as the training set. Five different classifiers are used in this study, including Random Forest (RF), Logistic Regression (LR), linear kernel Support Vector Machine (lSVM), radial basis function kernel SVM (rSVM) and multiple layer perceptron based neural network (NN). Neural network was designed to have a single layer consisting of neurons similar to the number of features in the dataset to keep the architecture of the neural network similar across different comparisons. The default settings of those models from the Sklearn package in Python [27] were used in the evaluation. Area Under the ROC Curve (AUC) [28] was used as the evaluation metric due to the unbalanced dataset.
TABLE 1:
Specification of the studies in ACES.
| Dataset | Geo accession no. | No. of poor | No. of good | Total |
|---|---|---|---|---|
| Desmedt | 7390 | 56 | 127 | 183 |
| Hatzis | 25066 | 102 | 48 | 150 |
| Ivshina | 4922 | 30 | 72 | 102 |
| Loi | 6532 | 24 | 33 | 57 |
| Pawitan | 1456 | 33 | 114 | 147 |
| Miller | 3494 | 21 | 68 | 89 |
| Minn | 2603 | 21 | 44 | 65 |
| Schmidt | 11121 | 24 | 145 | 169 |
| Symmans | 17705 | 37 | 187 | 224 |
| WangY | 5327 | 10 | 42 | 52 |
| WangYE | 2034 | 88 | 169 | 257 |
| Zhang | 12093 | 9 | 112 | 121 |
| ACES | 455 | 1161 | 1616 |
To evaluate relevancy of the interpretation methods, we used another dataset, NKI [29], to test the classification models trained on ACES gene clusters. This dataset was not used for any kind of model development to avoid potential information leak. In NKI, there were 295 patients among which 78 patients were metastatic. There were 11,658 gene probes in NKI. To enable using the classifier learned from the ACES dataset to make predictions for patients in the NKI dataset, genes from the clusters of ACES were mapped to the genes in the NKI dataset. A compatible expression dataset constructed from the average expression of the genes within the ACES clusters was created using NKI data without the missing genes.
2.4.1. Availability
A python implementation of PCI and the associated data for testing the algorithm are available at: https://github.com/nahimadnan/PCI.
2.5. Competing interpretation methods
We compared PCI with two well-known methods, LIME, and SHAP, for both descriptive accuracy and relevancy of the interpretations. For dataset-level interpretation comparison, two more approaches, Permutation Importance (PermImp) and Classifier feature importance score (Classifier_FIS), were included. Details on these interpretation approaches are given as follows.
2.5.1. LIME
LIME [9] aims to locally approximate an accurate but complex model, which is hard to interpret intrinsically. For a given test instance and a trained model, LIME generates a new dataset by accumulating different perturbations on that test instance and also the predictions from the trained model for the perturbations on the test instance. Then, LIME trains an interpretable model (i.e., linear regression model) on the new dataset and the coefficients from the trained linear regression model are the individual-level interpretation for that particular test instance. LIME implementation was downloaded from Github (https://github.com/marcotcr/lime).
2.5.2. SHAP
SHAP is a model agnostic interpretation method based on the optimal shaply values obtained from a game theoretical approach [10]. Each feature plays a role into the final prediction for an instance where the game theory comes into play. For LR and RF models, the whole training data was used as the background for SHAP computation. For kernel-based models (lSVM and rSVM models), we used kmeans on the training data for background to accelerate the processing of the individual-level interpretations. Dataset-level interpretation was generated by averaging the individual-level interpretations similar to PCI. SHAP implementation was downloaded from Github (https://github.com/slundberg/shap).
2.5.3. Permutation Importance (PermImp)
PermImp provides only dataset-level FIS. The feature importance is measured as the decrease in the prediction error when the feature values are permuted. We used the PermImp implemented in SKlearn [27].
2.5.4. Classifier feature importance score (Classifier_FIS)
For LR and lSVM, the coefficients from the classification model were directed used as FIS. Random Forest provides a FIS score for each feature by aggregating across all decision trees the feature’s contributions in decreasing the impurity in the dataset.
3. RESULTS AND DISCUSSION
3.1. Robust and biologically interpretable features
To construct interpretable classification models for breast cancer metastasis, we first aimed at obtaining a small number of comprehensible features which can provide comparable prediction performance as more complex models. To this end, we employed a community discovery algorithm to identify gene clusters as features (see Methods). Applying the algorithm to the ACES dataset, we identified a total of 328 clusters and more than 7000 singletons. As the majority of the clusters have only two or three genes, we further filtered out clusters with fewer than 10 genes, resulting in only 37 clusters with between 10 and 913 genes, covering about 4500 genes in total (Fig 1). To estimate the robustness of these gene clusters as features, we performed two additional clustering experiments. First, to comply with our classification model evaluation scheme, we obtained clustering results by randomly removing 10% of patients and repeated 100 times. In the second experiment, we repeatedly removed each of the 12 cohorts from the ACES dataset and performed clustering on the resulting patients. In both cases, the clustering results are very similar to the clustering results from the whole dataset, with an average adjusted rand index similarity score 0.925 and 0.916, respectively. The average expression levels for gene clusters from these perturbed clusters are also extremely similar to the values in the whole dataset-based clusters. Therefore, the 37 gene clusters obtained from the whole dataset are used as features in this paper, unless noted otherwise.
Fig. 1: Gene expression levels in the 37 gene clusters found from the ACES dataset, ordered by sizes.
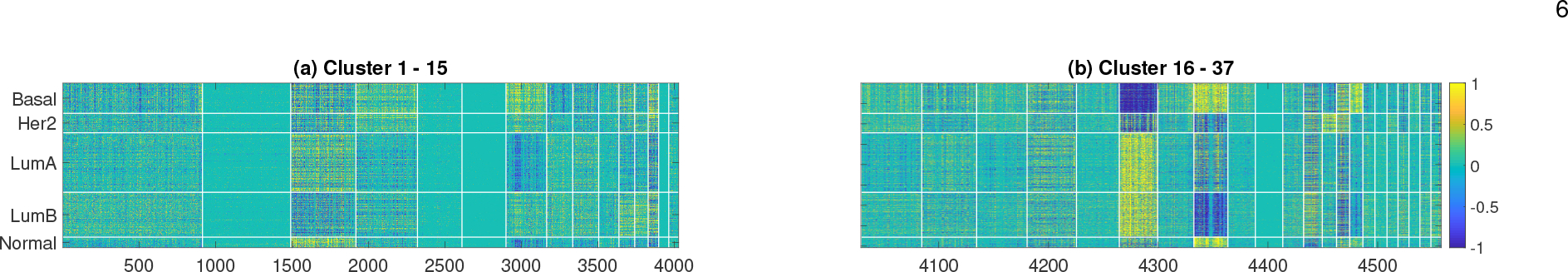
For clarity, data is re-scaled and values between [−0.5, 0.5] are converted to 0. Patients are grouped by breast cancer intrinsic subtypes (information not used for clustering), Colorbar is the same for both figure.
