

Abstract
The ERCC1 gene is essential for the repair of UV-induced DNA damage. Unlike most genes in the nucleotide excision repair (NER) pathway, ERCC1 is also involved in recombinational repair. Perhaps for this reason, ERCC1 knockout mice are not a model for the human NER deficiency disorder, xeroderma pigmentosum. Instead, ERCC1 null mice are severely runted and die before weaning from liver failure with accelerated hepatocyte polyploidy that is more reminiscent of a premature ageing disorder. To permit study of the role of ERCC1 in other tissues we have corrected the liver ERCC1 deficiency with a transgene under the control of a liver-specific promoter. The transgene alleviated runting and extended the lifespan. The elevated level of oxidative DNA damage and premature liver polyploidy were reversed and liver function was corrected. A widespread mitochondrial dysfunction was identified and an essential role for ERCC1 in the kidney was also revealed with transgene-containing ERCC1-deficient animals going on to die of renal failure. The nuclei of kidney proximal tubule cells became polyploid in a similar way to the premature liver polyploidy observed in younger ERCC1-deficient animals. We believe that this is a response to the accumulation of endogenous DNA damage in these particularly susceptible tissues which cannot be repaired in ERCC1-deficient animals.
INTRODUCTION
Nucleotide excision repair (NER) is one of a variety of DNA repair pathways that serve to protect cells from the deleterious consequences of DNA damage. NER deals primarily with UV-induced lesions and is also active against a broad range of endogenously generated oxidative lesions, although base excision repair is generally considered to play the key role in removing endogenous oxidative damage (reviewed in 1). The NER pathway consists of an initial damage recognition step, followed by dual incisions to either side of the lesion. The damaged site is then removed on a short oligonucleotide before repair synthesis takes place (reviewed in 2,3). Components of the NER pathway are known to be defective in the human inherited disease xeroderma pigmentosum (XP), which is characterised by UV hypersensitivity, pigmentation abnormalities and a 1000-fold increased risk of skin cancer (4). XP patients also have an increased incidence of internal tumours and, in some cases, neurological abnormalities, suggesting the importance of NER in the repair of endogenously generated as well as UV-induced DNA damage (4). ERCC1 was the first mammalian NER gene to be cloned (5), but it failed to complement any of the seven complementation groups for classical XP (6), suggesting that ERCC1 mutations may be lethal in man. Of course it is also possible that a human inherited disorder caused by ERCC1 deficiency remains to be identified.
ERCC1 is essential for NER, in which it acts in a complex with XPF to make the incision 5′ to the lesion site. Unlike the other NER proteins, ERCC1 and XPF are also involved in homologous recombination, double-strand break repair and the repair of interstrand crosslinks. Their role in recombination was deduced originally from studies on the Saccharomyces cerevisiae homologues, RAD10 (ERCC1) and RAD1 (XPF), which are involved in both NER and mitotic recombination (7). In addition, ERCC1-deficient or XPF-deficient mammalian cells are characteristically hypersensitive to the interstrand crosslinking agent mitomycin C. The key role, which explains the involvement of the ERCC1/XPF complex in these different recombination–repair pathways, is the ability to cleave single- stranded 3′-tails projecting from DNA duplexes (8–10).
The consequences of ERCC1 deficiency in man could thus extend beyond the characteristic features of the NER deficiency disease XP. To study the consequences of ERCC1 deficiency in a whole animal system we used gene targeting to inactivate the mouse ERCC1 gene (11). ERCC1-deficient mice were viable, but severely runted and died before weaning at the age of 3 weeks. Detailed analysis suggested that the most likely cause of death was hepatic failure. At birth ERCC1-deficient hepatocyte nuclei were already enlarged and polyploid; by 3 weeks of age increasingly polyploid and aneuploid nuclei were evident. Blood plasma analysis indicated that liver function was compromised. Since polyploidy is normally only observed in adult liver, the symptoms in the ERCC1-deficient mice were more reminiscent of premature ageing than a NER deficiency disease. Furthermore, elevated levels of p53 protein were detected in the liver, brain and kidneys of the ERCC1 nulls, indicating persistence of DNA damage in these tissues. Despite the detection of p53, we failed to see any increased incidence of apoptosis in these tissues. Subsequently, independently generated ERCC1 knockout mice have been reported with the same liver pathology and runted phenotype (12).
ERCC1-deficient CHO cells and mouse embryonic fibroblasts derived from ERCC1-deficient mice have a 100-fold elevated spontaneous mutation rate at the hypoxanthine phosphoribosyltransferase locus in addition to a higher level of genome instability, indicated by an increased frequency of micronuclei (13) and recombination-dependent genome rearrangements (14). We would like to study the consequences of ERCC1 deficiency, particularly cancer susceptibility, in a range of mouse tissues to assess the importance of ERCC1 in recombinational repair pathways in vivo. The premature death of our null mice has effectively precluded such studies. We speculated that by restoring ERCC1 gene function in the liver, the lifespan of the nulls would be extended sufficiently to enable further studies of the consequences of ERCC1 deficiency in other adult tissues and systems. Here we describe the restoration of ERCC1 function to the liver of our knockout mice by conventional transgenesis with an ERCC1 minigene construct under the control of a liver-specific promoter, thereby demonstrating correction of the severe liver phenotype and revealing the consequences of ERCC1 deficiency in some other tissues.
MATERIALS AND METHODS
Generation of transgenic mice
To direct liver-specific ERCC1 expression, a fragment containing exons 1–10 of the mouse ERCC1 cDNA was cloned (blunt-ended) into a StuI site in exon 2 of the vector pTTR1 ExV3 (15) (kindly provided by Dr T. Van Dyke, University of North Carolina, Chapel Hill). The resulting construct (pTTR/ERCC1; Fig. 1A) was purified as a 5.4 kb HindIII fragment and used for pronuclear microinjection as described (16). Transgene-positive offspring were identified by PCR. The transgene was crossed onto our ERCC1 null background by matings with ERCC1 heterozygotes. Genotyping was performed using a triplex PCR that identified both the ERCC1 wild-type and null alleles in addition to the transgene (Fig. 1B). Primers for exon 4 (CCAGTGTTGAAGTTTGTGCG, mouse ERCC1 cDNA sequence 429–448, GenBank accession no. X07414) and exon 5 (CGAAGGGCGAATTCTTCCC, mouse ERCC1 cDNA sequence 598–579) gave a 0.6 kb product specific for the wild-type allele and a 0.17 kb product specific for the transgene. The targeted allele was detected as a 0.8 kb product from the exon 5 primer and a primer from the neo cassette (GGTTCGAAATGACCGACCAAGCG, 958–980, GenBank accession no. V00618). Transgene copy number was determined by Southern blot analysis of genomic DNA prepared from tail biopsies. To assess the transgene copy number, 3 µg wild-type mouse embryonic stem cell DNA was spiked with various amounts of the transgene fragment, equivalent to 1 (5.4 pg), 2, 5, 10, 20 and 40 copies per mouse genome. Southern blots were first probed using a mouse ERCC1 cDNA fragment (11) to determine the transgene copy number and then reprobed using a mouse PrP coding region probe (17) to allow this estimate to be corrected for any errors in DNA loading. Quantification was performed using a phosphorimager (Molecular Dynamics, Chesham, UK).
