

Abstract
The incredible potential for fluorescent proteins to revolutionize biology has inspired the development of a variety of design strategies to address an equally broad range of photophysical characteristics, depending on potential applications. Of these, fluorescent proteins that simultaneously exhibit high quantum yield, red-shifted emission, and wide separation between excitation and emission wavelengths (Large Stokes Shift, LSS) are rare. The pursuit of LSS systems has led to the formation of a complex, obtained from the marriage of a rationally engineered protein (human cellular retinol binding protein II, hCRBPII) and different fluorogenic molecules, capable of supporting photobase activity. The large increase in basicity upon photoexcitation leads to protonation of the fluorophore in the excited state, dramatically red-shifting its emission, leading to an LSS protein/fluorophore complex. Essential for selective photobase activity is the intimate involvement of the target protein structure and sequence that enables Excited State Proton Transfer (ESPT). The potential power and usefulness of the strategy was demonstrated in live cell imaging of human cell lines.
Graphical Abstract
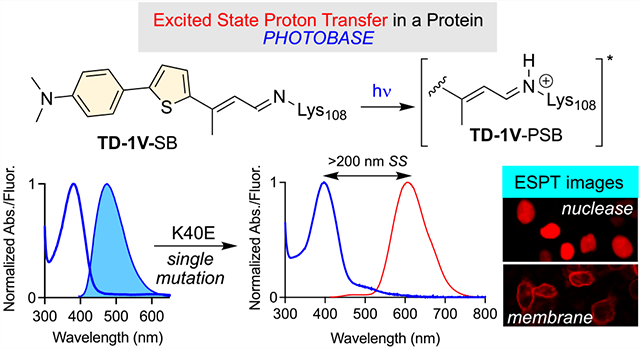
INTRODUCTION
Fluorescence microscopy has revolutionized many areas of biomedical research. These range from fundamental studies of in vivo protein–protein interactions, protein trafficking and single molecule nanoscopy to a wealth of other biological processes including whole body imaging and clinical applications in companion diagnostics such as ISH (in situ hybridization).1 The available toolkits that enable various technologies within the field include fluorescent proteins (FPs)1a,g,2 and proteins that are post-translationally modified by synthetic dyes.1e,3 Together, these FPs provide emission spectra spanning the visible spectrum from the blue to the near-infrared (NIR).1c,2d,4 The spectral diversity of fluorescent reporters enables multicolor imaging, wherein multiple target proteins and/or organelles can be visualized in a single microscopic experiment.4a,5
The evolving field of fluorescence microscopy has demanded newer, more sophisticated FPs to address deficiencies of the early generation candidates. While the ideal system might be case dependent, optimization of a set of photochemical characteristics is common to all; namely, photostability, red-shifted emission, high fluorescence quantum yield (QY), high extinction coefficient (ε), and large Stokes shift (≥100 nm). Recently, the design of FPs with “apparent” large Stokes Shift (LSS) has garnered attention.6 These proteins display a large gap between the excitation and the emission maxima. Although Stokes shift is defined as the electronic transition between the excited and ground state of the same chemical entity, LSS-FPs achieve their apparent large Stokes shift by a chemical conversion of the excited state to a new bathochromic species, with a corresponding red-shifted emission. The large Stokes shift of these FPs is advantageous for their application in fluorescence imaging, since the wide gap between the excitation and emission maxima diminishes self-absorption. This not only minimizes the inner-filter effect, but is also beneficial in multicolor imaging experiments, wherein potential cross-talk between multiple emitters is reduced. If properly designed, a LSS emitter can be excited in the visible range of the electromagnetic spectrum while it emits in the near-infrared region. This additional feature further benefits imaging experiments by reducing light scattering, increasing signal depth in tissues, and eliminating autofluorescence. Although large Stokes shift and high fluorescence efficiency are not mutually exclusive, they are generally not encountered together in synthetic probes.
Fluorochromes with certain features are known to have increased Stokes shifts. Examples are molecules that can undergo intramolecular charge transfer (ICT) upon excitation,7 form excimers/exciplexes,8 or those that are affected by the Stark effect.9 The application of these molecules as fluorescence imaging probes, however, is challenging in the practical sense—nonspecific targeting of ICT emitters leads to broad emission in the biological milieu, while incorporating an imaging trigger for the exciplex can be challenging. Alternatively, molecules that undergo excited state transformations circumvent these limitations; the absorption of species A can lead to a transformation of the excited state to form species B, the consequent emission of which does not constitute the same electronic transition; thus a large “apparent” Stokes shift is achieved (Figure 1a). This design strategy gives more flexibility to the selection of fluorochromes as well as an easier incorporation into host–guest systems such as a protein–ligand complex that mimics fluorescent proteins or other post-translationally modified tags. Perceivably, this will allow for an easier imaging application, welcomed by a broader community of cell biologists.
Figure 1.
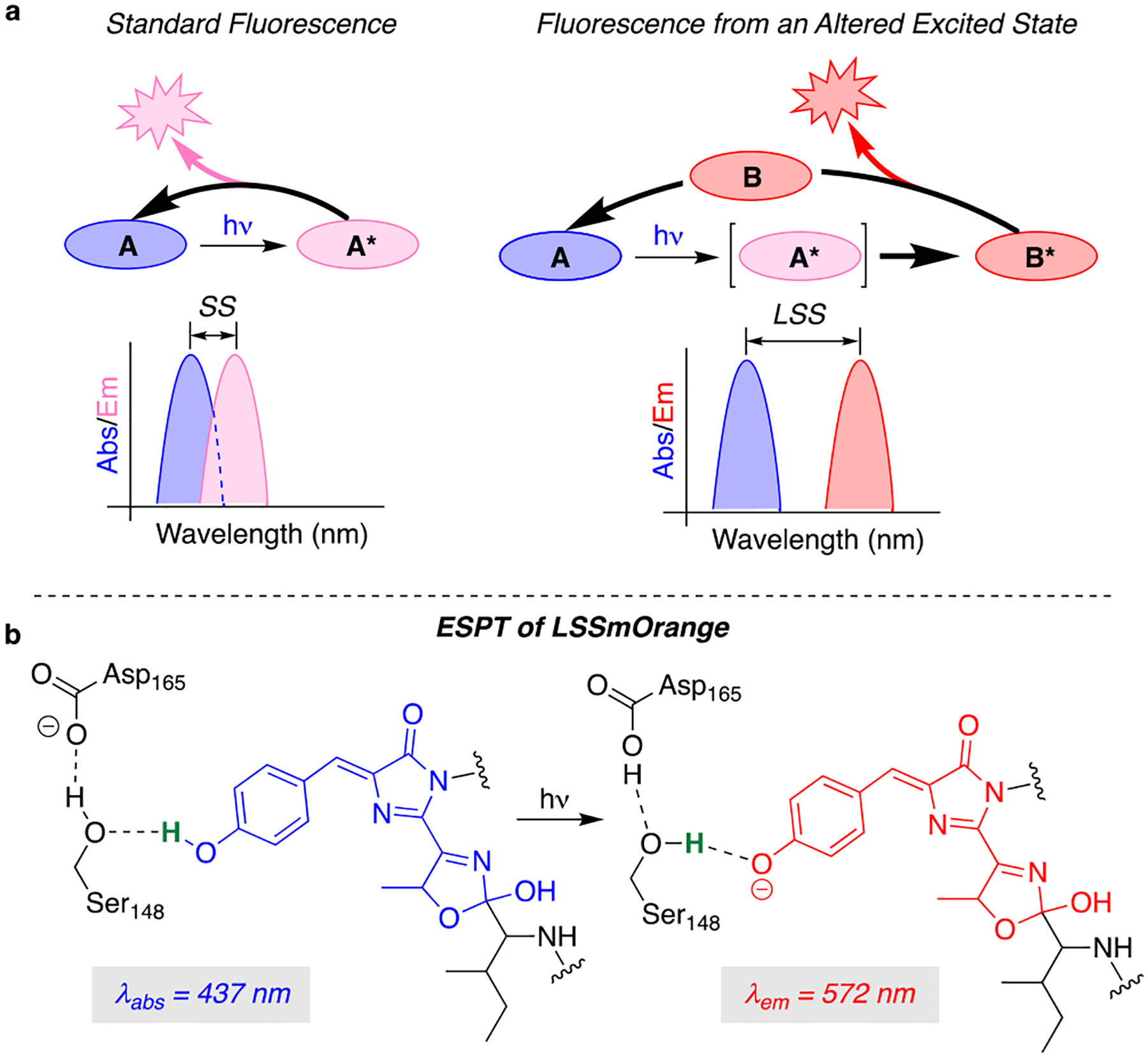
(a) Model for a large Stoke shift (LSS) system, as compared to standard fluorescence. Conversion of the excited state species A* to a bathochromically distinct molecule B* leads to the emission from a red state, resulting in a large Stoke shift; (b) The ESPT mechanism for LSSmOrange as a photoacid, excited at 437 nm, with emission observed at 572 nm.
