

Abstract
There are many short-read variant-calling tools, with different strengths and weaknesses. We present a tool, Minos, which combines outputs from arbitrary variant callers, increasing recall without loss of precision. We benchmark on 62 samples from three bacterial species and an outbreak of 385 Mycobacterium tuberculosis samples. Minos also enables joint genotyping; we demonstrate on a large (N=13k) M. tuberculosis cohort, building a map of non-synonymous SNPs and indels in a region where all such variants are assumed to cause rifampicin resistance. We quantify the correlation with phenotypic resistance and then replicate in a second cohort (N=10k).
Supplementary Information
The online version contains supplementary material available at (10.1186/s13059-022-02714-x).
Background
The use of whole genome short-read sequence data to study cohorts of bacterial genomes from a single species is now a standard practice (e.g., [1, 2]). There are a multitude of variant callers, which analyze reads from a sample and make statements about where it differs from a fixed reference genome. However, there is no one best variant caller, or even approach—all have strengths and weaknesses. The single-sample-inference problem is well studied and understood—mapping to a reference works well where the sample and reference are reasonably close, and primarily for SNPs (SAMtools [3]), but fares progressively worse as the reference and sample diverge [4]. On the other hand, methods based on local assembly (GATK [5], Octopus [6]) are better able to detect small indels, and those based on global assembly (Cortex [7], McCortex [8]) are exceptionally specific, robust to reference-choice, and better at accessing clustered SNPs or indels up to a few kb in size, but at a cost in sensitivity. Ideally, it would be valuable to be able to combine the output of two different methods (“callsets”) in some rigorous manner, resulting in a product better than either. Simply using the union of callsets gives no control over false discovery rate, and the intersection is too conservative, losing the benefit of callers with different strengths. This “variant adjudication” problem of rigorously combining callsets, where discordances between input variants are resolved and then variant sites are genotyped, is the first challenge we address in this study. In doing so we separate two processes which are typically bound together within a single variant-caller: the discovery of genetic variants, and the genotyping of these variants. In our schema, we allow different callers to do discovery, and then use our new method to adjudicate (i.e., genotype).
Moving beyond single samples, there are many use-cases where one needs to jointly analyze a cohort, producing a matrix of variants versus samples and making binary or probabilistic statements (genotype calls) at all positions that are segregating in the cohort. This is more tricky, primarily because the density of variation increases with cohort size, and inevitably there are situations where SNPs and indels overlap. This is typically described as joint genotyping [5], and itself is a form of adjudication problem—once we have the full list of segregating sites and alleles, we can revisit all samples and genotype each one. This is the second main problem we address in this study.
Finally, underlying both, there is an important technical challenge: how to combine Variant Call Format (VCF) [9] files, either for the same sample from different variant callers, or from multiple samples in a cohort when collecting a list of segregating sites. In both cases, we want a clean VCF file with a set of non-overlapping records, each representing a segregating site with alternate alleles, which can be independently genotyped (see Fig. 1). This likely entails combining independent overlapping variants from the input VCF files into some consistent multi-allelic record. This particular problem is technically awkward and can in theory get arbitrarily ugly—in the pathological worst case, there could be overlapping records that gradually tile across the whole genome.
Fig. 1.

Variant adjudication pipeline implemented by Minos. Input variants in one or more VCF file(s) are merged to make a deduplicated set of variants. When running on a single sample, the input VCF files could be from different tools. When joint genotyping across samples, there is one VCF file originating from each sample. Next, overlapping variants are clustered together—for example the variants at positions 7 and 8—allowing the construction of a non-nested variation graph. Genotype calls are made using read mapping to the graph
Our motivation was the desire to study tens of thousands of Mycobacterium tuberculosis genomes for the CRyPTIC project [10]. This project sequenced and phenotyped over 15,000 isolates for resistance to 13 different drugs using a 96-well microtitre plate [11], and then applied methods such as Genome-Wide Association Studies (GWAS) to analyze the genetic basis for drug resistance. M. tuberculosis has relatively low levels of diversity by bacterial standards, with no recombination, relatively few mobile genetic elements, and a small pan-genome [12]. However, despite this, almost 17% of the 4.4Mb genome was variable within the cohort, and in some regions almost every single base harbored a multiallelic SNP or indel. Typical approaches tuned for high precision callsets would refuse to make calls in such dense regions, but for our purposes we needed to be able to both represent and correctly genotype these regions.
There has been prior work on these problems, with recent tools focussed on human data. Joint genotyping is available in GATK; however, it relies on machine-learning-based filtering (VQSR) generated from human-specific truth-data. Applying GATK to non-human species required considerable efforts to train a black box VQSR for each new species (e.g., see [13] for Plasmodium). At the time of writing, GATK explicitly does not support bacterial data, although a new version for this is in development. There are also graph-based genotypers for structural variants that operate on similar principles to ours, although with different graph structures and genotyping models, such as Paragraph [14] and vg [15].
Two other graph-mapping based tools are available: BayesTyper [16] maps reads to a directed acyclic graph of informative kmers, and GraphTyper [17] maps to local graphs of SNPs and indels from pre-mapped reads. BayesTyper is set up to take VCFs from different callers, combine them and genotype, much like Minos. GraphTyper’s intended use is to genotype large human cohorts, either with SNP/indels it has discovered, or using a predefined VCF of structural variants. We compare Minos with both below.
Our approach was to build a pure adjudicator, able to run a single command that can take multiple VCF files, handle all overlapping variants, and output a single accurate callset with no inconsistencies. Intuitively, read pileup can be used to test goodness-of-fit. Reads should map perfectly to a reference containing the correct allele, so comparing pileups on alternate alleles can resolve disagreements between callsets. Of course, it would be prohibitively expensive to remap all reads to every input allele independently and then compare the pileup on each allele. Instead, we build a genome graph of the combined alleles from all callers and map once to that, using gramtools [18]. Reads naturally align to the correct allele, and we can genotype using the resulting coverage and ambiguity information (described below). We implemented this, plus a workflow for joint genotyping cohorts, in our new tool, Minos.
We first use 62 high-quality polished long-read assemblies from three bacterial species and benchmark Minos against BayesTyper and GraphTyper. We then apply all three tools to an M. tuberculosis outbreak (N=385) in the UK in 2013, evaluating precision for both reference and non-reference calls.
Finally, we apply our method to our motivating problem—studying antimicrobial resistance in large cohorts of M. tuberculosis. We first joint-genotype the CRyPTIC global cohort of M. tuberculosis genomes (N=15,215) which contains around 700,000 variants (roughly one SNP every 5bp). We focus on the 81bp rifampicin-resistance determining region (RRDR) in the rpoB gene, which bears an enormous level of variation. The WHO-endorsed Xpert® MTB/RIF assay assumes any non-synonymous SNP or indel in this region causes resistance to rifampicin [19], although as we discuss below, the story is a little more complex. We restrict to non-synonymous SNPs and indels and give an unprecedented map of dense variation in the RRDR and how strongly each variant correlates with resistance. We find the five known “borderline” mutations [20] but also show that there are more. We then joint-genotype a second, independent, cohort of 13,411 M. tuberculosis genomes which have also been phenotyped for rifampicin, and replicate the finding. We consider the significance of these findings in the Discussion.
Results
We developed a new tool called Minos, which takes putative variant calls as input, adjudicates between all of the calls, and reports a final accurate callset. It uses the standard Variant Call Format (VCF) for its input and output. It can accept VCF files from any source, using all records where the genotype (GT) field is present and has a non-reference call (records without this field are ignored). Additionally, Minos includes a Nextflow [21] pipeline to joint genotype large numbers (tens of thousands) of samples, producing a set of calls at the same variant sites across all samples. See Fig. 1 for an overview of the pipeline, and the “Methods” section for a complete description.
Existing tools to assess the accuracy of call sets, such as hap.py (https://github.com/Illumina/hap.py) and RTG vcfeval [22], were developed for human diploid data and require truth variant calls in a VCF file. Such evaluations typically need to cope with uncertain phasing in the “truth data”. However in our case, as is typical in bacterial genomics, the truth data is a polished (haploid) whole genome assembly assumed to contain no errors. We therefore developed a tool called Varifier to meet the need for a tool that uses such a truth sequence to determine the precision and recall of a call set. As described in full in the “Methods” section, Varifier evaluates each allele call by aligning the allele plus flanking sequence to the truth genome. This method is robust to complex variants, which can have more than one correct VCF representation. We found other tools could make errors around these types of variants—an example is given in Additional file 1. In this study, for each sample, we used a truth genome assembled from long reads (PacBio or Oxford Nanopore), and polished using the same Illumina reads that were used for variant calling. We used Varifier and these truth genomes to benchmark Minos against GraphTyper and BayesTyper with simulated and real data.
Single-sample benchmarking
We performed an initial sanity check that BayesTyper, GraphTyper, and Minos all work as expected, using simulated reads made with ART [23] and simulated variants in the M. tuberculosis H37Rv reference genome [24]. All tools performed near perfectly on this simple data set, affirming that they make no major errors (Additional file 1: Fig. S1, Additional files 2, 3, 4 and 5: Tables S1–S4). The default Minos filters dropped the recall of SNPs and short indels slightly (Additional file 1: Fig. S1), due to unrealistically low variation in read depth in the simulations, which caused the “MIN_GCP” filter (see the “Methods” section) to fail true positive calls. However, as shown later, this is not an issue in real sequencing read data. Having confirmed all tools passed a basic test, we move on from the simulations to empirical data.
Next, the tools were compared using real data from M. tuberculosis, Staphylococcus aureus, and Klebsiella pneumoniae, using samples which each had a high-quality polished long read assembly to act as truth, and matched Illumina data (see Methods). We selected reference genomes for each species (1 for M. tuberculosis, 2 for S. aureus and 5 for K. pneumoniae) to reflect the diversity of the species.
For each Illumina data set, reads were trimmed using Trimmomatic [25], mapped to the reference genome with BWA MEM [26], and PCR duplicate reads were removed with SAMtools. Variants were called independently using two variant callers with orthogonal strengths: SAMtools/BCFtools is pileup-based, with high sensitivity for SNPs and low precision for indels; Cortex is assembly-based, with high precision for SNPs and indels, but lower recall. The SAMtools/BCFtools and Cortex callsets were input to BayesTyper, GraphTyper, and Minos, resulting in a single set of calls from each tool (BayesTyper and GraphTyper required additional processing of the SAMtools/Cortex VCF files, described in Additional file 1). In order to maximize recall, and because the adjudication tools should remove false-positive variants, the unfiltered callsets from SAMtools and Cortex were used. All results shown are using the default variant call filters for each tool, except where noted, and with unreliable regions of the genomes masked.