Figure 1.

Molecular analysis of TTR/ERCC1 transgenic mice. (A) Schematic diagram of the TTR/ERCC1 transgene. TTR promoter sequences are shown by the diagonally striped box and the first intron is shown by the vertically striped box. TTR exons are numbered and shown by open boxes. The ERCC1 cDNA sequence is represented by the smaller open box. The SV40 derived 3′-end is shown by the grey box. Translational start (ATG) and polyadenylation (AATAAA) sites are indicated. Restriction sites used in cloning and subsequent analysis are shown: B, BamHI; E, EcoRI; H, HindIII; K, KpnI; N, NcoI; P, PstI; S, SstII. Probes a (ERCC1) and b (TTR) used in Southern and northern blot analysis are indicated, along with the 4.8 kb transgene band detected by probe a in Southern blots. (B) Detection of the transgene, wild-type and targeted ERCC1 alleles by PCR. Genomic DNA, isolated from mouse tail biopsies, was the template for a triplex PCR. The 800 bp product (Δ) is specific for the targeted allele; the 600 bp product (WT) is specific for the wild-type allele; the 170 bp product (TG) is specific for the transgene. (C) Transgene copy number determination by Southern blot analysis. Genomic DNA was prepared from tail biopsies of the founder animal (TG#17) and four of the F1 offspring. DNA from the mouse ES cell line HM-1 was spiked with the purified transgene fragment, equivalent to 1, 2, 5, 10, 20 and 40 copies of the transgene per genome. (Upper) ERCC1 probe, BamHI-restricted DNA (3 µg) hybridised with probe a. (Lower) Blot reprobed using a PrP probe to control for any variations in loading between tracks. Phosphorimagery was used to determine transgene copy number, which is indicated below the blots. (D) Northern blot analysis of transgene expression. Total RNA (30 µg) from a range of tissues was analysed by northern blotting. (Upper) ERCC1 wild-type animal, ERCC1 cDNA probe a; (middle) ERCC1 wild-type with transgene, ERCC1 cDNA probe a; (lower) ERCC1 null animal with transgene, TTR probe b. (E) Western blot analysis of transgene expression. Total protein (80 µg) from a range of tissues was analysed by western blotting using an antibody raised against a fragment of mouse ERCC1. (Upper) ERCC1 wild-type animal; (lower) ERCC1 null animal with transgene.
RNA and protein analysis
Total RNA was obtained from mouse tissues using the guanidine isothiocyanate-based reagent RNAzol (Biogenesis, Poole, UK) according to the manufacturer’s protocol. RNA samples were subjected to northern analysis as described (11). Detection of ERCC1 protein was achieved using a rabbit polyclonal antiserum raised against a His-tagged recombinant protein containing a central fragment (amino acids 36–175) of mouse ERCC1 (K.-T.Hsia and D.W.Melton, unpublished results). Western blot analysis was carried out as described (13), except that 12.5% SDS–PAGE gels were used and detection of the primary ERCC1 antibody was achieved with a horseradish peroxidase-conjugated donkey anti-rabbit IgG secondary antibody, followed by enhanced chemiluminescence (Amersham Pharmacia Biotech, Little Chalfont, UK).
Histology and FACS analysis
Tissues were fixed for 24–48 h in buffered formalin (4%) and embedded in paraffin. Sections were cut at 5 µm and stained with haematoxylin and eosin. FACS analysis was performed on fresh liver and kidney tissue as described (11).
Analysis of mouse plasma
Plasma from killed animals was collected and analysed to assess liver, kidney and mitochondrial functions as described (11).
HPLC analysis of liver DNA
DNA from fresh and frozen liver samples was prepared and hydrolysed as described (18). HPLC was performed with a Dionex Summit system (Camberley, UK) equipped with a GP-50 pump, Gina 50 chilling autosampler, a UV/Vis UVD170S detector, operated at 254 nm, and an ESA Coulochem II electrochemical detector (Anachem, Luton, UK) at 400 mV, 2 nA with a filter time of 5 s and AUFS of 1 V. The chromatography was done on a 3 µm Hypersil BDS C18 column (Anachem). The mobile phase consisted of filtered and degassed 0.05 M sodium phosphate, pH 5.5, containing 8% methanol delivered under pressure from a Dionex E01 eluent organiser at a flow rate of 0.5 ml/min. An aliquot of 100 µl of hydrolysed DNA sample was injected for analysis. Data analysis was carried out with Chromeleon software (Dionex).
RESULTS
Transgenic mice express ERCC1 in a liver-specific manner
To rescue the lethal liver phenotype in our ERCC1 null animals we generated mice carrying a transgene (Fig. 1A) in which ERCC1 expression was under the control of transthyretin (TTR) regulatory sequences that have previously been shown to bring about gene expression specifically in the liver (15). TTR is a thyroid hormone transport protein that is normally secreted by hepatocytes into serum and by the choroid plexus epithelium of the brain into the cerebrospinal fluid. Following pronuclear DNA injection a single transgene-positive female (TG#17) was identified from the 23 mice that survived to weaning. All subsequent TTR/ERCC1 transgenic lines were established by breeding from this animal. The founder was crossed with a wild-type (MF1) male to expand the number of transgene-positive animals for preliminary expression analysis and subsequent breeding. Transgene-positive animals were then crossed with heterozygotes from our ERCC1 knockout colony. Transgene-positive ERCC1 heterozygotes identified from these matings were subsequently crossed together to generate transgene-positive ERCC1 nulls (Fig. 1B).