An example of the latter strategy is the case of excited state proton transfer (ESPT). This strategy has been successfully implemented in the GFP lineage, as well as for small synthetic fluorescent molecules. The former is represented by a series of LSS-FPs such as LSSmOrange, LSSmKates, mKeima, CyOFP1, and mBeRFP, to name a few.10 Operationally, the intrinsic fluorophores for all these FPs, the core p-HBI, remains neutral in the ground state, while nearby amino acid residues are engineered to engage in hydrogen bonding with the phenolic hydroxyl group (Figure 1b). Upon light irradiation, the acidity of the phenolic species in the excited state increases by orders of magnitude (lowering the pKa), enabling the proximal amino acid residue(s) to initiate a proton abstraction, which may or may not occur through a proton relay. The resultant phenolate, with its enhanced conjugation, relaxes through a red-shifted emission that leads to an apparent large Stokes shift. Critical for its success is the placement of a hydrogen bond acceptor in proximity to the phenolic hydroxyl to facilitate the ESPT. Reports have shown Asp, Glu, Asn, and Lys are good hydrogen bond acceptors for this purpose.10
Small molecules that possess an appropriate hydrogen bonding donor and acceptor, oriented in a suitable position and distance within the same molecular framework, can also yield a large apparent Stokes shift through internal ESPT, or ESiPT (Excited State intramolecular Proton Transfer).7a,11 The first reported example is salicylic acid, described by Weller in the 1950s.12 Since the original report, a variety of molecular families capable of the ESiPT process have been reported. Common hydrogen bond acceptors in these systems include benzimidazoles, benzoxazoles, benzothiazoles, quinolines, flavones, chromenes, and anthraquinones.13 To furnish the ESiPT process, a carbonyl or other relevant heteroatom hydrogen bond acceptor is proximal to a phenol moiety within the same molecule. A number of ESiPT capable molecules are constituent elements for various chemosensors and chemidosimeters to probe biologically or environmentally important chemical species.11a
To date, all reported ESPT-capable FPs and ESiPT-capable small molecules function as photoacids—molecules that experience decreased pKa and lose a proton in the excited state. In fact, most reported ESPT small molecules are photoacids as well, with only a few known photobases.14 In contrast to photoacids, photobases are able to extract a proton in their excited states. In our own work, we have previously reported a fluorene-based super photobase, FR0-SB, which is capable of deprotonating a primary or secondary alcohol, exhibiting a ΔpKa greater than 14 units. The protonation of the photobase leads to an Intramolecular Charge Transfer (ICT) system, red-shifted in the excited state, leading to a > 250 nm apparent Stokes shift (Figure 2a).15 The latter discovery piqued our interest for the possibility of incorporating a fluorophoric photobase into a rationally engineered protein carrier, with the goal of generating a photoactivatable LSS-FP. The plan would require appropriate positioning of amino acid side chains to facilitate the required proton source for the photobase. To the best of our knowledge, this design strategy, which utilizes photobases instead of photoacids, is unique and could have advantages in generating highly conjugated polar ICT systems in the excited state that are well red-shifted in emission, with high QY.
Figure 2.
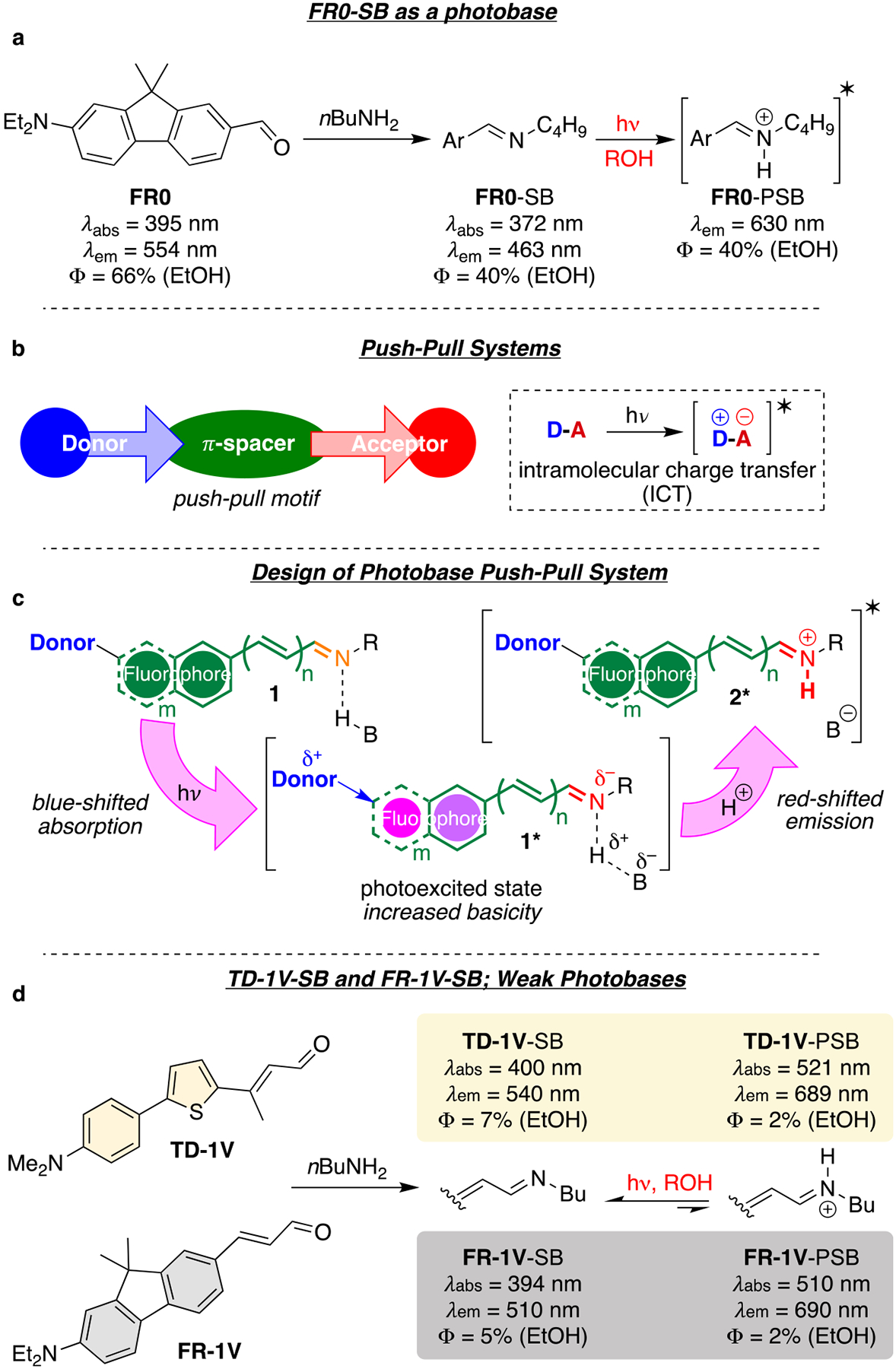
(a) The Schiff base of FR0 (FR0-SB) is a strong photobase, emitting as FR0 protonated Schiff base (FR0-PSB) in the excited state; (b) photoexcitation of push–pull systems can lead to dramatic redshift absorptions as a result of internal charge transfer (ICT); (c) illustrated is the design principle for a photobase capable of ICT. The aromatic spacers behave as “circuit breakers”, which are triggered open by photoexcitation. The charge redistribution increases the basicity of the imine, leading to a red-shifted ICT system; (d) the imine of TD-1V (TD-1V-SB) and FR-1V (FR-1V-SB), are weak photobases ideal for the proposed studies.
Herein, we demonstrate the engineering of a small protein receptor, which forms an imine through the reaction of specifically designed aldehydic ligands with an active site lysine residue. Photoexcitation of the protein/fluorophore complex leads to ESPT, presumably as a result of the protonation of the imine nitrogen atom via a nearby residue, judiciously placed to deliver the proton. The subsequent generation of an iminium in the excited state, evident from the apparent large Stokes shift in emission, highlights the photobasic ability of the protein/fluorophore complex. The following describes the design of the protein binding pocket, as well as the structural requirements of the chromophore, to yield a protein/fluorophore complex that exhibits a large apparent Stokes shift in excess of 200 nm, and fluorescence quantum efficiencies >70%. To the best of our knowledge this is the first example of a photobase active protein system. The photophysical characteristics of an LSS fluorescent protein complex would lay the groundwork for a new protein-labeling paradigm for imaging and detection.
RESULTS AND DISCUSSION
General Consideration for an ESPT-Capable Chromophore.
We envisioned that the photoexcitation of an appropriately engineered protein/fluorophore complex could lead to an ICT species. Photobasicity would result from localizing electron density on the electron-withdrawing (acceptor) moiety in the excited state. With an appropriately long excited state lifetime, the increased electron density, and thus increased basicity, could result in protonation of the acceptor. The protonation event would yield a red-shifted fluorophore, hence an LSS system. As depicted in Figure 2b, an ICT system is composed of an electron donor, a π-spacer, and an electron acceptor. We opted for the scenario shown in Figure 2c, where the effective electron acceptor (the iminium) is not present in the ground state. In an operative system, photoexcitation of the protein complex 1 will lead to an increase of the imine’s basicity (1*) in the excited state, and the subsequent protonation would generate an excited state iminium (2*), producing the requisite electron acceptor for a functional ICT system.
On the basis of our prior protein engineering work on rhodopsin protein mimics, we opted for an aldehydic molecule that would form the requisite imine upon reaction with an active site lysine residue in the protein host.16 Our work with the super photobase FR0-SB (vide supra) provided the structural requirements for generating an ICT system; an electron donor appendage (diethylamino group), a π-spacer (fluorene), and an electron acceptor unit (imine). Yet, FR0-SB was not considered as a promising candidate because of its strong activity as an ESPT capable fluorophore, which we deemed detrimental in achieving selectivity and low background. Thus, we opted for an imine system that exhibited weak ESPT in protic solvents. This may seem counterintuitive; however, it is imperative that the iminium is generated in the excited state, and only in the binding cavity of the engineered target protein; namely, the protein host should play a critical role in the proton transfer event. This ensures that nonspecific imine formation in the biological milieu is inconsequential, since ESPT is only possible in the designed protein environment. In this manner, it should be possible to achieve selectivity, as emission of the iminium is only observed in the target protein.