Note that GraphTyper can be run in two genotyping modes: default and “sv” (described in [27]). The “sv” mode resulted in significantly worse results in many cases (Additional file 6: Table S5); therefore, it is not discussed further in this manuscript. All GraphTyper results in this manuscript refer to running in default mode.
The results are summarized in Table 1 (also Additional file 1: Fig. S2, S3). Minos achieved the best F-score and recall across seven of the eight data sets. The biggest variation between tools was seen in the recall. Although GraphTyper had the highest precision, Minos was equally precise in three of the data sets, and the biggest difference in mean precision between Minos and GraphTyper was 0.05%.
Table 1.
Mean precision, recall, and F-score on each empirical data set with each reference genome. Numbers in bold show the best precision, recall, and F-score for each species and reference genome
| Species | Number of samples | Reference genome | Tool | Mean precision | Mean recall | Mean F-score |
|---|---|---|---|---|---|---|
| M. tuberculosis | 17 | H37Rv | BayesTyper | 0.9995 | 0.9217 | 0.9579 |
| GraphTyper | 0.9997 | 0.8938 | 0.9422 | |||
| Minos | 0.9997 | 0.9181 | 0.9559 | |||
| S. aureus | 28 | TW20 | BayesTyper | 0.9984 | 0.8669 | 0.9279 |
| GraphTyper | 0.9990 | 0.7530 | 0.8545 | |||
| Minos | 0.9988 | 0.8786 | 0.9347 | |||
| USA300 | BayesTyper | 0.9993 | 0.8671 | 0.9283 | ||
| GraphTyper | 0.9995 | 0.7506 | 0.8534 | |||
| Minos | 0.9994 | 0.8792 | 0.9353 | |||
| K. pneumoniae | 17 | GCF_000784945.1 | BayesTyper | 0.9990 | 0.9052 | 0.9495 |
| GraphTyper | 0.9999 | 0.9063 | 0.9505 | |||
| Minos | 0.9999 | 0.9143 | 0.9550 | |||
| GCF_001952915.1 | BayesTyper | 0.9995 | 0.8800 | 0.9346 | ||
| GraphTyper | 0.9999 | 0.8788 | 0.9340 | |||
| Minos | 0.9994 | 0.8922 | 0.9417 | |||
| GCF_003073315.1 | BayesTyper | 0.9996 | 0.9267 | 0.9617 | ||
| GraphTyper | 0.9999 | 0.9297 | 0.9634 | |||
| Minos | 0.9998 | 0.9367 | 0.9672 | |||
| GCF_003076555.1 | BayesTyper | 0.9994 | 0.9397 | 0.9686 | ||
| GraphTyper | 0.9999 | 0.9387 | 0.9683 | |||
| Minos | 0.9999 | 0.9438 | 0.9710 | |||
| GCF_011006575.1 | BayesTyper | 0.9995 | 0.9078 | 0.9511 | ||
| GraphTyper | 0.9999 | 0.9075 | 0.9513 | |||
| Minos | 0.9998 | 0.9238 | 0.9602 |
We also investigated the effect of including more variant callers as input to BayesTyper, GraphTyper, and Minos. In principle, a new caller adds value if it finds variants which are missed by the other callers. The above analysis was repeated, but with calls from Snippy (https://github.com/tseemann/snippy) included together with those from SAMtools and Cortex. The results were almost identical: the mean precision and recall across all data for each tool was the same to two decimal places (Additional file 7: Table S6, Additional file 1: Fig. S4), indicating that for these data, Snippy offers no additional benefit once Samtools and Cortex are combined.
Performance
On the real bacterial data, all tools had a relatively fast run time and low RAM usage (Additional file 8: Table S7, Additional file 1: Fig. S5). On each data set, GraphTyper had the shortest run time and smallest RAM usage, followed by Minos and then BayesTyper. The median wall clock time for Minos across the data sets ranged from approximately 2.5 min per sample (M. tuberculosis) to 6 min (K. pneumoniae). K. pneumoniae required the highest RAM, where the median peak usage across all runs for Minos was 2.5GB.
Joint genotyping of cohorts
We wrote a Nextflow pipeline, included in the Minos code repository, to easily joint genotype large numbers of samples. It outputs a single VCF file containing all samples genotyped at the same sites, and the same information in per-sample VCF files. We tested this pipeline, together with BayesTyper and GraphTyper, to re-analyze 385 M. tuberculosis [28] samples from an outbreak in the UK, which we call the “Walker 2013” data set. Note that BayesTyper and GraphTyper are not set up for this use case, and therefore, processing the data was required to attempt to use these tools in this manner, as described in the Methods section.
Our final analysis was to joint genotype two large M. tuberculosis cohorts of more than ten thousand samples each, using the results to analyze the phenotypes and corresponding genotypes in the 81bp rifampicin resistance determining region (RRDR) of the rpoB gene. The first large data set (“CRyPTIC”) consisted of 15,215 samples released by the CRyPTIC project [10], phenotyped using a microtitre plate assay [11, 29], and the second (“Mykrobe”) data set consisted of 13,411 samples previously published [30] and phenotyped using traditional culture-based DST (drug susceptibility testing).
Variants were called on each sample independently with the same methods as above using Cortex and SAMtools, and then calls adjudicated with Minos. Each of these per-sample Minos VCF files was used as input to each of the three tools. The result was a callset for each tool, at the same variant sites for all samples for that tool (variant sites were not the same between tools). Minos is the only tool of the three that is set up to consistently merge all input VCF records, so that no two sites in the output contain reference positions in common. Furthermore, this means Minos does not, unlike BayesTyper and GraphTyper, output two separate VCF records with incompatible genotype calls. This is discussed in more detail in Additional file 1.
A summary of the data sets and variants output by Minos is given in Table 2. A typical M. tuberculosis genome might be around 1000 to 2000 SNPs distant from the H37Rv reference genome. Since these are spread across a 4.4Mb genome, excessive density of variants is not a problem for the single-sample adjudication problem. However when joint genotyping the larger cohorts, we are adjudicating all segregating variation, covering 17–18% of the genome, including some regions of hundreds of base-pairs where almost every single base is a multiallelic SNP or indel. These dense regions present a scaling challenge to graph genome algorithms, depending on implementation and indexing strategy.
Table 2.
Summary of M. tuberculosis data sets used for joint genotyping. “Genome inside sites” is the total length of all reference alleles across all sites after clustering. It is reported as the total number of base pairs, and in parentheses as a percentage of the 4.4Mbp H37Rv reference genome. SNP sites is the number of sites where all alleles have length 1
| Data set | Number of samples | Unique variants | Excluded variants | Sites after clustering | Genome inside sites (bp(%)) | Total alleles | SNP sites |
|---|---|---|---|---|---|---|---|
| Walker 2013 | 385 | 31,548 | 231 | 30,621 | 41,437 (1%) | 62,690 | 27,639 |
| Mykrobe | 13,411 | 699,484 | 6,259 | 593,584 | 756,003 (17%) | 1,414,723 | 552,543 |
| CRyPTIC | 15,215 | 718,863 | 6,576 | 611,269 | 778,949 (18%) | 1,469,100 | 568,224 |
Outbreak analysis
We evaluated performance of Minos and alternate tools on genomes in the Walker 2013 data set, from an outbreak of M. tuberculosis. To measure the precision and recall of the three tools before and after joint genotyping, the 17 M. tuberculosis samples used earlier were added into the outbreak data set. Joint genotyping the samples generally had a negligible effect on precision and recall (Additional file 9: Table S8). BayesTyper recall increased by 0.07%, whereas the recall of GraphTyper and Minos dropped by 4.2% and 0.6% respectively. BayesTyper precision fell by 0.13%, and both GraphTyper and Minos increased by 0.01%.
Up to this point in the manuscript, precision has always been calculated by considering only non-reference allele calls, focussing on the differences between a given sample and the reference genome. However, joint genotyping involves genotyping every sample at every variant site. As a result, the majority of calls have the reference genotype, and correctly genotyping these cases is critical for applications such as building a phylogenetic tree, computing a genetic distance matrix or for GWAS. The difference between excluding or including reference calls for each tool is shown in Fig. 2 and Additional file 9: Table S8. After joint genotyping, and including reference genotype calls in the calculation, Minos achieved a precision of 99.95%, compared with 92.87% and 88.85% for BayesTyper and GraphTyper respectively.
Fig. 2.
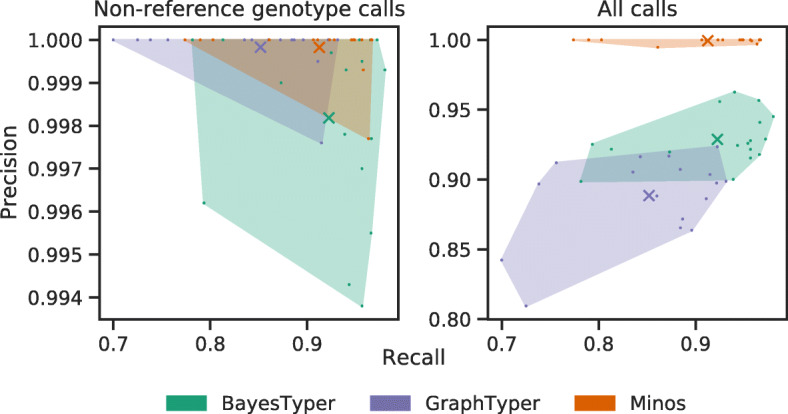
Precision and recall when joint genotyping M. tuberculosis outbreak data. The left plot considers non-reference allele calls only, i.e., the variant sites that are genotyped to be different from the reference genome. The right plot shows the results when all allele calls are included. Individual samples are marked as dots, and the mean precision and recall for each tool is shown as a cross. The convex hull of the data points for each caller is shaded with an associated color
Association of rifampicin resistance with the RRDR region of rpoB
Returning to the motivating problem for which Minos was developed, we applied Minos to two large M. tuberculosis cohorts (“CRyPTIC” and “Mykrobe” data sets) for which we have associated resistance phenotype data for the first-line drug rifampicin. We sought first to confirm that Minos would indeed function at this scale, and then to build a detailed map of variation in the RRDR (rifampicin resistance determining region) of the rpoB gene. Minos output genotype calls at 593,584 and 611,269 variant sites for the Mykrobe and CRyPTIC data sets respectively (Table 2). These sites cover approximately 17–18% of the 4.4Mb H37Rv reference genome.
The total turnaround time of the pipeline, which was limited to a maximum of 2000 concurrent tasks, was approximately 23 h for the Mykrobe data set and 26 h for CRyPTIC. These represent real-world times, since the compute cluster we used was shared with numerous other users. A breakdown of the run times and maximum RAM usage is provided in Additional file 10: Table S9. The total CPU time was 787 core-days for the Mykrobe data set and 905 core-days for CRyPTIC. The pipeline is optimized to minimize memory, with the peak memory used when merging and clustering variants at less than 8GB. The majority of the run time comprises running Minos on each sample, which required less than 2GB of RAM per sample.