The number of transgene arrays present in the founder was examined using Southern blot analysis. Genomic DNA from TG#17 was restricted with a range of enzymes known not to cut within the transgene. The result indicated a single transgene array in the founder (data not shown). It has been reported that TTR promoter-controlled transgene expression is detected in tissues other than the liver if the transgene is present in more than six copies (15). Southern blot analysis was used to determine the number of transgene copies in the genome of the founder animal and in offspring, each hemizygous for the transgene array, from crosses onto the knockout background (Fig. 1C). The analysis showed the presence of the endogenous 8.3 and (very weak) 4.3 kb wild-type ERCC1 bands in each track. The transgenic sample, TG#9, was an ERCC1 heterozygote and, consequently, an additional band from the null allele was evident at 6.7 kb (19). In addition, a previously identified restriction fragment length polymorphism for the wild-type 8.3 kb band was evident in TG#55. The 4.8 kb transgene-specific band was evident in each of the transgenic sample lanes and, at appropriately varying intensities, in the copy number titration lanes. Since the endogenous ERCC1 bands were unsuitable as internal standards for DNA loading because of the polymorphisms, the blot was stripped and reprobed with a PrP gene probe. Phosphorimagery was used to quantify the level of the transgene signal relative to the 8.5 kb PrP band. The transgene copy number estimates were: TG#17, two copies; TG#9, three copies; TG#26, 54 and 55, four copies. The consistently higher copy number estimates for the F1 offspring than for the founder (TG#17) suggests that the founder may have been a transgenic mosaic.
The normal pattern of ERCC1 mRNA expression in a range of wild-type mouse tissues was determined by northern analysis (Fig. 1D, upper). The 1.1 kb ERCC1 mRNA was expressed at the highest level in testes and at the lowest level in liver. Western blotting, using an antibody raised against a central fragment of the mouse ERCC1 protein, revealed a different pattern of protein expression in the same wild-type tissues (Fig. 1E, upper). The 33 kDa ERCC1 protein was again most abundant in testes, but the protein was now least abundant in heart. The liver also showed two additional, slower migrating ERCC1 protein species, presumably resulting from a liver-specific post-translational modification. We have found that our standard loading controls for blots, which involve reprobing for housekeeping genes such as GAPDH and actin, are inappropriate when different tissues are being compared. For this reason, ethidium bromide staining of northern gels and Coomassie blue staining of duplicate western gels was used to confirm that equal loadings were achieved (data not shown).
The liver-specific expression of the transgene on a wild-type background was revealed by northern analysis using an ERCC1 probe (Fig. 1D, middle). Expression of the 1.5 kb TTR/ERCC1 transcript was strong in the liver, much higher than expression in the liver of the wild-type 1.1 kb ERCC1 mRNA. However, the transgene transcript could not be detected in other tissues (but see below). The proximity of the wild-type 1.1 kb ERCC1 mRNA made it impossible to exclude the possibility of low level transgene expression in other tissues. The same complication also extended to expression of the transgene on an ERCC1-deficient background because we have previously reported low levels of ERCC1 transcripts of greater than normal size in tissues from ERCC1-deficient mice (11). Consequently, we used a TTR probe to monitor transgene expression on an ERCC1-deficient background (Fig. 1D, lower). The probe detected the endogenous 0.7 kb TTR mRNA at a high level in liver and a much lower level in brain. Expression of the 1.5 kb TTR/ERCC1 transcript was specific to the liver.
Despite the presence of ERCC1 transcripts, our antibody to mouse ERCC1 did not detect any ERCC1 protein in tissues from ERCC1-deficient mice (data not shown). Protein expression from the transgene on the ERCC1-deficient background was strong in the liver, higher than, but showing the same multiple species, as the endogenous protein (Fig. 1E, lower). In some animals, including the one shown in Figure 1E, a low level of transgene expression was also detected in the brain and testes (<5% of the liver expression level). Thus, although all animals showed the expected high level of transgene expression in the liver, expression was not absolutely liver specific.
As the ultimate aim of this work was to increase the lifespan of our ERCC1 nulls, we considered it prudent to confirm that transgene expression was maintained. Northern blot analysis of liver RNA from animals (aged from 4 to 24 weeks) of four different generations confirmed that it was. Although there was some variation in the absolute level of transgene expression between liver samples (ranging from 29 to 105% of the endogenous TTR mRNA), there was no correlation with the age of the animal or the generation from which the sample was taken.
Expression of the ERCC1 transgene in the liver of ERCC1-deficient mice alleviates runting
The severe runting associated with ERCC1 deficiency has been reported previously (11,12). The body weights of TTR/ERCC1 transgene-positive ERCC1-deficient mice and their littermates were measured at weaning (age 21–28 days). The average weight of a transgene-positive ERCC1-deficient mouse was 7.75 ± 0.56 g, compared to 13.25 ± 0.3 g for wild-type littermates. The body weight, as a percentage of the wild-type, was 58% for the transgene-positive ERCC1 nulls, compared to the previously reported 20% for our knockout mice (11). This size difference meant that transgene-positive nulls could be readily distinguished from transgene-negative null littermates by eye (Fig. 2A).
Figure 2.
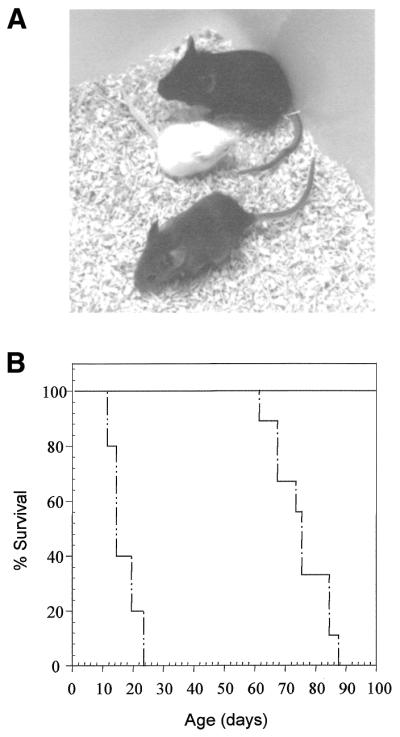
Phenotypic rescue by expression of the TTR/ERCC1 transgene in the liver. (A) Transgene-positive ERCC1 null mouse (bottom) with a runted transgene-negative ERCC1 null (middle) and a wild-type littermate (top). (B) Increased lifespan of transgene-positive ERCC1 null mice. The percentage of surviving mice for each genotype is plotted against age. Wild-type, solid line, n = 9; transgene-positive ERCC1 null, single dotted line, n = 9; transgene-negative ERCC1 null, double dotted line, n = 5.
Expression of the ERCC1 transgene in the liver extends the lifespan of ERCC1-deficient mice
We have previously reported that our ERCC1-deficient mice die prematurely before weaning at 3 weeks (11). This is confirmed in Figure 2B, where all (n = 5) of the transgene-negative ERCC1 nulls died within 24 days of birth. The transgene-positive ERCC1 nulls (n = 9) showed 100% survival up to 61 days, with all mice dying between 61 and 88 days. No wild-type littermates (n = 9) died within this period. Thus, the lifespan of the transgene-positive nulls has been extended to three times that of their transgene-negative ERCC1 null siblings.