As shown in Fig. 1, many of the 37 gene clusters clearly exhibit subtype-specific expression patterns, and there are noticeable difference among the expression patterns for different subtypes. To identify the biological relevance of these gene clusters, we performed Gene Ontology (GO) enrichment analysis using David Bioinformatics Resources web application [30]. GO analysis results are shown in Table 2. Most clusters have statistically significantly enriched GO terms, including many well known cancer-related biological processes and signaling pathways, such as cell cycle, DNA repair, antigen binding, focal adhesion, defense response, histone core, T-cell receptor, FC-epsilon, inferon-gamma mediated, Wnt, NIK/NF-kappaB and PI3K-Akt [31]. Several significantly enriched GO terms are less well known, but are not without evidence, to be related to cancer cells. For example, oxidative phosphorylation and translation initiation have been reported to be dysregulated in cancer cells [32], [33]. Therefore, the gene clusters identified from the patients appear to be highly relevant, and can be potentially used for predicting cancer outcomes including metastasis. As many of these gene clusters represent well-known biological processes or pathways, the reduced number of features easily aids the ability for domain expert to interpret the classification models / results.
TABLE 2:
Top enriched Gene Ontology biological processes terms in gene clusters.
| Go term | FDR | Go term | FDR |
|---|---|---|---|
|
| |||
| Cluster 1 (913 genes) | Cluster 2 (567 genes) | ||
|
| |||
| Oxidative phosphorylation | 6.5E-23 | G-protein coupled receptor activity | 6.8E-10 |
| NIK/NF-kappaB signaling | 2.5E-7 | Cytokine-cytokine receptor interaction | 8.4E-3 |
| Wnt signaling pathway | 6.6E-5 | Natural killer cell activation involved in immune response | 1.4E-3 |
|
| |||
| Cluster 3 (427 genes) | Cluster 4 (403 genes) | ||
|
| |||
| Focal adhesion | 2.6E-16 | T cell receptor signaling pathway | 2.1E-12 |
| calcium ion binding | 3.1E-14 | Cell adhesion molecules (CAMs) | 2.2E-11 |
| ECM-receptor interaction | 4.1E-12 | Antigen processing and presentation | 8.2E-11 |
| PI3K-Akt signaling pathway | 2.9E-10 | Jak-STAT signaling pathway | 6.5E-3 |
|
| |||
| Cluster 7 (268 genes) | Cluster 10 (132 genes) | ||
|
| |||
| Cell cycle | 1.3E-60 | Translational initiation | 4.4E-70 |
| cell division | 4.1E-40 | SRP-dependent cotranslational protein targeting to membrane | 5.0E-69 |
| DNA repair | 5.9E-9 | viral transcription | 1.3E-66 |
|
| |||
| Cluster 13 (69 genes) | Cluster 27 (17 genes) | ||
|
| |||
| Defense response to virus | 9.6E-32 | Histone core | 2.9E-33 |
| Immunity | 1.4E-32 | Extracellular exosome | 6.5E-7 |
|
| |||
| Cluster 29 (12 genes) | Cluster 30 (12 genes) | ||
|
| |||
| Antigen binding | 1.6E-10 | Cell adhesion | 1.4E-2 |
To further analyze the clustering stability among the clusters of fold specific training data, we computed the mean adjusted rand index between every pair for 10-FCV (i.e., 100 folds) and LOSO-CV (i.e., 12 folds). The heatmap of the adjusted rand index between clusters of each pair of folds of 10-FCV and LOSO-CV are shown in Fig. 2. respectively. The mean adjusted rand index of the upper triangle for both the 10-FCV and LOSO-CV is 0.89 and 0.87 respectively, which indicates that the training specific clusters are stable among themselves demonstrates that the clustering algorithm is robust.
