

Abstract
Renal cell carcinomas are common genitourinary tumors characterized by high vascularization and strong reliance on glycolysis. Despite the many available therapies for renal cell carcinomas, first-line targeted therapies, such as cabozantinib, and durable reaponses are seen in only a small percentage of patients. Yet, little is known about the mechanisms that drive response (or lack thereof). This dearth of knowledge can be explained by the dynamic and complex microenvironment of renal carcinoma, which remains challenging to recapitulate in vitro. Here, we present a microphysiological model of renal cell carcinoma, including a tubular blood vessel model of induced pluripotent stem cell-derived endothelial cells and an adjacent 3D carcinoma model. Our model recapitulated hypoxia, glycolic metabolism, and sprouting angiogenesis. Using our model, we showed that cabozantinib altered cancer cell metabolism and decreased sprouting angiogenesis but did not restore barrier function. This microphysiological model could be helpful to elucidate, through multiple endpoints, the contributions of the relevant environmental components in eliciting a functional response or resistance to therapy in renal cell carcinoma.
Introduction
Renal cell carcinoma (RCC) is a common genitourinary disease with poor prognosis despite treatment[1–5]. Unfortunately, no single treatment elicits responses in all RCC patients. Many targeted therapies against vascular endothelial growth factor (VEGF) pathways have been developed with the aim of reducing tumor microvessel density (e.g., sunitinib, cabozantinib, axitinib)[6], but treatment response is varied, and most patients eventually develop distant metastases[7]. Thereby, it remains a pressing need to better understand the mechanisms underlying response or resistance to therapy to improve outcome in RCC[8].
Cabozantinib, a small molecule tyrosine kinase inhibitor, is a targeted agent that blocks tumor angiogenesis thorugh the VEGF receptor and limits tumor growth, dissemination, and metastasis through the HGF-MET pathway. Cabozantinib has shown increased objective response rate and progression-free survival compared to other drugs in the same category (e.g., sunitinib)[9], and is now prescribed as first or second-line therapy, either alone or in combination with immune checkpoint inhibitor therapy[10]. Recently, the development of clinically relevant disease models has been suggested as a strategy to overcome the absence of prognostic markers for RCC. Although there has been much interest in reproducing normal kidney function in vitro, few efforts have focused on reproducing the microenvironment of kidney cancer. Specifically for RCC, the characteristic high vascularization and strong reliance on glycolysis [11] (i.e., Warburg effect or uncoupling of glycolysis and TCA cycle in aerobic conditions in cancer cells)[12–14] are traits that remain challenging to reproduce in in vitro models. While some studies have aimed to study tumor angiogenesis and vascularization for anti-angiogenic therapy [15, 16], RCC-targeted drugs and their effects on the RCC tumor microenvironment (TME) remain unexplored. Therefore, there is a demand for in vitro models to investigate the effects of novel targeted therapies in the RCC TME [17].
Microphysiological systems (MPS) are 3D microfluidics-based models that aim to reproduce in vivo characteristics of tissues in vitro. MPS present advantages over traditional platforms to model many relevant aspects of the TME, including angiogenesis, hypoxia, tumor growth, migration, and altered response to therapy [18]. Within MPS, the use of both molecular and functional readouts (i.e., direct measurements of treatment efficacy on cells) have been useful in assessing drug efficacy of other cancer types in vitro[19]. Here, we developed a reproducible MPS model using a tubular vessel model of induced pluripotent stem cell (iPSC)-derived endothelial cells (iECs) and a 3-D compact mass of RCC epithelial cells to recapitulate the different steps of tumor angiogenesis in vitro.
Molecular and functional readouts revealed that our model captures key elements of the RCC TME dynamics, which is characterized by hypoxia and nutrient starvation, followed by tumor-induced angiogenesis. Additionally, we leveraged our platform to evaluate the effects of cabozantinib in the TME. Our observations revealed a switch of tumor metabolism and molecular pathways to a less glucose-dependent metabolism. Further, we observed a partial restoration of normal vascular function, consisting of a decrease in angiogenic sprouting but not a recovery of normal permeability. Our model also recapitulated differential signaling pathway activation for the RCC cell lines, consistent with in vivo observations[20].
Reproducing more physiological cell organizations and responses, such as cell-induced hypoxia or angiogenic sprouting, presents important challenges in traditional in vitro platforms. Therefore, our findings highlight the utility of these models to reproduce complex tumor microenvironments to investigate targeted therapies. To our knowledge, this is the first in vitro model simulating tumor-endothelium interactions in the RCC microenvironment and studying the effects of clinically relevant drugs on both the tumor and the surrounding vessels. In addition, we anticipate that MPS models of the TME will allow us to decipher inter-and intracellular signaling relevant to drug response in hopes of optimizing therapeutic approaches against RCC.
Materials and Methods
Cell culture and maintenance
Induced pluripotent stem cell-derived endothelial cells (iECs, Cellular Dynamics International, R1022) were used between passages of 2–5. iECs were maintained in Vasculife Basal maintenance media kit (LifeLine Cell Technologies, LL-0003), supplemented with iCell Endothelial Cell Medium Supplement (Cellular Dynamics International, M1019)[21](hereafter called iEC media). iECs were cultured on conventional cell culture flasks, coated with bovine fibronectin (Sigma-Aldrich, F1141) (diluted to 30 μg/mL in distilled water) for 30 min at room temperature. Cells were grown to an 80% confluence before trypsinization with 0.25% trypsin with EDTA (Thermo Fisher, Gibco, R001100).
A498 (ATCC, HTB-44) and Caki-2 kidney carcinoma cells (a gift from the Lang Lab, UW-Madison) were cultured in iEC media as detailed above and grown until 85% confluence on conventional cell culture flasks and trypsinized as described for iECs.
While A498 is a typical clear cell RCC (ccRCC) cell line, the Caki-2 RCC subtype is less certain[22]. Caki-2 have clear cell histology, but there is debate on their von Hippel Lindau gene status and the lack of expression of HIF2a, which is atypical in ccRCC[23].
Primary cell culture extraction and maintenance
Primary RCC cells were grown in Minimum Essential Medium (MEM) with L-Glutamine, supplemented with 10% of FBS (Corning, 45000–734), 1% Penicillin/Streptomycin (Gibco, 15140122), 1% MEM vitamin mixture 100× (Lonza, 13–607C), 0.5% ITS (Roche, 11074547001)) Sodium Pyruvate 1 mM (Lonza, 13–115E), 100 μM of MEM NEAA (ThermoFisher, 11140–050), 0.4 μg/ml of Hydrocortisone (VWR, 101095–150), 5 ng/ml hEGF (Thermo-Fisher Scientific, PHG0311). The research protocol to obtain tumor and normal adjacent tissue following surgery at the University of Wisconsin Hospital (Madison, WI) was approved by the Institutional Review Board (2011–0719). Informed consent to use residual tissue was obtained from patients before surgery.
Primary cell isolation was performed as previously described[24, 25]. Briefly, a pathologist confirmed ccRCC diagnosis and tissue samples (ca. 3 cm3) were finely minced and digested. Digestion media was a mixture of MEM (Corning, 10–010-CV), 5 mg/ml collagenase (Thermo-Fisher 17100017), 5 mg/ml hyaluronidase (Sigma, H3506), 1 mg/ml of DNase I (Roche, 04716728001), dispase 1% (Worthington, LS02100), and 1% Penicillin-streptomycin. Samples were incubated in digestion media for 1 h at 37 °C. CD31 (i.e., Platelet/endothelial cell adhesion molecule-1, also called PECAM-1) a classical marker of endothelial cells was used for the removal of endothelial cells[26, 27] of this highly vascularized tissue, using anti-CD31-conjugated magnetic microbeads (Miltenyi Biotech, 130-091-935) and separation columns (Miltenyi Biotech, 130-042-401). CD31− cells were seeded in 75 cm2 flasks (Corning, CLS430641U) at 3·104 cells/cm2 and were characterized via immunofluorescence (Figure S8). Cell viability was high in iEC media, which enabled us to co-culture them with iECs in the microdevices.
Microdevice fabrication
The microfluidic organotypic culture platform was adapted from the LumeNEXT approach previously reported[28, 29], with ports designed for passive pumping for the cell loading and media exchange. Briefly, we used standard polydimethylsiloxane (PDMS) micromolding techniques to create a microdevice consisting of two primary components: a culture chamber and a removable PDMS rod. Rods are drawn from 23-gauge needles (340 μm inner diameter). The chamber is formed by oxygen plasma bonding two distinct PDMS layers patterned using SU-8 on silicon master molds (SU8–100, MicroChem) onto a glass-bottom dish (MatTek, P35G-0.170–14-C). Certain variability (≤ 10%) is expected in vessel diameter due to differences in the internal diameter of the needles, but no impact on readouts has been observed with this size variation. PDMS layers were made using the Sylgard 184 silicone elastomer kit (Dow Corning) with a 10:1 curing agent ratio. Layers were incubated in EtOH 70% overnight to remove uncured PDMS oligomers. Then, PDMS layers were aligned using fine tweezers and plasma bonded using a Diener Electronic Femto Plasma Surface System.
Hydrogel preparation in microdevice
After assembly and UV-sterilization, devices were treated for 10 min with 1% poly(ethyleneimine) (Sigma-Aldrich, 03880) in water and with 0.1% glutaraldehyde (Sigma-Aldrich, G6257) in water for 30 min to enhance hydrogel attachment to the PDMS. Devices were washed four times with water and thoroughly dried before hydrogel injection. Type I rat tail collagen hydrogels were prepared at 6 mg/ml in ice to prevent premature gelation (detailed recipe in Table S1). Using a chilled tip, a mixture of collagen type I (Corning, 354249); NaOH 0.5 M (Fisher Scientific, S318); PBS 10x (Fisher Scientific, BP3991) and dH2O was then incubated on ice for 20 min. Provided the pH of the mixture be approximately 7.4 (Capitol Scientific, PH1170–7), the mixture was injected into the LumeNEXT device and polymerized for 15 min at room temperature, followed by 20 min at 37 °C. Afterward, the PDMS rods were gently pulled out of the polymerized collagen gel from the output port resulting in two lumen structures in the collagen gel.
Cell culture and drug treatment in the microdevice
After rod removal, the lumen structure destined to seed the iECs was coated with fibronectin as described in previous sections and washed thrice with iEC media. iECs are then trypsinized and resuspended to a concentration of 15,000 cell/μL and loaded into the open lumen via passive pumping. Devices were then incubated for 1 h at 37°C to allow iECs to adhere, and then washed thrice with relevant media to remove unadhered cells before seeding the A498 cells.
A498 and Caki-2 cells were routinely trypsinized and resuspended at a concentration of 80,000 cell/μL. A second collagen hydrogel (4.5 mg/ml) was then prepared to embed the A498 cells (detailed recipe in Table S1). 2 μL of this cell-hydrogel mixture was injected per lumen and left to polymerize as described in previous sections. After polymerization, a 15-μl droplet of iEC media was added per device and replenished daily after 3 media washes.
Cabozantinib (Selleckchem, S1119) was resuspended in DMSO to a final concentration of 50 mg/ml. Aliquots were stored at −20°C until used. Cabozantinib was diluted in iEC media to a final concentration of 1 μM to treat the cells within the microdevices.