Expression of the ERCC1 transgene in the liver corrects elevated levels of oxidative DNA damage associated with ERCC1 deficiency
Although NER deals primarily with UV-induced lesions, it also removes a broad range of endogenously generated oxidative lesions. The level of oxidative DNA damage in liver DNA was assessed by measuring the commonest oxidised base, 8-oxoG, by HPLC analysis with electrochemical detection. Levels were calculated as mol 8-oxoG/105 mol deoxyguanosine (Table 1). The mean value obtained for 10–21-day-old ERCC1 null liver was 9-fold higher than control littermates. This increase in 8-oxoG levels in ERCC1 null liver was statistically significant (P = 0.005 by Student’s t-test). The mean value for 6–10-week-old transgene-positive ERCC1 null littermates was not significantly different from the age-matched control group (P = 0.89 by Student’s t-test). Thus, the level of oxidative DNA damage is elevated in ERCC1 null liver and this elevation is corrected by the ERCC1 transgene.
Table 1. Levels of 8-oxoG in the liver and kidney of ERCC1 null and transgene-positive ERCC1 null mice.
| Genotype | Age | Tissue | 8-oxoG levela SD. | Probability for null hypothesise |
|---|---|---|---|---|
| Controlb (7) | 10–21 days | Liver | 0.27 ± 0.35 | |
| Nullc (7) | 10–21 days | Liver | 2.57 ± 1.71 | P = 0.005 |
| Control (6) | 6–10 weeks | Liver | 0.10 ± 0.04 | |
| Null + TGd (6) | 6–10 weeks | Liver | 0.10 ± 0.07 | P = 0.89 |
| Control (2) | 7–8 weeks | Kidney | 0.06 ± 0.04 | |
| Null + TG (2) | 7–8 weeks | Kidney | 0.28 ± 0.02 | P = 0.02 |
aMean values expressed as mol 8-oxoG/105 mol deoxyguanosine ±
bControl genotypes were ERCC1 wild-type and heterozygote littermates, with or without an ERCC1 transgene. Figures in parentheses indicate the number of independent samples assayed for each genotype.
cERCC1-deficient.
dERCC1-deficient with an ERCC1 transgene.
eBy Student’s t-test compared to the control group.
Expression of the ERCC1 transgene in the liver corrects the nuclear abnormalities associated with ERCC1 deficiency
Liver sections were taken from 3-week-old ERCC1 nulls, transgene-positive nulls and wild-type littermates (five animals of each genotype; representative sections are shown in Fig. 3A). ERCC1 nulls consistently showed the characteristic variable nuclear size, including extremely large nuclei, indicative of increased ploidy, that we have characterised previously (11,20). All wild-type and transgene-positive ERCC1 null sections showed the typical appearance of young mouse livers, with uniformly small hepatocyte nuclei. In parallel, the DNA content of liver cell nuclei was determined by FACS analysis (Fig. 3B). The wild-type FACS profiles showed a major (G1) peak, representing the normal diploid cell population prior to DNA replication, and a much smaller (G2) peak, corresponding to cells that had undergone replication. This profile was essentially duplicated in all transgene-positive ERCC1 nulls. (The sub-G1 material in the example shown reflects damage to some of the fragile hepatocyte nuclei during preparation for FACS analysis.) These profiles contrasted sharply with the profiles obtained from all the ERCC1 null livers. In addition to the major peak, representing normal diploid hepatocyte nuclei in G1, there was a much larger peak corresponding to nuclei with double (4N) the normal DNA content. From centromeric staining we have previously found that most of these 4N cells are tetraploid cells in G1 (20). Both the diploid and tetraploid G1 peaks have distinct shoulders, possibly indicating the existence of cells with reduced, aneuploid DNA content. A discrete peak corresponding to nuclei with still higher DNA content (∼8N) was also apparent. In the representative profiles shown 9.3% of nuclei from the ERCC1 nulls fell into this category, compared to only 0.3 and 1% from wild-type and transgene-positive nulls. Thus, the premature polyploidy seen in ERCC1 nulls has been effectively corrected by the transgene.
Figure 3.

Correction of the nuclear abnormalities in the livers of transgene-positive ERCC1 nulls. (A) Haematoxylin and eosin stained liver sections from 3-week-old wild-type (top), ERCC1 null (middle) and transgene-positive ERCC1 null mice (bottom). Normal size hepatocyte nuclei are indicated by arrowheads in the wild-type and transgene-positive ERCC1 null; enlarged nuclei in the null are indicated by arrows. (B) DNA content of liver cell nuclei. FACScan analysis of liver biopsies from the same animals are shown. For each profile the percentage of nuclei with elevated (8N) DNA content is indicated.
Plasma analysis shows correction of hepatic function in transgene-positive ERCC1 nulls
Having shown that the transgene corrected the liver phenotype at the histological level it remained necessary to demonstrate that liver function was indeed restored. Plasma samples were collected from 3-week-old ERCC1 nulls, transgene-positive nulls and control littermates and from 7-week-old transgene-positive nulls and littermate controls (Table 2). A 2.5-fold increase in plasma alkaline phosphatase (ALP) activity, normally indicative of reduced liver excretory function, was observed in the 3-week-old ERCC1 nulls compared to control littermates. In the 3- and 7-week-old transgene-positive nulls ALP activity was not significantly elevated compared to control littermates, demonstrating that this aspect of liver dysfunction in ERCC1 nulls had indeed been corrected by the transgene. A second measure of liver excretory function, plasma levels of bilirubin, was below the level of detection in all samples analysed.