Fig. 2:
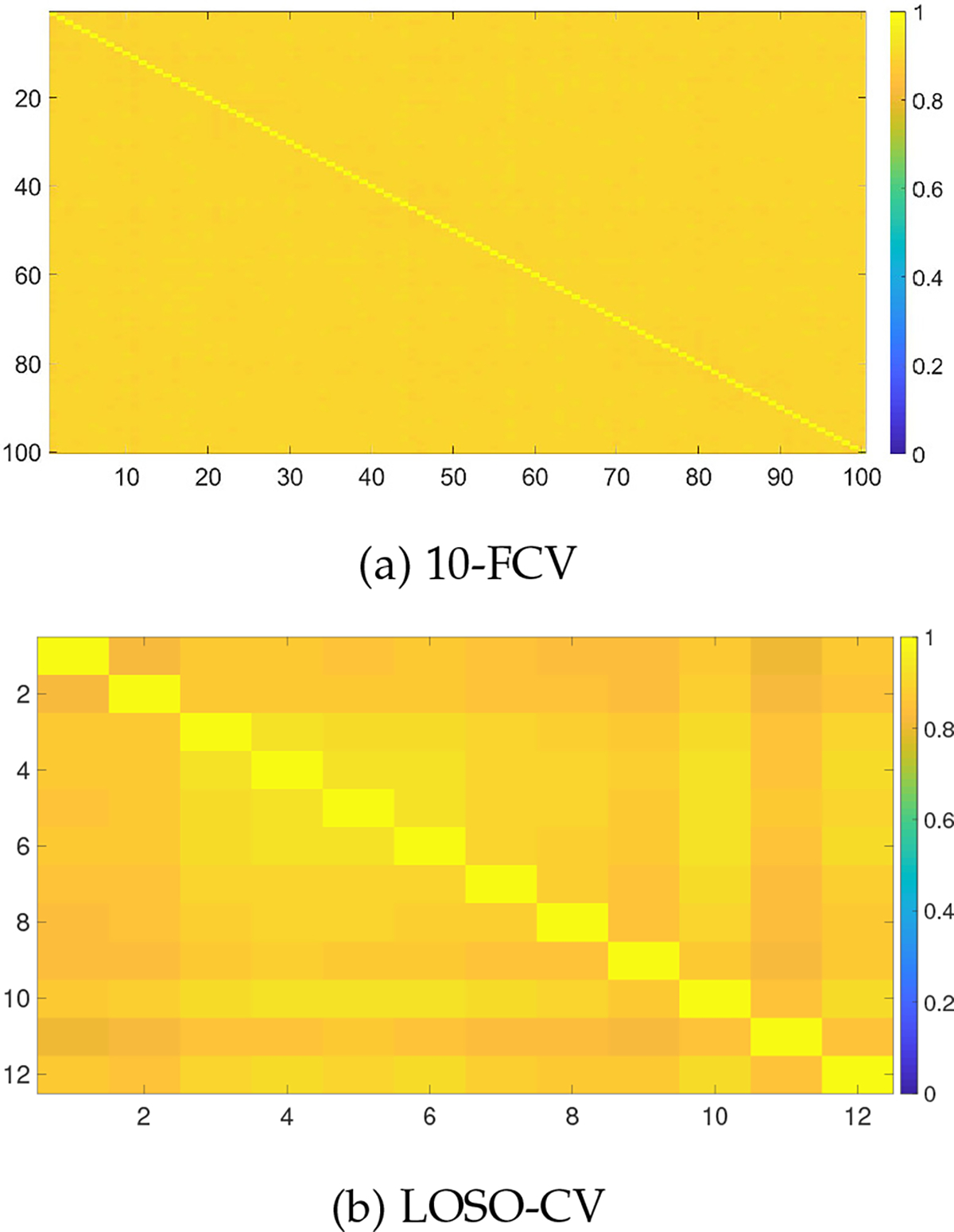
Clustering stability among the clusters from different folds of the training data in two cross-validation schemes: 10-FCV (a) and LOSO-CV (b).
3.2. Predictive accuracy of gene cluster-based classifiers
While interpretablity plays an increasingly important role in the development of ML models, it must be emphasized that this needs to be done without sacrificing prediction accuracy. Therefore, we evaluated the prediction performance of various types of classifiers using gene clusters as features. For comparison, a baseline model was also constructed with all genes from ACES dataset as features. Performance was measured with AUC using 10-fold cross validation (10-FCV) as well as leave-one-study-out cross validation (LOSO-CV) settings. In addition, to avoid potential bias caused by gene clustering using all instances including testing instances, we also evaluated the prediction performance of classifiers trained on the clusters obtained from training data alone, as well as classifiers trained on the largest 37 clusters from training data alone.
The prediction performance of five classification algorithms using different feature types are given in Fig 3. The results clearly show that models trained on features from the 37 gene clusters are able to obtain similar (RF and rSVM models) or even better (LR and lSVM models) and poor (NN model) prediction accuracy than the models trained on gene features, in both 10-FCV and LOSO-CV schemes. To keep the NN architecture similar across different feature types, one middle layer of neurons was kept equal to the number of features in the feature type. The good accuracy of gene features indicates that the layer with large number of neurons (i.e., 12750 neurons) were able to obtain better accuracy compared to 37 gene clusters where there was one layer with 37 neurons. In addition, the classifiers trained on the 37 gene clusters from training patients alone have almost identical prediction performance as the classifiers based on the 37 gene clusters from all patients, suggesting that there is minimum information leak in using all patients to derive gene clusters. On the other hand, the classifiers trained on the gene clusters from training data alone achieved similar performance as the classifiers based on all genes, which is understandable, as a large number of clusters contain less than three genes. Finally, non-linear models, i.e., RF, rSVM and NN, achieved slightly better performance than the two linear models, LR and lSVM, indicating the complex no-linear relationship of transcriptional regulation in cancer progression.
Fig. 3:
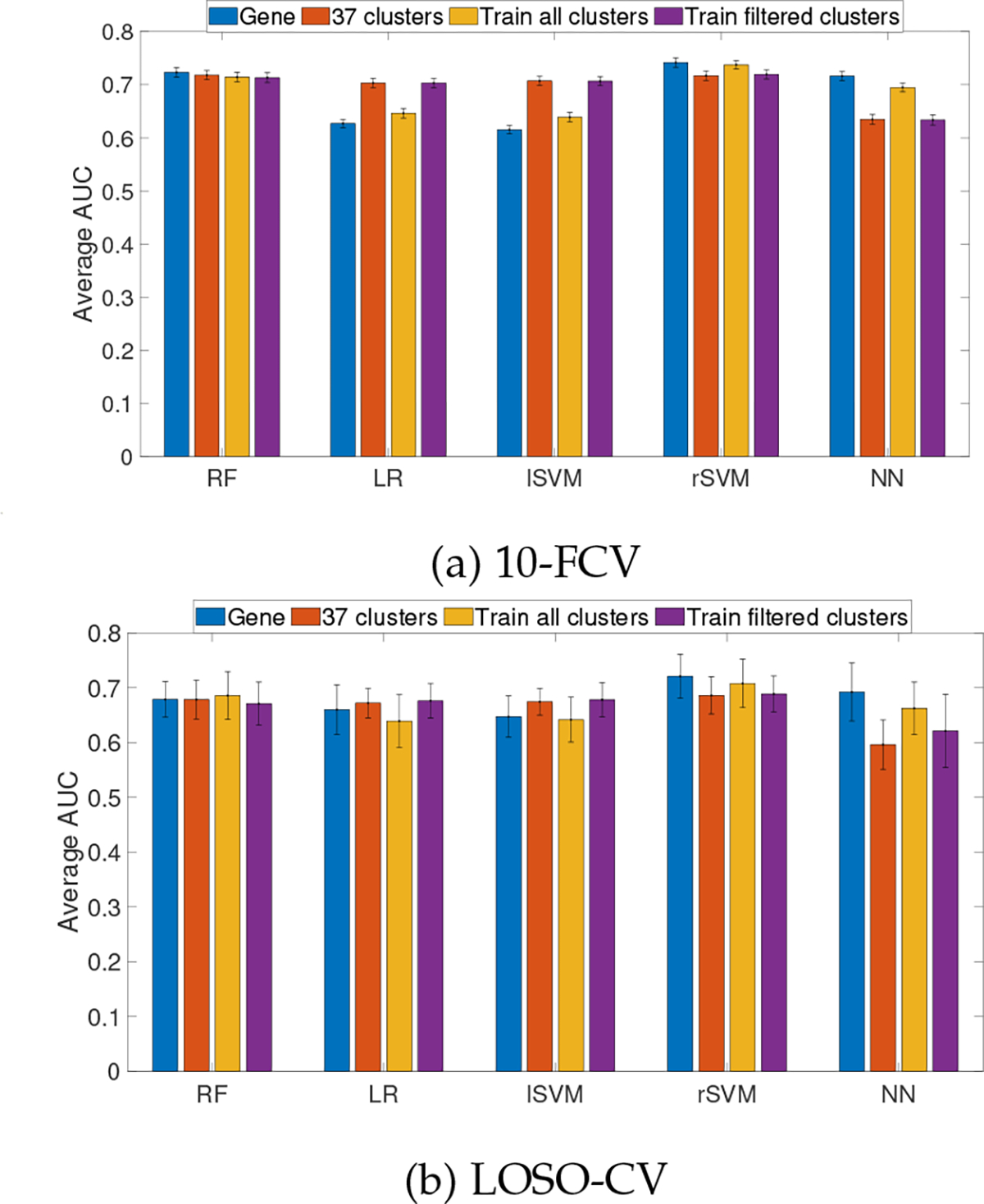
AUC from different feature types for RF, LR, lSVM, rSVM and NN in (a) 10-FCV and (b) LOSO-CV. Error bars denote the 95% confidence interval. Train all clusters: all clusters obtained from training data. Train filtered clusters: clusters with larger than 10 genes.