Cell viability assays
Cell viability was evaluated in response to different doses of cabozantinib using the CellTiter-Glo® 2.0 Assay (Promega, G9241). Briefly, for 2D assays, cells were plated on a glass-bottom well plate (Corning, 3601) at a 3000 cells/well density. After 24 h, cabozantinib was added at the studied concentration. Subsequently, CellTiter-Glo® reagent was added directly to the cells (or hydrogels, in their case) in a 1:1 ratio. CellTiter-Glo lyses cells and performs a semi-quantitative analysis of ATP levels. ATP-dependent luminescence is known to correlate with cell numbers linearly. Samples were incubated for 60 min in the dark, and cell-induced luminescence was evaluated in a standard plate reader. Background intensity (no cells) was subtracted from the experimental conditions, and values were then normalized to vehicle controls. 2D dose-response curves were plotted with GraphPad Prism v7 using the inhibitor concentration vs response function.
Real-time fluorescent detection studies
Lumens were treated with Premo™ FUCCI Cell Cycle Sensor (Thermo, P36237) to study iEC proliferation in the lumens). Premo FUCCI contains two different reporters coupled to TagRFP and EmGFP, expressed alternatively during the G1 or the S/G2/M phases. Premo FUCCI was added to the lumens at 1:10 in the cell culture media overnight to label proliferating cells in green, whereas non-proliferating cells appear red. Cells were imaged daily over 72 h to correlate dividing cells with the appearance of angiogenic sprouts in the lumens.
Real-time oxygen levels were assessed using Image-iT™ Red Hypoxia Reagent (Thermo-Fisher Scientific H10498) per the manufacturer’s instructions. The stain was supplemented in the hydrogels and culture media to maintain adequate levels in culture.
To study cell death in the A498 3D culture, The systems were stained with 1:1000 propidium iodide and 1:1000 Caspase 3/7 reagent (Image-iT™ LIVE Red Caspase-3 and -7, Thermo-Fisher Scientific I35102). Staining was incubated in both control and cabozantinib-treated samples for at least 5 hours to diffuse in the microdevice and then was imaged using confocal microscopy.
Permeability studies
The barrier function of the iEC vessel models was assessed by measuring dextran diffusion across the vessel endothelium as previously described[30]. A 1 μM solution of dextran (FITC-conjugated 40 kDa, D1845; TRITC-conjugated 70 kDa, D1818, both ThermoFisher Scientific) was prepared in iEC media. For each vessel, 5 μL of dextran solution was injected through the small lumen port. Dextran diffusion was imaged with the Nikon TI® Eclipse inverted microscope every 5 min over 15 min. Effective permeability coefficients (expressed in cm/s) were calculated using the following equation[31]:
| Eq.1 |
where Io is the total initial intensity outside the vessel, If is the total intensity outside the vessel at 15 min, to is the initial time point, tf is the final time point of 15 minutes, and D is vessel diameter.
Image analysis
Lumen confluency was calculated on phalloidin images after a binarization using a set threshold as previously described[24, 32]. Visual inspection of each threshold confirmed the accuracy in representing the initial image. Then, a consistent Region of Interest was defined to include only the lumen (i.e., not surrounding areas or sprouts). Finally, the percentage of pixels where the phalloidin signal was present was automatically measured for each lumen.
Immunofluorescence staining
Microdevices were fixed after four days of culture by perfusing 4% paraformaldehyde in PBS through the lumen for 15 min. Lumens were washed three times with 0.1% Tween in PBS for 30 min between every step. First, cells were permeabilized with 0.2% Triton® X-100 for 30 min and blocked with 3% BSA in PBS overnight at 4 °C. Next, cells were incubated with the required primary antibody (references and concentrations in Table S2) diluted in 3% BSA, 0.1% Triton® X-100 in PBS (called “antibody dilution buffer”) overnight at 4°C. Cells were then stained with Texas Red®-X Phalloidin, corresponding secondary antibodies, and DAPI in antibody dilution buffer + 10% Goat Serum for 2 h at 4 °C. Finally, the lumens were washed to remove excess staining and minimize the background.
qPCR
mRNA extraction
Device layers were separated to retrieve the lumens for mRNA extraction. Lumens in co-culture were manually separated using a razor and fine tweezers to section the hydrogel and selectively retrieve each cell type. Hydrogels were fragmented to ensure sample homogeneity by passing the sample repeatedly through a 23G needle until no hydrogel fragments were observable before the RNA isolation protocol. Then, RNA was extracted using the Dynabeads™ mRNA DIRECT™ Purification Kit (Thermo Fisher, 61,011) according to the manufacturer’s instructions[25]. At least 2 collagen hydrogels containing iEC lumens, A498 or Caki-2 were pooled per condition.
RT-qPCR
RNA reverse transcription was performed with the RT2 PreAMP cDNA Synthesis Kit (Qiagen, 330451) before qPCR analysis. iECs were analyzed using a Qiagen RT2 profiler Angiogenesis panel (Qiagen, PAHS-024ZA) and RT2 SYBR® Green qPCR Mastermix (Qiagen, 330500) in a Roche 96–384 thermal cycler. Independent qPCR reactions were set for each primer, using the manufacturer’s cycling conditions.
Data were analyzed using 4 reference (housekeeping) genes provided for normalization using the Qiagen online software (http://pcrdataanalysis.sabiosciences.com/pcr/arrayanalysis.php) for the arrays. We normalized the primer assays using the reference genes included in the arrays (B2M and ACTB).
Magpix
Multiplexed protein secretion analysis was performed on iEC vessels and co-cultures with A498 or Caki-2 cells using the MagPix Luminex Xmap (Luminex Corporation) with the “Milliplex human angiogenesis growth factor panel bead kit” (R&D Systems, LXSAHM-10) per the manufacturer’s protocol. Conditioned media was pooled from sets of 4 iPSC-derived vessels (and at least 2 independent experiments) using a p10 pipette over 2 days, yielding 120 μL per set. Data were collected with xPonent software (Luminex). Soluble factor concentrations in media were calculated using mean fluorescence intensities (MFI) by creating a standard curve for each analyte using a five-parameter logistic (5-PL) curve-fit.
Imaging
Bright-field and fluorescent images were obtained using a Nikon TI Eclipse inverted microscope and processed using Nikon Elements and the National Institutes of Health ImageJ software. Confocal images were acquired using a Leica SP8 in the University of Wisconsin, Madison Optical Imaging Core.
NAD(P)H/FAD imaging
Fluorescence lifetime images and intensity were taken on a custom-built inverted multiphoton microscope (Bruker Fluorescence Microscopy, Middleton, WI), as described previously[33–35]. Briefly, the system consists of a Ti:sapphire laser (Spectra Physics, Insight DS-Dual), an inverted microscope (Nikon, Eclipse Ti), and a 40× water immersion (1.15NA, Nikon) objective. NAD(P)H and FAD images were acquired sequentially for the same field of view. NAD(P)H fluorescence was isolated using an excitation wavelength of 750 nm and an emission bandpass filter of 440/80 nm. FAD fluorescence was isolated using an excitation wavelength of 890 nm and an emission bandpass filter of 550/100 nm. Fluorescence lifetime images were collected using time-correlated single-photon counting electronics (SPC-150, Becker, and Hickl) and a GaAsP photomultiplier tube (H7422P-40, Hamamatsu). A pixel dwell time of 4.8 μs was used to acquire 512 × 512-pixel images over 60 s total integration time. For intensity imaging, a pixel dwell time of 4.8 μs was used to collect 1024 × 1024 images. The photon count rates were maintained at 1–2 × 105 photons/s to ensure adequate photon observations for lifetime decay fits and no photobleaching. The instrument response function was measured from the second harmonic generation of urea crystals excited at 900 nm, and the full width at half maximum (FWHM) was calculated to be 244 ps. A Fluoresbrite YG microsphere (Polysciences Inc.) was imaged as a daily standard for fluorescence lifetime. The lifetime decay curves fit a single exponential decay, and the fluorescence lifetime was measured to be 2.1 ns (n = 7), which is consistent with published values.
Quantification of fluorescence lifetime components
NAD and NADPH (indicated as NAD(P)H) and FAD fluorescence lifetime images of cells were analyzed using SPCImage software (Becker & Hickl) as described previously[34, 36]. Briefly, at each pixel, the fluorescence lifetime decay curve was deconvolved with the instrument response function and fit a two-component exponential decay model,
| Eq. 2 |
where I(t) is the fluorescence intensity at time t after the laser excitation pulse, α represents the fractional contribution from each component, C accounts for background light, and τ represents the fluorescence lifetime of each component. A two-component model was used because NAD(P)H and FAD can exist in two conformational states, bound or unbound to enzymes[37]. For NAD(P)H the short and long lifetime components correspond with the unbound and bound conformations, respectively[37]. While the opposite is true for FAD, the short and long lifetime components reflect the bound and unbound conformations, respectively[37]. The mean lifetimes were calculated using the following equation,
| Eq. 3 |
for both NAD(P)H and FAD. The optical redox ratio was calculated from the NAD(P)H and FAD lifetime data by summing the photons detected at each pixel in the image to compute the total intensity. The intensity of NAD(P)H was then divided by the intensity of FAD for each pixel. Redox ratio values were normalized to the high-density (HD) condition taken at the same time point.
An automated cell segmentation pipeline was created in Cell Profiler. Briefly, a customized threshold code identified pixels belonging to nuclear regions. Cells were identified by propagating out from the nuclei within the image. An Otsu Global threshold was used to improve the propagation and prevent it from continuing into background pixels. The cell cytoplasm was defined as the cell borders minus the nucleus. Values for the τm, τ1, τ2, α1, and intensities of NAD(P)H and FAD, as well as the redox ratio, were averaged for all pixels within each cell cytoplasm.
Results
We engineered a microphysiological model to mimic RCC tumor development, tumor pro-angiogenic signaling, and tumor angiogenesis typically observed in nearby renal vessels (Figure 1A). Our model leverages PDMS and mold-casting of luminal structures (Figure 1B–C). One of the resulting tubular structures was lined with iECs to model a blood vessel, while the other lumen was filled with hydrogel-embedded RCC epithelial cells to mimic the tumor mass development in the renal tubule (Figure 1D). Few commercial options are available to model kidney endothelial cells. We chose a commercial cell source that could be used for general in vitro research: iPSC-ECs (iEC’s). Our lab and others[38] have previously used these cells due to their advantages for in vitro modeling: they undergo angiogenic sprouting and have a lower barrier function than other models[21]. The higher permeability is more consistent with renal capillaries, which are fenestrated to facilitate the solute exchange with adjacent renal tubules [39].
Figure 1: Renal cell carcinoma on a chip.