Table 2. Liver, kidney and mitochondrial function assays on mouse plasma.
| Genotype | Age (weeks) | Alkaline phosphatase (U l–1) | Probability for null hypothesisd | Bilirubin (µmol l–1) | Lactate (mmol l–1) | Probability for null hypothesisd | Creatinine (µmol l–1) | Probability for null hypothesisd |
|---|---|---|---|---|---|---|---|---|
| The mean ± SD is given for each assay. Figures in parentheses indicate the number of independent samples assayed for each genotype. | ||||||||
| Controla (14) | 3 | 583 ± 191 | <1 | 3.2 ± 1.6 | 28.4 ± 3.2 | |||
| Nullb (9) | 3 | 1409 ± 49 | P = 4.2 × 10–6 | <1 | 7.1 ± 1.7 | P = 6.6 × 10–5 | 31.7 ± 4.7 | P = 0.16 |
| Null + TGc (10) | 3 | 744 ± 206 | P = 0.21 | <1 | 3.45 ± 0.81 | P = 0.61 | 28.0 ± 4.0 | P = 0.87 |
| Control (7) | 7 | 437 ± 97 | <1 | 5.0 ± 1.0 | 40.0 ± 5.0 | |||
| Null+TG (7) | 7 | 527 ± 162 | P = 0.26 | <1 | 6.9 ± 1.6 | P = 0.04 | 47.4 ± 7.6 | P = 0.05 |
aControl genotypes were ERCC1 wild-type and heterozygote, with or without an ERCC1 transgene.
bERCC1 deficient. The low blood volume obtained from the nulls necessitated pooling the nine samples into groups of three before measurement.
cERCC1 deficient with an ERCC1 transgene. The low blood volume obtained from the 3-week-old animals necessitated pooling the 10 samples into three groups before measurement.
dBy Student’s t-test compared to the control group.
Reduced mitochondrial and renal function in ERCC1-deficient mice
Plasma lactate levels were 2-fold elevated in 3-week-old ERCC1 nulls compared to control littermates. This increase, which is indicative of reduced mitochondrial function, was highly significant. Lactate levels in 3-week-old transgene-positive nulls were not significantly different from controls. A small (40%) but significant increase in lactate levels was observed in the 7-week-old transgene-positive nulls compared to control littermates. Plasma creatinine levels were equivalent in all the 3-week-old samples. There was a small (20%) but significant increase in creatinine levels in 7-week-old transgene-positive nulls compared to controls. An increase in plasma creatinine is indicative of reduced renal excretory function.
Transgene-positive ERCC1 null mice exhibit kidney abnormalities
The extended lifespan of our transgene-positive nulls has enabled us to study the consequences of ERCC1 deficiency in adult tissues, other than the liver, for the first time. As expected, the level of oxidative DNA damage was elevated in the kidneys of 7–8-week-old transgene-positive ERCC1 nulls compared to control littermates (Table 1). The 4-fold increase in 8-oxoG levels was statistically significant (P = 0.02 by Student’s t-test). Comparison of tissue sections from transgene-positive nulls (aged 6 weeks and over) with wild-type littermates revealed no clear differences between brain, spleen, skin or liver. Plasma creatinine levels from 7-week-old transgene-positive nulls indicated a developing dysfunction in renal excretion. This was confirmed on post mortem examination of five 10-week-old transgene-positive nulls, which all revealed an accumulation of ascites fluid in the peritoneal cavity. Compared to wild-type controls (Fig. 4A and C), kidney sections from these animals (Fig. 4B, D and E) showed partially sclerosing glomeruli and tubular distention by protein casts, although inflammatory changes were essentially absent. There was considerable nuclear pleomorphism throughout, concentrated particularly in the proximal tubules, where many of the nuclei were also enlarged. The DNA content of kidney cell nuclei was determined by FACS analysis. FACS profiles for 10-week-old wild-type kidneys (Fig. 4F) showed the typical major (G1) peak, with only 4% of the population in G2. Cells with greater than this G2 (4N) DNA content comprised <1% of the population. The profile from transgene-positive ERCC1 null kidneys was very different, being shifted towards nuclei with higher ploidy levels (Fig. 4G). Nineteen per cent of this population had a 4N DNA content. In addition, 3% of nuclei had a higher ploidy status (6N or 8N). Thus, having corrected the liver phenotype with an ERCC1 transgene that is expressed in a liver-specific fashion, it seems likely that these animals are now succumbing to the consequences of ERCC1 deficiency in the kidney.
Figure 4.

Transgene-positive ERCC1 nulls develop kidney pathology. Haematoxylin and eosin stained kidney sections from 10-week-old ERCC1 wild-type (A and C) and transgene-positive ERCC1 null mice (B, D and E). Enlarged proximal tubule cell nuclei in transgene-positive ERCC1 null mice are indicated by arrowheads. g, glomerulus; dg, damaged (partially sclerosed) glomerulus; dpt, distended proximal tubule; pc, protein cast; pt, proximal tubule. (F and G) FACScan analysis of kidney cortex biopsies from animals of the same genotype. For each profile the percentage of nuclei with 4N and >6N DNA content is indicated.
DISCUSSION
The lethal liver phenotype of our ERCC1 knockout mice has prevented us from studying the consequences of ERCC1 deficiency, particularly cancer susceptibility, in a range of adult mouse tissues to assess the importance of ERCC1 in recombinational repair pathways in vivo. We show here that expression of an ERCC1 transgene in the liver corrects the liver phenotype and significantly extends the lifespan of these DNA repair-deficient mice. Liver-specific ERCC1 expression was achieved using a transgene under the control of the TTR gene promoter. The promoter fragment used was known to elicit high levels of hepatocyte-specific transgene expression, with expression detectable in the choroid plexus of the brain only when six or more copies of the transgene were present (15). Characterisation of the founder transgenic animal and F1 offspring indicated that the founder was most likely mosaic for the transgene, with an average of one or two copies of the transgene present per cell in a single array, whilst the F1 animals had between three and four copies of the transgene.
Whilst it was unlikely that the transgene array here would be subject to repeat-induced silencing, as is often seen in animals with high copy number transgene arrays (21), it remained possible that other factors, such as the chromosomal integration site, might repress transgene expression over time or in subsequent generations. For instance, age-dependent silencing of a β-globin driven lacZ reporter gene has previously been described (22). TTR-regulated ERCC1 transgene expression was maintained at high levels (29–105% of endogenous TTR levels) in the livers of all mice through the four generations analysed and was stable throughout the life of the animals. This was of particular importance to ensure that any phenotypic correction achieved by transgene expression was maintained.
The level of transgene mRNA greatly exceeded the normal level of ERCC1 transcripts in the liver. ERCC1 protein was also present at higher levels in the livers of transgene-positive ERCC1 nulls than in wild-type livers. Both transgene-encoded and endogenous ERCC1 protein showed liver-specific post-translational modifications. Although the significance of these novel protein species is uncertain, they could be involved in some liver-specific ERCC1 function which might provide an explanation for the particular sensitivity of the liver to ERCC1 deficiency.
Over-expression of ERCC1 in CHO cells resulted in hypersensitivity to a DNA crosslinking agent (23). An attempt to over-express ERCC1 in CHO cells showed only a 4-fold increase at the protein level despite a 1000-fold increase in gene copy number and a dramatic increase in the level of mRNA (24). These results raise the complication for transgenic studies that over-expression of ERCC1 may be detrimental to the survival of a cell. The livers in transgene-containing ERCC1 wild-type and null mice were normal by all our assay criteria. Evidently, ERCC1 expression in our transgenic liver was below the level at which adverse consequences are encountered.