3.3. Evaluation of descriptive accuracy using inter-classifier stability
The inter-classifier stabilities (ICS) of several algorithms are given in Table 3. As can be seen, PCI outperformed LIME and SHAP significantly on signed individual-level ICS for all classifier pairs. Measured by unsigned individual-level ICS, PCI achieved better stability in 6 out of 10 model combinations compared to LIME and SHAP, respectively. Although the prediction accuracy of 37 gene clusters of NN model was poor compared to the other features, both the signed and unsigned individual-level interpretations are consistent with the other classification algorithms suggesting the potential of PCI in providing robust individual-level interpretations regardless of the prediction accuracy of the classification algorithms. Therefore, it is evident that PCI can provide more robust interpretations compared to the existing methods; later, we will show that the individual-level stability can be utilized to improve the classification model for better performance in cross-cohort prediction. On dataset level, PCI achieved three highest ICS score, LIME achieved four highest ICS score and the other three methods achieved three highest ICS score suggesting that no method is globally accurate in this comparison. Although PCI wins in three comparisons it provides highest ICS average score indicating the robustness of the PCI method in ICS on dataset level.
TABLE 3:
Inter-classifier Stability of different interpretation methods.
| Individual-level interpretation with sign | Individual-level interpretation without sign | Dataset-level interpretation | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| PCI | LIME | SHAP | PCI | LIME | SHAP | PCI | LIME | SHAP | PermImp | Classifier_FIS | |
| RF-LR | 0.773 | 0.664 | 0.447 | 0.384 | 0.441 | 0.325 | 0.526 | 0.446 | 0.426 | 0.492 | 0.21 |
| RF-lSVM | 0.773 | 0.617 | 0.406 | 0.384 | 0.375 | 0.311 | 0.319 | 0.393 | 0.235 | 0.314 | −0.037 |
| RF-rSVM | 0.772 | 0.749 | 0.544 | 0.385 | 0.504 | 0.346 | 0.490 | 0.515 | 0.57 | 0.275 | NaN |
| RF-NN | 0.765 | 0.524 | 0.137 | 0.357 | 0.307 | 0.184 | 0.489 | 0.362 | 0.441 | 0.385 | NaN |
| LR-lSVM | 0.968 | 0.942 | 0.919 | 0.893 | 0.839 | 0.886 | 0.851 | 0.84 | 0.838 | 0.848 | 0.86 |
| LR-rSVM | 0.860 | 0.793 | 0.67 | 0.515 | 0.508 | 0.571 | 0.378 | 0.513 | 0.49 | 0.089 | NaN |
| LR-NN | 0.844 | 0.493 | 0.118 | 0.444 | 0.218 | 0.237 | 0.293 | 0.233 | 0.205 | 0.111 | NaN |
| lSVM-rSVM | 0.847 | 0.751 | 0.642 | 0.468 | 0.361 | 0.503 | 0.208 | 0.347 | 0.307 | 0.045 | NaN |
| lSVM-NN | 0.842 | 0.416 | 0.119 | 0.437 | 0.105 | 0.206 | 0.060 | 0.101 | 0.031 | 0.021 | NaN |
| rSVM-NN | 0.865 | 0.58 | 0.14 | 0.526 | 0.324 | 0.356 | 0.826 | 0.393 | 0.766 | 0.939 | NaN |
| AVG | 0.831 | 0.653 | 0.414 | 0.479 | 0.398 | 0.392 | 0.444 | 0.414 | 0.431 | 0.352 | 0.369 |
We also evaluated PermImp which is a model agnostic method that gives only dataset-level interpretations. We also used the feature importance scores (FIS) from three classifiers (LR, lSVM, and RF) directly as dataset-level interpretations. Interestingly, while the dataset-level interpretations of PCI, LIME and SHAP were obtained by averaging individual-level interpretation vectors, they provided much better inter-classifier stability on the dataset level than PermImp and classifier FIS. This result suggests that the former three methods are able to correctly identify different features important for individual instances, independent of the underlying classifiers. In contrast the interpretations from PermImp and the Classifier FIS cannot distinguish the difference between individual instances and as a result, their dataset-level interpretations may have been biased by different training instances in different classifiers, leading to much lower inter-classifier stability. Using FIS from classifiers, good stability was achieved between LR and lSVM, which is expected as both classifiers use linear models and thus have similar coefficients. On the other hand, FIS provides very little stability when comparing the two linear models (LR, lSVM) with the no-linear model RF, even though they have similar prediction performance. This result therefore strongly advocates the use of interpretation methods for non-linear models, even when the model itself may provide some dataset-level importance measure.