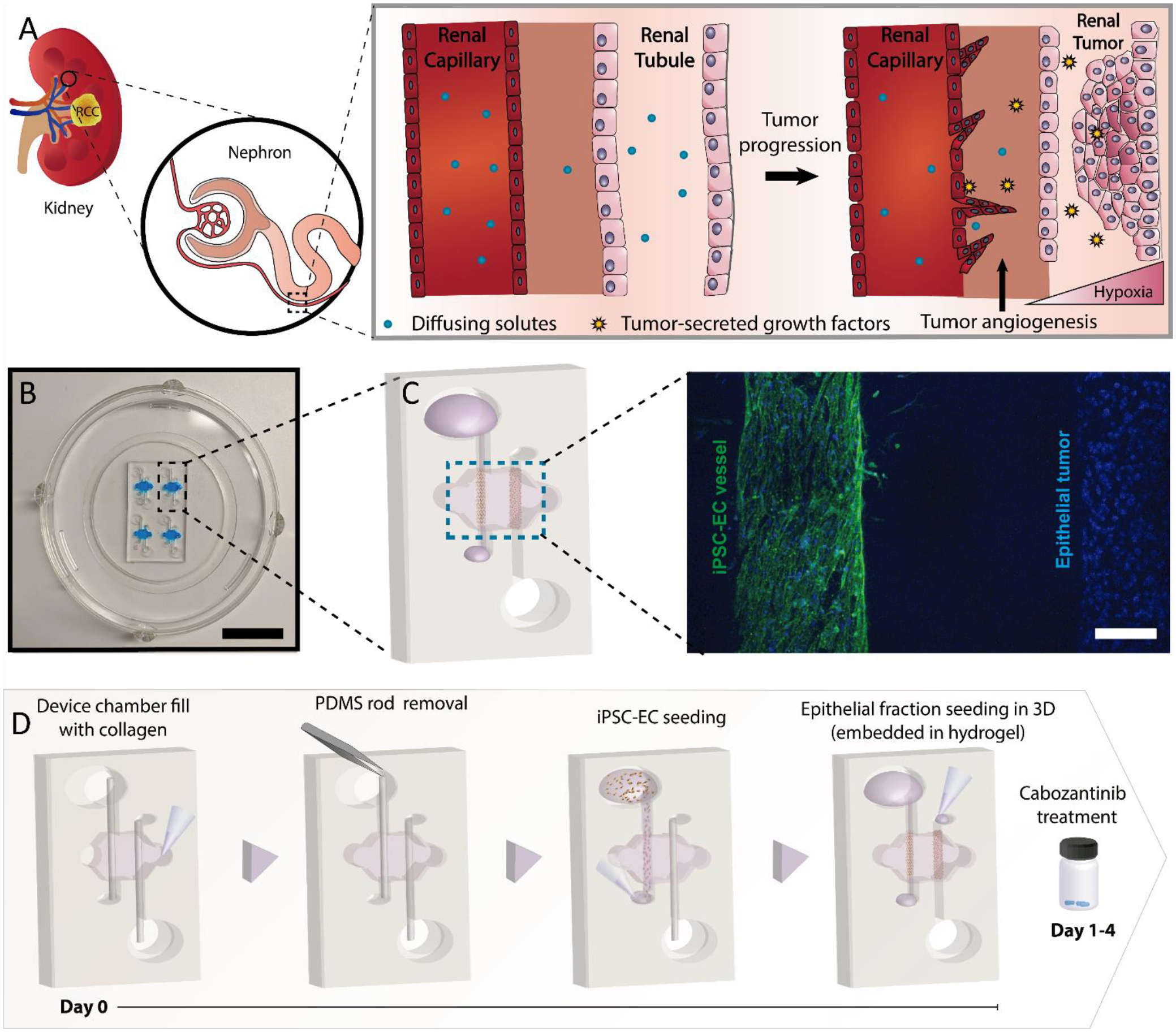
Schematic illustrating the biological processes reproduced in the microdevice. A) Illustration of the tumor progression and angiogenesis model. The model includes a renal capillary and epithelial tubule as a renal tumor develops, along with the increase in angiogenesis in the nearby vasculature. B) Picture of the microdevice and schematic of the co-culture organization in the device. C) Projected confocal image of an iEC-Epithelial co-culture stained with CD31 (green, iEC) and DAPI (blue, all nuclei). D) Schematic depicting the co-culture procedure in the microdevice and timeline for the cabozantinib treatment. Cells shown on the right are epithelial cells.
We began characterizing our model with an iEC endothelial vessel model in monoculture. This model showed little angiogenic sprouting, with most invading cells detached (Figure 2A). Bigger sprouts were hollow, consistent with in vivo observations (Figure 2B), and cell proliferation occurred in the same timeline as the sprouting process (Figure 2C). Angiogenic sprouts were visible after 72 h (Figure 2C, right), consistent with early steps of the angiogenic cascade[40].
Figure 2: iPSC-EC (iEC) endothelial vessel model establishment and characterization.
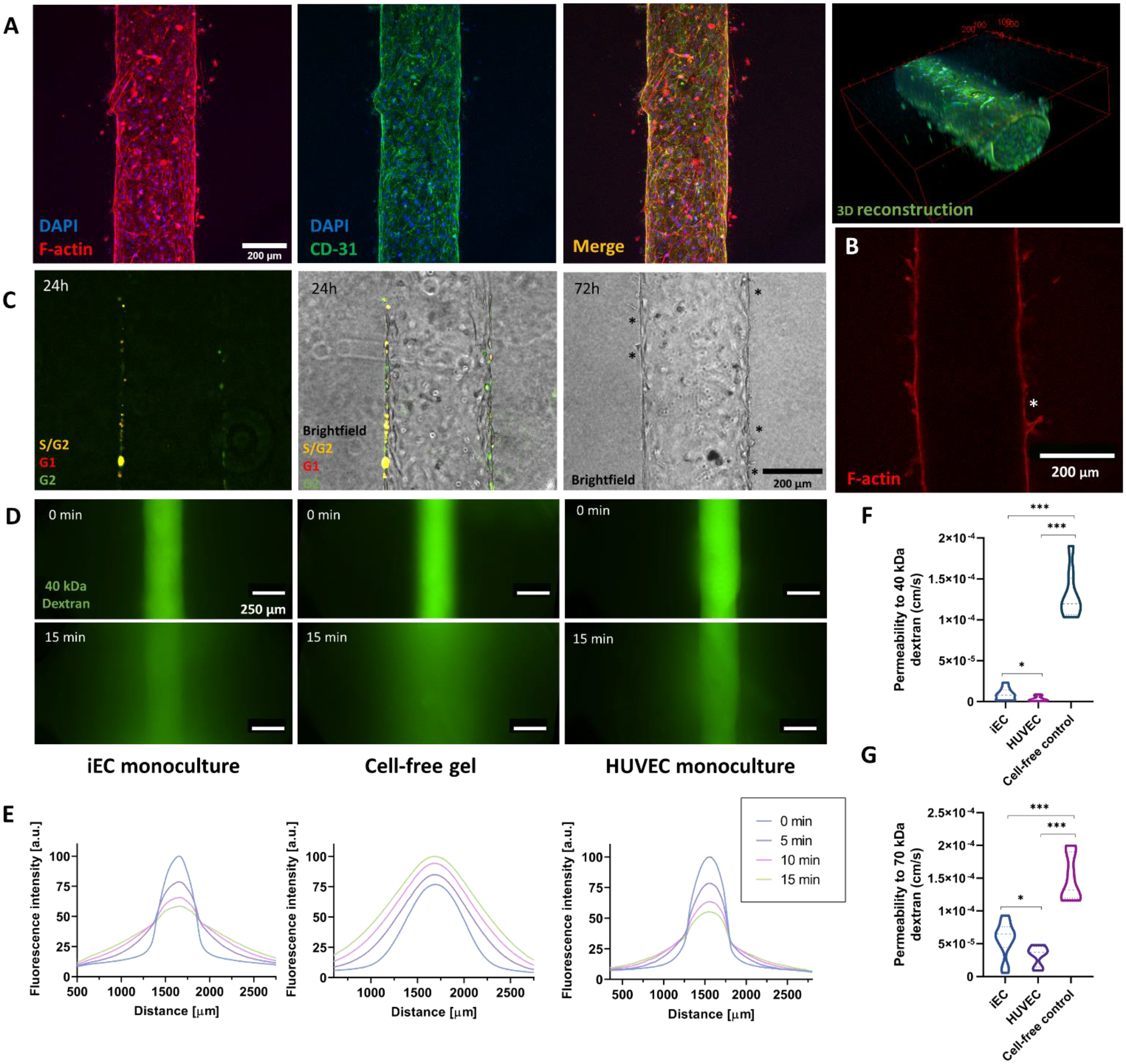
A) iEC lumen stained with F-actin, CD31, and DAPI. 3D reconstruction of the tubular lumen using the CD31 staining. B) Cross-section of an iEC lumen stained with F-actin. The asterisk highlights a hollow sprout. C) Proliferation occurs within 24 – 72h in the lumen close to sprouting events (depicted with black asterisks). D) iEC vessel barrier function assessment via 40 kDa-FITC diffusion assays through the lumen compared to cell-free tubular structures (cell-free gel) and HUVEC vessels. Images are taken every 5 min for 15 min. E) Fluorescence plot profiles at 0 and 15 min are shown for each condition. F) Permeability coefficient comparison for 40 kDa and G) 70 kDa dextran. N ≥ 4 replicates per experiment from ≥ 3 independent experiments. Bars represent average ± S.E.M. *p < 0.05; **p < 0.01; ***p<0.001.
We then compared iEC vessel barrier function as compared to a common control (HUVEC lumen model) and a cell-free negative control (a cell-free tubular collagen structure), using two fluorescently labeled dextrans differing in molecular weight (40 and 70 kDa) (Representative diffusion images in Figure 2D). iEC lumens exhibited lower barrier function than HUVEC lumen models, but both were significantly more efficient at retaining the dextran than cell-free hydrogel controls (9.09 ± 2.02 · 10−6 cm/s for iEC, 3.70 ± 0.73· 10−6 cm/s for HUVEC, 1.22 ± 0.19 · 10−4 cm/s for the cell-free control) (Figure 2F–G).
We then evaluated the crosstalk between kidney tumor cells and nearby blood vessels in the MPS. We co-cultured our iEC vessel model with a high-density (hereafter called HD model) 3D dispersion of kidney cancer cells (A498 or Caki-2) within the adjacent luminal structure. The layout allows a constant distance between the luminal structures, and the seeding density was optimized to recreate the high density and levels of necrosis of RCC (Fig S1) [41]. The gradient of dead cells peaking in central clusters was consistent with the appearance of a necrotic core [42, 43] at 40 million/ml for A498 and Caki-2.
Next, we validated the tumor-like properties of the high-density tumor model (HD) and compared it to the lowest concentration assessed during our optimizations (Fig S1 C) (10 million/ml, hereafter called LD) (Figure S1A phase contrast), where no cell death gradient was observed. We evaluated hypoxia, hypoxia-related gene expression, metabolic activity, and growth factor secretion – all known hallmarks of RCC tumors.
An oxygen-sensitive dye revealed an increase in hypoxia over time for both cell types (Figure 3A–C). Next, via qPCR, we analyzed the changes in expression of hypoxia-related genes, such as carbonic anhydrase ix (CA9) and hypoxia-inducible factor 2A (EPAS2, also called HIF2A), which we found to be overexpressed in our tumor models (HIF2A only in A498) after 48 h (Figure 3D).
Figure 3: Establishment and characterization of the ccRCC epithelial tumor portion of the co-culture model.