We then used the joint genotyping output to analyze the genotype-phenotype relationship within the RRDR of the rpoB gene, by restricting to the 13,259 samples of the Mykrobe data set, and the 8955 samples of the CRyPTIC data set with a high quality rifampicin phenotype [10] (from 12,099 CRyPTIC samples with any quality rifampicin phenotype). Figure 3 shows all the identified amino acid variants—substitutions, insertions, and deletions—plotted along the RRDR, with their prevalence and proportion of resistant samples for the CRyPTIC data. The same plots for the Mykrobe data and all CRyPTIC data are given in Additional file 1: Fig. S6, and the raw data are in Additional file 11: Table S10.
Fig. 3.

All amino acid variants identified in the RRDR of the rpoB gene by joint genotyping 8,955 samples from the CRyPTIC M. tuberculosis data set. Each plot shows the RRDR region from left to right. Single amino acid variants are shown in the upper grid, with the y axis corresponding to the variant amino acid. The lower area shows deletions and insertions, with the inserted sequence given in the colored boxes. For example, the leftmost deletion of amino acids TS at position 427-428 is found in one sample, which is resistant. The leftmost insertion adds R after the S at position 431 (found in one resistant sample). The plots show the same variants, but with different color schemes. In a each variant is colored by the number of samples possessing that variant. Plot b colors the variants by the percent of samples with that variant that are rifampicin resistant
As expected the variant S450L, found in 1919 of the samples, dominates. The next most common variant D435V appears in 345 samples. Several rare indels were identified, most of which appear to cause rifampicin resistance. In the CRyPTIC samples, two insertions were identified (R inserted at 431–432 and F at 433–434), and all nine samples with either of these are rifampicin resistant. Six of the eight deletions arise only in resistant samples. In the Mykrobe data, there were seven deletions and three insertions, all of which were only found in resistant samples.
We find a total of 72 distinct amino acid mutations and indels within the 81bp RRDR in the CRyPTIC samples with high-quality phenotypes, including the 5 mutations classified as “borderline resistant” by the WHO [20] (H445L, H445N, D435Y, L452P, L430P). There are 13 variants in the CRyPTIC samples where the number of susceptible samples is greater than the number of resistant samples. The most common are among the borderlines listed above—L430P, where 45/67 samples are susceptible (and 9/19 susceptible in the Mykrobe data), and H445N, where 16/22 samples are susceptible (2/6 in Mykrobe samples). The remaining such variants are rare, each seen in up to five samples. Full counts can be seen in Additional file 1: Fig. S6. Even if the known borderline mutations are excluded, Fig. 3 (right panel) shows a large number of moderate to low frequency variants with a range of correlations with resistance. We discuss these results further below.
Discussion
Variant analysis from short read sequence data is by now a mature field, and there are many tens of different tools for detecting SNPs and short indels [31]. Setting aside performance issues and focussing entirely on completeness and correctness of the inferred SNPs and indels, it is clear that there is no single best tool. The relative weight given to mapping, assembly, paired-end information, and species-specific optimization (eg via machine learning) results in different strengths and weaknesses. This leads to two rational choices: first, to benchmark for your chosen application and choose the best tool, and second to find a way to combine the strengths of different callers. When setting up the CRyPTIC project, we observed that there was no off-the-shelf solution to this problem, and set out to produce an easy-to-use tool that would do this in a rigorous manner. Minos was the result, which we incorporated into a workflow for analysis of M. tuberculosis genomes called Clockwork. We found that in terms of single genome analysis, performance was relatively similar to other benchmarked tools—Minos generally had the best recall, and GraphTyper had marginally the best precision. However, only Minos would out of the box ingest two (or more) VCF files and output results; the other tools forced users to write code to prepare input data and glue together their processing stages. Combining VCF files with SNPs and indels is particularly challenging for cohorts, where a large proportion of the genome can be variable (17% in our CRyPTIC cohort for example). When analyzing the Walker 2013 M. tuberculosis outbreak and including reference/wild-type calls, Minos had much higher precision (7–10% higher) than the other tools, and on scaling up, only Minos could process the 12k and 13k CRyPTIC and Mykrobe cohorts.
Rifampicin is a bactericidal drug which is a critical component of the antitubercular arsenal, resistance to which is typically used as an epidemiological proxy for multi-drug resistance (defined as having resistance to both rifampicin and isoniazid), particularly in PCR-based rapid diagnostics such as the Xpert® assay. The latest WHO technical report [20] showed that there were 6 known borderline mutations in rpoB (of which 5 were in the RRDR) [20, 32–34], and carefully reported the MIC (minimal inhibitory concentration) distributions for isolates with these mutations. In essence, the distribution of MICs (“the level of resistance”) overlapped with the distribution for wild-type (susceptible) M. tuberculosis, which leads to poor reproducibility of binary classification when the threshold between resistant and susceptible (ECOFF) lies in that overlap. Nevertheless, in the light of various reports of worse patient outcome associated with some of these mutations [35–40], the WHO expert group decided that all non-synonymous mutations and indels, even previously unseen ones, should be treated as causing resistance for the purposes of diagnostics and therapy. Our analysis of this, the largest consistently phenotyped cohort to date [10], reveals that the level of resistance caused by mutations in the RRDR is indeed heterogeneous, and reveals further candidate borderline mutations and indels, including a cascade of overlapping rare indels at position 431. This general picture is replicated in the second cohort (Mykrobe data set). For a more nuanced analysis, it is necessary to look at the MIC distribution associated with specific mutations (rather than using a binary resistant/susceptible classification), which has been done in [41].
The main limitation to this study is that for joint genotyping we set a deletion length limit of 50bp. This reflects an underlying design decision: Minos is a tool for combining VCF files from different callers or samples, adjudicating, and outputting an improved VCF file. However VCF is really not an appropriate file format for handling many overlapping small variants and large indels—for example, a 5-kb deletion covering 20 SNPs. We address this question of how best to genotype and encode multiscale variation (such as SNPs on top of long alternate haplotypes or SNPs under deletions) in a separate study [18].
Conclusions
We have presented a new tool, Minos, that enables users to combine results from their preferred variant callers, integrating their strengths, to reach closer to the underlying truth. It also provides a method for joint genotyping SNPs and indels in bacterial genomes. As genomic analysis is now ubiquitous in bacteriology, we believe Minos will be of wide utility.
Methods
Minos pipeline
First we describe the methods used by Minos, which is implemented in Python and available under the MIT license at https://github.com/iqbal-lab-org/minos. The pipeline is outlined in Fig. 1. The first two stages process the input variant calls, which must be in one or more VCF files, to produce a single set of calls that can be used to generate a reference graph for read mapping. Initially, calls are normalized and deduplicated to make a single “merged” set of calls. These calls are then “clustered” into variant sites that define the variant graph used for read mapping. The merging and clustering are described below.
VCF merging
Each VCF file is processed individually as follows. Variant alleles to be retained for further processing are extracted, where for each record if the genotype (GT) field is present then only called the alleles are kept; otherwise, all alleles are used. The remaining records and their alleles are written to a new VCF file. Variants are decomposed into unique SNPs and indels using the commands vcfbreakmulti and vcfallelicprimitives -l 10000 from vcflib (https://github.com/vcflib/vcflib), followed by the normalize function from vt [42], and finally the vcfuniq command from vcflib. These VCF files are loaded into a single data structure containing all the unique variants, plus the origin (i.e., which VCF file) of each variant.
Variant clustering
The merged variants are used to produce a single VCF file of “clustered” variants that is compatible with gramtools, which in turn is used to generate a variant graph and map reads to that graph. Although gramtools supports more complex situations (SNPs on alternate haplotypes separate from the reference genome, SNPs underneath long deletions) [18], we restrict to “non-nested” variation to maintain compatibility with VCF. Therefore, overlapping variants must be converted into a single variant site (i.e., line in a VCF file) containing multiple alleles (an example is given in Additional file 1: Fig. S7). This is straightforward when processing a single sample with a few input VCF files, such as the SAMtools and Cortex VCF files used in this study when benchmarking Minos against other tools. Where possible, all combinations of alleles are generated and included in the graph. However, this is not always feasible when genotyping a large number of samples (hundreds or thousands) because the number of theoretically possible alleles at one site could be very large.
To process a large number of samples, heuristics are used to simplify the variant graph. First, the number of alleles is limited by only allowing deletions of length (by default) ≤50bp. This prevents a combinatorial explosion where SNPs underneath the deletion can cause impractically large numbers of alleles: n biallelic SNPs generates 2n alleles. Second, the number of alleles in a variant site is limited to (by default) 500. If generating all allele combinations at a site results in too many alleles, then only combinations of alleles actually seen in each sample are used. This happened at 1252 sites in the Mykrobe data set, covering 53,632bp of the reference genome, and at 1195 sites (covering 50,324bp) in the CRyPTIC data set. Third, it is possible for the graph to contain the same sequence more than once across multiple variant sites, by choosing different paths through the graph (an example is given in Additional file 1: Fig. S8—roughly this can happen in low complexity sequence where two alternate deletions of some repetitive sequence can lead to the same final sequence). As each new variant site is added, the previous seven sites are checked and any sites generating duplicate sequences are merged into a single deduplicated site. Removing these duplications is necessary to prevent downstream read mapping issues caused by ambiguous mapping to different paths in the graph that are really the same sequence. Finally, to reduce RAM usage and run time, there is an option to split the graph into chunks, with read mapping run separately on each of these chunks. Using this option requires the reads to be in a sorted indexed BAM file, so that the reads for each chunk can be efficiently extracted for mapping.
Graph mapping and genotyping
The clustered VCF file made in the previous stage is input to the build command of gramtools [18] to make a variant graph for read mapping. Reads are mapped to the graph using the gramtools command quasimap. Each variant site is genotyped using the output from gramtools, which reports the number of reads mapped to each allele, and the read depth across each position of each allele. Minos supports haploid genotyping calling only, using the model described below. Minos and gramtools use similar models—the gramtools model was based on that of Minos, but was modified to handle nested genotyping.
At each variant site, the aim is to choose the correct, i.e., most likely, allele from a set of alleles A. We have the following information from gramtools:
A function (where P(A) is the power set of A), defined by γ(X)= the number of reads that map to all alleles in X (and map to no other alleles). Since gramtools only reports the combinations of alleles that it sees, we define γ by assuming that all elements of P(A) not reported by gramtools have zero reads. Note also that gramtools does exact matching of the full read length only - clipping the ends of the reads or mismatches between the read and graph are not allowed;
For each allele a belonging to A, the read depth at each position in a, where reads are allowed to be multiply mapped. This means that for each allele gramtools matches a read to, a per-base coverage counter of each allele’s matching bases is incremented. Thus, if a read maps to the middle 100bp of two long alternative alleles, then a counter is incremented at each position of those 100bp in each allele.