Another route to heighten selectivity is to accelerate protein/fluorophore maturation kinetics relative to nonspecific imine formations. A properly oriented protic amino acid residue may not only facilitate ESPT to the imine, but could also accelerate imine formation through hydrogen bonding with the aldehydic oxygen. In fact, we have previously shown that the incorporation of a single acidic residue can lead to >26 fold rate enhancement in imine formation.16c The rapid imine formation with the protein target differentiates it from nonspecific reactions in the biological milieu, such that the fluorescence from the target protein dominates, hence resulting in negligible background. Finally, the ability to control the pKa of the resultant imine in the binding pocket of the target protein is key, since ESPT is impossible if the imine is already protonated in the ground state.
In line with the structural requisite for an ESPT-capable ICT dye, we selected a panel of fluorophores that contained a push–pull scaffold for ligand/protein complex engineering. These included a Dapoxyl analog with red-shifted emission and high quantum yield (TD-1 V, Thio-Dapoxyl with 1 vinyl appendage, Figure 2d),17 and derivatives of FR0 with reduced photobasic ability (see below for structures, and Table S6 of the Supporting Information, SI, for photochemical details). Early evaluation focused our attention on TD-1 V, as well as FR-1 V, a homologue of FR0, both of which demonstrated spectroscopic characteristics that were on par with those described above, i.e., fluorophoric molecules capable of forming an ICT system upon Schiff base formation, yet with depressed photobase activity in protic solvents. Comparing the emission of FR0-SB with that of FR-1 V-SB in ethanol, it is quite evident that the former leads largely to the emission from the iminium via ESPT, while the latter displays only a small portion of fluorescence resulting from ESPT (Figure S1, see trace in EtOH, Table S7). A further check on TD-1 V-SB shows an even smaller portion of its fluorescence arising from ESPT (see Figure S1, trace in EtOH). The project now required a protein capable of binding the ligands as an imine with an active site lysine residue. Further engineering of the protein’s active-site cavity would be required to not only ensure a low pKa for the imine (to avoid protonation in the ground state), but also judiciously place amino acids relative to the position of the fluorophore to promote high levels of ESPT within the binding pocket.
Protein Reengineering.
TD-1V was chosen as the starting ligand for protein reengineering. As described in detail below, our efforts focused on the reengineering of human Cellular Retinol Binding Protein II (hCRBPII), a small (136 aa), cytosolic protein with high expression yields and excellent structural resiliency, as a host for the fluorogenic dyes.16a,18 Our prior work in this area has provided the required insight for reengineering this family of proteins.16a–d,19 Most critical for the success of any protein engineering effort, is the robustness of the protein structure to numerous mutations. In our studies with over 500 mutants, hCRBPII has proven itself as a durable and structurally robust protein.
Introduction of a Glutamic Acid Residue as Proton Donor.
Investigations to find an appropriate protein host for TD-1V began with the Q108K:K40L double mutant of hCRBPII. Prior studies have shown that these two mutations are critical for binding an aldehydic chromophore,16a albeit requiring optimization for each ligand family. Q108K serves as the active-site lysine for reaction with the aldehyde, while the K40L mutation presumably removes interactions between Q108K and Lys40 that is detrimental to imine formation. Addition of TD-1V to Q108K:K40L-hCRBPII led to the formation of the iminium, although the protein/fluorophore complex was not stable and its UV–vis spectrum was broad and featureless (Figure S2). After extensive structure-guided screening (see SI and Figure S3 for details), we arrived at the Q108K:K40L:T53A:R58L:Q38F:Q4F-hCRBPII hexamutant (M1), which in part had followed parallel reengineering efforts utilizing retinal as a ligand.16a,20 The three hydrophobic mutations (T53A, R58L, and Q38F) were introduced to increase binding affinity and enhance the rate of pigment maturation. The Q4F mutation typically increases soluble protein expression, but more importantly, aids in suppressing the pKa of the iminium, ensuring that the ground state system is populated solely as the imine.
M1 binds TD-1V as a SB in PBS buffer (pH = 7.2), as evident by its blue-shifted absorption (λabs = 380 nm, pKa = 5.1) Excitation at 380 nm led to the predominant emission of the imine at 474 nm (Figure 3a). Incubation of TD-1V/M1 complex in acidified PBS buffer (pH 3.3) yields the protonated SB, with absorption at 621 nm. Excitation at the latter wavelength leads to emission at 692 nm. The absorption and emission values for the ground state SB and PSB for M1 define the anticipated spectroscopic regions for each state. We surmised that a nearby acidic residue could provide the necessary proton for a weak photobase, and thus, 19 residues proximal to the putative imine nitrogen atom were individually mutated to Glu (Q4, W8, Q38, K40, I42, F49, T51, T53, V62, F64, V86, L93, C95, W106, L115, L117, L119, Q128, and F130, Figure 3b). Residues as far as 10 Å from the imine were examined, owing to the possibility that ESPT could occur as a result of remote hydrogen bonded water networks (through proton relays). Unfortunately, a number of the mutants led to insoluble protein expression. Nonetheless, soluble protein was obtained after placement of Glu at nine positions (K40, T51, V62, I42, L117, T53, L119, Q38, and Q128) in the parent mutant M1.
Figure 3.
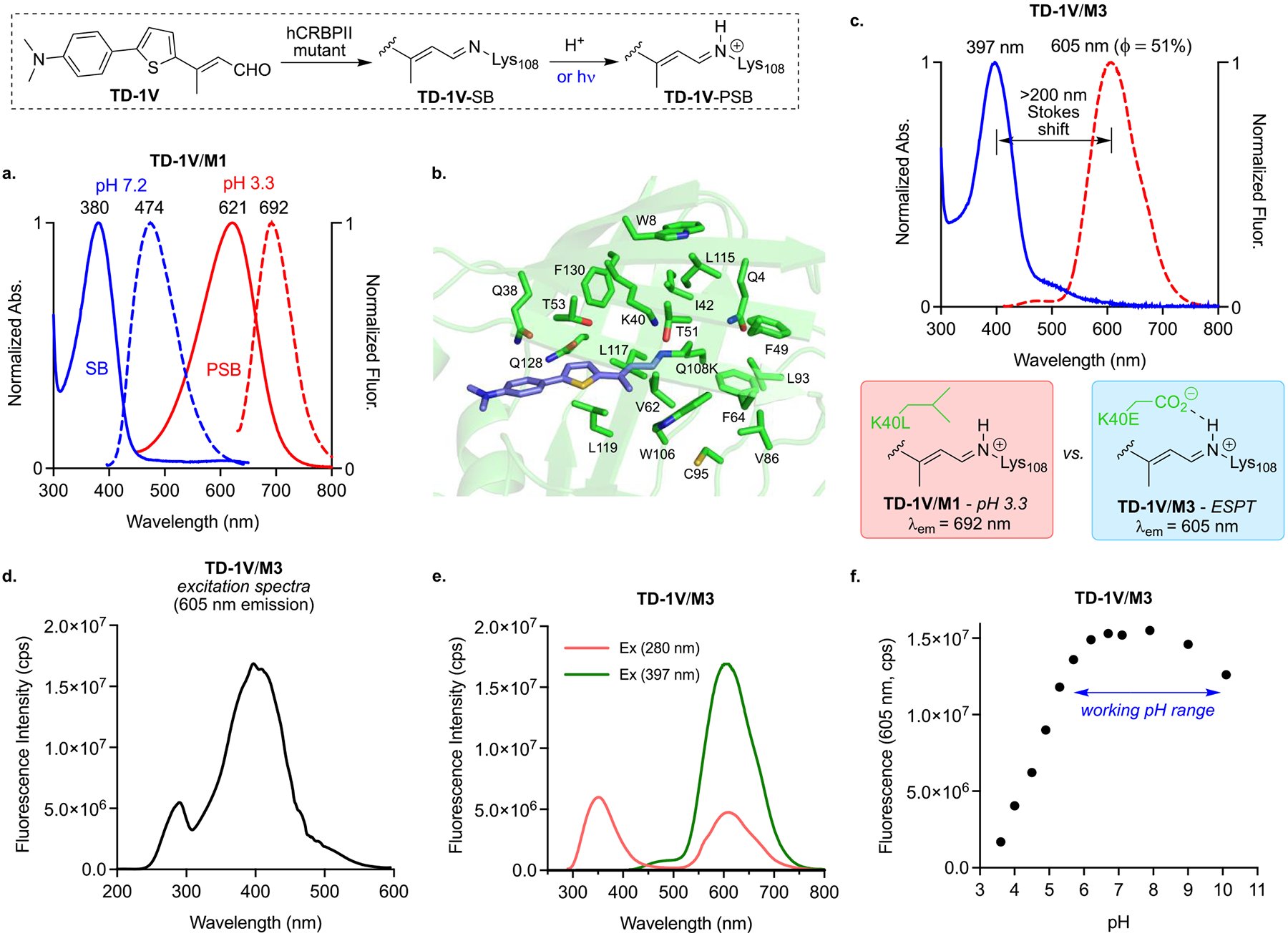
(a) UV–vis and fluorescence spectra (380 nm excitation) of TD-1V/M1 complex at pH 7.2 leads to SB emission at 474 nm, while the acidified sample (pH 3.3) is red-shifted to 621 nm (PSB form), with emission at 692 nm; (b) energy minimized structure of TD-1V/Q108K-hCRBPII complex with selected residues within 10 Å of the imine nitrogen atom highlighted; (c) UV–vis and fluorescence spectra (397 nm excitation) of TD-1V/M3 complex, exhibiting ESPT emission with >200 nm Stokes shift. Comparison below figure highlights the reason why the ESPT generated iminium is blue-shifted relative to the iminium generated via acidification of the media; (d) excitation spectrum of the TD-1V/M3 complex (emission observed at 605 nm); (e) emission spectra of TD-1V/M3 complex, excited at 280 and 397 nm; and (f) emission of the TD-1V/M3 complex incubated at various pH values, exhibiting a pH working range (~5–10) of the pigment with high emission output.