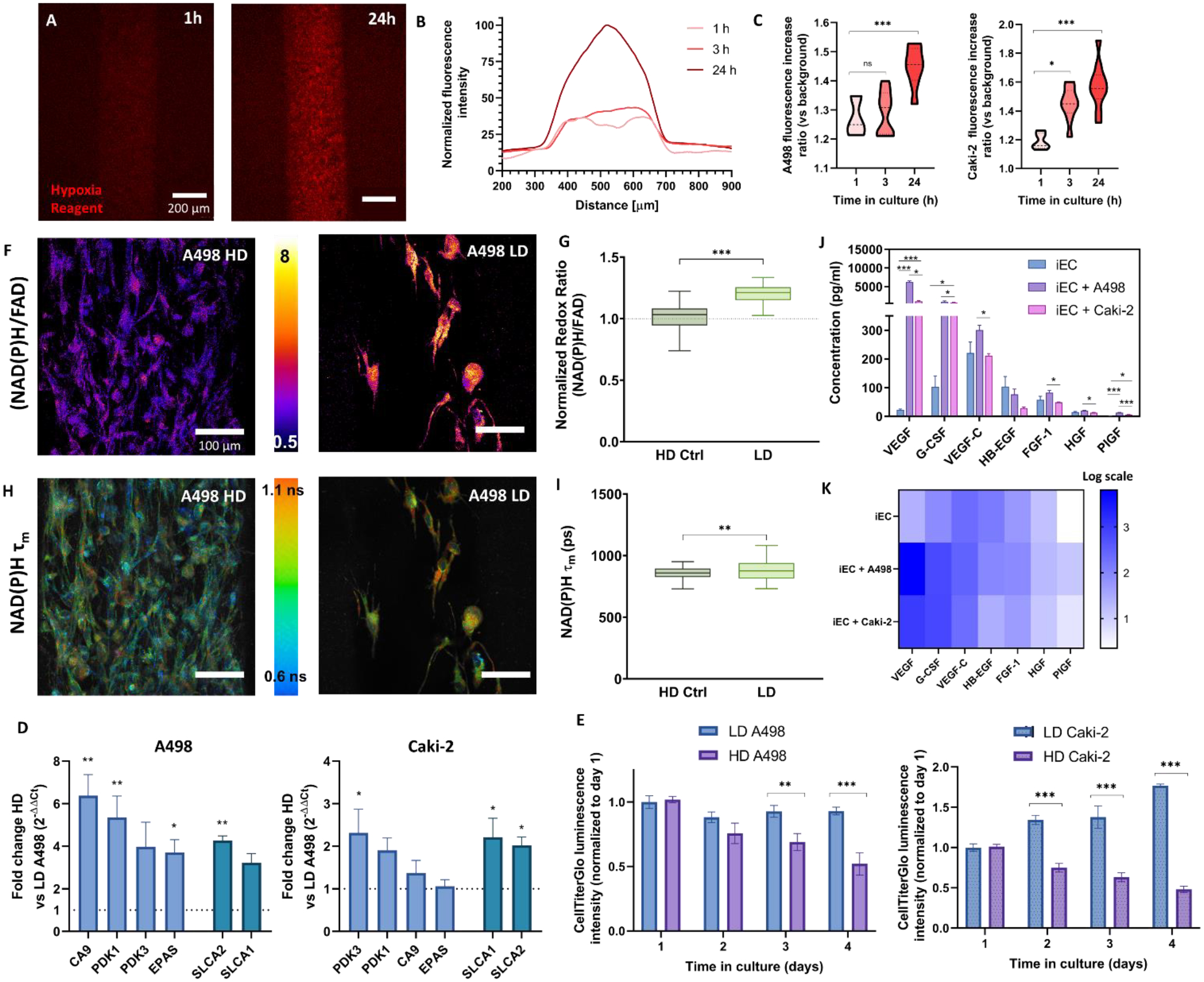
A) Projected Z-stack of images of the hypoxia dye of the HD (high density) A498 model in real-time. The fluorescence of the dye is inversely proportional to the oxygen tension. B) Line profile representation of the hypoxic response of the A498 tumor model within the first 24h. C) Quantification of the hypoxia dye intensity over time for A498 (left) and Caki-2 (right). D) Representative redox ratio images of A498 resulting from an increase in A498 density (HD vs LD) and E) Tukey box plot of redox ratio normalized to HD control condition. F) Representative NAD(P)H mean fluorescence lifetime (τm) images. Scalebar = 100 μm. G) Tukey box plot of τm quantification. H) Bead-based ELISA panel of secreted pro-angiogenic factors by the iEC monoculture model compared to A498 and Caki-2 co-culture models. ANOVA + Tukey test was used to determine significance. I) Results from (F) shown as a logarithmic scale heatmap for improved visualization. J) ATP levels of LD and HD of A498 (left) and Caki-2 (right) tumor models over 4 days. K) Gene expression changes in the HD A498 (left) and Caki-2 (right) tumor models compared to the LD. Genes are grouped left to right in metabolism (blue) and glucose transporters (cyan). I-J) N ≥ 4 replicates per experiment from ≥ 3 independent experiments. Paired t-tests were used to determine significance in D-E. Kruskal-Wallis and post-hoc Dunn tests were used to determine significance in C, G, and I. Bars represent average ± S.E.M. *p < 0.05; **p < 0.01; ***p<0.001.
Finally, we assessed the changes in ATP concentration compared to the LD control (Figure 2E) over 4 days. Since ATP-dependent luminescence linearly correlates with cell number[44], the decreases in normalized ATP luminescence point to lower live cell numbers of Caki-2 and A498 than the LD control. Heterogeneity in cell proliferation in the model was verified in Caki-2 via ki-67 staining (Figure S6), where we found ki67+ cells to be predominantly located in outermost regions of the model.
Overexpression of pyruvate dehydrogenase kinase (PDK)-1 and 3 in A498 indicated a shift towards an anaerobic glucose metabolism, consistent with the upregulation of SLC2A1 and 2 (which codify glucose transporters Glut1 and 2)(Figure 2D). Next, we corroborated these results using multiphoton optical metabolic imaging (OMI) on our platform’s A498 model as a functional readout of cell metabolism. OMI is a technique that leverages the autofluorescence of NADH, NADPH (which are spectrally overlapping and hereafter denoted NAD(P)H), and FAD. The NAD(P)H/FAD ratio is commonly known as the redox ratio and is affected by the intracellular metabolism and microenvironment[36, 45]. The redox ratio significantly increased in our A498 tumor model, indicating a shift to more glycolytic metabolism (Figure 3F–G). We complemented the redox ratio with NAD(P)H fluorescence lifetime imaging microscopy (FLIM) and report the NAD(P)H mean lifetime (τm). FLIM is a measurement of the fluorescence decay time of the molecule NAD(P)H, which is known to vary according to the binding state of NAD(P)H: slower when NAD(P)H is bound to protein or faster when it is free in the cytoplasm[46]. HD conditions showed a decreased NAD(P)H τm compared with our LD control (Figure 3H–I). These lifetime results are consistent with decreased protein-binding activity for NAD(P)H, while the redox ratio results are consistent with decreased NAD(P)H concentration relative to FAD, in line with our previous qPCR and CellTiterGlo results. Although we did not look at FLIM measurements in Caki-2, the increases in PDK-1 and SLC2A1 and 2 points to the same effects (Figure 3D).
Hypoxic tissues secrete pro-angiogenic growth factors to, in turn, increase the supply of oxygen and nutrients. Multiplexed bead-based ELISA (Figure 3 J–K) revealed significant increases in VEGF-A, G-CSF and PIGF (compared to iEC monocultures).
Next, we assessed the effects of the tumor cells in the endothelial model. First, CD31 (an endothelial junctional marker, Figure 4A) staining revealed increased angiogenic sprouting compared with iEC alone. Specifically, in co-cultures with A498 both the number and length of angiogenic sprouts increased (Figure 4H–I), whereas with Caki-2 only the sprout length increased. Additionally, both co-cultures showed significantly increased vessel permeability (Figure 4C–F), accompanied with decreased vessel confluency (18.8% for the co-culture with A498 and 38.5% with Caki-2) (Figure 4G)). Furthermore, the expression of CD105 (a marker of endothelial resistance to TGFβ signaling, anti-angiogenic mediator and suggested stemness marker)[47, 48] decreased overall in iECs in the A498 co-culture model (Figure 4B). Some staining was observed in A498s, consistent with previous reports of CD105 expression in RCC[48, 49]. A qPCR array revealed many significant gene upregulations in the endothelium (Figure 4J), including several pro-inflammatory genes (i.e., IL-6, TGF-A), which could explain the increase in permeability; as well as traditional pro-angiogenic cascade mediators (i.e., VEGFA, FGF2, MDK, VEGFB, NRP1, MMP14).
Figure 4: Co-culture effects in the endothelial vessel model.
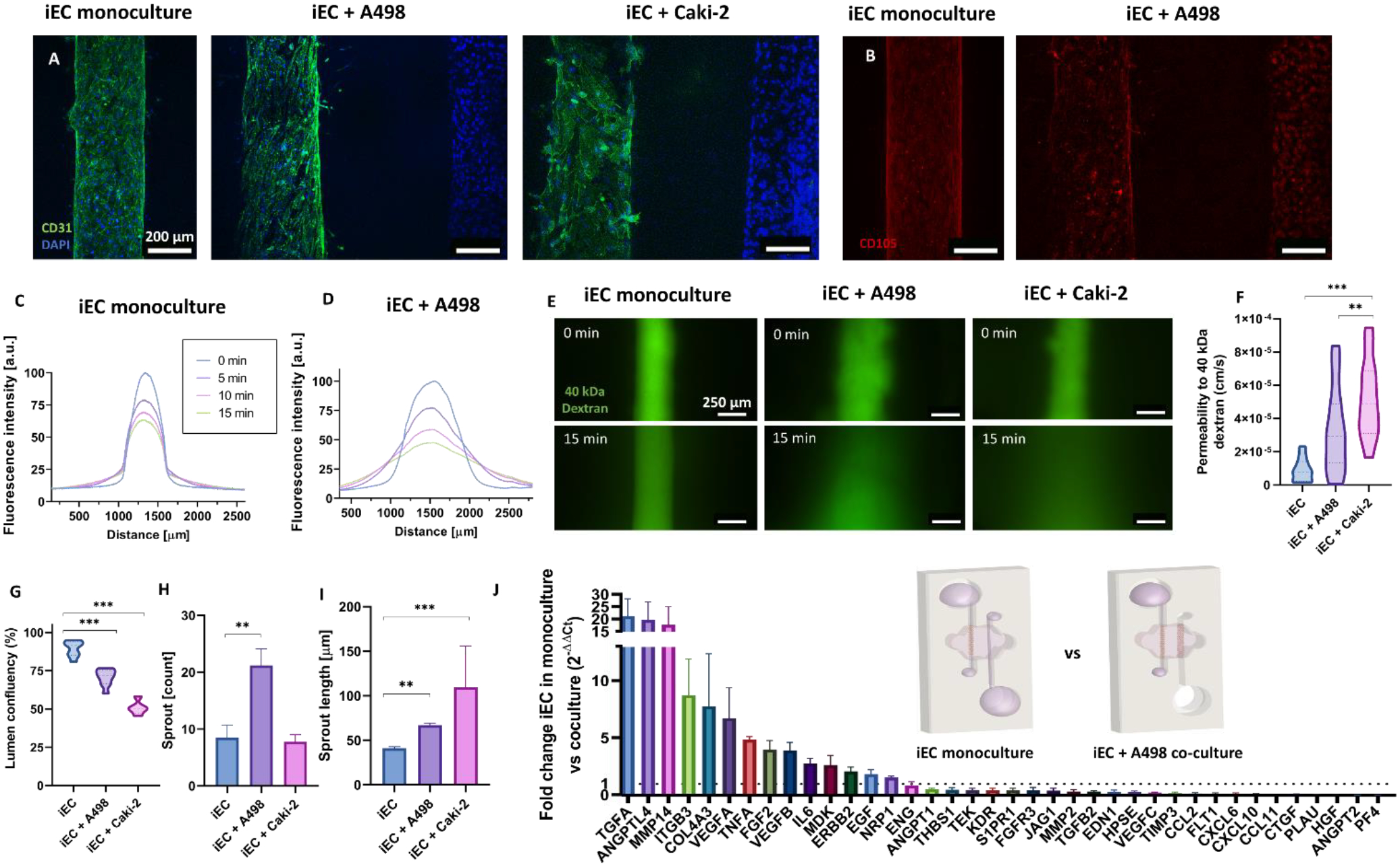
A) Projected z-stack images of CD31 and DAPI-stained monoculture and co-culture models. B) CD105 expression decreases in the A498 co-culture model. C-D) Plot profile of the diffusion of a 40 kDa-FITC conjugated dextran through the (A) monoculture vessel model and (B) A498 co-culture model. E) Images of the dextran diffusion for the monoculture and co-cultures at times 0- and 15-min. F) Confluency of the iEC lumens calculated as the area occupied by F-actin staining. G) Permeability coefficient calculated from 40 kDa diffusion data. H) Quantification of the sprout number increase and I) sprout length increase in the co-cultures as compared to the monocultures. J) Waterfall plot of the significant gene expression changes between monoculture and A498 co-culture conditions. N ≥ 4 replicates per experiment from ≥ 3 independent experiments. Bars represent average ± S.E.M. One way-ANOVA test and Tukey post-hoc test were used to determine significance in all graphs except J, where FDR-corrected t-test was used. *p < 0.05; **p < 0.01; ***p<0.001.