Let c be the total coverage at a site, given by
Let a be an allele belonging to A. Define the coverage ca of a to be
Let ε be the error rate in the reads, for which a default value of 0.002 is used and can be changed by the user. Let d be the expected read depth and σ2 the read depth variance, which are estimated using the read depth reported by gramtools at each variant site. We assume that the read depth follows a negative binomial distribution NB(n,r), where the parameters n and r are given by
This requires σ2>d. If this is not the case, then we set σ2 to be double the read depth d. This is only expected to happen in rare circumstances and was only seen in the simulated data sets where the read depth was very even, unlike real data. The genotyping model used by Minos comprises the three terms:
“Correct” coverage: NB(n,r,ca);
Coverage due to read errors: ;
A gap (i.e., zero coverage) penalty: , where p=1−NB(n,r,0) is the probability that a given position has zero depth, ℓ is the length of allele a, and b is the number of positions in a with non zero coverage.
The log likelihood is then calculated by summing the natural logarithm of these three terms. The allele with the greatest log likelihood is chosen, with genotype confidence of the difference in log likelihoods of that allele and the second greatest log likelihood. The genotype confidence is reported in the Minos output VCF file using the tag GT_CONF.
Variant call filtering
The FILTER column of the VCF file made by Minos is implemented using four filters. The first requires a read depth of at least two, called MIN_DP in the output VCF file. The second is a read depth no more than the mean depth plus three standard deviations, called MAX_DP. The third filter identifies apparent heterozygous calls (for example, caused by contamination), requiring by default at least 90% of the reads to support the called allele. It is called MIN_FRS (“minimum fraction of read support”, can be set by the user). The final filter removes low confidence genotype calls. Since the genotyping model is dependent on read depth, the confidence score is not directly comparable between different sets of reads. This is accounted for by normalising the confidence score as follows. 10,000 SNPs are simulated by sampling read depths from a negative binomial distribution, defined by the observed mean depth and variance—this is the same distribution as used in the genotyping model. Incorrect read depth is sampled from a binomial distribution with n = observed read depth, and p = read error rate. These simulated SNPs are genotyped using the same method as used when variant calling, generating an expected distribution of genotyping confidence scores specific to this run. When genotyping the real variant sites, any call with a confidence score in the first 0.5% of the simulated genotype score distribution fails the filter (the threshold can be set by the user). This filter is called MIN_GCP (minimum genotype confidence percentile) in the output VCF file.
Joint variant calling
The Minos Nextflow pipeline for joint genotyping large sets of samples is conceptually very similar to running on a per-sample basis and proceeds as follows. The starting point is a VCF file of variant calls for each sample. These VCF files are clustered and merged, as described above, to produce a single gramtools graph for variant calling all samples. This graph should encapsulate all variants found across all of the input VCF files (except for deletions longer than 50bp). Each sample is genotyped using its reads mapped to the gramtools graph, resulting in a VCF file for each sample, where the variant sites are identical across all samples. The pipeline also produces a single multi-sample VCF file, combining the files using ivcfmerge (https://github.com/iqbal-lab-org/ivcfmerge). Finally, a distance matrix is calculated by defining the distance between any two samples to be the number of variant sites where those samples have different genotype calls.
Variant call evaluation
The variant call evaluation with Varifier was implemented in Python and is available under the MIT license at https://github.com/iqbal-lab-org/varifier. The required input is as follows: (1) a VCF file of variant calls to be evaluated; (2) a “mapping genome” FASTA file, which is the reference sequence corresponding to the VCF file; and (3) a “truth genome,” which is the sequence assumed to be correct. The basic idea is to assign a score from zero (meaning false-positive) to one (true-positive) to each variant call, where fractional scores indicate partially correct calls. The score is determined by mapping probe sequences generated from the reference and alternative alleles to the truth genome.
Precision
Each variant is processed using the following method. First, variants with no genotype call (GT field) or a heterozygous genotype are ignored. This method is designed for haploid organisms only since it essentially looks for perfect allele matches to the truth genome, which does not work for heterozygous genotype calls. A probe sequence is generated comprising the called allele, plus the (by default) 100 flanking nucleotides from the mapping reference before and after the allele—we call this the “alt probe”. Similarly, a “ref probe” is generated that uses the reference allele instead of the called allele. The alt probe is mapped to the truth genome using minimap2 [43], and mappings that do not include the allele in the alignment (i.e., if the start and end positions of the allele in the probe do not lie completely inside the start and end positions of the mapping because of soft-clipping) or have mapping quality equal to zero are ignored. If there are no remaining mappings, then the variant is classified as a false positive and assigned a score of zero. Otherwise, the minimap2 mapping that has the greatest number of allele positions matching the mapping genome is chosen as the “best” match. Similarly, the ref probe is mapped to the mapping genome and the best mapping is chosen, but with the additional requirement that the alignment start position in the mapping genome must be equal to that of the best alt probe mapping. If this results in no ref probe mapping, but with the alt probe mapped, then the variant is classified as a true positive and assigned a score of 1.
If both probe sequences have a best match identified, then edit distances between sequences are used to define a score for the variant call. For motivation, consider the following relatively simple example allele sequences:
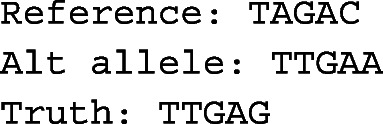
Although the called allele is incorrect because it missed the C to G SNP, it does include the other A to T SNP and is 80% correct (4/5 of the sequence matches the truth). However, the called allele only contains half of the correct variation between the reference and the truth (1/2 SNPs are called), and we would like to account for this. To avoid long insertions or deletions dominating results, we score an insertion or deletion of any length as 1 (i.e., the same as a SNP) when calculating edit distance. Let d(t,r) be the edit distance between the truth and reference alleles, and d(t,a) the edit distance between the truth and alternative alleles. We define the score as zero if d(t,r) is zero, otherwise:
In the example above, the score is 1−(1/2)=0.5. Note that in the simple case of a single SNP, a false-positive scores zero and a true positive scores one. This edit distance-based measure is designed to handle indels and other complex variants. Although the score is usually between zero and one inclusive, in rare cases where the called allele is very distant from the truth, it is possible to have a negative score. The overall precision is calculated by dividing the total of the numerators by the total of the denominators, summed over all variants under consideration.
Recall
Recall is determined using the following method (a flow chart is provided in Additional file 1: Fig. S9). First, a VCF file of all expected calls must either be supplied by the user, or alternatively is made by comparing the mapping genome to the truth genome. Two separate expected callsets are made: one using MUMmer [44] and the other using minimap2 and PAFtools. MUMmer is used by running the commands from its dnadiff pipeline. minimap2 is run with the options -c –cs, and the output is piped into unix sort -k6,6 -k8,8n, and then into the call command of PAFtools with the options -l50 -L50. Taking the union gives a set of variant calls between the mapping and truth genomes, which we expect to contain false positives that need to be removed. The final truth variant callset is made using the same probe mapping method described in the above precision section, to remove false-positive calls. Each variant call from MUMmer and minimap2 is kept if probe mapping to the truth genome results in a true-positive call where the called allele matches perfectly (in other words, the variant has a score of one). In the rare case where both tools call at the same position but with conflicting calls, the calls are not used.
To recap, at this point in the recall pipeline, we have a set of variants to be evaluated, a set of truth variant calls, and the mapping and truth genomes. The truth calls are in a VCF file with respect to the mapping genome. Next, all variants in the VCF file of calls to evaluate are applied to the mapping genome, to produce a new “mutated” genome. Determining recall is now the same as answering: how many truth variants are found in the mutated genome? This is answered using probe mapping using the same method as for precision. The VCF file of truth calls is evaluated, where the mutated reference takes the place of the “truth” genome.
Benchmarking
Truth genomes
The S. aureus truth genomes were generated from PacBio and Illumina reads as follows. The PacBio raw reads were assembled using canu v1.6 [45] to produce the draft assembly. The contigs from the draft assembly were aligned to the respective reference genomes using the nucmer utility from the MUMmer3 [46] package. The contigs were oriented to match the reference and trimmed based on the nucmer alignments and circularized using minimus2 [47]. The assemblies were polished using the Illumina reads by iteratively running Pilon v1.23 [48] until no more corections were made, up to a maximum of 10 runs. Reads were mapped using BWA MEM version 0.7.17 to make input for each Pilon iteration.
The 17 M. tuberculosis truth genomes were from [18], which already had the same Pilon polishing process applied to them as used on the S. aureus genomes. We used K. pneumoniae assemblies from [49] for the truth genomes, which we note already had Pilon run on them as part of their assembly process.
Genome masks
A genome mask was available for M. tuberculosis H37Rv, which is used routinely by Public Health England. The plasmids were masked from the K. pneumoniae and S. aureus mapping genomes. A mask was generated for each truth genome by excluding positions where there was not a majority agreement between Illumina mapped Illumina reads and that genome, as described in Additional file 1. These masks were used by Varifier, which ignores all variants intersecting any masked region of the truth or mapping genomes.
K. pneumoniae reference genomes
The five K. pneumoniae genomes used as references for variant calling were chosen as follows. The average nucleotide identity (ANI) was calculated between each truth genome and all K. pneumoniae genomes in RefSeq using FastANI [50]. RefSeq [51] genomes with a minimum ANI less than 97.5% or a maximum ANI of 100% were excluded. The remaining genomes were listed in order of the minimum ANI, and 5 the genomes were chosen evenly spaced from this list, to obtain a range of ANI between the truth and mapping genomes.
Joint genotyping
The nextflow pipeline included in the Minos github repository was used to run joint genotyping on all three data sets. A nextflow configuration file is included that contains preset profiles to set sensible default parameters for “medium” and “large” size data sets. The “medium” profile was used for the Walker 2013 data set, and “large” was used for the CRyPTIC and Mykrobe data. The pipeline needs an input TSV file, listing sample names and paths to VCF and reads files, plus the reference genome in FASTA format, and optionally a reference genome mask in BED format. The single command line used to run the complete pipeline is provided in Additional file 1. The pipeline is set up to handle large cohorts by saving RAM where possible. It splits the genome into slices, each containing approximately the same number of alleles, and processes each slice in series. Each slice overlaps by a read length to remove end effects from mapping. The Walker 2013 set was split into 100 slices, and the two large sets into 300 slices. The slicing option requires that the input reads for each sample are in a sorted, indexed BAM file, so that the reads for each slice can be efficiently extracted. Such BAM files are typically generated during most variant calling pipelines, and so this requirement is unlikely to create further work.