3.4. Evaluation of relevancy of dataset-level interpretation via gene ontology analysis
Fig 4a shows the individual-level interpretation from PCI for an LR model on the ACES dataset. It can be observed that the interpretations for patients belonging to different subtypes have distinct patterns, which share some similarity with but are not identical to the expression patterns shown in Fig. 1. In addition, several features are common across almost all subtypes, but with opposite signs, which reflects the combination of the differential expression level of the gene cluster as well as the different metastasis potentials of different subtypes. To investigate whether the top features found by PCI are directly associated with metastasis, we aggregated the dataset-level interpretations from all five classifiers. As shown in Fig 4b, Cluster 21, 7, 29, 4, 30 10, and 8 have the highest aggregated feature importance scores from RF, LR, lSVM, rSVM and NN models. Among these, Cluster 7 and Cluster 4 are significantly enriched in well-known cancer cell hallmark pathways such as cell cycle and DNA repair, Cell adhesion molecules, and Jak-STAT signaling pathway [31]. In addition, Cluster 4 and Cluster 29 are enriched with several immune-related pathways, such as antigen binding, antigen processing and presentation, and T cell receptor signaling pathways. Immune cells have known strong connections with cancer progression and metastasis [34], [35]. Cluster 10 is enriched in translational initiation, which is regulated in response to nutrient availability and mitogenic stimulation and is coupled with cell cycle progression and cell growth [36]. Recent studies have demonstrated that translation deregulation contributes to the metastatic phenotype through selective effects on the translation of mRNAs whose products are involved in various steps of metastasis including migration, invasion, angiogenesis, homing, and activation of survival loops at distal sites [33], [37].
Fig. 4:
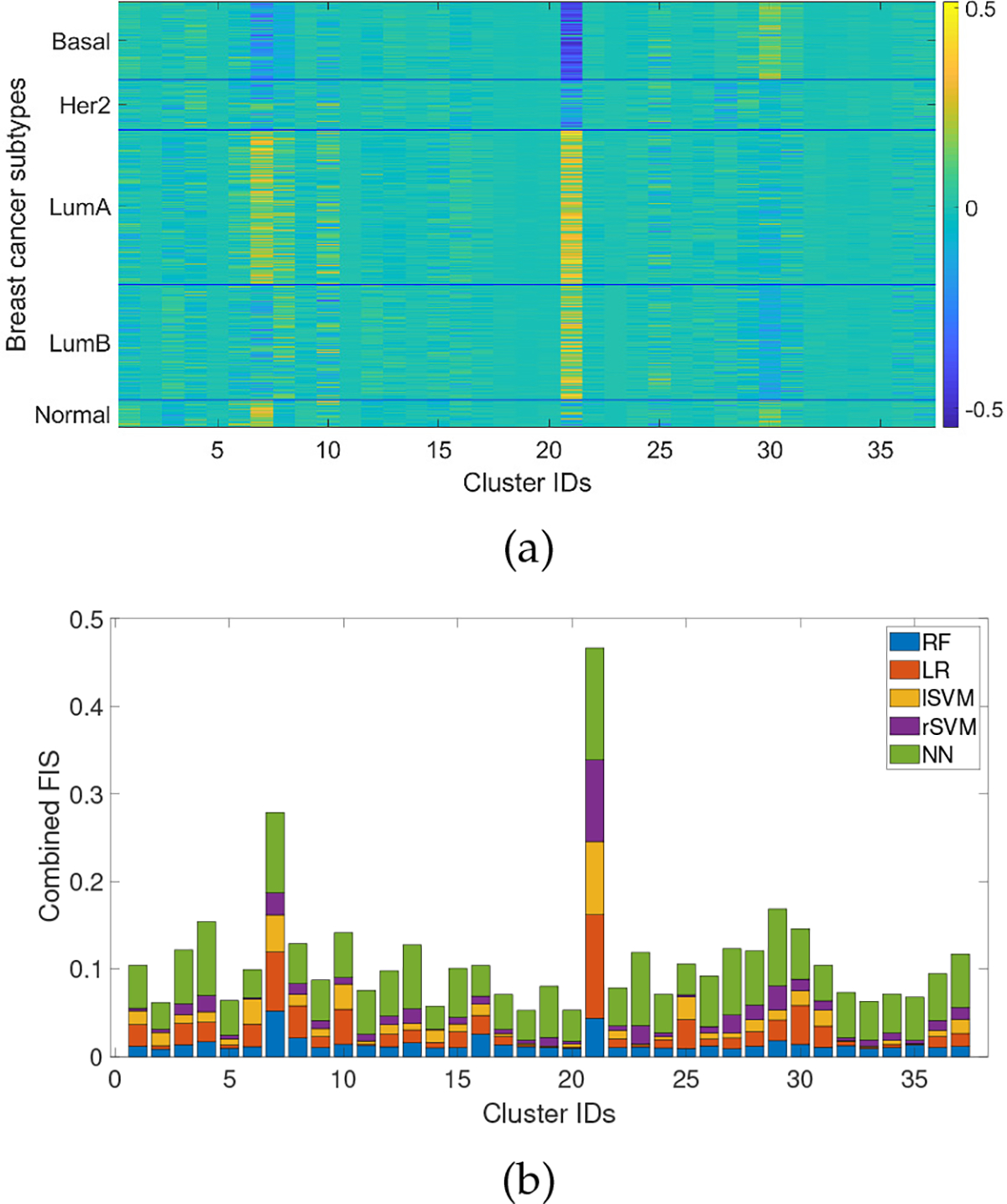
(a) Individual-level interpretation grouped by breast cancer intrinsic subtypes from LR model. (b) Dataset-level interpretation from four models stacked together.
Interestingly, while cluster 8 does not have any highly enriched pathways, four genes in the cluster has function in cilium (p-value = 0.01, Fisher’s exact test), which is an antenna-like organelle that protrudes from most mammalian cells. Recent studies have shown that dysregualtion of cilium genes, such as EZH2, leads to metastasis [38], [39]. Cluster 21 and 30 are both small, with 35 and 12 genes respectively. In particular, cluster 21 is consistently shown to be the most important features by PCI with all four classifiers. The most enriched pathways include chromatin remodelling (p-value = 0.0009, Fisher’s exact test) and angiogenesis (p-value = 0.01, Fisher’s exact test), both are well known to be associated with cancer progression. Angiogenesis, the recruitment of new blood vessels, is an essential component of the metastatic pathway [40]. Among the 12 genes in cluster 30, 5 are involved in cell adhesion (p-value = 0.0002, Fisher’s exact test), which is one of the most well-known pathways involved in tumor migration and metastasis [41]. Therefore, these biological processes are strongly connected with breast cancer metastasis progression. One limitation of the gene set-based approach is that the identified important gene sets can be relatively large which makes it difficult to pinpoint to the key individual genes or designing personalized intervention strategies. In theory, it should be possible to further analyze the most important gene sets to identify individual genes (or smaller subsets of genes) for each patient, in a hierarchical manner. We will explore this possibility in future work.