Cabozantinib is a small tyrosine-kinase inhibitor targeting: VEGFR-1,-2 and -3, Tie-2, FLT-3 (expressed by vascular cells), and RCC tumor overexpressed molecule MET. Cabozantinib is thought to block tumor angiogenesis, proliferation, dissemination, and metastasis. Our model is ideal for studying the functional effects and crosstalk resulting from cabozantinib treatment in the TME in vitro.
A dose-response curve showed 1 μM cabozantinib preserved cell viability (around 80%)(Figure S2). High viability enabled studying treatment effects on cell death and metabolism in surviving cells. Further, pharmacodynamic studies show cabozantinib concentrations in patient serum range similarly[50].
First, we assessed phenotypic changes in the iEC vessel resulting from cabozantinib treatment. CD31 expression was less localized in cell-cell junctions and more disorganized (Figure 5A). However, sprout number and length were restored iEC monoculture-like levels (Figure 5B–C). However, vessel permeability to 40 kDa dextran (Figure 5 D–F) and vessel confluency (Figure 5G) were not restored. Notably, iEC permeability values showed a significant increase in the Caki-2 co-cultures after treatment, parallel to the significant increase in vessel confluency (Figure 5I). Next, we compared iEC monoculture to cabozantinib iEC-A498 models via qPCR to assess the recovery of normal vascular function (Figure 5H) and found significant downregulation of pro-angiogenic and inflammatory markers (e.g., VEGFA, VEGFC, TGFA, TNF), and migration-related genes (e.g., MMP2, ITGAV). MDK, a pro-angiogenic and pro-metastatic gene, and ITGB3, a known regulator of endothelial cell migration [51], were the only significantly upregulated genes.
Figure 5: Cabozantinib partially restores endothelial vessel functionality to the monoculture baseline.
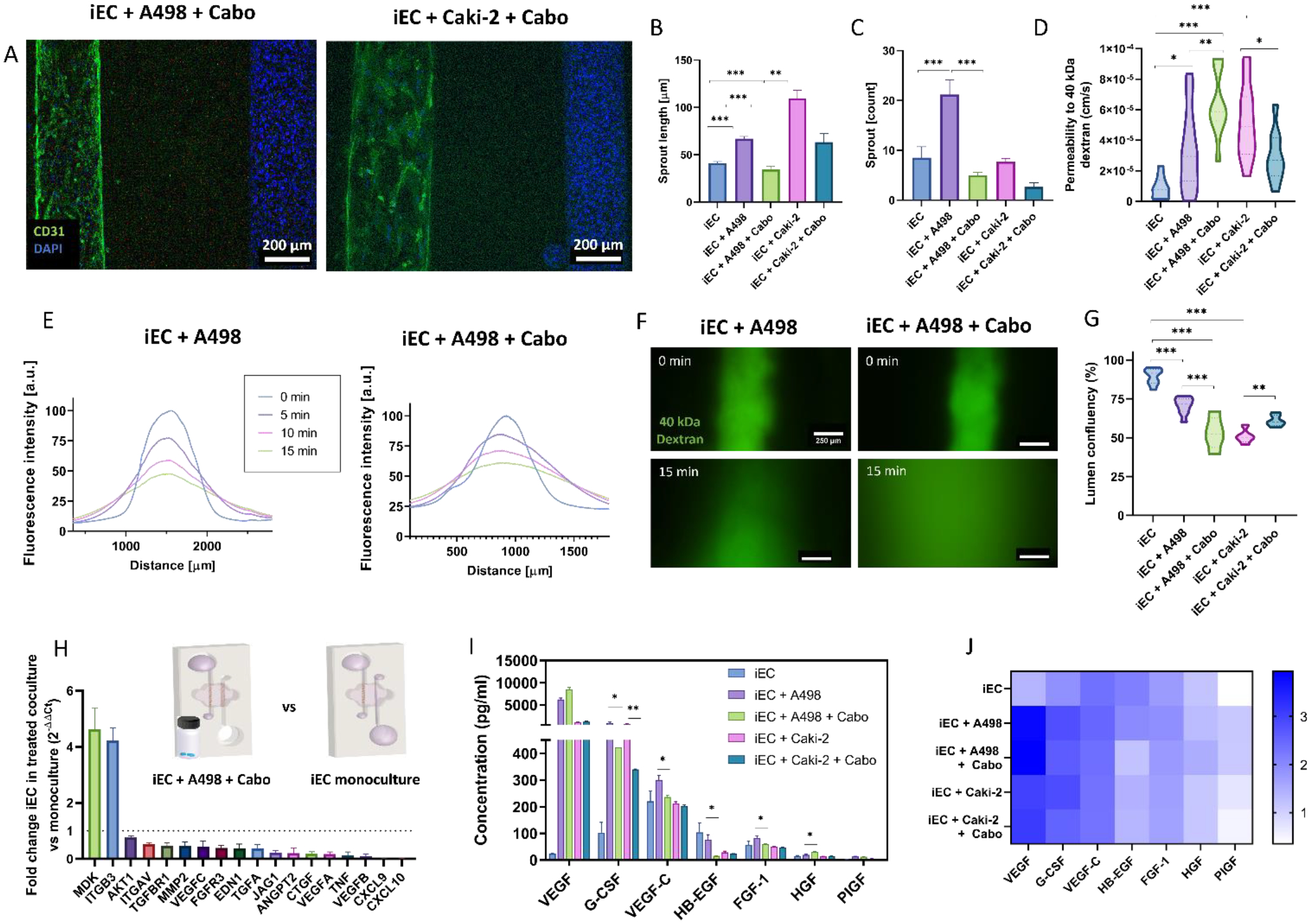
A) Projected z-stack images of CD31 and DAPI-stained treated co-culture models. B-C) Quantification of angiogenic sprouting changes after cabozantinib treatment. Sprout numbers (B) and sprout length (C) in the iEC vessel. D) Permeability coefficient calculated from 40 kDa diffusion data. E) Representative plot profile of the diffusion of a 40 kDa-FITC conjugated dextran through the control (left) and cabozantinib treated (right) A498 co-culture vessel model. F) Representative images of the dextran diffusion through the iEC vessel for the untreated and cabozantinib-treated A498 co-cultures at times 0- and 15-min. G) iEC lumen confluency percentage quantified as area occupied by F-actin staining. H) Waterfall plot of iECs in treated A498 co-culture compared to monoculture conditions. I) Bead-based ELISA panel of secreted pro-angiogenic factors by the iEC monoculture model compared to A498 and Caki-2 untreated and cabozantinib-treated co-culture models. J) Results from (I) shown as a logarithmic scale heatmap for improved visualization. N ≥ 4 replicates per experiment from ≥ 3 independent experiments. Bars represent average ± S.E.M. One way-ANOVA test and Tukey post-hoc test was used to determine significance in all graphs except H, where FDR-corrected t-test was used. *p < 0.05; **p < 0.01; ***p<0.001.
Next, (Figure 5I–J) secreted factor analyses after treatment showed significant decreases, mostly in the treated A498 co-culture models: VEGF-C, G-CSF (both A498 and Caki-2), HB-EGF, and FGF-1. However, the most upregulated factors, VEGF, and G-CSF, remained much higher in the treated co-cultures than in the monocultures, indicating that the downstream signaling pathways may remain activated after treatment.
Overall, similar effects were observed when using primary cells in co-culture with iPSC-ECs in the system (Figure S7): increased sprout length, increased permeability in co-culture and lower lumen confluency than in monocultures, an effect that worsened after cabozantinib treatment.
In addition, since cabozantinib targets molecular pathways involved in tumor metabolism, proliferation, and dissemination, we assessed molecular and functional effects of cabozantinib on tumor cells in our MPS (Figure 6A), using qPCR (Figure 6C for A498, Figure S3 for Caki-2) and the readouts described in previous sections (i.e., metabolic imaging, proliferation, cell fate). Despite the treatment, we observed numerous A498 and Caki-2 cells invading the surrounding 3D matrix. Interestingly, A498 cells migrated individually (Figure 6A left), whereas Caki-2 cells exhibited a collective movement, showing spheroid-like clusters of cells protruding into the matrix (Figure 6A right) after 72h. Overall, migration was still observed after cabozantinib treatment in both tumor cell lines.
Figure 6: Cabozantinib induces metabolic changes and cell death in the epithelial tumor model.