Joint genotyping with BayesTyper and GraphTyper both required a single sorted VCF file from concatenating (taking account of VCF headers) all input VCF files. This file was sorted, compressed with bgzip, and indexed with tabix for GraphTyper. For BayesTyper, bcftools norm was run on the VCF file. Then the same commands used for running on individual samples were used, as described in Additional file 1. These were successful on all samples in the Walker 2013 data set. On the Mykrobe data set both tools failed on the first sample, ERR025833. For BayesTyper, the combine function ran successfully, but the cluster command failed. GraphTyper failed when running the genotype function, after processing approximately 10% of the genome, with a peak RAM usage of 76GB.
After joint genotyping the CRyPTIC and Mykrobe data sets, the RRDR region was analyzed as follows. To avoid ambiguity, we note that the RRDR region is the 27 amino acid sequence at 426-452 in the rpoB gene in the H37Rv genome (it is often alternatively described with E. coli numbering as 507-533). In H37Rv genome coordinates, this is 759807-763325.
To analyze the RRDR region, only samples that had a high-quality rifampicin phenotype of resistant or susceptible were used. This information is provided in the supplementary tables of [30] for the Mykrobe data. For the CRyPTIC data, we used the samples from [10]. Each of these samples was processed as follows. Its variants contained in the whole rpoB gene were extracted and applied to the genome sequence and translated, making a mutated amino acid sequence. The amino acid variants of the sample were deduced from aligning the mutated amino acid sequence to the reference amino acid sequence. Then the variants in the RRDR were extracted for each sample. In this way, combinations of nucleotide variants were accounted for (for example, two consecutive SNPs could cause a single amino acid change).
Supplementary Information
Additional file 1 Supplementary text and supplementary Figures S1-S9.
Additional file 2 Supplementary Table S1. Raw results generated using calls from Cortex and SAMtools.
Additional file 3 Supplementary Table S2. Raw results generated using calls from Cortex, SAMtools and Snippy.
Additional file 4 Supplementary Table S3. Results of calling simulated SNPs, insertions and deletions in the M. tuberculosis genome.
Additional file 5 Supplementary Table S4. Results of calling simulated complex variants in the M. tuberculosis genome. Total length is the length of the ref allele. Columns 2–5 show the number of each type of variant added to make the complex variant. For example, the first set of variants was 3 SNPs in a window of 10bp, and no insertions or deletions. The second set was 3 SNPs, one insertion of 2bp, and one deletion of 2bp in a window of 10bp.
Additional file 6 Supplementary Table S5. Results of all tools evaluated on the empirical bacteria data set. Results are using the default filter of each tool, which means only taking VCF records where the FILTER column was equal to PASS. The “unfiltered” results are from ignoring the FILTER column and using all records.
Additional file 7 Supplementary Table S6. Summary of effect of including variants from Snippy, in addition to Cortex and SAMtools, as input to BayesTyper, GraphTyper, and Minos. The values are the mean change in precision and recall calculated across all simulated data and all bacteria data.
Additional file 8 Supplementary Table S7. Run time and memory summary for each tool on each data set. Values are taken from the output of the Unix command time -v.
Additional file 9 Supplementary Table S8. Change in accuracy before and after running the joint genotyping pipeline on the Walker 2013 M. tuberculosis data. Joint genotyped precision is calculated in two ways: using just the non-reference allele calls, and using all calls. Note that this does not apply to the recall because in that case we only look for non-reference calls, and so including reference calls has no effect.
Additional file 10 Supplementary Table S9. Joint genotyping Minos memory usage and run times. Values were taken from the LSF reports for each Nextflow task (“Max Memory” and “Run time”). The first three stages VCF merge, VCF cluster, and Gramtools build used 10, 20 and 20 CPUs respectively. The run times are quoted here, not total CPU time. 1Minos is run on each sample in parallel. The mean run time per sample and maximum RAM across all samples is shown. 2This process in an intermediate stage used when producing the final merged VCF file. It merges batches of 300 VCF files each into one VCF file. The merged VCF files are then input to the last task VCF final merge. The mean run time for each batch and maximum RAM across all batches is shown.
Additional file 11 Supplementary Table S10. Counts of resistant and susceptible phenotypes for each amino acid variant seen in the RRDR region of the rpoB gene for the CRyPTIC and Mykrobe data sets. Counts are shown for all CRyPTIC samples with a rifampicin phenotype (CRyPTIC all) and only for those with a high quality phenotype (CRyPTIC high qual). Asterisk (*) shows borderline resistant mutations identified by the WHO.
Additional file 12 Supplementary Table S11. M. tuberculosis accessions.
Additional file 13 Supplementary Table S12. K. pneumoniae accessions.
Additional file 14 Supplementary Table S13. S. aureus accessions.
Acknowledgements
The authors thank Robyn Ffrancon for software engineering in gramtools, and Sara Goodwin from Cold Spring Harbour for assistance with PacBio sequencing.
CRyPTIC consortium
Members of the CRyPTIC consortium (in alphabetical order): Ivan Barilar1, Simone Battaglia2, Emanuele Borroni2, Angela Pires Brandao 3,4, Alice Brankin5, Andrea Maurizio Cabibbe2, Joshua Carter6, Darren Chetty7, Daniela Maria Cirillo2, Pauline Claxton8, David A Clifton5, Ted Cohen9, Jorge Coronel10, Derrick W Crook5, Viola Dreyer1, Sarah G Earle5, Vincent Escuyer11, Lucilaine Ferrazoli4, Philip W Fowler5, George Fu Gao12, Jennifer Gardy13, Saheer Gharbia14, Kelen Teixeira Ghisi4, Arash Ghodousi 2,15, Ana Luíza Gibertoni Cruz5, Louis Grandjean16, Clara Grazian17, Ramona Groenheit18, Jennifer L Guthrie 19,20, Wencong He12, Harald Hoffmann 21,22, Sarah J Hoosdally5, Martin Hunt 23,5, Zamin Iqbal23, Nazir Ahmed Ismail24, Lisa Jarrett25, Lavania Joseph24, Ruwen Jou26, Priti Kambli27, Rukhsar Khot27, Jeff Knaggs 23,5, Anastasia Koch28, Donna Kohlerschmidt11, Samaneh Kouchaki 5,29, Alexander S Lachapelle5, Ajit Lalvani30, Simon Grandjean Lapierre31, Ian F Laurenson8, Brice Letcher23, Wan-Hsuan Lin26, Chunfa Liu12, Dongxin Liu12, Kerri M Malone23, Ayan Mandal32, Mikael Mansjö18, Daniela Matias25, Graeme Meintjes28, Flávia de Freitas Mendes4, Matthias Merker1, Marina Mihalic22, James Millard7, Paolo Miotto2, Nerges Mistry32, David Moore 33,10, Kimberlee A Musser11, Dumisani Ngcamu24, Hoang Ngoc Nhung34, Stefan Niemann 1,35, Kayzad Soli Nilgiriwala32, Camus Nimmo16, Max O’Donnell36, Nana Okozi24, Rosangela Siqueira Oliveira4, Shaheed Vally Omar24, Nicholas Paton37, Timothy EA Peto5, Juliana Maira Watanabe Pinhata4, Sara Plesnik22, Zully M Puyen38, Marie Sylvianne Rabodoarivelo39, Niaina Rakotosamimanana39, Paola MV Rancoita15, Priti Rathod25, Esther Robinson25, Gillian Rodger5, Camilla Rodrigues27, Timothy C Rodwell 40,41, Aysha Roohi5, David Santos-Lazaro38, Sanchi Shah32, Thomas Andreas Kohl1, Grace Smith 25,14, Walter Solano10, Andrea Spitaleri 2,15, Philip Supply42, Adrie JC Steyn7, Utkarsha Surve27, Sabira Tahseen43, Nguyen Thuy Thuong Thuong34, Guy Thwaites 34,5, Katharina Todt22, Alberto Trovato2, Christian Utpatel1, Annelies Van Rie44, Srinivasan Vijay45, Timothy M Walker 5,34, A Sarah Walker5, Robin Warren46, Jim Werngren18, Maria Wijkander18, Robert J Wilkinson 47,48,30, Daniel J Wilson5, Penelope Wintringer23, Yu-Xin Xiao26, Yang Yang5, Zhao Yanlin12, Shen-Yuan Yao24, Baoli Zhu49.
1Research Center Borstel, Borstel, Germany 2IRCCS San Raffaele Scientific Institute, Milan, Italy 3Oswaldo Cruz Foundation, Rio de Janeiro, Brazil 4Institute Adolfo Lutz, São Paulo, Brazil 5University of Oxford, Oxford, UK 6Stanford University School of Medicine, Stanford, USA 7Africa Health Research Institute, Durban, South Africa 8Scottish Mycobacteria Reference Laboratory, Edinburgh, UK 9Yale School of Public Health, Yale, USA 10Universidad Peruana Cayetano Heredia, Lima, Perú 11Wadsworth Center, New York State Department of Health, Albany, USA 12Chinese Center for Disease Control and Prevention, Beijing, China 13Bill & Melinda Gates Foundation, Seattle, USA 14UK Health Security Agency, London, UK 15Vita-Salute San Raffaele University, Milan, Italy 16University College London, London, UK 17University of New South Wales, Sydney, Australia 18Public Health Agency of Sweden, Solna, Sweden 19The University of British Columbia, Vancouver, Canada 20Public Health Ontario, Toronto, Canada 21SYNLAB Gauting, Munich, Germany 22Institute of Microbiology and Laboratory Medicine, IMLred, WHO-SRL Gauting, Germany 23EMBL-EBI, Hinxton, UK 24National Institute for Communicable Diseases, Johannesburg, South Africa 25UK Health Security Agency, Birmingham, UK 26Taiwan Centers for Disease Control, Taipei, Taiwan 27Hinduja Hospital, Mumbai, India 28University of Cape Town, Cape Town, South Africa 29University of Surrey, Guildford, UK 30Imperial College, London, UK 31Université de Montréal, Canada 32The Foundation for Medical Research, Mumbai, India 33London School of Hygiene and Tropical Medicine, London, UK 34Oxford University Clinical Research Unit, Ho Chi Minh City, Viet Nam 35German Center for Infection Research (DZIF), Hamburg-Lübeck-Borstel-Riems, Germany 36Colombia University Irving Medical Center, New York, USA 37National University of Singapore, Singapore 38Instituto Nacional de Salud, Lima, Perú 39Institut Pasteur de Madagascar, Antananarivo, Madagascar 40FIND, Geneva, Switzerland 41University of California, San Diego, USA 42Univ. Lille, CNRS, Inserm, CHU Lille, Institut Pasteur de Lille, U1019 - UMR 9017 - CIIL - Center for Infection and Immunity of Lille, F-59000 Lille, France 43National TB Reference Laboratory, National TB Control Program, Islamabad, Pakistan 44University of Antwerp, Antwerp, Belgium 45University of Edinburgh, Edinburgh, UK 46Stellenbosch University, Cape Town, South Africa 47Wellcome Centre for Infectious Diseases Research in Africa, Cape Town, South Africa 48Francis Crick Institute, London, UK 49Institute of Microbiology, Chinese Academy of Sciences, Beijing, China
Peer review information
Wenjing She was the primary editor of this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Review history
The review history is available as Additional file 15.