3.5. Evaluation of relevancy of individual-level interpretation on its utility to improve model generalizability
While one of the main usability of interpretation is to understand the contribution of features in the final prediction for a specific test instance by an ML model, another exciting and challenging goal could be the improvement of prediction accuracy of the underlying model utilizing the interpretation method. We hypothesize that the interpretation method can help identify “bad” training instances that may limit the generalization of the model, the removal of which could result in better classifier in an independent test data. Our intuition is that a “good” instance would have very similar interpretations from an interpretation method among different classifiers. Based on this idea, Pearson correlation coefficient is calculated between individual-level interpretations from two classifiers (e.g., RF and LR) for each instance in the ACES dataset. The average correlation coefficient across the six combinations of the four classifiers (i.e., RF, LR, lSVM and rSVM) is used as a measure of the interpretation stability of the instance. As shown in Figure 5, most instances have relatively high interpretation stability, while some instances have much lower stability. After removing 100 patients from the ACES dataset based on the lowest interpretation stability score, new classifiers were trained on the remaining instances using different classification algorithms. To observe the AUC improvement resulted in the classifiers, AUC is measured on an independent NKI dataset for models trained on all of the instances of ACES and models trained on the filtered instances of ACES. As shown in Table 4, improved performance was observed in all four classification models utilizing the filtered training data for PCI. As a comparison, we also removed 100 instances with the lowest average ICS for LIME and SHAP in the same way as for PCI. Results revealed that both LIME and SHAP also resulted in better AUC than the original classifiers except LIME in RF and rSVM classifiers. These results strongly support that instance-level interpretation methods such as PCI, LIME and SHAP can be used, among other possibilities, as diagnostic tools for identifying bad instances in the training data which have an adverse effect on the generalization of the underlying ML model, and that the extent to achieve can be a meaningful measure of interpretation relevancy.
Fig. 5: Distribution of average correlation between PCI interpretations from six pairwise classifier combinations for each instance in ACES.
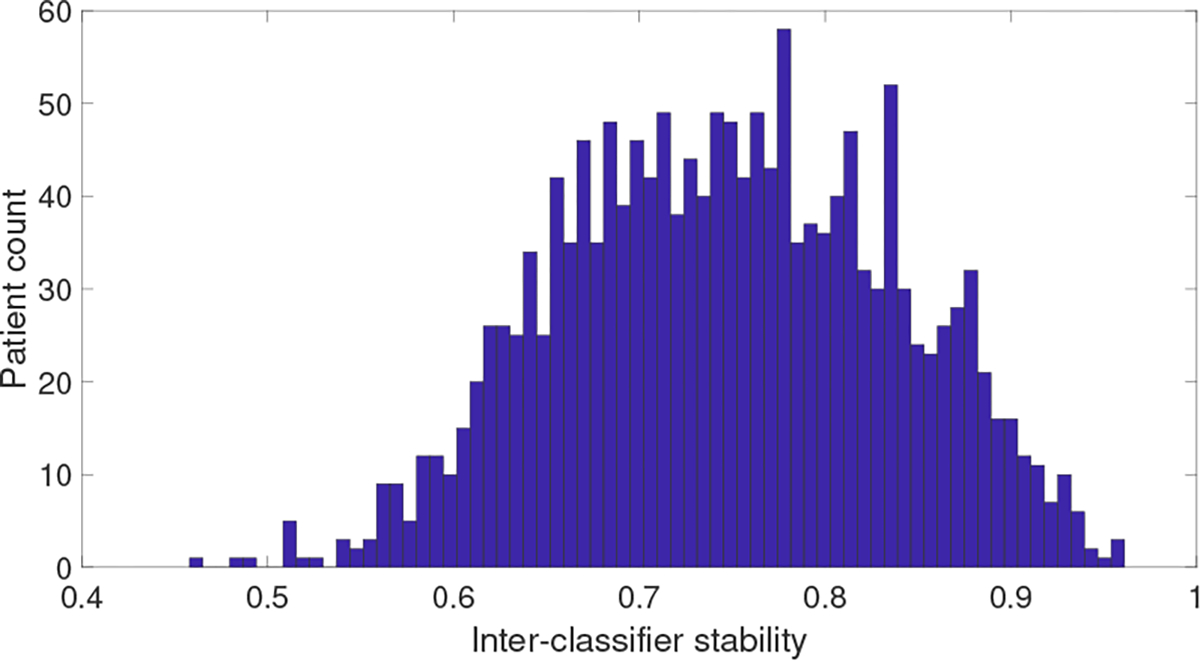
TABLE 4:
Classifiers are retrained on filtered instances of ACES dataset and tested on NKI. The AUC gain of the classifiers on NKI dataset after filtering ACES instances for PCI, LIME and SHAP.
| AUC for all ACES instances | AUC for filtered instances | |||
|---|---|---|---|---|
| Method | PCI | LIME | SHAP | |
| RF | 0.732 | 0.738 | 0.721 | 0.734 |
| LR | 0.730 | 0.731 | 0.731 | 0.735 |
| lSVM | 0.732 | 0.736 | 0.735 | 0.736 |
| rSVM | 0.715 | 0.716 | 0.714 | 0.720 |
4. CONCLUSION
In this article, we proposed a set of ideas to construct and evaluate interpretable machine learning models, and demonstrated its application in breast cancer metastasis prediction. The application has led to improved classification models as well as more stable and understandable interpretations of the models revealing some interesting biology. Using a small number of highly compact and biologically interpretable gene clusters, we were able to construct classifiers with similar or better prediction accuracy compared to classifiers built with many more individual genes as features. We also proposed a model-agnostic post hoc interpretation method, which have achieved better inter-classifier stability than state-of-the-art interpretation methods. Moreover, employing the inter-classifier stability concept introduced in this work, we proposed an idea to identify “bad” instances within the training dataset and resulted in improved prediction accuracy on an independent test dataset. The whole pipeline to construct and evaluate interpretable models can be easily applied to other omics-based medical applications. In addition, the various components proposed in this work, including the clustering-based feature reduction, probability change-based interpretation, as well as inter-classifier stability measure to evaluate model interpretation and to remove “bad” instances, may find applications in other domains to address common ML challenges such as the need to reducing model complexity and improving model interpretability / generalizability.