While expression of the transgene was high in liver it was not absolutely liver specific. Low levels of transgene transcripts and, particularly, transgene-encoded ERCC1 protein (<5% of the level in liver) were observed in other tissues (particularly brain and testes) in some of the animals tested. This was not unexpected given previous reports of low levels of ectopic transgene expression from the same TTR promoter fragment (15,25).
Our TTR/ERCC1 transgene-positive ERCC1 nulls survived 61–88 days, compared to the transgene-negative null littermates that died prior to weaning at 21 days. We conclude that liver-specific transgene expression results in an increased lifespan for our ERCC1 nulls. The ERCC1 knockout mice reported by Weeda et al. (12) had a lifespan of 38–78 days, depending on the genetic background, whilst mice carrying a subtle alteration lived for 2–6 months, possibly due to some residual ERCC1 activity. In addition to the desired effect of increasing the lifespan of our nulls, there was also alleviation of the runted phenotype associated with ERCC1 deficiency. The weight of the transgene-positive nulls was 60% of their wild-type littermates, compared to only 20% for the transgene-negative nulls. Whilst we believe that runting arises from the overall ERCC1-deficient status, this work may suggest that runting is predominantly a consequence of the effect of this DNA repair defect in the liver. If liver function is impaired in ERCC1 nulls the synthesis of growth hormones, such as insulin-like growth factor (IGF-I), which takes place primarily in the liver, will also be affected and may result in growth retardation as a consequence. Wistar rats with chemically induced liver damage showed a general reduction in body weight, which was alleviated by administration of IGF-I (26).
Levels of the commonest oxidised base, 8-oxoG, were 9-fold elevated in ERCC1-deficient liver and 4-fold elevated in transgene-positive ERCC1 null kidney. This is the first in vivo demonstration of increased levels of endogenous DNA damage in ERCC1 deficiency. Moreover, the elevated DNA damage levels in the liver were corrected by the ERCC1 transgene. Traditionally base excision repair was considered to have the key role in removing 8-oxoG, which is strongly mutagenic and also acts as a block to transcription by RNA polymerase II (27). However, the discovery of transcription-coupled repair of 8-oxoG and the observation that this process continues to operate in OGG1 null cells (28) has, belatedly, led to the recognition that NER may also have an important role to play in the repair of 8-oxoG (discussed in 28) and of another form of oxidative damage (purine cyclodeoxynucleosides; 29). Nine-fold higher levels of 8-oxoG in ERCC1-deficient liver compared to control littermates demonstrate that ERCC1 is important for the repair of 8-oxoG, particularly considering that the levels of 8-oxoG in the livers of OGG1 null mice were only 1.7-fold higher than controls (30). Most likely the increase in 8-oxoG levels results from the lack of the NER function of ERCC1. However, in a pathway distinct from NER, XPG is involved in the repair of oxidised pyrimidines (31). Perhaps ERCC1 might be required for transcription-coupled repair of 8-oxoG in a similar NER-independent manner. We cannot be sure that the increased levels of oxidative damage observed are responsible for the ERCC1-deficient liver phenotype; other lesions (such as interstrand crosslinks) which are also recognised by ERCC1 could also be responsible. The role of NER and of ERCC1 in particular in the repair of 8-oxoG and the significance of oxidative DNA damage to the phenotype in ERCC1-deficient liver could be determined by measuring 8-oxoG levels in XPA and XPC knockout mice, which do not show the liver phenotype (32–34).
Liver nuclear abnormalities have been seen in all ERCC1 knockout and mutant mice reported to date (11,12). We believe that the premature polyploidy in hepatocytes is a protective response to the accumulation of DNA damage that is normally repaired by ERCC1, although it could also result from the lack of another, non-repair-related function of ERCC1 that is peculiar to the liver. Expression of the liver-specific ERCC1 transgene corrected the liver phenotype in our nulls. Despite this observation, it remained necessary to confirm that transgene expression also resulted in the restoration of hepatic function. The reduced liver excretory function in 3-week-old ERCC1 nulls, detected as a 2.5-fold increase in plasma ALP activity, was corrected in 3- and 7-week-old transgene-positive nulls. Plasma levels of bilirubin, a second measure of liver excretory function, were below the levels of detection in all samples analysed. In humans elevated bilirubin levels are only expected when the liver has lost at least half of its excretory capacity.
The plasma analysis also provided evidence for reduced mitochondrial function in 3-week-old ERCC1 nulls in the form of a highly significant 2-fold increase in lactate levels compared to control littermates. Lactate levels in 3-week-old transgene-positive nulls were not significantly different from controls. The increase in lactate levels in the 7-week-old transgene-positive nulls compared to control littermates could indicate that the mitochondrial dysfunction associated with ERCC1 deficiency is progressive and not just confined to the liver. The link between deficiency of a nuclear DNA repair enzyme and mitochondrial dysfunction remains to be established.
In addition to the liver, our original ERCC1 knockout mice showed stabilisation of p53 in both brain and kidney (11). However, there was no detectable pathology in these organs immediately prior to death at 21 days. Correction of the liver phenotype has allowed us to study the developing consequences of ERCC1 deficiency in these tissues in older animals. Although there was no indication of any pathology in the brains of 7-week-old transgene-positive nulls, the small (20%) but significant increase in creatinine levels compared to controls provided the first indication of reduced renal excretory function. By 10 weeks transgene-positive nulls had excessive accumulation of ascites fluid, with damaged glomeruli and dilated or blocked proximal tubules. Similar kidney changes have been reported in an independent ERCC1 knockout line with greater longevity than our own line (12). We found that nuclei from proximal tubule cells were pleomorphic and many were also enlarged. Significantly, these are the same cells where p53 stabilisation was previously detected in our 3-week old ERCC1 nulls, although no pathology was evident at that early stage. FACS analysis revealed the existence of a large polyploid cell population in kidneys from 10-week-old transgene-positive nulls, similar to the polyploidy seen in the liver of 3-week-old ERCC1 nulls. Although progressively increasing ploidy is a normal feature of liver development, it is not usually seen in the kidney. However, the kidney is particularly susceptible to genotoxic agents, such as ionising radiation (35), cisplatin (36) and cadmium (37), which all induce the appearance of enlarged proximal tubule cell nuclei. Elevated levels of oxidative DNA damage were found in the kidneys of transgene-positive ERCC1 nulls. Having restored DNA repair capacity to the liver it seems likely that these animals now succumb to kidney failure. This is perhaps not unexpected given that the physiological role fulfilled by the kidneys would expose tubule cells to toxic and potentially DNA damaging compounds. Much of the resulting damage could not be repaired in the absence of ERCC1.