The absorption spectra of TD-1V/hCRBPII mutant complexes can have two maxima, depending on the pKa of its corresponding imine. The engineered sequences favored mutants with low pKa, such that the ground state species could be protonated in the excited state. The excitation of the SB in these complexes can lead to two emission maxima, corresponding to fluorescence from the neutral (~480 nm) or the protonated imine (>600 nm) in the excited state as a result of ESPT. Figure 3c depicts the UV–vis spectrum of TD-1V/M3 complex with absorption maximum at 397 nm, corresponding to its SB form (pKa 5.2, see Figure S4 for pKa measurements). Excitation of the complex at 397 nm leads to fluorescence centered at 605 nm, red-shifted far beyond the anticipated region for emission of the SB. Apparently, Glu40 is incapable of protonating the imine in the ground state to any appreciable extent, yet, upon photoexcitation, the enhanced basicity of the imine triggers proton transfer to yield the iminium, which relaxes partly through fluorescence, followed by proton loss.
The blue-shifted emission of the TD-1V/M3 complex (605 nm) relative to the protonated TD-1V/M1 complex (692 nm) warrants explanation. The change in emission reflects the interaction of the Glu40 anion, generated after proton transfer, leading to a charge stabilized ion-pair that localizes the cationic charge on the nitrogen atom. This reduces the conjugation of the positive charge along the π-system, and consequently yields the blue-shifted emission. This speculation arises from our prior work that demonstrated the same phenomena in absorption regulation of rhodopsin protein mimics, where a salt bridge interaction at the iminium led to blue-shifted absorptions.16a In comparison, the TD-1V/M1 complex, acidified to a pH of 3.3, leads to the same iminium, yet lacking the anionic interaction that would localize charge on the nitrogen atom. The observed red-shift is due to the greater delocalization of the positive charge along the polyene.
Emission data, obtained upon excitation of the SB at its absorption maximum for the nine mutants discussed above, are listed in Table 1. We defined ΦESPT as the fraction of the total fluorescence that originates from the excited state iminium. Briefly, the fluorescence spectrum was deconvoluted to give separate traces representing the emission from the neutral and protonated imine, respectively. The area under each deconvoluted peak was integrated to provide the relative percentage of total fluorescence quantum yield. The value derived from the iminium is defined as ΦESPT (see Figure S5 for a representative example for spectral deconvolution and calculation of ΦESPT). Consequently, larger ΦESPT values denote a more facile ESPT process.
Table 1.
Spectroscopic Properties TD-1V/hCRBPII Mutant Complexes
| mutant | hCRBPII mutanta | λ abs | λ em (SB) | λ em (PSB) | Φb | ΦESPT |
|---|---|---|---|---|---|---|
| M1 | Q108 K:K40L:T53A:R58L:Q38F:Q4F | 379 | 474 | n.d. | 0.03 | <0.02 |
| M2 | Q108 K:K40L:T53A:R58L:Q38E:Q4F | 389 | 485 | n.d. | 0.05 | <0.02 |
| M3 | Q108 K:K40E:T53A:R58L:Q38F:Q4F | 397 | n.d. | 605 | 0.51 | >0.99 |
| M4 | Q108 K:K40L:T53A:R58L:Q38F:Q4F:I42E | 406 | n.d. | 603 | 0.41 | 0.97 |
| M5 | Q108 K:K40L:T53A:R58L:Q38F:Q4F:T51E | 399 | n.d. | 602 | 0.12 | 0.86 |
| M6 | Q108 K:K40L:T53E:R58L:Q38F:Q4F | 387 | 480 | n.d. | 0.04 | <0.02 |
| M7 | Q108 K:K40L:T53A:R58L:Q38F:Q4F:V62E | 386 | 474 | 598 | 0.08 | 0.27 |
| M8 | Q108 K:K40L:T53A:R58L:Q38F:Q4F:L117E | 391 | 486 | 653 | 0.13 | 0.73 |
| M9 | Q108 K:K40L:T53A:R58L:Q38F:Q4F:L119E | 384 | 479 | n.d. | 0.05 | 0.13 |
| M10 | Q108 K:K40L:T53A:R58L:Q38F:Q4F:Q128E | 385 | 473 | n.d. | 0.05 | <0.02 |
| M11 | Q108 K:K40E:T53A:R58W:Q38F:Q4F:Y19W | 392 | n.d. | 621 | 0.43 | >0.99 |
| M12 | Q108 K:K40D:T53A:R58L:Q38F:Q4F | 393 | 481 | 639 | 0.34 | 0.81 |
20 μM protein and 0.5 equiv TD-1V at pH 7.2.
Absolute quantum yield was measured on a Quantaurus-QY. Not detected (n.d.).
High ΦESPT values were obtained for hCRBPII/TD-1V complexes that possess either K40E, I42E, or T51E mutations (Table 1, M3, M4, and M5), highlighting efficient ESPT of the imine (ΦESPT ≥ 0.86). Among the latter mutants, high fluorescence quantum yields were observed for M3 and M4 (0.51 and 0.41, respectively), while a pronounced reduction was observed for M5 (0.12). On the basis of numerous crystal structures of this series of proteins in the PDB, the putative positions of all three residues are in close proximity (~5 Å) to the imine nitrogen atom. Variants with V62E and L117E (M7 and M8, respectively) lead to a significantly lower ΦESPT, with emission that results from both the chromophore’s SB and PSB forms in the excited state; the remaining mutants led to typical SB emission as the major fluorescing event.
The excitation spectrum of the TD-1V/M3 complex, collected at 605 nm, verifies that the observed fluorescence originates from the SB (Figure 3d). Interestingly, the excitation spectrum also shows an absorption at ~280 nm, which presumably belongs to the Trp residues in hCRBPII. As a confirmation, excitation of the protein complex at 280 nm leads to peaks at both the expected SB and ESPT emissions of the bound chromophore. This observation suggests the existence of an energy transfer process from tryptophan to the bound TD-1V-SB (Figure 3e).
ESPT for the TD-1VM3 complex is observed over a wide range of pH values, as shown in Figure 3f. Fluorescence emission is maximal at pH 6–9 and drops at more acidic or more basic pH values. At pH 10, the blue-shifted peak (emission of the SB) appears at <500 nm, presumably as a result of the deprotonation of the glutamic acid at position 40, and/or a less active bound water molecule as a proton donor. One would assume that a system with a stronger ICT would yield a more basic excited state capable of ESPT. At pH values below 5, the imine is protonated, and thus incapable of ESPT. If necessary, the working window can be adjusted based on the pKa of the ground state iminium. We have previously shown that the pKa can be adjusted through sequence alterations of the binding pocket in a rational manner.19
Structural Determinants of Optimal ESPT Variants.
Sequence comparison of M1 (not capable of ESPT) with M3 (full ESPT) leads to the conclusion that the glutamic acid at position 40 must be responsible for providing the proton to the photobase upon photoactivation. Unfortunately, crystallization of the TD-1V/M3 complex was not fruitful. Previous investigations with retinal as ligand had shown that the inclusion of two additional Trp residues at positions 19 and 58 results in a more ordered chromophore in the binding pocket. This inspired the Q108K:K40E:T53A:R58W:Q38F:Q4-F:Y19W (M11) variant, which gave similar ESPT spectroscopic results (Table 1) and readily produced well-diffracting crystals suitable for structural analysis. The structure shows that the Glu40 carboxylate oxygen atoms are ~5 Å from the imine nitrogen atom (Figure 4a), too far for direct interaction. A direct imine carboxylate interaction would likely lead to an iminium at physiological pH, which would be incompetent for ESPT. The conformation of Glu40 is defined by water-mediated interactions with the side chain of Thr51 and the mainchain carbonyl of Tyr60 (Figure 4b). We surmised that both Thr51 and Tyr60 act as hydrogen bond acceptors to the water molecule, since Thr51 is also hydrogen-bonded to its own mainchain carbonyl. This would suggest that Glu40 is protonated and participates as a hydrogen bond donor for the water molecule. More importantly, the ground state structure corroborates the lack of any observable PSB at pH 7.2, since Glu40 maintains its proton as a result of the latter interactions In fact, the same interactions could also stabilize Glu40 as it donates a proton during the ESPT process. The intervening region between the SB and the carboxylate is surrounded by hydrophobic residues, apparently increasing the Glu40 pKa and excluding water from the region. Though this discussion assumes ESPT emanates from the fluorophore conformation observed crystallographically, other ESPT-competent fluorophore conformations cannot be ruled out.