Authors’ contributions
The study was conceived by ZI. MH developed Minos and Varifier and ran all benchmarking. BL developed gramtools functionality used by Minos and contributed to the genotyping model and Minos code. GN wrote the ivcfmerge code, used by joint genotyping. MBH and LL helped design Varifier, and MBH wrote parts of the Varifier code. RC contributed to the genotyping model. MS and SR assembled and quality-checked the S. aureus assemblies. MH and ZI drafted the manuscript. KM helped with the RRDR analysis and edited the manuscript. All authors reviewed and approved the manuscript.
Funding
The CRyPTIC consortium is supported by the Bill & Melinda Gates Foundation Trust (OPP1133541) and the Wellcome Trust/Newton Fund-MRC Collaborative Award (200205/Z/15/Z). BL is funded by an EMBL predoctoral fellowship. MS and SR are funded under US NSF award DBI-1350041. MH is funded by the National Institute for Health Research (NIHR) Health Protection Research Unit in Healthcare Associated Infections and Antimicrobial Resistance (NIHR200915), a partnership between the UK Health Security Agency (UKHSA) and the University of Oxford. The views expressed are those of the author(s) and not necessarily those of the NIHR, UKHSA, or the Department of Health and Social Care. For the purpose of open access, the author has applied a Creative Commons Attribution (CC BY) licence to any Author Accepted Manuscript version arising. Open Access funding enabled and organized by Projekt DEAL.
Availability of data and materials
The reference genomes used for variant calling were as follows.
• M. tuberculosis: H37Rv version 3 reference genome NC_000962.3 [24].
• S. aureus: USA300 genome GCA_000013465.1 [52] and the TW20 genome GCA_000027045.1 [53].
• K. pneumoniae: GCF_000784945 [54], GCF_001952915 [55], GCF_003073315, GCF_003076555 [56], and GCF_011006575 [57].
A data download is available from Figshare [58] that contains FASTA files of all truth genomes and BED files of all genome masks. Accessions for reads and assemblies are provided in Additional files 12, 13 and 14: Tables S11–S13 and are also included in the data download in tab-delimited format. The Mykrobe data set accessions are in the Supplementary file from [30], available at [59]. Details of the CRyPTIC data can be found in [10].
The pipeline to call variants, run BayesTyper, GraphTyper, and Minos, and run Varifier was written in Python and is available under the MIT license from Github [60]. A copy of the two singularity [61] containers used for this study are available from Figshare: [62] was used for all benchmarking except for [63], which was used for analysis that used Snippy calls. All software versions and command lines used are in Additional file 1.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
E.R. is employed by the UK Health Security Agency and holds an honorary contract with Imperial College London. I.F.L. is Director of the Scottish Mycobacteria Reference Laboratory. S.N. receives funding from German Center for Infection Research, Excellenz Cluster Precision Medicine in Chronic Inflammation, Leibniz Science Campus Evolutionary Medicine of the LUNG (EvoLUNG)tion EXC 2167. P.S. is a consultant at Genoscreen. T.R. is funded by NIH and DoD and receives salary support from the non-profit organization FIND. T.R. is a co-founder, board member and shareholder of Verus Diagnostics Inc, a company that was founded with the intent of developing diagnostic assays. Verus Diagnostics was not involved in any way with data collection, analysis, or publication of the results. T.R. has not received any financial support from Verus Diagnostics. UCSD Conflict of Interest office has reviewed and approved T.R.’s role in Verus Diagnostics Inc. T.R. is a co-inventor of a provisional patent for a TB diagnostic assay (provisional patent #: 63/048.989). T.R. is a co-inventor on a patent associated with the processing of TB sequencing data (European Patent Application No. 14840432.0 & USSN 14/912,918). T.R. has agreed to “donate all present and future interest in and rights to royalties from this patent” to UCSD to ensure that he does not receive any financial benefits from this patent. S.S. is working and holding ESOPs at HaystackAnalytics Pvt. Ltd. (Product: Using whole genome sequencing for drug susceptibility testing for Mycobacterium tuberculosis). G.F.G. is listed as an inventor on patent applications for RBD-dimer-based CoV vaccines. The patents for RBD-dimers as protein subunit vaccines for SARS-CoV-2 have been licensed to Anhui Zhifei Longcom Biopharmaceutical Co. Ltd, China.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Harris SR, Feil EJ, Holden MTG, Quail MA, Nickerson EK, Chantratita N, Gardete S, Tavares A, Day N, Lindsay JA, Edgeworth JD, de Lencastre H, Parkhill J, Peacock SJ, Bentley SD. Evolution of MRSA during hospital transmission and intercontinental spread. Science. 2010;327(5964):469–74. doi: 10.1126/science.1182395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Holt KE, Wertheim H, Zadoks RN, Baker S, Whitehouse CA, Dance D, Jenney A, Connor TR, Hsu LY, Severin J, Brisse S, Cao H, Wilksch J, Gorrie C, Schultz MB, Edwards DJ, Nguyen KV, Nguyen TV, Dao TT, Mensink M, Minh VL, Nhu NTK, Schultsz C, Kuntaman K, Newton PN, Moore CE, Strugnell RA, Thomson NR. Genomic analysis of diversity, population structure, virulence, and antimicrobial resistance in Klebsiella pneumoniae, an urgent threat to public health. Proc Natl Acad Sci. 2015;112(27):3574–81. doi: 10.1073/pnas.1501049112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bush SJ, Foster D, Eyre DW, Clark EL, De Maio N, Shaw LP, Stoesser N, Peto TEA, Crook DW, Walker AS. Genomic diversity affects the accuracy of bacterial single-nucleotide polymorphism–calling pipelines. GigaScience. 2020;9(2):007. doi: 10.1093/gigascience/giaa007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Poplin R, Ruano-Rubio V, DePristo MA, Fennell TJ, Carneiro MO, Van der Auwera GA, Kling DE, Gauthier LD, Levy-Moonshine A, Roazen D, Shakir K, Thibault J, Chandran S, Whelan C, Lek M, Gabriel S, Daly MJ, Neale B, MacArthur DG, Banks E. Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv. 2017. 10.1101/201178.
- 6.Cooke DP, Wedge DC, Lunter G. A unified haplotype-based method for accurate and comprehensive variant calling. Nat Biotechnol. 2021;39(7):885–92. doi: 10.1038/s41587-021-00861-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iqbal Z, Turner I, McVean G. High-throughput microbial population genomics using the Cortex variation assembler. Bioinformatics. 2013;29(2):275–6. doi: 10.1093/bioinformatics/bts673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Turner I, Garimella KV, Iqbal Z, McVean G. Integrating long-range connectivity information into de Bruijn graphs. Bioinformatics. 2018;34(15):2556–65. doi: 10.1093/bioinformatics/bty157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, McVean G, Durbin R, Group GPA. The variant call format and VCFtools. Bioinformatics. 2011;27(15):2156–8. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brankin A, Malone KM, Barilar I, Battaglia S, Borroni E, Brandao AP, Cabibbe AM, Carter J, Cirillo DM, Claxton P, Clifton DA, Cohen T, Coronel J, Crook DW, Dreyer V, Earle SG, Escuyer V, Ferrazoli L, Fowler PW, Gao GF, Gardy J, Gharbia S, Ghisi KT, Ghodousi A, Cruz ALG, Grandjean L, Grazian C, Groenheit R, Guthrie JL, He W, Hoffmann H, Hoosdally SJ, Hunt M, Iqbal Z, Ismail NA, Jarrett L, Joseph L, Jou R, Kambli P, Khot R, Knaggs J, Koch A, Kohlerschmidt D, Kouchaki S, Lachapelle AS, Lalvani A, Lapierre SG, Laurenson IF, Letcher B, Lin W-H, Liu C, Liu D, Mandal A, Mansjö M, Matias D, Meintjes G, de Freitas Mendes F, Merker M, Mihalic M, Millard J, Miotto P, Mistry N, Moore D, Musser KA, Ngcamu D, Nhung HN, Niemann S, Nilgiriwala KS, Nimmo C, Okozi N, Oliveira RS, Omar SV, Paton N, Peto TE, Pinhata JMW, Plesnik S, Puyen ZM, Rabodoarivelo MS, Rakotosamimanana N, Rancoita PM, Rathod P, Robinson E, Rodger G, Rodrigues C, Rodwell TC, Roohi A, Santos-Lazaro D, Shah S, Kohl TA, Smith G, Solano W, Spitaleri A, Supply P, Surve U, Tahseen S, Thuong NTT, Thwaites G, Todt K, Trovato A, Utpatel C, Van Rie A, Vijay S, Walker TM, Sarah Walker A, Warren R, Werngren J, Wijkander M, Wilkinson RJ, Wilson DJ, Wintringer P, Xiao Y-X, Yang Y, Yanlin Z, Yao S-Y, Zhu B. A data compendium of Mycobacterium tuberculosis antibiotic resistance. bioRxiv. 2021. 10.1101/2021.09.14.460274.