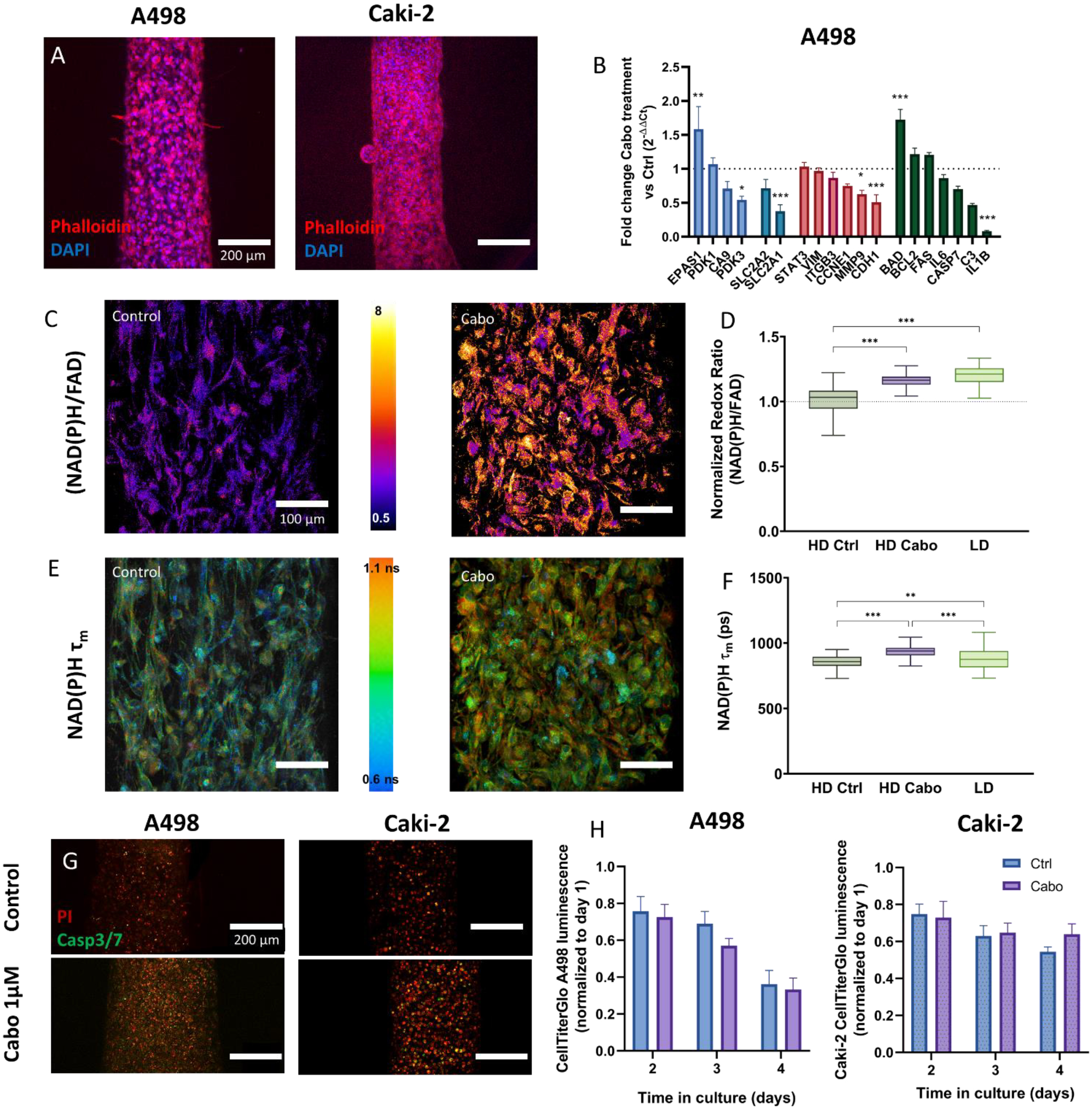
A) Z-projected confocal image of the A498 (left) and Caki-2 (right) tumor model. Scalebar = 200 μm. B) qPCR changes in the A498 tumor model after treatment. Genes are grouped left to right in categories: metabolism (blue), glucose transporters (cyan), adhesion and migration (red), and cell survival (green). C-F) Metabolic imaging changes in A498 resulting from cabozantinib treatment. (C) Representative redox ratio images and (D) Tukey box plot of redox ratio normalized to HD control condition, (E) Representative NAD(P)H mean fluorescence lifetime (τm) and (F) Tukey box plot of τm quantification. Scalebar = 100 μm. G) Changes in A498 and Caki-2 viability as a result to treatment. Z-projected confocal images of A498 stained with propidium iodide (red) and Caspase3/7 live staining reagent (green). H) Normalized CellTiterGlo measurements of untreated and treated A498 (left) and Caki-2 (right) tumor models’ overtime. n ≥ 4 replicates per experiment from ≥ 3 independent experiments. Bars represent average ± S.E.M. FDR-corrected t-test was used for (B), whereas a Kruskal-Wallis test and Dunn post-hoc test was used for (D and F). *p < 0.05; **p < 0.01; ***p<0.001.
Interestingly, qPCR analysis of genes related to cell migration (Figure 6B red) revealed a decrease in CDH1, a key player in epithelial to mesenchymal transition (i.e., a pro-migratory phenotype), in A498, and a reduction in MMP-3 would suggest less ECM remodeling typically required for cell migration. Since CDH1 expression in Caki-2 was low (i.e., Ct >30), leading to higher uncertainty of results, we decided not to report changes in this gene. Conversely, MMP3 was not expressed at all by Caki-2, therefore pointing to a different mechanism of cell migration, consistent with our observations in the model.
Next, we found upregulation of hypoxia-related genes after treatment: EPAS1 in A498 and CA9 in Caki-2(Figure 6B blue). Significant dysregulations in PDK expression were also found: PDK3 was downregulated in A498 and PDK1 in Caki-2. Notably, we did not detect PDK3 in Caki-2. Further, the decrease of SLC2A1 expression in A498 pointed to a less oxidation-reliant metabolism (Figure 6B cyan), consistent with the increases observed in redox ratio and NAD(P)H τm. The increase in redox ratio indicates a reduced oxidation-reduction state, whereas the increase in NAD(P)H τm indicates increased NAD(P)H protein-binding activity after treatment. Both increases suggested a metabolic shift to a low density (LD)-like phenotype.
The expression of proliferation marker CCNE1 (Figure 6B red) did not change due to cabozantinib treatment. Consistent with these results, cabozantinib had no significant effect on normalized ATP levels (Figure 6H), suggesting stable cell numbers. At this point, we considered that cell viability could be decreasing due to the treatment (Figure 6G). While cabozantinib-treated A498s showed an increase in apoptosis consistent with the increase in expression of BAD (an inhibitor of anti-apoptotic proteins), propidium iodide-stained cells also increased after treatment, pointing to increases via both caspase-dependent and -independent cell death. Conversely, Caki-2 cells showed an increase in propidium iodide staining and a decrease in RNA expression of Caspase7 (Figure 6B green). These results point to increases in cell death in treated cancer cells through different mechanisms.
Finally, we looked at inflammatory mediators via qPCR, which increase endothelial permeability and promote angiogenic responses (Figure 6B green). Inflammatory mediators decreased in both tumor models after treatment, although the specific mediators were different: IL1B decreased in A498 tumor models after treatment while IL-6 decreased in Caki-2. IL-6 is a key regulator of endothelial cell adhesion and permeability, so the reduced levels after treatment are consistent with endothelial function recovery.
Discussion
Over the years, numerous therapies have been approved for RCC, including many tyrosine-kinase inhibitors (TKI). However, most therapies show heterogeneous responses across patients, making clinical treatment decisions challenging. This clinical heterogeneity, along with frequently observed resistance to TKI suggests that our understanding of the RCC TME, and its influence in TKI response (or resistance) mechanisms is limited[52, 53].
The most frequent RCC drivers are mutations in the tumor suppressor gene Von Hippel Lindau (VHL), which plays a key role in regulating molecular pathways related to hypoxia and angiogenesis. Targeting RCC tumor-associated vasculature using TKIs has been the most popular therapeutic approach for decades. Microphysiological systems (MPS) have demonstrated high potential in recapitulating numerous complex tumor microenvironments (TME). Despite the advantages of MPS in recapitulating sprouting angiogenesis, hypoxia, and nutrient gradients, little attention has been brought to developing MPS of RCC[17].
In this paper, we developed an MPS of RCC to assess the efficacy of anti-angiogenic therapies on key tumor hallmarks: vascular integrity, angiogenic sprouting, tumor-vascular signaling, metabolism, and proliferation. To our knowledge, this is the first MPS to assess functional effects of therapies. Particularly, sprouting angiogenesis assays and relevant luminal structures, which alters cell behavior[54, 55], are challenging to recapitulate using traditional cell culture techniques[24]. Our model appears to provide a more physiologically relevant structure and environment to recapitulate RCC more closely than traditional in vitro models. Interestingly, we observed that CD105, a marker of cell stemness[56], decreases in the endothelial cells in our model as a result of the co-culture, suggesting an adaptation of our iECs to a more differentiated phenotype due to TME created in our model.
Our epithelial models (high density or HD) recapitulated a central necrotic area and overall low proliferation due to a depleted environment. Further these models showed a hypoxic response that developed within 24h, as shown via qPCR and live-cell oxygen-sensitive staining, consistent with the highly hypoxic RCC[57]. While RCC cells are known to have the molecular hypoxia response pathways constitutively activated, our results point to an increase in the activity of this pathway in a nutrient- and oxygen-depleted microenvironment, which are known initiators of angiogenesis in nearby vessels[58]. Interestingly, the nutrient and oxygen-depleted environment is consistent with the heterogeneity in proliferation shown via ki-67 staining for the Caki-2 model.
Next in the angiogenic cascade is an increase in glucose consumption and transport due to a decoupling of glycolysis and TCA cycle. Both effects were observed in our HD model acompared to the same cells at lower density (LD) via gene expression changes and metabolic imaging. Metabolic imaging provided real-time quantitative measurements of the redox ratio and metabolic phenotype at a single-cell level. Overall, molecular analysis and metabolic imaging demonstrated that tumor cells adapted to the nutrient-depleted microenvironment by shifting to a more glucose-dependent metabolism. Interestingly, the two cell lines we chose have stark differences between each other, starting with the VHL mutational status and 2D cell proliferation rate. Yet, the similarities in behavior and microenvironmental contributions to angiogenesis of both tumor models (A498 and Caki-2) are striking.
Tumor angiogenesis cascades respond to highly hypoxic nearby tissues (or tumors) secreting relevant growth factors and activating the angiogenesis cascade. Angiogenic cascade initiators VEGF and G-CSF were high in our models. Next, our model exhibited the main hallmarks of tumor angiogenesis: increased permeability, vessel leakiness, and increased angiogenic sprouting in the endothelial vessel.
Cabozantinib – targeting VEGFR1, 2, 3, and MET amongst other kinases –approved by the FDA in 2015, has demonstrated increased objective response and progression-free survival in clinical trials against other agents[9, 59]. However, in vitro studies of cabozantinib effects in the RCC TME are scarce. Here, we evaluated cabozantinib effects in each angiogenic cascade hallmark. In the vessel, the primary target of cabozantinib[60], angiogenic sprouting was suppressed in both co-culture models, suggesting that cabozantinib effectively blocks tumor angiogenesis. iEC migration detached from the lumen increased after cabozantinib treatment, which would point to an endothelial cell disconnection from the sprouts[61].
Cabozantinib blocks the VEGF pathway, key for endothelial cell survival, which could explain the decrease in vessel confluency and increased permeability in A498 treated co-cultures. Surprisingly, treated vessels in the Caki-2 co-culture partially recovered their confluency and permeability. Further, the iEC gene expression profile in treated co-cultures resembled the iEC monoculture condition more than the untreated co-culture. Finally, while A498 co-cultures showed a higher secretion of pro-angiogenic factors (e.g., VEGFA), Caki-2 responded better to VEGF targeted therapeutic Cabozantinib. This result highlights the importance of the TME and functional readouts in assessing the effectiveness of anti-cancer therapeutics among RCC subtypes, as recent literature highlights[6, 20, 62, 63].
Cabozantinib had no effect on tumor cell proliferation or at preventing migration outside of the model. Gene expression did not detect clear changes in metabolic activity of the tumor cells due to treatment. Several genes were significantly downregulated, and others were upregulated as a potential compensatory mechanism. Metabolic imaging results showed lower dependence on glycolysis and decreased free NAD(P)H in the cytosol, pointing to a recovery of the low-density A498 tumor cell phenotype. Treated cells are not as nutrient and oxygen-deprived, which points to cabozantinib being effective at suppressing aggressive tumor metabolism pro-angiogenic activity. Decreased metastasis[13] and improved overall prognosis[64] have both been reported for cabozantinib. A decrease in pro-angiogenic activity in our model can be inferred from the decrease in pro-angiogenic growth factors after treatment, which remained higher from that observed in the monoculture conditions. These results highlight the value of functional reports as a complement to molecular readouts for a full picture of drug efficacy in vitro.
Finally, treatment affected tumor cell viability; although, surprisingly, the pathways were different for both cell types, recapitulating in vivo observations[20]. These results are encouraging since cabozantinib decreased cell viability independently of the genetic profile of the tumor cells[22]. CellTiterGlo results revealed substantial cell viability after treatment. Future studies could investigate longer-term effects of cabozantinib on the tumor to evaluate potential developments of resistance and their underlying mechanisms.