Figure 4.
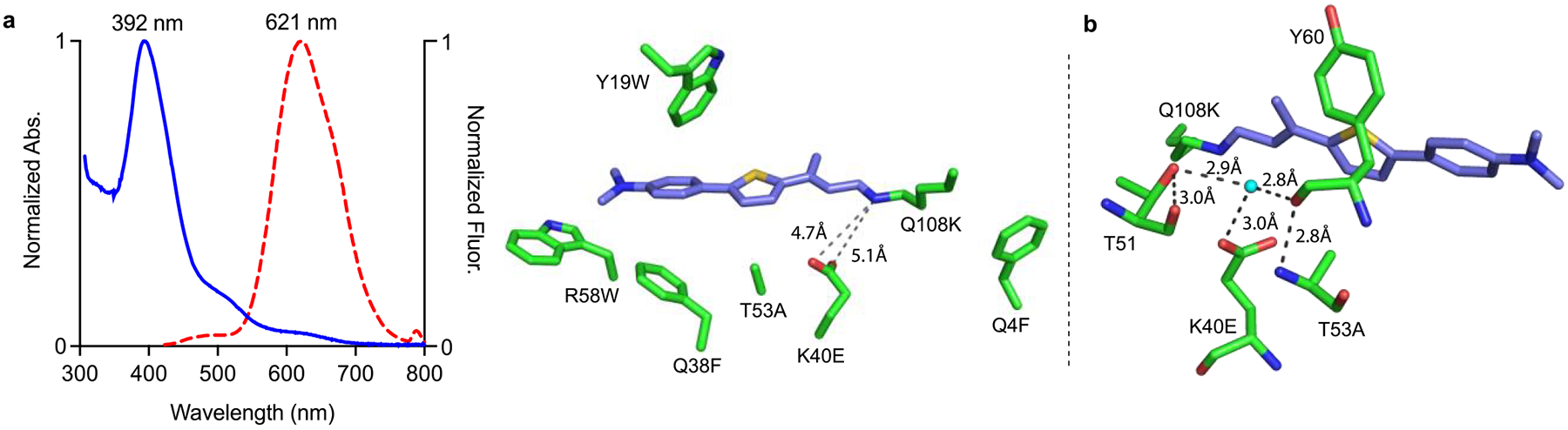
(a) UV–vis, fluorescence (>99% ϕESPT), and crystal structure of TD-1V/M11 complex. The imine nitrogen atom is ~5 Å from Glu40, which is presumably the proton source for the formation of the iminium upon photoexcitation; (b) highlighted is the water molecule that interacts with K40E and the main chain carbonyl of Y60 and T51. These interactions lead to the proposition that the K40E side chain is protonated in the ground state, poised to deliver the proton to the imine upon excitation.
The analogous Asp40 mutant also supports protonation of the excited state species, but with reduced efficiency (~80% transfer) and a higher imine pKa (6.4 as compared to 5.2 for Glu40). The smaller SB population leads to less available imine for ESPT. At first glance, it seems counterintuitive that the shorter Asp side chain would lead to a higher pKa of the bound TD-1V-PSB, as the longer Glu side chain should be closer to the imine, and thus enjoy enhanced electrostatic interactions. This piqued our interest in factors that not only affect the pKa of the SB, but also could lead to more facile ESPT. Crystallographic analysis of the K40D variant indicates that the Asp40 side chain maintains a similar 5 Å distance to the imine, but does not make the water mediated interaction with Thr51 and Tyr60 seen with Glu40 (Figure S6). In contrast to Glu40, Asp40 can donate its proton to the imine, stabilizing the ensuing charge through ionic interactions, which leads to a higher pKa. The pKa’s of the carboxylic acid and the iminium are coupled due to their proximity, thus the pKa of the iminium is governed by its interactions with the carboxylate side chain. The reduced ESPT efficiency may be related to the absence of interactions that would stabilize the position of Asp40 (Figure S6). Furthermore, the K40D mutant M12 produced a less stable protein product, and was prone to dimerization. Together these results are illustrative of how intricate water-mediated interactions lead to the low iminium pKa and high ESPT efficiency required to optimize an LSS fluorescent protein.
Evaluation of Other Ionizable Residues.
Because introduction of Glu or Asp at residue 40 leads to ESPT, we hypothesized that other residues may be able to support the ESPT process as well. Hence, as listed in Table 2, Glu40 was replaced in M3 with other amino acids with proton-bearing side chains. Neither cysteine (M13) nor threonine (M14) led to ESPT upon excitation of the SB at physiological pH (Figure S7), and produced spectra similar to that observed for M1 (K40L, see Figure 3a). However, substitution of basic residues histidine (M15), lysine (M16), and arginine (M17) in position 40 resulted in partial ESPT (Figure S8). Presumably, they either directly protonate the imine in the excited state, or they polarize a hydrogen bonded water molecule, which would function as the proton donor. Nonetheless, their Φfl were much lower than that observed with K40E (M3). A potential explanation is that the carboxylate counteranion, produced as a result of ESPT from Glu, has a stronger interaction with the iminium as compared to the iminium/amine pair, which results from ESPT with His, Lys or Arg. The ionic interaction may lead to chromophore rigidification and a higher quantum yield. Similar to the basic residues, glutamine (M18) and tyrosine (M19) showed dual fluorescence bands (blue-shifted emission from the SB and red-shifted emission from the protonated excited state of the SB) with suppressed quantum yields (Figure S9).
Table 2.
ESPT of M3 Mutants at Position 40
| mutant | residue 40a | λ abs | λ em (SB) | λ em (PSB) | Φb | ΦESPT |
|---|---|---|---|---|---|---|
| M1 | K40L | 379 | 474 | n.d. | 0.03 | <0.02 |
| M3 | K40E | 397 | n.d. | 605 | 0.51 | >0.99 |
| M13 | K40C | 383 | 480 | n.d. | 0.07 | <0.02 |
| M14 | K40T | 386 | 487 | n.d. | 0.13 | <0.02 |
| M15 | K40H | 393 | 477 | 642 | 0.09 | 0.50 |
| M16 | K40 | 389 | 491 | 661 | 0.08 | 0.40 |
| M17 c | K40R | 392 | 492 | 653 | - | 0.43 |
| M18 | K40Q | 388 | 488 | 663 | 0.11 | 0.11 |
| M19 | K40Y | 393 | 495 | 674 | 0.04 | 0.14 |
20 μM protein and 0.5 equiv TD-1V at pH 7.2.
Absolute quantum yield was measured on a Quantaurus-QY.
The QY of M17 could not be obtained due to its inherent instability. Not detected (n.d.).
Expansion of the Ligand Scope for ESPT.
Inspired by the ESPT results obtained with TD-1V, we explored the efficiency of other ligands (FR0, FR-1V, FR-2V, and FR-Th) to elicit ESPT when complexed with M3 (Figure 5). The characteristic ESPT long wavelength emissions were observed to various extent with FR-1V, FR-2V, and FR-Th, although the FR-Th/M3 complex was not ideal, with low ΦESPT (0.14). Interestingly, the FR0/M3 complex was not ESPT active, yet it exhibits full ESPT with an alternate mutant that contains K40D and V62E as potential proton donors (see Figure S10). This highlights the obligatory role of the protein host in providing a path for proton transfer, which also depends on the structure and trajectory of the bound molecule in the active site. Similar to TD-1V, the FR-1V/M3 complex exhibits complete ESPT- with its long wavelength emission band centered at 595 nm (ΦESPT > 0.99), along with a surprisingly effective fluorescence QY (72%), and an apparent Stokes shift of over 200 nm (Figure 6a). Its excitation spectrum also shows double maxima at 280 and 595 nm, respectively, as observed with TD-1V/M3 (see Figure S11). This spectroscopic resemblance may be attributed to the similarity between FR-1V and TD-1V in terms of their molecular geometries, conformations within the protein cavity, and electronic structures. Not surprisingly, FR-2V, with its increased conjugation, led to a more red-shifted absorption for its SB and its corresponding ESPT emission when complexed with M3 (405 and 663 nm, respectively). Nonetheless, ESPT emission suffered somewhat (ΦESPT = 0.82), likely as a result of its increased flexibility.
Figure 5.