- 11.Rancoita PMV, Cugnata F, Gibertoni Cruz AL, Borroni E, Hoosdally SJ, Walker TM, et al.Validating a 14-Drug Microtiter Plate Containing Bedaquiline and Delamanid for Large-Scale Research Susceptibility Testing of Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2018;62(9). 10.1128/AAC.00344-18. [DOI] [PMC free article] [PubMed]
- 12.Sanoussi CN, Coscolla M, Ofori-Anyinam B, Otchere ID, Antonio M, Niemann S, Parkhill J, Harris S, Yeboah-Manu D, Gagneux S, Rigouts L, Affolabi D, de Jong BC, Meehan CJ. Mycobacterium tuberculosis complex lineage 5 exhibits high levels of within-lineage genomic diversity and differing gene content compared to the type strain H37Rv. Microb Genomics. 2021;7(7). 10.1099/mgen.0.000437. [DOI] [PMC free article] [PubMed]
- 13.Miles A, Iqbal Z, Vauterin P, Pearson R, Campino S, Theron M, Gould K, Mead D, Drury E, O’Brien J, Ruano Rubio V, MacInnis B, Mwangi J, Samarakoon U, Ranford-Cartwright L, Ferdig M, Hayton K, Su X. -z., Wellems T, Rayner J, McVean G, Kwiatkowski D. Indels, structural variation, and recombination drive genomic diversity in Plasmodium falciparum. Genome Res. 2016;26(9):1288–99. doi: 10.1101/gr.203711.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen S, Krusche P, Dolzhenko E, Sherman RM, Petrovski R, Schlesinger F, Kirsche M, Bentley DR, Schatz MC, Sedlazeck FJ, Eberle MA. Paragraph: a graph-based structural variant genotyper for short-read sequence data. Genome Biol. 2019;20(1):291. doi: 10.1186/s13059-019-1909-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hickey G, Heller D, Monlong J, Sibbesen JA, Sirén J, Eizenga J, Dawson ET, Garrison E, Novak AM, Paten B. Genotyping structural variants in pangenome graphs using the vg toolkit. Genome Biol. 2020;21(1):35. doi: 10.1186/s13059-020-1941-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sibbesen JA, Maretty L, Danish Pan-Genome Consortium. Krogh A. Accurate genotyping across variant classes and lengths using variant graphs. Nat Genet. 2018;50(7):1054–9. doi: 10.1038/s41588-018-0145-5. [DOI] [PubMed] [Google Scholar]
- 17.Eggertsson HP, Jonsson H, Kristmundsdottir S, Hjartarson E, Kehr B, Masson G, Zink F, Hjorleifsson KE, Jonasdottir A, Jonasdottir A, Jonsdottir I, Gudbjartsson DF, Melsted P, Stefansson K, Halldorsson BV. Graphtyper enables population-scale genotyping using pangenome graphs. Nat Genet. 2017;49(11):1654–60. doi: 10.1038/ng.3964. [DOI] [PubMed] [Google Scholar]
- 18.Letcher B, Hunt M, Iqbal Z. Gramtools enables multiscale variation analysis with genome graphs. Genome Biol. 2021;22(1):259. doi: 10.1186/s13059-021-02474-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.World Health Organization . Xpert MTB/RIF Implementation Manual: Technical and Operational ‘how-to’; Practical Considerations. Geneva: World Health Organization; 2014. [PubMed] [Google Scholar]
- 20.World Health Organization. Technical Report on Critical Concentrations for Drug Susceptibility Testing of Isoniazid and the Rifamycins (rifampicin, Rifabutin and Rifapentine): World Health Organization; 2021. https://www.who.int/publications-detail-redirect/technical-report-on-critical-concentrations-for-drugsusceptibility-testing-of-isoniazid-and-therifamycins-(rifampicin-rifabutin-and-rifapentine). Accessed 2 Aug 2021.
- 21.Di Tommaso P, Chatzou M, Floden EW, Barja PP, Palumbo E, Notredame C. Nextflow enables reproducible computational workflows. Nat Biotechnol. 2017;35(4):316–9. doi: 10.1038/nbt.3820. [DOI] [PubMed] [Google Scholar]
- 22.Cleary JG, Braithwaite R, Gaastra K, Hilbush BS, Inglis S, Irvine SA, Jackson A, Littin R, Rathod M, Ware D, Zook JM, Trigg L, Vega FMDL. Comparing Variant Call Files for Performance Benchmarking of Next-Generation Sequencing Variant Calling Pipelines. bioRxiv. 2015. https://www.biorxiv.org/content/10.1101/023754v2. Accessed 2 Aug 2021.
- 23.Huang W, Li L, Myers JR, Marth GT. ART: a next-generation sequencing read simulator. Bioinformatics (Oxford, England) 2012;28(4):593–4. doi: 10.1093/bioinformatics/btr708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE, Tekaia F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentles S, Hamlin N, Holroyd S, Hornsby T, Jagels K, Krogh A, McLean J, Moule S, Murphy L, Oliver K, Osborne J, Quail MA, Rajandream M-A, Rogers J, Rutter S, Seeger K, Skelton J, Squares R, Squares S, Sulston JE, Taylor K, Whitehead S, Barrell BG. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393(6685):537–44. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 25.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–20. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv:1303.3997 [q-bio]. http://arxiv.org/abs/1303.3997. Accessed 2021-01-21.
- 27.Eggertsson HP, Kristmundsdottir S, Beyter D, Jonsson H, Skuladottir A, Hardarson MT, Gudbjartsson DF, Stefansson K, Halldorsson BV, Melsted P. GraphTyper2 enables population-scale genotyping of structural variation using pangenome graphs. Nat Commun. 2019;10(1):5402. doi: 10.1038/s41467-019-13341-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Walker TM, Ip CL, Harrell RH, Evans JT, Kapatai G, Dedicoat MJ, Eyre DW, Wilson DJ, Hawkey PM, Crook DW, Parkhill J, Harris D, Walker AS, Bowden R, Monk P, Smith EG, Peto TE. Whole-genome sequencing to delineate Mycobacterium tuberculosis outbreaks: a retrospective observational study. Lancet Infect Dis. 2013;13(2):137–46. doi: 10.1016/S1473-3099(12)70277-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.The CRyPTIC Consortium, Fowler PW. Epidemiological cutoff values for a 96-well broth microdilution plate for high-throughput research antibiotic susceptibility testing of M. tuberculosis. Technical report. 2021. https://www.medrxiv.org/content/10.1101/2021.02.24.21252386v1. Accessed 9 Sept 2021.
- 30.Hunt M, Bradley P, Lapierre SG, Heys S, Thomsit M, Hall MB, Malone KM, Wintringer P, Walker TM, Cirillo DM, Comas I, Farhat MR, Fowler P, Gardy J, Ismail N, Kohl TA, Mathys V, Merker M, Niemann S, Omar SV, Sintchenko V, Smith G, Soolingen DV, Supply P, Tahseen S, Wilcox M, Arandjelovic I, Peto TEA, Crook DW, Iqbal Z. Antibiotic resistance prediction for Mycobacterium tuberculosis from genome sequence data with Mykrobe. Wellcome Open Res. 2019;4:191. doi: 10.12688/wellcomeopenres.15603.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bush SJ, Foster D, Eyre DW, Clark EL, De Maio N, Shaw LP, Stoesser N, Peto TEA, Crook DW, Walker AS. Genomic diversity affects the accuracy of bacterial single-nucleotide polymorphism–calling pipelines. GigaScience. 2020;9(2):007. doi: 10.1093/gigascience/giaa007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miotto P, Tessema B, Tagliani E, Chindelevitch L, Starks A, Emerson C, Hanna D, Kim PS, Liwski R, Zignol M, Gilpin C, Niemann S, Denkinger CM, Fleming J, Warren RM, Crook D, Posey J, Gagneux S, Hoffner S, Rodrigues C, Comas I, Engelthaler DM, Murray M, Alland D, Rigouts L, Lange C, Dheda K, Hasan R, Ranganathan UDK, McNerney R, Ezewudo M, Cirillo D, Schito M, Köser CU, Rodwell TC. A standardised method for interpreting the association between mutations and phenotypic drug resistance in Mycobacterium tuberculosis. Eur Respir J. 2017;50(6):1701354. doi: 10.1183/13993003.01354-2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miotto P, Cabibbe AM, Borroni E, Degano M, Cirillo DM. Role of Disputed Mutations in the rpoB Gene in Interpretation of Automated Liquid MGIT Culture Results for Rifampin Susceptibility Testing of Mycobacterium tuberculosis. J Clin Microbiol. 2018;56(5). 10.1128/JCM.01599-17. [DOI] [PMC free article] [PubMed]
- 34.Torrea G, Ng KCS, Van Deun A, André E, Kaisergruber J, Ssengooba W, Desmaretz C, Gabriels S, Driesen M, Diels M, Asnong S, Fissette K, Gumusboga M, Rigouts L, Affolabi D, Joloba M, De Jong BC. Variable ability of rapid tests to detect Mycobacterium tuberculosis rpoB mutations conferring phenotypically occult rifampicin resistance. Sci Rep. 2019;9(1):11826. doi: 10.1038/s41598-019-48401-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Williamson DA, Roberts SA, Bower JE, Vaughan R, Newton S, Lowe O, Lewis CA, Freeman JT. Clinical failures associated with rpoB mutations in phenotypically occult multidrug-resistant Mycobacterium tuberculosis. Int J Tuberc Lung Dis Off J Int Union Against Tuberc Lung Dis. 2012;16(2):216–20. doi: 10.5588/ijtld.11.0178. [DOI] [PubMed] [Google Scholar]
- 36.Ho J, Jelfs P, Sintchencko V. Phenotypically occult multidrug-resistant Mycobacterium tuberculosis: dilemmas in diagnosis and treatment. J Antimicrob Chemother. 2013;68(12):2915–20. doi: 10.1093/jac/dkt284. [DOI] [PubMed] [Google Scholar]
- 37.Van Deun A, Aung KJM, Bola V, Lebeke R, Hossain MA, de Rijk WB, Rigouts L, Gumusboga A, Torrea G, de Jong BC. Rifampin drug resistance tests for tuberculosis: challenging the gold standard. J Clin Microbiol. 2013;51(8):2633–40. doi: 10.1128/JCM.00553-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pang Y, Ruan Y-Z, Zhao J, Chen C, Xu C-H, Su W, Huan S-T, Li R-Z, Zhao Y-L, Chin DP, Wang L-X. Diagnostic dilemma: treatment outcomes of tuberculosis patients with inconsistent rifampicin susceptibility. Int J Tuberc Lung Dis Off J Int Union Against Tuberc Lung Dis. 2014;18(3):357–62. doi: 10.5588/ijtld.13.0459. [DOI] [PubMed] [Google Scholar]
- 39.Van Deun A, Aung KJM, Hossain A, de Rijk P, Gumusboga M, Rigouts L, de Jong BC. Disputed rpoB mutations can frequently cause important rifampicin resistance among new tuberculosis patients. Int J Tuberc Lung Dis Off J Int Union Against Tuberc Lung Dis. 2015;19(2):185–90. doi: 10.5588/ijtld.14.0651. [DOI] [PubMed] [Google Scholar]
- 40.Shah NS, Grace Lin SY, Barry PM, Cheng Y-N, Schecter G, Desmond E. Clinical Impact on Tuberculosis Treatment Outcomes of Discordance Between Molecular and Growth-Based Assays for Rifampin Resistance, California 2003-2013. Open Forum Infect Dis. 2016;3(3):150. doi: 10.1093/ofid/ofw150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.The CRyPTIC Consortium, Carter JJ. Quantitative measurement of antibiotic resistance in Mycobacterium tuberculosis reveals genetic determinants of resistance and susceptibility in a target gene approach. bioRxiv. 2021. 10.1101/2021.09.14.460353. [DOI] [PMC free article] [PubMed]