Additional cell types are found and play important roles in the RCC TME, mainly immune cell infiltrates[65], which are especially important due to the rise of immunotherapies[66]. These cell types should be considered to develop a more comprehensive model to try and predict treatment efficacy of contemporary therapeutic regimens and evolving combination therapies[6, 67]. Our model has shown potential for interrogating drug efficacy at a mechanistic level, so in the future it could be leveraged to study resistance mechanisms [42, 68]. Further, this model excels at recapitulating and revealing heterogeneous behaviors in the tumor microenvironment, such as the effect of nutrient and oxygen-deprived environments. This crucial advantage, along with the amenability of this model for histology (Figure S4) and micromanipulation (e.g., microdissection)(Figure S5), provides a robust platform to investigate emerging properties of the TME that may escape traditional in vitro models in future studies using techniques like RNAseq or spatial transcriptomics. Notably, we have also demonstrated the ability to interrogate primary tumor samples using this model, which opens the door to personalized medicine applications of this model. Although this application would require further characterization of these samples and validation with clinical markers, the more physiologically relevant TME model presented here is uniquely suited to identify novel biomarkers and improve clinical decisions.
Supplementary Material
Acknowledgements
The authors would like to thank the University of Wisconsin Carbone Cancer Center (UWCCC) for the use of its Shared Services to complete this research. NIH grants R33 CA225281‐–01.
Footnotes
Declaration of conflict of interests
D.J.B. holds equity in Bellbrook Labs LLC, Tasso Inc., Turba LLC, Salus Discovery LLC, Stacks to the Future LLC, Lynx Biosciences Inc., Flambeau Diagnostics, and Onexio Biosystems.
Data availability
All raw data used in this study is available on request to the authors.
References
- [1].Frank I, Blute ML, Cheville JC, Lohse CM, Weaver AL, Zincke H, An outcome prediction model for patients with clear cell renal cell carcinoma treated with radical nephrectomy based on tumor stage, size, grade and necrosis: the SSIGN score, The Journal of urology 168(6) (2002) 2395–2400. [DOI] [PubMed] [Google Scholar]
- [2].Ljungberg B, Albiges L, Abu-Ghanem Y, Bensalah K, Dabestani S, Fernández-Pello S, Giles RH, Hofmann F, Hora M, Kuczyk MA, Kuusk T, Lam TB, Marconi L, Merseburger AS, Powles T, Staehler M, Tahbaz R, Volpe A, Bex A, European Association of Urology Guidelines on Renal Cell Carcinoma: The 2019 Update, European Urology 75(5) (2019) 799–810. [DOI] [PubMed] [Google Scholar]
- [3].Meskawi M, Sun M, Trinh Q-D, Bianchi M, Hansen J, Tian Z, Rink M, Ismail S, Shariat SF, Montorsi F, A review of integrated staging systems for renal cell carcinoma, European urology 62(2) (2012) 303–314. [DOI] [PubMed] [Google Scholar]
- [4].Wolff I, May M, Hoschke B, Zigeuner R, Cindolo L, Hutterer G, Schips L, De Cobelli O, Rocco B, De Nunzio C, Do we need new high-risk criteria for surgically treated renal cancer patients to improve the outcome of future clinical trials in the adjuvant setting? Results of a comprehensive analysis based on the multicenter CORONA database, European Journal of Surgical Oncology (EJSO) 42(5) (2016) 744–750. [DOI] [PubMed] [Google Scholar]
- [5].Patard J-J, Kim HL, Lam JS, Dorey FJ, Pantuck AJ, Zisman A, Ficarra V, Han K-R, Cindolo L, De La Taille A, Use of the University of California Los Angeles integrated staging system to predict survival in renal cell carcinoma: an international multicenter study, Journal of Clinical Oncology 22(16) (2004) 3316–3322. [DOI] [PubMed] [Google Scholar]
- [6].Hsieh JJ, Purdue MP, Signoretti S, Swanton C, Albiges L, Schmidinger M, Heng DY, Larkin J, Ficarra V, Renal cell carcinoma, Nature Reviews Disease Primers 3(1) (2017) 17009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Motzer RJ, Hutson TE, Cella D, Reeves J, Hawkins R, Guo J, Nathan P, Staehler M, de Souza P, Merchan JR, Pazopanib versus sunitinib in metastatic renal-cell carcinoma, New England Journal of Medicine 369(8) (2013) 722–731. [DOI] [PubMed] [Google Scholar]
- [8].Chan JY, Choudhury Y, Tan M-H, Predictive molecular biomarkers to guide clinical decision making in kidney cancer: current progress and future challenges, Expert review of molecular diagnostics 15(5) (2015) 631–646. [DOI] [PubMed] [Google Scholar]
- [9].Choueiri TK, Halabi S, Sanford BL, Hahn O, Michaelson MD, Walsh MK, Feldman DR, Olencki T, Picus J, Small EJ, Dakhil S, George DJ, Morris MJ, Cabozantinib Versus Sunitinib As Initial Targeted Therapy for Patients With Metastatic Renal Cell Carcinoma of Poor or Intermediate Risk: The Alliance A031203 CABOSUN Trial, Journal of clinical oncology : official journal of the American Society of Clinical Oncology 35(6) (2017) 591–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Choueiri TK, Escudier B, Powles T, Tannir NM, Mainwaring PN, Rini BI, Hammers HJ, Donskov F, Roth BJ, Peltola K, Lee JL, Heng DYC, Schmidinger M, Agarwal N, Sternberg CN, McDermott DF, Aftab DT, Hessel C, Scheffold C, Schwab G, Hutson TE, Pal S, Motzer RJ, Cabozantinib versus everolimus in advanced renal cell carcinoma (METEOR): final results from a randomised, open-label, phase 3 trial, The lancet oncology 17(7) (2016) 917–927. [DOI] [PubMed] [Google Scholar]
- [11].Courtney KD, Bezwada D, Mashimo T, Pichumani K, Vemireddy V, Funk AM, Wimberly J, McNeil SS, Kapur P, Lotan Y, Isotope tracing of human clear cell renal cell carcinomas demonstrates suppressed glucose oxidation in vivo, Cell metabolism 28(5) (2018) 793–800. e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Chan DA, Sutphin PD, Nguyen P, Turcotte S, Lai EW, Banh A, Reynolds GE, Chi JT, Wu J, Solow-Cordero DE, Bonnet M, Flanagan JU, Bouley DM, Graves EE, Denny WA, Hay MP, Giaccia AJ, Targeting GLUT1 and the Warburg effect in renal cell carcinoma by chemical synthetic lethality, Science translational medicine 3(94) (2011) 94ra70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Lu J, The Warburg metabolism fuels tumor metastasis, Cancer and Metastasis Reviews 38(1) (2019) 157–164. [DOI] [PubMed] [Google Scholar]
- [14].Singer K, Kastenberger M, Gottfried E, Hammerschmied CG, Buttner M, Aigner M, Seliger B, Walter B, Schlosser H, Hartmann A, Andreesen R, Mackensen A, Kreutz M, Warburg phenotype in renal cell carcinoma: high expression of glucose-transporter 1 (GLUT-1) correlates with low CD8(+) T-cell infiltration in the tumor, Int J Cancer 128(9) (2011) 2085–95. [DOI] [PubMed] [Google Scholar]
- [15].Sobrino A, Phan DT, Datta R, Wang X, Hachey SJ, Romero-Lopez M, Gratton E, Lee AP, George SC, Hughes CC, 3D microtumors in vitro supported by perfused vascular networks, Sci Rep 6 (2016) 31589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Miller CP, Tsuchida C, Zheng Y, Himmelfarb J, Akilesh S, A 3D Human Renal Cell Carcinoma-on-a-Chip for the Study of Tumor Angiogenesis, Neoplasia (New York, N.Y.) 20(6) (2018) 610–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Virumbrales-Muñoz M, Ayuso JM, Gong MM, Humayun M, Livingston MK, Lugo-Cintron KM, McMinn P, Alvarez-Garcia YR, Beebe DJ, Microfluidic lumen-based systems for advancing tubular organ modeling, Chem Soc Rev 49(17) (2020) 6402–6442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Sontheimer-Phelps A, Hassell BA, Ingber DE, Modelling cancer in microfluidic human organs-on-chips, Nature Reviews Cancer (2019). [DOI] [PubMed] [Google Scholar]
- [19].Morgan MM, Johnson BP, Livingston MK, Schuler LA, Alarid ET, Sung KE, Beebe DJ, Personalized in vitro cancer models to predict therapeutic response: Challenges and a framework for improvement, Pharmacology & therapeutics 165 (2016) 79–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Brugarolas J, Rajaram S, Christie A, Kapur P, The Evolution of Angiogenic and Inflamed Tumors: The Renal Cancer Paradigm, Cancer cell 38(6) (2020) 771–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ingram PN, Hind LE, Jiminez-Torres JA, Huttenlocher A, Beebe DJ, An Accessible Organotypic Microvessel Model Using iPSC-Derived Endothelium, Advanced healthcare materials 7(2) (2018) 1700497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Brodaczewska KK, Szczylik C, Fiedorowicz M, Porta C, Czarnecka AM, Choosing the right cell line for renal cell cancer research, Mol Cancer 15(1) (2016) 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Rosa R, Damiano V, Nappi L, Formisano L, Massari F, Scarpa A, Martignoni G, Bianco R, Tortora G, Angiogenic and signalling proteins correlate with sensitivity to sequential treatment in renal cell cancer, British journal of cancer 109(3) (2013) 686–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Jimenez-Torres JA, Virumbrales-Muñoz M, Sung KE, Lee MH, Abel EJ, Beebe DJ, Patient-specific organotypic blood vessels as an in vitro model for anti-angiogenic drug response testing in renal cell carcinoma, EBioMedicine 42 (2019) 408–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Virumbrales-Munoz M, Chen J, Ayuso J, Lee M, Abel EJ, Beebe DJ, Organotypic primary blood vessel models of clear cell renal cell carcinoma for single-patient clinical trials, Lab Chip 20 (2020) 4420–4432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Pusztaszeri MP, Seelentag W, Bosman FT, Immunohistochemical expression of endothelial markers CD31, CD34, von Willebrand factor, and Fli-1 in normal human tissues, Journal of Histochemistry & Cytochemistry 54(4) (2006) 385–395. [DOI] [PubMed] [Google Scholar]
- [27].Lertkiatmongkol P, Liao D, Mei H, Hu Y, Newman PJ, Endothelial functions of platelet/endothelial cell adhesion molecule-1 (CD31), Curr Opin Hematol 23(3) (2016) 253–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Jiménez‐Torres JA, Peery SL, Sung KE, Beebe DJ, LumeNEXT: a practical method to pattern luminal structures in ECM gels, Advanced healthcare materials 5(2) (2016) 198–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ayuso JM, Gillette A, Lugo-Cintron K, Acevedo-Acevedo S, Gomez I, Morgan M, Heaster T, Wisinski KB, Palecek SP, Skala MC, Beebe DJ, Organotypic microfluidic breast cancer model reveals starvation-induced spatial-temporal metabolic adaptations, EBioMedicine 37 (2018) 144–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Ingram PN, Hind LE, Jiminez-Torres JA, Huttenlocher A, Beebe DJ, An Accessible Organotypic Microvessel Model Using iPSC-Derived Endothelium, Adv Healthc Mater 7(2) (2018) 10.1002/adhm.201700497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Huxley VH, Curry FE, Adamson RH, Quantitative fluorescence microscopy on single capillaries: alpha-lactalbumin transport, The American journal of physiology 252(1 Pt 2) (1987) H188–97. [DOI] [PubMed] [Google Scholar]
- [32].Virumbrales-Muñoz M, Ayuso JM, Olave M, Monge R, de Miguel D, Martinez-Lostao L, Le Gac S, Doblare M, Ochoa I, Fernandez LJ, Multiwell capillarity-based microfluidic device for the study of 3D tumour tissue-2D endothelium interactions and drug screening in co-culture models, Sci Rep 7(1) (2017) 11998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Cannon T, Shah A, Skala M, Autofluorescence imaging captures heterogeneous drug response differences between 2D and 3D breast cancer cultures, Biomedical optics express 8(3) (2017) 1911–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Walsh AJ, Cook RS, Skala MC, Functional optical imaging of primary human tumor organoids: development of a personalized drug screen, Journal of Nuclear Medicine 58(9) (2017) 1367–1372. [DOI] [PubMed] [Google Scholar]
- [35].Pasch CA, Favreau PF, Yueh AE, Babiarz CP, Gillette AA, Sharick JT, Karim MR, Nickel KP, DeZeeuw AK, Sprackling CM, Patient-derived cancer organoid cultures to predict sensitivity to chemotherapy and radiation, Clinical Cancer Research 25(17) (2019) 5376–5387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Ayuso JM, Gillette A, Lugo-Cintrón K, Acevedo-Acevedo S, Gomez I, Morgan M, Heaster T, Wisinski KB, Palecek SP, Skala MC, Organotypic microfluidic breast cancer model reveals starvation-induced spatial-temporal metabolic adaptations, EBioMedicine 37 (2018) 144–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Teale F, Principles of Fluorescence Spectroscopy-Lakowicz, Jr, MACMILLAN MAGAZINES LTD PORTERS SOUTH, 4 CRINAN ST, LONDON, ENGLAND N1 9XW, 1984.