ACKNOWLEDGEMENT
This research was supported in part by NSF grant ABI-1565076 and NIH grant U54CA217297. The funding body played no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Biographies
Nahim Adnan is a Ph.D. candidate in the Computer Science Department at the University of Texas at San Antonio. He received both his Bachelor’s degree and Master’s degree in Computer Science and Engineering from University of Dhaka, Bangladesh, in 2012 and 2014, respectively. His current research area is in machine learning and biomedical data science, with applications in cancer prognosis and treatment.
Maryam Zand, Ph.D. received her doctoral degree in Computer Science from the University of Texas at San Antonio in 2021. She received her Bachelor of Science degree in Computer Science from Shahid Bahonar University of Kerman in 2008. She also obtained a Master of Science degree in Computer Science from University of Tehran, Iran, in 2012. She has recently joined J. Craig Venter Institute as a Senior Bioinformatics Analyst. Her research interests include Bioinformatics, machine learning and data science.
Tim TH Huang, Ph.D., is Professor and Chair in the Department of Molecular Medicine at the UT Health San Antonio and Deputy Director of the NCI-designated Cancer Therapy and Research Center. He is also the holder of Alice P. McDermott Distinguished University Chair. He has been conducting studies on cancer epigenetics for the last 25 years and has pioneered the development of microarray technologies for the detection of promoter DNA methylation in solid tumors, and he has been a mentor to many successful investigators in the field of Cancer Systems Biology.
Jianhua Ruan, Ph.D., is currently Professor in the Department of Computer Science at the University of Texas at San Antonio. He received his Ph.D. in Computer Science from Washington University in St Louis (2007), M.S. in Computer Science from California State University San Bernardino (2002), and B.S. in Biology from the University of Science and Technology of China (1998). His research interests lie in the broad area of bioinformatics, computational biology, and machine learning. His research is sponsored by the National Institutes of Health, and the National Science Foundation.
Contributor Information
Nahim Adnan, Department of Computer Science, University of Texas at San Antonio, TX, 78249.
Maryam Zand, Department of Computer Science, University of Texas at San Antonio, TX, 78249.
Tim HM Huang, Department of Molecular Medicine, University of Texas Health Science Center, San Antonio, TX 78230.
Jianhua Ruan, Department of Computer Science, University of Texas at San Antonio, TX, 78249.
REFERENCES
- [1].Caruana R, Lou Y, Gehrke J, Koch P, Sturm M, and Elhadad N, “Intelligible Models for HealthCare: Predicting Pneumonia Risk and Hospital 30-day Readmission,” in Proc of the 21th ACM SIGKDD Conference on Knowledge Discovery and Data Mining, 2015, Sydney, NSW, Australia, aug 2015, pp. 1721–1730. [Google Scholar]
- [2].Kim B, Khanna R, and Koyejo O, “Examples are not enough, learn to criticize! criticism for interpretability,” in Proceedings of the 30th International Conference on Neural Information Processing Systems, ser. NIPS’16. Red Hook, NY, USA: Curran Associates Inc., Dec. 2016, pp. 2288–2296. [Google Scholar]
- [3].Miller T, “Explanation in artificial intelligence: Insights from the social sciences,” Artificial Intelligence, vol. 267, pp. 1–38, 2019. [Google Scholar]
- [4].Montavon G, Samek W, and Müller K-R, “Methods for interpreting and understanding deep neural networks,” Digital Signal Processing, vol. 73, pp. 1–15, 2018. [Google Scholar]
- [5].Shrikumar A, Greenside P, and Kundaje A, “Learning Important Features Through Propagating Activation Differences,” arXiv:1704.02685 [cs], Oct. 2019. [Online]. Available: http://arxiv.org/abs/1704.02685 [Google Scholar]
- [6].Bau D, Zhou B, Khosla A, Oliva A, and Torralba A, “Network Dissection: Quantifying Interpretability of Deep Visual Representations,” in 2017 IEEE Conference on Computer Vision and Pattern Recognition (CVPR), Jul. 2017, pp. 3319–3327. [Google Scholar]
- [7].Shrikumar A, Greenside P, and Kundaje A, “Learning Important Features Through Propagating Activation Differences,” ICML, 2017. [Google Scholar]
- [8].Oh JH, Choi W, Ko E, Kang M, Tannenbaum A, and Deasy JO, “Pathcnn: interpretable convolutional neural networks for survival prediction and pathway analysis applied to glioblastoma,” Bioinformatics, vol. 37, no. Supplement_1, pp. i443–i450, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ribeiro MT, Singh S, and Guestrin C, “”Why Should I Trust You?”: Explaining the Predictions of Any Classifier,” in KDD ‘16. San Francisco, California, USA: ACM Press, 2016, pp. 1135–1144. [Google Scholar]
- [10].Lundberg SM and Lee S-I, “A unified approach to interpreting model predictions,” in Advances in Neural Information Processing Systems 30, Guyon I, Luxburg UV, Bengio S, Wallach H, Fergus R, Vishwanathan S, and Garnett R, Eds., 2017, pp. 4765–4774. [Google Scholar]
- [11].Fisher A, Rudin C, and Dominici F, “All models are wrong, but many are useful: Learning a variable’s importance by studying an entire class of prediction models simultaneously.” J. Mach. Learn. Res, vol. 20, no. 177, pp. 1–81, 2019. [PMC free article] [PubMed] [Google Scholar]
- [12].Goldstein A, Kapelner A, Bleich J, and Pitkin E, “Peeking inside the black box: Visualizing statistical learning with plots of individual conditional expectation,” journal of Computational and Graphical Statistics, vol. 24, no. 1, pp. 44–65, 2015. [Google Scholar]
- [13].Apley DW and Zhu J, “Visualizing the effects of predictor variables in black box supervised learning models,” Journal of the Royal Statistical Society: Series B (Statistical Methodology), vol. 82, no. 4, pp. 1059–1086, 2020. [Google Scholar]