- 42.Tan A, Abecasis GR, Kang HM. Unified representation of genetic variants. Bioinformatics. 2015;31(13):2202–4. doi: 10.1093/bioinformatics/btv112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34(18):3094–100. doi: 10.1093/bioinformatics/bty191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marçais G, Delcher AL, Phillippy AM, Coston R, Salzberg SL, Zimin A. MUMmer4: A fast and versatile genome alignment system. PLOS Comput Biol. 2018;14(1):1005944. doi: 10.1371/journal.pcbi.1005944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Phillippy AM. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017;27(5):722–36. doi: 10.1101/gr.215087.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL. Versatile and open software for comparing large genomes. Genome Biol. 2004;5(2):12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sommer DD, Delcher AL, Salzberg SL, Pop M. Minimus: a fast, lightweight genome assembler. BMC Bioinforma. 2007;8(1):64. doi: 10.1186/1471-2105-8-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, Cuomo CA, Zeng Q, Wortman J, Young SK, Earl AM. Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLOS ONE. 2014;9(11):112963. doi: 10.1371/journal.pone.0112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gorrie CL, Mirceta M, Wick RR, Judd LM, Wyres KL, Thomson NR, Strugnell RA, Pratt NF, Garlick JS, Watson KM, Hunter PC, McGloughlin SA, Spelman DW, Jenney AWJ, Holt KE. Antimicrobial-resistant Klebsiella pneumoniae carriage and infection in specialized geriatric care wards linked to acquisition in the referring hospital. Clin Inf Dis Off Publ Inf Dis Soc Am. 2018;2:161–70. doi: 10.1093/cid/ciy027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jain C, Rodriguez-R LM, Phillippy AM, Konstantinidis KT, Aluru S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat Commun. 2018;9(1):5114. doi: 10.1038/s41467-018-07641-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.O’Leary NA, Wright MW, Brister JR, Ciufo S, Haddad D, McVeigh R, Rajput B, Robbertse B, Smith-White B, Ako-Adjei D, Astashyn A, Badretdin A, Bao Y, Blinkova O, Brover V, Chetvernin V, Choi J, Cox E, Ermolaeva O, Farrell CM, Goldfarb T, Gupta T, Haft D, Hatcher E, Hlavina W, Joardar VS, Kodali VK, Li W, Maglott D, Masterson P, McGarvey KM, Murphy MR, O’Neill K, Pujar S, Rangwala SH, Rausch D, Riddick LD, Schoch C, Shkeda A, Storz SS, Sun H, Thibaud-Nissen F, Tolstoy I, Tully RE, Vatsan AR, Wallin C, Webb D, Wu W, Landrum MJ, Kimchi A, Tatusova T, DiCuccio M, Kitts P, Murphy TD, Pruitt KD. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016;44(D1):733–45. doi: 10.1093/nar/gkv1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Diep BA, Gill SR, Chang RF, Phan TH, Chen JH, Davidson MG, Lin F, Lin J, Carleton HA, Mongodin EF, Sensabaugh GF, Perdreau-Remington F. Complete genome sequence of USA300, an epidemic clone of community-acquired meticillin-resistant Staphylococcus aureus. Lancet. 2006;367(9512):731–9. doi: 10.1016/S0140-6736(06)68231-7. [DOI] [PubMed] [Google Scholar]
- 53.Holden MTG, Lindsay JA, Corton C, Quail MA, Cockfield JD, Pathak S, Batra R, Parkhill J, Bentley SD, Edgeworth JD. Genome Sequence of a Recently Emerged, Highly Transmissible, Multi-Antibiotic- and Antiseptic-Resistant Variant of Methicillin-Resistant Staphylococcus aureus, Sequence Type 239 (TW) J Bacteriol. 2010;192(3):888–92. doi: 10.1128/JB.01255-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Conlan S, Thomas PJ, Deming C, Park M, Lau AF, Dekker JP, Snitkin ES, Clark TA, Luong K, Song Y, Tsai Y-C, Boitano M, Dayal J, Brooks SY, Schmidt B, Young AC, Thomas JW, Bouffard GG, Blakesley RW, NISC Comparative Sequencing Program. Mullikin JC, Korlach J, Henderson DK, Frank KM, Palmore TN, Segre JA. Single-molecule sequencing to track plasmid diversity of hospital-associated carbapenemase-producing Enterobacteriaceae. Sci Transl Med. 2014;6(254):254ra126. doi: 10.1126/scitranslmed.3009845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huang W, Wang G, Sebra R, Zhuge J, Yin C, Aguero-Rosenfeld ME, Schuetz AN, Dimitrova N, Fallon JT. Emergence and Evolution of Multidrug-Resistant Klebsiella pneumoniae with both blaKPC and blaCTX-M Integrated in the Chromosome. Antimicrob Agents Chemother. 2017;61(7). 10.1128/AAC.00076-17. [DOI] [PMC free article] [PubMed]
- 56.Wyres KL, Hawkey J, Hetland MAK, Fostervold A, Wick RR, Judd LM, Hamidian M, Howden BP, Löhr IH, Holt KE. Emergence and rapid global dissemination of CTX-M-15-associated Klebsiella pneumoniae strain ST307. J Antimicrob Chemother. 2019;74(3):577–81. doi: 10.1093/jac/dky492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li J, Xu Q, Ogurek S, Li Z, Wang P, Xie Q, Sheng Z, Wang M. Efflux Pump AcrAB Confers Decreased Susceptibility to Piperacillin–Tazobactam and Ceftolozane–Tazobactam in Tigecycline-Non-Susceptible Klebsiella pneumoniae. Infect Drug Resist. 2020;13:4309–19. doi: 10.2147/IDR.S279020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hunt M. Minos supplementary fasta and bed files. Figshare. 2021. 10.6084/m9.figshare.16613398.
- 59.Hunt M. Mykrobe sample data. Figshare. 2019. 10.6084/m9.figshare.7556789.
- 60.Hunt M. Minos paper benchmarking. Github. 2022. https://github.com/iqbal-lab-org/minos-paper-benchmarking. Accessed 23 Nov 2021.
- 61.Kurtzer GM, Sochat V, Bauer MW. Singularity: Scientific containers for mobility of compute. PLOS ONE. 2017;12(5):0177459. doi: 10.1371/journal.pone.0177459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hunt M. Singularity container for minos paper benchmarking. Figshare. 2021. 10.6084/m9.figshare.16613383.
- 63.Hunt M. Singularity container for minos paper benchmarking including Snippy. Figshare. 2022. 10.6084/m9.figshare.19130396.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1 Supplementary text and supplementary Figures S1-S9.
Additional file 2 Supplementary Table S1. Raw results generated using calls from Cortex and SAMtools.
Additional file 3 Supplementary Table S2. Raw results generated using calls from Cortex, SAMtools and Snippy.
Additional file 4 Supplementary Table S3. Results of calling simulated SNPs, insertions and deletions in the M. tuberculosis genome.
Additional file 5 Supplementary Table S4. Results of calling simulated complex variants in the M. tuberculosis genome. Total length is the length of the ref allele. Columns 2–5 show the number of each type of variant added to make the complex variant. For example, the first set of variants was 3 SNPs in a window of 10bp, and no insertions or deletions. The second set was 3 SNPs, one insertion of 2bp, and one deletion of 2bp in a window of 10bp.
Additional file 6 Supplementary Table S5. Results of all tools evaluated on the empirical bacteria data set. Results are using the default filter of each tool, which means only taking VCF records where the FILTER column was equal to PASS. The “unfiltered” results are from ignoring the FILTER column and using all records.
Additional file 7 Supplementary Table S6. Summary of effect of including variants from Snippy, in addition to Cortex and SAMtools, as input to BayesTyper, GraphTyper, and Minos. The values are the mean change in precision and recall calculated across all simulated data and all bacteria data.
Additional file 8 Supplementary Table S7. Run time and memory summary for each tool on each data set. Values are taken from the output of the Unix command time -v.
Additional file 9 Supplementary Table S8. Change in accuracy before and after running the joint genotyping pipeline on the Walker 2013 M. tuberculosis data. Joint genotyped precision is calculated in two ways: using just the non-reference allele calls, and using all calls. Note that this does not apply to the recall because in that case we only look for non-reference calls, and so including reference calls has no effect.
Additional file 10 Supplementary Table S9. Joint genotyping Minos memory usage and run times. Values were taken from the LSF reports for each Nextflow task (“Max Memory” and “Run time”). The first three stages VCF merge, VCF cluster, and Gramtools build used 10, 20 and 20 CPUs respectively. The run times are quoted here, not total CPU time. 1Minos is run on each sample in parallel. The mean run time per sample and maximum RAM across all samples is shown. 2This process in an intermediate stage used when producing the final merged VCF file. It merges batches of 300 VCF files each into one VCF file. The merged VCF files are then input to the last task VCF final merge. The mean run time for each batch and maximum RAM across all batches is shown.
Additional file 11 Supplementary Table S10. Counts of resistant and susceptible phenotypes for each amino acid variant seen in the RRDR region of the rpoB gene for the CRyPTIC and Mykrobe data sets. Counts are shown for all CRyPTIC samples with a rifampicin phenotype (CRyPTIC all) and only for those with a high quality phenotype (CRyPTIC high qual). Asterisk (*) shows borderline resistant mutations identified by the WHO.
Additional file 12 Supplementary Table S11. M. tuberculosis accessions.
Additional file 13 Supplementary Table S12. K. pneumoniae accessions.
Additional file 14 Supplementary Table S13. S. aureus accessions.
Data Availability Statement
The reference genomes used for variant calling were as follows.
• M. tuberculosis: H37Rv version 3 reference genome NC_000962.3 [24].
• S. aureus: USA300 genome GCA_000013465.1 [52] and the TW20 genome GCA_000027045.1 [53].
• K. pneumoniae: GCF_000784945 [54], GCF_001952915 [55], GCF_003073315, GCF_003076555 [56], and GCF_011006575 [57].
A data download is available from Figshare [58] that contains FASTA files of all truth genomes and BED files of all genome masks. Accessions for reads and assemblies are provided in Additional files 12, 13 and 14: Tables S11–S13 and are also included in the data download in tab-delimited format. The Mykrobe data set accessions are in the Supplementary file from [30], available at [59]. Details of the CRyPTIC data can be found in [10].
The pipeline to call variants, run BayesTyper, GraphTyper, and Minos, and run Varifier was written in Python and is available under the MIT license from Github [60]. A copy of the two singularity [61] containers used for this study are available from Figshare: [62] was used for all benchmarking except for [63], which was used for analysis that used Snippy calls. All software versions and command lines used are in Additional file 1.