- [38].Belair DG, Whisler JA, Valdez J, Velazquez J, Molenda JA, Vickerman V, Lewis R, Daigh C, Hansen TD, Mann DA, Thomson JA, Griffith LG, Kamm RD, Schwartz MP, Murphy WL, Human vascular tissue models formed from human induced pluripotent stem cell derived endothelial cells, Stem Cell Rev Rep 11(3) (2015) 511–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Atkins JL, O’Morchoe C, Pinter G, Total lymphatic clearance of protein from the renal interstitium, Kidney and Proteins in Health and Disease, Karger Publishers; 1988, pp. 238–244. [DOI] [PubMed] [Google Scholar]
- [40].Carmeliet P, Collen D, Molecular basis of angiogenesis: role of VEGF and VE‐cadherin, Annals of the New York Academy of Sciences 902(1) (2000) 249–264. [DOI] [PubMed] [Google Scholar]
- [41].Delahunt B, McKenney JK, Lohse CM, Leibovich BC, Thompson RH, Boorjian SA, Cheville JC, A novel grading system for clear cell renal cell carcinoma incorporating tumor necrosis, The American journal of surgical pathology 37(3) (2013) 311–322. [DOI] [PubMed] [Google Scholar]
- [42].Ayuso JM, Virumbrales-Munoz M, McMinn PH, Rehman S, Gomez I, Karim MR, Trusttchel R, Wisinski KB, Beebe DJ, Skala MC, Tumor-on-a-chip: a microfluidic model to study cell response to environmental gradients, Lab Chip 19(20) (2019) 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Virumbrales-Muñoz M, Ayuso JM, Lacueva A, Randelovic T, Livingston MK, Beebe DJ, Olivan S, Pereboom D, Doblare M, Fernandez L, Ochoa I, Enabling cell recovery from 3D cell culture microfluidic devices for tumour microenvironment biomarker profiling, Sci Rep 9(1) (2019) 6199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Ayuso JM, Vitek R, Swick AD, Skala MC, Wisinski KB, Kimple RJ, Lambert PF, Beebe DJ, Effects of culture method on response to EGFR therapy in head and neck squamous cell carcinoma cells, Sci Rep 9(1) (2019) 12480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Skala MC, Riching KM, Gendron-Fitzpatrick A, Eickhoff J, Eliceiri KW, White JG, Ramanujam N, In vivo multiphoton microscopy of NADH and FAD redox states, fluorescence lifetimes, and cellular morphology in precancerous epithelia, Proceedings of the National Academy of Sciences 104(49) (2007) 19494–19499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Datta R, Heaster T, Sharick J, Gillette A, Skala M, Fluorescence lifetime imaging microscopy: fundamentals and advances in instrumentation, analysis, and applications, Journal of biomedical optics 25(7) (2020) 071203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Li C, Hampson IN, HAMPSON L, KUMAR P, Bernabeu C, Kumar S, CD105 antagonizes the inhibitory signaling of transforming growth factor βl on human vascular endothelial cells, The FASEB Journal 14(1) (2000) 55–64. [DOI] [PubMed] [Google Scholar]
- [48].Shi D, Che J, Yan Y, Peng B, Yao X, Guo C, Expression and clinical value of CD105 in renal cell carcinoma based on data mining in The Cancer Genome Atlas, Exp Ther Med 17(6) (2019) 4499–4505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Saroufim A, Messai Y, Hasmim M, Rioux N, Iacovelli R, Verhoest G, Bensalah K, Patard J, Albiges L, Azzarone B, Tumoral CD105 is a novel independent prognostic marker for prognosis in clear-cell renal cell carcinoma, British journal of cancer 110(7) (2014) 1778–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Lacy SA, Miles DR, Nguyen LT, Clinical Pharmacokinetics and Pharmacodynamics of Cabozantinib, Clin Pharmacokinet 56(5) (2017) 477–491. [DOI] [PubMed] [Google Scholar]
- [51].Zhu C, Kong Z, Wang B, Cheng W, Wu A, Meng X, ITGB3/CD61: a hub modulator and target in the tumor microenvironment, Am J Transl Res 11(12) (2019) 7195–7208. [PMC free article] [PubMed] [Google Scholar]
- [52].Jiang X, Wang J, Deng X, Xiong F, Zhang S, Gong Z, Li X, Cao K, Deng H, He Y, Liao Q, Xiang B, Zhou M, Guo C, Zeng Z, Li G, Li X, Xiong W, The role of microenvironment in tumor angiogenesis, Journal of Experimental & Clinical Cancer Research 39(1) (2020) 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Ma S, Pradeep S, Hu W, Zhang D, Coleman R, Sood A, The role of tumor microenvironment in resistance to anti-angiogenic therapy, F1000Res 7 (2018) 326–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Bischel LL, Sung KE, Jiménez-Torres JA, Mader B, Keely PJ, Beebe DJ, The importance of being a lumen, The FASEB Journal 28(11) (2014) 4583–4590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Dessalles CA, Leclech C, Castagnino A, Barakat AI, Integration of substrate- and flow-derived stresses in endothelial cell mechanobiology, Communications Biology 4(1) (2021) 764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Nassiri F, Cusimano MD, Scheithauer BW, Rotondo F, Fazio A, Yousef GM, Syro LV, Kovacs K, Lloyd RV, Endoglin (CD105): a review of its role in angiogenesis and tumor diagnosis, progression and therapy, Anticancer research 31(6) (2011) 2283–2290. [PubMed] [Google Scholar]
- [57].Ozcan A, Shen SS, Zhai QJ, Truong LD, Expression of GLUT1 in primary renal tumors: morphologic and biologic implications, Am J Clin Pathol 128(2) (2007) 245–54. [DOI] [PubMed] [Google Scholar]
- [58].Bagri A, Kouros-Mehr H, Leong KG, Plowman GD, Use of anti-VEGF adjuvant therapy in cancer: challenges and rationale, Trends in molecular medicine 16(3) (2010) 122–32. [DOI] [PubMed] [Google Scholar]
- [59].Choueiri TK, Escudier B, Powles T, Mainwaring PN, Rini BI, Donskov F, Hammers H, Hutson TE, Lee JL, Peltola K, Roth BJ, Bjarnason GA, Geczi L, Keam B, Maroto P, Heng DY, Schmidinger M, Kantoff PW, Borgman-Hagey A, Hessel C, Scheffold C, Schwab GM, Tannir NM, Motzer RJ, Cabozantinib versus Everolimus in Advanced Renal-Cell Carcinoma, N Engl J Med 373(19) (2015) 1814–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Yakes FM, Chen J, Tan J, Yamaguchi K, Shi Y, Yu P, Qian F, Chu F, Bentzien F, Cancilla B, Orf J, You A, Laird AD, Engst S, Lee L, Lesch J, Chou YC, Joly AH, Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth, Molecular cancer therapeutics 10(12) (2011) 2298–308. [DOI] [PubMed] [Google Scholar]
- [61].Wang WY, Lin D, Jarman EH, Polacheck WJ, Baker BM, Functional angiogenesis requires microenvironmental cues balancing endothelial cell migration and proliferation, Lab Chip 20(6) (2020) 1153–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Rini BI, Atkins MB, Resistance to targeted therapy in renal-cell carcinoma, The lancet oncology 10(10) (2009) 992–1000. [DOI] [PubMed] [Google Scholar]
- [63].Tran TA, Leong HS, Pavia-Jimenez A, Fedyshyn S, Yang J, Kucejova B, Sivanand S, Spence P, Xie XJ, Pena-Llopis S, Power N, Brugarolas J, Fibroblast Growth Factor Receptor-Dependent and -Independent Paracrine Signaling by Sunitinib-Resistant Renal Cell Carcinoma, Molecular and cellular biology 36(13) (2016) 1836–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Lidgren A, Bergh A, Grankvist K, Rasmuson T, Ljungberg B, Glucose transporter-1 expression in renal cell carcinoma and its correlation with hypoxia inducible factor-1 alpha, BJU Int 101(4) (2008) 480–4. [DOI] [PubMed] [Google Scholar]
- [65].Singer K, Kastenberger M, Gottfried E, Hammerschmied CG, Büttner M, Aigner M, Seliger B, Walter B, Schlösser H, Hartmann A, Warburg phenotype in renal cell carcinoma: High expression of glucose‐transporter 1 (GLUT‐1) correlates with low CD8+ T‐cell infiltration in the tumor, International journal of cancer 128(9) (2011) 2085–2095. [DOI] [PubMed] [Google Scholar]
- [66].Aparicio LM, Fernandez IP, Cassinello J, Tyrosine kinase inhibitors reprogramming immunity in renal cell carcinoma: rethinking cancer immunotherapy, Clin Transl Oncol (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Atkins MB, Plimack ER, Puzanov I, Fishman MN, McDermott D, Cho DC, Vaishampayan U, George S, Olencki T, Tarazi J, Rosbrook B, Fernandez K, Keefe S, Choueiri T, Axitinib in combination with pembrolizumab in patients (pts) with advanced renal cell carcinoma (aRCC): Preliminary safety and efficacy results, Annals of Oncology 27(suppl_6) (2016) 773PD–773PD. [Google Scholar]
- [68].Crusz SM, Tang YZ, Sarker S-J, Prevoo W, Kiyani I, Beltran L, Peters J, Sahdev A, Bex A, Powles T, Heterogeneous response and progression patterns reveal phenotypic heterogeneity of tyrosine kinase inhibitor response in metastatic renal cell carcinoma, BMC medicine 14(1) (2016) 185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Barkal LJ, Procknow CL, Álvarez-García YR, Niu M, Jiménez-Torres JA, Brockman-Schneider RA, Gern JE, Denlinger LC, Theberge AB, Keller NP, Microbial volatile communication in human organotypic lung models, Nature communications 8(1) (2017) 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Lugo-Cintrón KM, Ayuso JM, Humayun M, Gong MM, Kerr SC, Ponik SM, Harari PM, Virumbrales-Muñoz M, Beebe DJ, Primary Head and Neck Tumour-Derived Fibroblasts Promote Lymphangiogenesis in a Lymphatic Organotypic Co-culture Model, EBioMedicine 73 (2021) 103634. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw data used in this study is available on request to the authors.