- [14].Murdoch WJ, Singh C, Kumbier K, Abbasi-Asl R, and Yu B, “Definitions, methods, and applications in interpretable machine learning,” Proceedings of the National Academy of Sciences, vol. 116, no. 44, pp. 22 071–22 080, Oct. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Yuan Y, Van Allen EM, Omberg L, Wagle N, Amin-Mansour A, Sokolov A, Byers LA, Xu Y, Hess KR, Diao L et al. , “Assessing the clinical utility of cancer genomic and proteomic data across tumor types,” Nature biotechnology, vol. 32, no. 7, pp. 644–652, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Jahid MJ, Huang TH, and Ruan J, “A personalized committee classification approach to improving prediction of breast cancer metastasis,” Bioinformatics, vol. 30, no. 13, pp. 1858–1866, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ruan J, Jahid MJ, Gu F, Lei C, Huang Y-W, Hsu Y-T, Mutch DG, Chen C-L, Kirma NB, and Huang TH-M, “A novel algorithm for network-based prediction of cancer recurrence,” Genomics, vol. 111, no. 1, pp. 17–23, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Adnan N, Liu Z, Huang TH, and Ruan J, “Comparative evaluation of network features for the prediction of breast cancer metastasis,” BMC Medical Genomics, vol. 13, no. 5, pp. 1–10, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Weigelt B, Peterse JL, and Van’t Veer LJ, “Breast cancer metastasis: markers and models,” Nature reviews cancer, vol. 5, no. 8, pp. 591–602, 2005. [DOI] [PubMed] [Google Scholar]
- [20].Siegel RL, Miller KD, and Jemal A, “Cancer statistics, 2016,” CA: a cancer journal for clinicians, vol. 66, no. 1, pp. 7–30, 2016. [DOI] [PubMed] [Google Scholar]
- [21].Zheng X, Amos C, and Frost H, “Comparison of pathway and gene-level models for cancer prognosis prediction,” BMC Bioinformatics, vol. 21, February 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ruan J, “A fully automated method for discovering community structures in high dimensional data,” in 2009 Ninth IEEE International Conference on Data Mining, Dec 2009, pp. 968–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Rudin C, “Stop explaining black box machine learning models for high stakes decisions and use interpretable models instead,” Nature Machine Intelligence, vol. 1, no. 5, pp. 206–215, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Doshi-Velez F and Kim B, “Towards A Rigorous Science of Interpretable Machine Learning,” arXiv:1702.08608 [cs, stat], Mar. 2017, arXiv: 1702.08608. [Google Scholar]
- [25].Lipton ZC, “The Mythos of Model Interpretability,” arXiv:1606.03490 [cs, stat], Mar. 2017. [Google Scholar]
- [26].Staiger C, Cadot S, Györffy B, Wessels L, and Klau G, “Current composite-feature classification methods do not outperform simple single-genes classifiers in breast cancer prognosis,” Frontiers in Genetics, vol. 4, p. 289, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Buitinck L, Louppe G, Blondel M, Pedregosa F, Mueller A, Grisel O, Niculae V, Prettenhofer P, Gramfort A, Grobler J, Layton R, VanderPlas J, Joly A, Holt B, and Varoquaux G, “API design for machine learning software: experiences from the scikit-learn project,” in ECML PKDD Workshop: Languages for Data Mining and Machine Learning, 2013, pp. 108–122. [Google Scholar]
- [28].Melo F, Area under the ROC Curve. New York, NY: Springer New York, 2013, pp. 38–39. [Google Scholar]
- [29].van ‘t Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AAM, Mao M, Peterse HL, van der Kooy K, Marton MJ, Witteveen AT, Schreiber GJ, Kerkhoven RM, Roberts C, Linsley PS, Bernards R, and Friend SH, “Gene expression profiling predicts clinical outcome of breast cancer,” Nature, vol. 415, p. 530, Jan. 2002. [DOI] [PubMed] [Google Scholar]
- [30].Huang DW, Sherman BT, and Lempicki RA, “Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists,” Nucleic Acids Research, vol. 37, no. 1, pp. 1–13, November 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Hanahan D and Weinberg RA, “The hallmarks of cancer,” cell, vol. 100, no. 1, pp. 57–70, 2000. [DOI] [PubMed] [Google Scholar]
- [32].Avagliano A, Maria Rosaria R, Aliotta F, Belviso I, Accurso A, Masone S, Montagnani S, and Arcucci A, “Mitochondrial flexibility of breast cancers: A growth advantage and a therapeutic opportunity,” Cells, vol. 8, p. 401, 04 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ruggero D, “Translational control in cancer etiology,” Cold Spring Harbor perspectives in biology, vol. 5, July 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kitamura T, Qian B-Z, and Pollard JW, “Immune cell promotion of metastasis,” Nature Reviews Immunology, vol. 15, no. 2, pp. 73–86, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Garner H and de Visser KE, “Immune crosstalk in cancer progression and metastatic spread: a complex conversation,” Nature Reviews Immunology, vol. 20, no. 8, pp. 483–497, 2020. [DOI] [PubMed] [Google Scholar]
- [36].Meric F and Hunt K, “Translation initiation in cancer: A novel target for therapy,” Molecular cancer therapeutics, vol. 1, pp. 971–9, October 2002. [PubMed] [Google Scholar]
- [37].Nasr Z and Pelletier J, “Tumor progression and metastasis: Role of translational deregulation,” Anticancer research, vol. 32, pp. 3077–84, August 2012. [PubMed] [Google Scholar]
- [38].Fabbri L, Bost F, and Mazure N, “Primary cilium in cancer hallmarks,” International Journal of Molecular Sciences, vol. 20, p. 1336, 03 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Zingg D, Debbache J, Peña-Hernández R, Antunes AT, Schaefer SM, Cheng PF, Zimmerli D, Haeusel J, Calçada RR, Tuncer E, Zhang Y, Bossart R, Wong K-K, Basler K, Dummer R, Santoro R, Levesque MP, and Sommer L, “Ezh2-mediated primary cilium deconstruction drives metastatic melanoma formation,” Cancer Cell, vol. 34, no. 1, pp. 69–84.e14, 2018. [DOI] [PubMed] [Google Scholar]
- [40].Folkman J, “Role of angiogenesis in tumor growth and metastasis,” Semin Oncol, vol. 29, pp. 8–15, December 2002. [DOI] [PubMed] [Google Scholar]
- [41].Behrens J, “The role of cell adhesion molecules in cancer invasion and metastasis,” Breast cancer research and treatment, vol. 24, no. 3, pp. 175–184, 1993. [DOI] [PubMed] [Google Scholar]