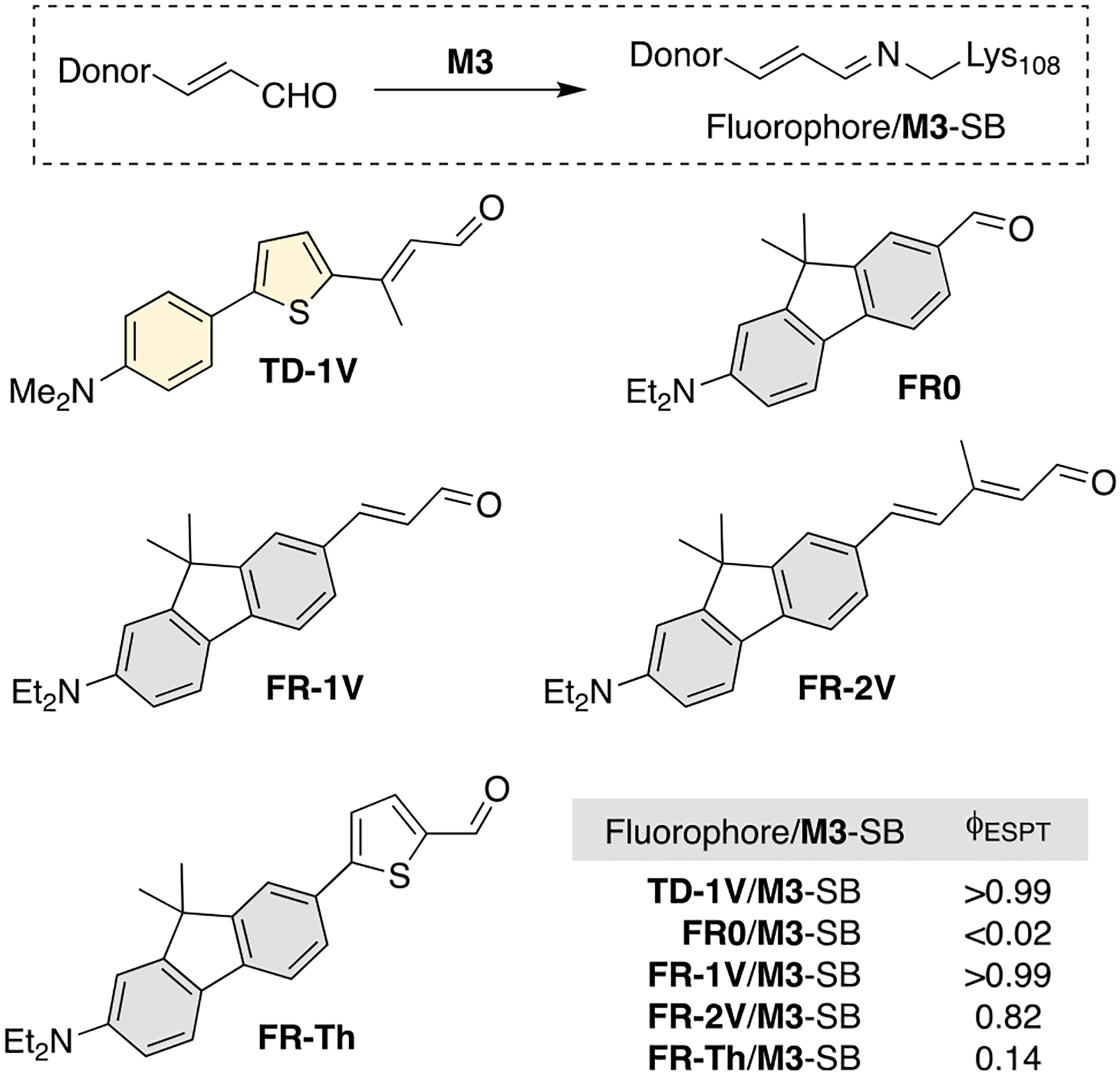
Structure of various ICT-capable fluorophores complexed with M3 as a Schiff base (SB). TD-1V and FR-1V stand out with high ϕESPT (>0.99).
Figure 6.
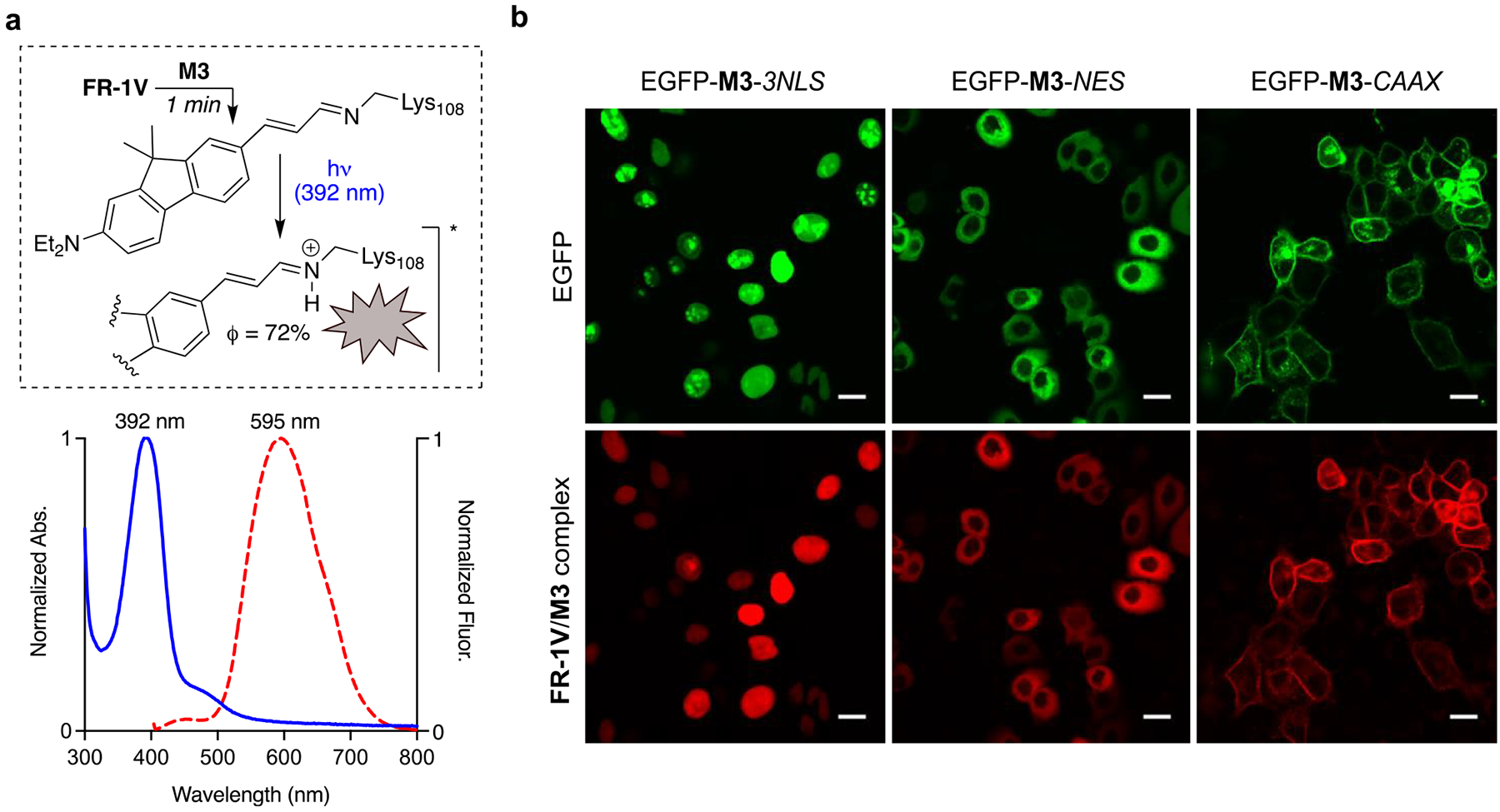
(a) FR-1V complexed with M3 yields a SB in ground state (λmax 392 nm, pKa = 5.2). ESPT of the complex leads to emission at 595 nm; (b) compartmentalized FR-1V/M3 imaging in live HeLa cells. NLS = nuclear localization sequence. NES = nuclear export sequence. CAAX = prenylation tag. Cells were stained with 500 nM FR-1V and incubated at 37 °C for 1 min. Cells were washed 3 times with DPBS before imaging. Scale bar, 10 μm.
Though structures could not be obtained for any of the FR fluorophores bound to M3, the FR-1V/M15 complex, differing from M3 only at position 40 (K40H versus K40E in M3), produced well diffracting crystals. The latter complex exhibits the anticipated ESPT behavior, albeit less robustly, with the most significant issue being the prevalence of the iminium state at pH 7.2 (Figure S12). A possible explanation for the spectral properties is inspired by the structure of the complex (Figure S13). Two distinct conformations of K40H are seen in the four molecules that comprise the asymmetric unit. In two chains (A and B), K40H makes a direct interaction with the iminium nitrogen atom (3.0 Å), while in the other two chains (C and D), K40H rotates away, and instead makes a water mediated hydrogen bonded network with Gln128 and Arg35. Likely, K40H’s direct interaction stabilizes the PSB (increased pKa), explaining the significant amount of the protonated imine. Alternatively, in chains C and D, where the K40H is rotated away from the imine, the pKa is suppressed, yet its proximity to the His residue accommodates ESPT. This is similar to that seen for M11-bound TD-1V, where Glu40 is nearby, but not directly interacting with the imine of the SB.
Live-Cell Imaging.
Our next aim was to demonstrate that the utilization of the photobase-initiated ESPT process can complement current photoacid–based LSS design approaches. We chose the hCRBPII hexamutant M3 for proof-of-principle live-cell imaging. As discussed earlier, it is critical to have a rapid binding event between the chosen aldehydic ligand and the protein host, such that its formation outcompetes other possible nonspecific binding within the biological milieu. This effectively minimizes background fluorescence from unintended sources. Initial screens with ligands was performed with a mixed order binding kinetic measurement. M3 (20 μM) was incubated with selected ligands (10 μM) at room temperature. TD-1V and FR-1V exhibited fast binding kinetics with half-time of 1.5 min and 0.16 s, respectively (see Figure S14). We further measured the second order binding rate constant of FR-1V with M3 (1.38 × 106 M−1 s−1, see Figure S15 for detail). Cellular imaging with far-red/near-infrared emitting pigments is preferred because of their cleaner optical window, deeper penetration depth, and minimal fluorescence background. With the latter consideration in mind, along its superior photochemical characteristics, and rapid pigment maturity (Figure 6a), FR-1V was chosen for the proof-of-concept cell imaging assays.