In summary, using conventional transgenesis we have succeeded in extending the lifespan of our DNA repair-deficient ERCC1 null mice by correcting the runting and the novel premature polyploidy in liver. The increase in the lifespan of these repair-deficient animals has enabled us to identify an essential role for ERCC1 in kidney. We are currently using these animals to study the role of ERCC1 (and the recombinational repair pathways that it is involved in) in spermatogenesis and in the response to UV-induced DNA damage in the skin. The lifespan of the animals remains too short to study the role of ERCC1 in protecting against cancer. Mice with conditional ERCC1 gene inactivation in specific tissues, induced by site-specific recombination (38), will be needed to undertake these studies.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Carolanne McEwan for mouse genotyping. K.-T.H. was supported by the National Science Council, Taiwan, ROC. This work was supported by a programme grant (SP2095/0301) from The Cancer Research Campaign to D.W.M.
REFERENCES
- 1.Lindahl T. (1993) Instability and decay of the primary structure of DNA. Nature, 362, 709–715. [DOI] [PubMed] [Google Scholar]
- 2.Wood R.D. (1996) DNA repair in eukaryotes. Annu. Rev. Biochem., 65, 135–167. [DOI] [PubMed] [Google Scholar]
- 3.Sancar A. (1996) DNA excision repair. Annu. Rev. Biochem., 65, 43–81. [DOI] [PubMed] [Google Scholar]
- 4.Friedberg E.C., Walker,G.C. and Siede,W. (1995) DNA Repair and Mutagenesis. ASM Press, Washington, DC.
- 5.Westerveld A., Hoeijmakers,J.H.J., van Duin,M., Dewit,J., Odijk,H., Pastink,A., Wood,R.D. and Bootsma,D. (1984) Molecular cloning of a human DNA repair gene. Nature, 310, 425–429. [DOI] [PubMed] [Google Scholar]
- 6.van Duin M., Vredeveldt,G., Mayne,L.V., Odijk,H., Vermeulen,W., Klein,B., Weeda,G., Hoeijmakers,J.H.J., Bootsma,D. and Westerveld,A. (1989) The cloned human DNA excision repair gene ERCC1 fails to correct xeroderma pigmentosum complementation group A through group I. Mutat. Res., 217, 83–92. [DOI] [PubMed] [Google Scholar]
- 7.Schiestl R.H. and Prakash,S. (1990) RAD10, an excision repair gene of Saccharomyces cerevisiae, is involved in the RAD1 pathway of mitotic recombination. Mol. Cell. Biol., 10, 2485–2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kuraoka I., Kobertz,W.R., Ariza,R.R., Biggerstaff,M., Essigmann,J.M. and Wood,R.D. (2000) Repair of an interstrand DNA cross-link initiated by ERCC1-XPF repair/recombination nuclease. J. Biol. Chem., 275, 26632–26636. [DOI] [PubMed] [Google Scholar]
- 9.Adair G.M., Rolig,R.L., Moore-Faver,D., Zabelshansky,M., Wilson,J.H. and Nairn,R.S. (2000) Role of ERCC1 in removal of long non-homologous tails during targeted homologous recombination. EMBO J., 19, 5552–5561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Silva I.U., McHugh,P.J., Clingen,P.H. and Hartley,J.A. (2000) Defining the roles of nucleotide excision repair and recombination in the repair of DNA interstrand cross-links in mammalian cells. Mol. Cell. Biol., 20, 7980–7990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McWhir J., Selfridge,J., Harrison,D.J., Squires,S. and Melton,D.W. (1993) Mice with DNA repair gene (ERCC1) deficiency have elevated levels of p53, liver nuclear abnormalities and die before weaning. Nature Genet., 5, 217–224. [DOI] [PubMed] [Google Scholar]
- 12.Weeda G., Donker,I., de Wit,J., Morreau,H., Janssens,R., Vissers,C.J., Nigg,A., van Steeg,H., Bootsma,D. and Hoeijmakers,J.H.J. (1997) Disruption of mouse ERCC1 results in a novel repair syndrome with growth failure, nuclear abnormalities and senescence. Curr. Biol., 7, 427–439. [DOI] [PubMed] [Google Scholar]
- 13.Melton D.W., Ketchen,A.M., Nunez,F., Bonatti-Abbondandolo,S., Abbondandolo,A., Squires,S. and Johnson,R.T. (1998) Cells from ERCC1-deficient mice show increased genome instability and a reduced frequency of S-phase-dependent illegitimate chromosome exchange but a normal frequency of homologous recombination. J. Cell Sci., 111, 394–404. [DOI] [PubMed] [Google Scholar]
- 14.Sargent R.G., Meservy,J.L., Perkins,B.D., Kilburn,A.E., Intody,Z., Adair,G.M., Nairn,R.S. and Wilson,J.H. (2000) Role of the nucleotide excision repair gene ERCC1 in formation of recombination-dependent rearrangements in mammalian cells. Nucleic Acids Res., 28, 3771–3778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yan C., Costa,R.H., Darnell,J.E., Chen,J. and Van Dyke,T. (1990) Distinct positive and negative elements control the limited hepatocyte and choroid plexus expression of transthyretin in transgenic mice. EMBO J., 9, 869–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hogan B., Costantini,F. and Lacy,E. (1986) Manipulating the Mouse Embryo. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 17.Moore R.C., Redhead,N.J., Selfridge,J., Hope,J., Manson,J.C. and Melton,D.W. (1995) Double replacement gene targeting for the production of a series of mouse strains with different prion protein gene alterations. Biotechnology, 13, 999–1004. [DOI] [PubMed] [Google Scholar]
- 18.Wood S.G., Gedik,C.M., Vaughan,N.J. and Collins,A.R. (1999) Measurement of 8-oxo-deoxyguanosine in lymphocytes, cultured cells and tissue samples by HPLC with electochemical detection. In Barnett,Y.A. and Barnett,C.R. (eds), Methods in Molecular Medicine, Vol. 38: Ageing Methods and Protocols. Humana Press, Totowa, NJ, pp. 171–178. [DOI] [PubMed]
- 19.Selfridge J., Pow,A.M., McWhir,J., Magin,T.M. and Melton,D.W. (1992) Gene targeting using a mouse HPRT minigene/HPRT-deficient embryonic stem cell system: inactivation of the mouse ERCC1 gene. Somat. Cell Mol. Genet., 18, 325–336. [DOI] [PubMed] [Google Scholar]
- 20.Nuñez F., Chipchase,M.D., Clarke,A.R. and Melton,D.W. (2000) Nucleotide excision repair gene (ERCC1) deficiency causes G2 arrest in hepatocytes and a reduction in liver binucleation: the role of p53 and p21. FASEB J., 14, 1073–1082. [DOI] [PubMed] [Google Scholar]
- 21.Garrick D., Fiering,S., Martin,D.I.K. and Whitelaw,E. (1998) Repeat-induced gene silencing in mammals. Nature Genet., 18, 56–59. [DOI] [PubMed] [Google Scholar]
- 22.Robertson G., Garrick,D., Wilson,M., Martin,D.I.K. and Whitelaw,E. (1996) Age-dependent silencing of globin transgenes in the mouse. Nucleic Acids Res., 24, 1465–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bramson J. and Panasci,L. (1993) Effect of ERCC1 overexpression on sensitivity of Chinese hamster ovary cells to DNA damaging agents. Cancer Res., 53, 3237–3240. [PubMed] [Google Scholar]
- 24.Sijbers A.M., van der Spek,P.J., Odijk,H., van den Berg,J., van Duin,M., Westerveld,A., Jaspers,N.G., Bootsma,D. and Hoeijmakers,J.H. (1996) Mutational analysis of the human nucleotide excision repair gene ERCC1. Nucleic Acids Res., 24, 3370–3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu H., Wade,M., Krall,L., Grisham,J., Xiong,Y. and Van Dyke,T. (1996) Targeted in vivo expression of the cyclin-dependent kinase inhibitor p21 halts hepatocyte cell-cycle progression, post-natal liver development and regeneration. Genes Dev., 10, 245–260. [DOI] [PubMed] [Google Scholar]
- 26.Castilla-Cortazar I., Garcia,M., Quiroga,J., Diez,N., Diez-Caballero,F., Calvo,A., Diaz,M. and Prieto,J. (2000) Insulin-like growth factor reverts testicular atrophy in rats with advanced cirrhosis. Hepatology, 31, 592–600. [DOI] [PubMed] [Google Scholar]
- 27.Le Page F., Kwoh,E.E., Avrutskaya,A., Gentil,A., Leadon,S.A., Sarasin,A. and Cooper,P.K. (2000) Transcription-coupled repair of 8-oxoguanine: requirement for XPG, TFIIH and CSB and implications for Cockayne syndrome. Cell, 101, 159–171. [DOI] [PubMed] [Google Scholar]
- 28.Le Page F., Klungland,A., Barnes,D.E., Sarasin,A. and Boiteux,S. (2000) Transcription coupled repair of 8-oxoguanine in murine cells: the Ogg1 protein is required for repair in nontranscribed sequences but not in transcribed sequences. Proc. Natl Acad. Sci. USA, 97, 8397–8402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuraoka I., Bender,C., Romieu,A., Cadet,J., Wood,R.D. and Lindahl,T. (2000) Removal of oxygen free-radical-induced 5′,8-purine cyclodeoxynucleosides from DNA by the nucleotide excision-repair pathway in human cells. Proc. Natl Acad. Sci. USA, 97, 3832–3837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Klungland A., Rosewell,I., Hollenbach,S., Larsen,E., Daly,G., Epe,B., Seeberg,E., Lindahl,T. and Barnes,D.E. (1999) Accumulation of premutagenic DNA lesions in mice defective in removal of oxidative base damage. Proc. Natl Acad. Sci. USA, 96, 13300–13305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klungland A., Höss,M., Gunz,D., Constantinou,A., Clarkson,S.G., Doetsch,P.W., Bolton,P.H., Wood,R.D. and Lindahl,T. (1999) Base excision repair of oxidative DNA damage activated by XPG protein. Mol. Cell, 3, 33–42. [DOI] [PubMed] [Google Scholar]
- 32.de Vries A., van Oostrom,C.T.M., Hofhuis,F.M.A., Dortant,P.M., Berg,R.J.W., de Gruijl,F.R., Wester,P.W., van Kreijl,C.F., Capel,P.J.A., van Steeg,H. and Verbeek,S.J. (1995) Increased susceptibility to ultraviolet-B and carcinogens of mice lacking the DNA excision-repair gene Xpa. Nature, 377, 169–173. [DOI] [PubMed] [Google Scholar]
- 33.Nakane H., Takeuchi,S., Yuba,S., Saijo,M., Nakatsu,Y., Murai,H., Nakatsuru,Y., Ishikawa,T., Hirota,S., Kitamura,Y., Kato,Y., Tsunoda,Y., Miyauchi,H., Horio,T., Tokunaga,T., Matsunaga,T., Nikaido,O., Nishimune,Y., Okada,Y. and Tanaka,K. (1995) High incidence of ultraviolet-B-induced or chemical carcinogen-induced skin tumors in mice lacking the xeroderma pigmentosum group-A gene. Nature, 377, 165–168. [DOI] [PubMed] [Google Scholar]
- 34.Sands A.T., Abuin,A., Sanchez,A., Conti,C.J. and Bradley,A. (1995) High susceptibility to ultraviolet-induced carcinogenesis in mice lacking Xpc. Nature, 377, 162–165. [DOI] [PubMed] [Google Scholar]
- 35.Inomata T., Itoh,S., Kariya,S., Mesaki,K., Nishioka,A., Ogawa,Y., Yoshida,S., Sonobe,H. and Ohtsuki,Y. (1999) Late pathologic changes in guinea pig kidneys irradiated with conventional fractionation and hyperfractionation. Int. J. Radiat. Oncol. Biol. Phys., 44, 171–177. [DOI] [PubMed] [Google Scholar]
- 36.Stewart F.A., Oussoren,Y. and Bartelink,H. (1989) The influence of cisplatin on the response of mouse kidneys to multifraction irradiation. Radiother. Oncol., 15, 93–102. [DOI] [PubMed] [Google Scholar]
- 37.Matsuura K., Takasugi,M., Kunifuji,Y., Horie,A. and Kuroiwa,A. (1991) Morphological effects of cadmium on proximal tubular cells in rats. Biol. Trace Elem. Res., 31, 171–182. [DOI] [PubMed] [Google Scholar]
- 38.Gu H., Marth,J.D., Orban,P.C., Mossmann,H. and Rajewsky,K. (1994) Deletion of a DNA polymerase beta gene segment in T cells using cell type-specific gene targeting. Science, 265, 103–106. [DOI] [PubMed] [Google Scholar]