Three different C-terminus appended constructs of M3 cloned into a pFlag-CMV2 vector were prepared: (1) 3 × NLS tag (three sequential nuclear localizing sequences); (2) NES (nuclear export signal) tag; and (3) CAAX motif, for plasma membrane localization. All constructs contained an EGFP gene inserted at the N-terminus of M3 to serve, not only as a control for transfection, but also as an indicator for colocalization with the ESPT fluorophore. HeLa cells transfected with the constructs described above were incubated with FR-1V (500 nM) for 1 min at 37 °C, washed 3 times to remove the dye containing media, and immediately subjected to imaging. The field of view was excited with a 405 nm LED laser, and the emission resulting from ESPT was collected through a 615 nm long-pass filter, non-ESPT based emitters. Even though the 615 nm long-pass filter (LP615 channel) essentially filters half of the ESPT emission band, the resultant images were bright, enabling fine structural visualization of labeled plasma membrane (see Figure 6b). To further examine the labeling and ESPT fluorescence signal specificity, the pixel profiles of EGFP and ESPT emissions from the two channels are superimposed in Figure 7. Since EGFP and M3 were appended together, any nonoverlapping peaks in these line profiles would be the result of off-target labeling and background fluorescence. Happily, images of all three constructs showed no observable background fluorescence or nonspecific labeling. To our knowledge, the FR-1V/M3 tag is brighter than any other LSS fluorescent proteins, and its labeling kinetics is faster than most of the known imaging tags/labels.21
Figure 7.
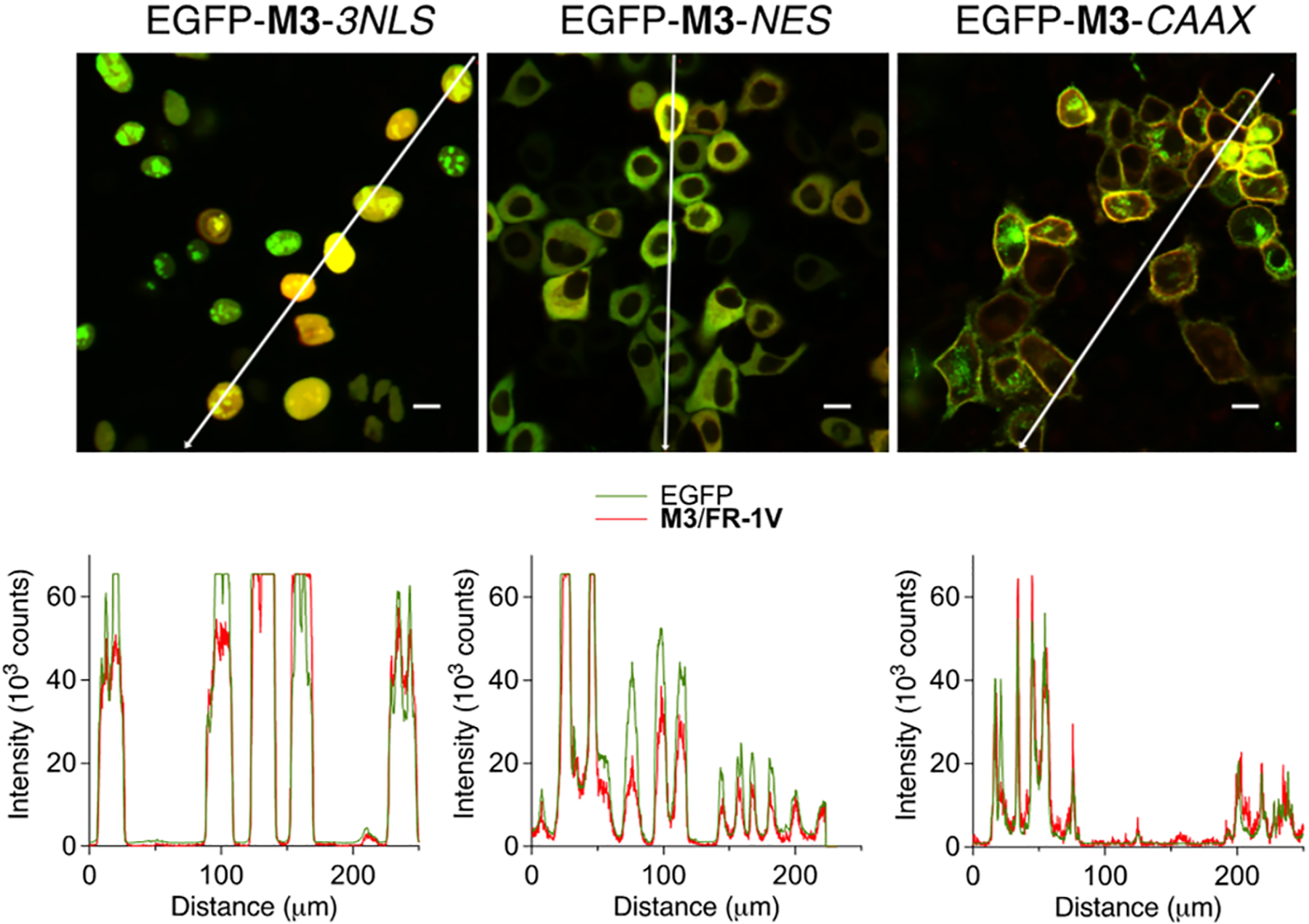
ESPT fluorescence specificity of FR-1V/M3. Top row shows the merged EGFP and LP615 channels of transfected live HeLa cells with M3 localized in cell nuclei, cytosol, and plasma membrane, respectively. Bottom row shows the line profiles of colocalized EGFP and ESPT signal along the white arrows in the top images. Scale bar, 10 μm.
To place this work in context, it is prudent to correlate the work presented here with our previously reported efforts with photoswitches.16b In prior work, the FR-1V fluorophore was used to conjugate with a different hCRBPII engineered mutant in a proof-of-principle demonstration of a no-wash live cell imaging in the near-infrared window. Comparable to the no-wash system, the ESPT system reported in this work also shows high specificity in our imaging model. Although both systems use the same fluorophore and mutants from the same protein scaffold, the underlying difference is in the photochemical mechanics. Critically, the previous no-wash system is realized via excited-state photoisomerization of a double bond, which leads to a positional change of the chromophore relative to the protein binding pocket, and subsequent ground-state proton transfer that yields the ICT system. Thus, the ground state is protonated, resulting in an emission with <100 nm Stokes shift. In contrast, the current rapid LSS system is realized via photobase-initiated excited-state proton transfer. The two systems together highlight the ability to synthetically tweak and control a given photochemical complex on the molecular level in a manner to alter its energy dissipation pathway on the surface of excited state and achieve desired functionalities (namely isomerization of a double bond or protonation in the excited state).
CONCLUSIONS
We demonstrate the conceptualization, rational design, and implementation of a new paradigm of LSS fluorescent probes utilizing photobasic fluorophores. Excited State Proton Transfer (ESPT) leads to emission of the protein host/fluorophore complex in the NIR region. The fluorogenic molecules undergo Intramolecular Charge Transfer (ICT) upon photoexcitation, leading to an increased electron density, and heightened basicity of the imine nitrogen atom. Engineered protein hosts are designed to not only bind the fluorogenic molecules as an imine, but also deliver a proton upon excitation to support the ESPT process. Such a photobase phenomenon is rarely seen, even in small molecules in solution. The ability to apply this phenomenon in a protein system opens new avenues in the development of photoactive and fluorescent proteins. Specifically, we have identified fluorescent ligands for engineered hCRBPII protein hosts capable of >200 nm apparent Stokes shift, maximal >70% fluorescence quantum yield, and close to unity ESPT emission ratio. Among these ligands, FR-1V was found to have superior fast-labeling kinetics and rapid cell permeation. In a proof-of-concept imaging assay, the FR-1V/M3 complex exhibits the ability to fluoresce mammalian cells, at a wavelength 200 nm from it is excitation, with short incubation periods, high brightness, and excellent staining specificity. We speculate that this concept and design is general, readily applicable to imaging tag designs, and can be extended to much broader fields, including smart light-responsive materials, functional probes, and optogenetics.
Supplementary Material
ACKNOWLEDGMENTS
All crystallographic data were collected at the Advanced Photon Source, an Office of Science User Facility operated for the US Department of Energy (DOE) Office of Science by Argonne National Laboratory, supported by the US DOE under contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation the Michigan Technology Tri-Corridor (Grant 085P1000817) and the MSU office of the Vice President for Research.
Funding
Generous support was provided by the NIH (GM101353).
ABBREVIATIONS
- QY
Quantum Yield
- FP
Fluorescent Protein
- NIR
Near InfraRed
- LSS
Large Stokes Shift
- ICT
Internal Charge Transfer
- ESPT
Excited State Proton Transfer
- ESIPT
Excited State Internal Proton Transfer
- SB
Schiff Base
- PSB
Protonated Schiff Base
- hCRBPII
human Cellular Retinol Binding Protein II
- Å
angstrom
- ε
extinction coefficient
- EGFP
enhanced GFP
- NLS
Nuclear Localization Sequence
- PBS
Phosphate Buffered Saline
- PDB
Protein Data Bank
- RFP
Red Fluorescent Protein
- NES
Nuclear Exclusion Sequence
- Abs
Absorption
- Em
Emission
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.1c05039.
Experimental data including synthesis of the fluorophores, protein expression and purification, UV–vis spectra, HPLC extraction, crystallization conditions, and X-ray data collection and refinement statics; crystallographic files such as atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes: 7LSQ, 7MFX, 7MFY, 7MFZ) (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.1c05039
The authors declare no competing financial interest.
Contributor Information
Elizabeth M. Santos, Michigan State University, Department of Chemistry, East Lansing, Michigan 48824, United States; Present Address: Dow Performance Silicones, 2200 W. Salzburg Rd, Midland, MI 48686.
Wei Sheng, Michigan State University, Department of Chemistry, East Lansing, Michigan 48824, United States;.
Rahele Esmatpour Salmani, Michigan State University Department of Chemistry, East Lansing, Michigan 48824, United States.
Setare Tahmasebi Nick, Michigan State University, Department of Chemistry, East Lansing, Michigan 48824, United States.
Alireza Ghanbarpour, Michigan State University, Department of Chemistry, East Lansing, Michigan 48824, United States;.
Hadi Gholami, Michigan State University, Department of Chemistry, East Lansing, Michigan 48824, United States;.
Chrysoula Vasileiou, Michigan State University, Department of Chemistry, East Lansing, Michigan 48824, United States.
James H. Geiger, Michigan State University, Department of Chemistry, East Lansing, Michigan 48824, United States;
Babak Borhan, Michigan State University, Department of Chemistry, East Lansing, Michigan 48824, United States;.
REFERENCES
- (1).(a) Chudakov DM; Matz MV; Lukyanov S; Lukyanov KA Fluorescent Proteins and Their Applications in Imaging Living Cells and Tissues. Physiol. Rev 2010, 90, 1103. [DOI] [PubMed] [Google Scholar]; (b) Dean KM; Palmer AE Advances in fluorescence labeling strategies for dynamic cellular imaging. Nat. Chem. Biol 2014, 10, 512. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Guo ZQ; Park S; Yoon J; Shin I Recent progress in the development of near-infrared fluorescent probes for bioimaging applications. Chem. Soc. Rev 2014, 43, 16. [DOI] [PubMed] [Google Scholar]; (d) Hilderbrand SA; Weissleder R Near-infrared fluorescence: application to in vivo molecular imaging. Curr. Opin. Chem. Biol 2010, 14, 71. [DOI] [PubMed] [Google Scholar]; (e) Kowada T; Maeda H; Kikuchi K BODIPY-based probes for the fluorescence imaging of biomolecules in living cells. Chem. Soc. Rev 2015, 44, 4953. [DOI] [PubMed] [Google Scholar]; (f) Luo SL; Zhang EL; Su YP; Cheng TM; Shi CM A review of NIR dyes in cancer targeting and imaging. Biomaterials 2011, 32, 7127. [DOI] [PubMed] [Google Scholar]; (g) Nienhaus K; Nienhaus GU Fluorescent proteins for live-cell imaging with super-resolution. Chem. Soc. Rev 2014, 43, 1088. [DOI] [PubMed] [Google Scholar]; (h) Schaferling M The Art of Fluorescence Imaging with Chemical Sensors. Angew. Chem., Int. Ed 2012, 51, 3532. [DOI] [PubMed] [Google Scholar]; (i) Specht EA; Braselmann E; Palmer AE In Annual Review of Physiology; Julius D, Ed.; Annual Reviews: Palo Alto, 2017; Vol. 79, p 93. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Yang ZG; Cao JF; He YX; Yang JH; Kim T; Peng XJ; Kim JS Macro-/micro-environment-sensitive chemosensing and biological imaging. Chem. Soc. Rev 2014, 43, 4563. [DOI] [PubMed] [Google Scholar]; (k) Yuan L; Lin WY; Zheng KB; He LW; Huang WM Far-red to near infrared analyte-responsive fluorescent probes based on organic fluorophore platforms for fluorescence imaging. Chem. Soc. Rev 2013, 42, 622. [DOI] [PubMed] [Google Scholar]; (l) Wang CS; Wang ZH; Zhao T; Li Y; Huang G; Sumer BD; Gao JM Optical molecular imaging for tumor detection and image-guided surgery. Biomaterials 2018, 157, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).(a) Day RN; Davidson MW The fluorescent protein palette: tools for cellular imaging. Chem. Soc. Rev 2009, 38, 2887. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Remington SJ Green fluorescent protein: A perspective. Protein Sci. 2011, 20, 1509. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Sample V; Newman RH; Zhang J The structure and function of fluorescent proteins. Chem. Soc. Rev 2009, 38, 2852. [DOI] [PubMed] [Google Scholar]; (d) Shaner NC; Steinbach PA; Tsien RY A guide to choosing fluorescent proteins. Nat. Methods 2005, 2, 905. [DOI] [PubMed] [Google Scholar]; (e) Wu B; Piatkevich KD; Lionnet T; Singer RH; Verkhusha VV Modern fluorescent proteins and imaging technologies to study gene expression, nuclear localization, and dynamics. Curr. Opin. Cell Biol 2011, 23, 310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).(a) Miller LW; Cai YF; Sheetz MP; Cornish VW In vivo protein labeling with trimethoprim conjugates: a flexible chemical tag. Nat. Methods 2005, 2, 255. [DOI] [PubMed] [Google Scholar]; (b) Sun Y; Weliky DP C-13-C-13 Correlation Spectroscopy of Membrane-Associated Influenza Virus Fusion Peptide Strongly Supports a Helix-Turn-Helix Motif and Two Turn Conformations. J. Am. Chem. Soc 2009, 131, 13228. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Griffin BA; Adams SR; Jones J; Tsien RY Fluorescent labeling of recombinant proteins in living cells with FlAsH. Methods Enzymol. 2000, 327, 565. [DOI] [PubMed] [Google Scholar]; (d) Hori Y; Kikuchi K Protein labeling with fluorogenic probes for no-wash live-cell imaging of proteins. Curr. Opin. Chem. Biol 2013, 17, 644. [DOI] [PubMed] [Google Scholar]; (e) Keppler A; Gendreizig S; Gronemeyer T; Pick H; Vogel H; Johnsson K A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat. Biotechnol 2003, 21, 86. [DOI] [PubMed] [Google Scholar]; (f) Loving GS; Sainlos M; Imperiali B Monitoring protein interactions and dynamics with solvatochromic fluorophores. Trends Biotechnol. 2010, 28, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Zhang J; Campbell RE; Ting AY; Tsien RY Creating new fluorescent probes for cell biology. Nat. Rev. Mol. Cell Biol 2002, 3, 906. [DOI] [PubMed] [Google Scholar]; (h) Dahlberg PD; Sartor AM; Wang JR; Saurabh S; Shapiro L; Moerner WE Identification of PAmKate as a Red Photoactivatable Fluorescent Protein for Cryogenic Super-Resolution Imaging. J. Am. Chem. Soc 2018, 140, 12310. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Fu YX; Han HH; Zhang JJ; He XP; Feringa BL; Tian H Photocontrolled Fluorescence “Double-Check” Bioimaging Enabled by a Glycoprobe-Protein Hybrid. J. Am. Chem. Soc 2018, 140, 8671. [DOI] [PubMed] [Google Scholar]; (j) Jun JV; Petersson EJ; Chenoweth DM Rational Design and Facile Synthesis of a Highly Tunable Quinoline-Based Fluorescent Small-Molecule Scaffold for Live Cell Imaging. J. Am. Chem. Soc 2018, 140, 9486. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Zhang Y; Song KH; Tang SC; Ravelo L; Cusido J; Sun C; Zhang HF; Raymo FM Far-Red Photoactivatable BODIPYs for the Super-Resolution Imaging of Live Cells. J. Am. Chem. Soc 2018, 140, 12741. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Murrey HE; Judkins JC; am Ende CW; Ballard TE; Fang Y; Riccardi K; Di L; Guilmette ER; Schwartz JW; Fox JM; Johnson DS Systematic Evaluation of Bioorthogonal Reactions in Live Cells with Clickable Halo Tag Ligands: Implications for Intracellular Imaging. J. Am. Chem. Soc 2015, 137, 11461. [DOI] [PMC free article] [PubMed] [Google Scholar]; (m) Xu W; Zeng ZB; Jiang JH; Chang YT; Yuan L Discerning the Chemistry in Individual Organelles with Small-Molecule Fluorescent Probes. Angew. Chem., Int. Ed 2016, 55, 13658. [DOI] [PubMed] [Google Scholar]
- (4).(a) Shcherbakova DM; Verkhusha VV Near-infrared fluorescent proteins for multicolor in vivo imaging. Nat. Methods 2013, 10, 751. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shcherbo D; Shemiakina II; Ryabova AV; Luker KE; Schmidt BT; Souslova EA; Gorodnicheva TV; Strukova L; Shidlovskiy KM; Britanova OV; Zaraisky AG; Lukyanov KA; Loschenov VB; Luker GD; Chudakov DM Near-infrared fluorescent proteins. Nat. Methods 2010, 7, 827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Sobhy MA; Elshenawy MM; Takahashi M; Whitman BH; Walter NG; Hamdan SM Versatile single-molecule multicolor excitation and detection fluorescence setup for studying biomolecular dynamics. Rev. Sci. Instrum 2011, 82, 113702. [DOI] [PubMed] [Google Scholar]
- (6).(a) Chen YH; Zhao JZ; Guo HM; Xie LJ Geometry Relaxation-Induced Large Stokes Shift in Red-Emitting Borondipyrromethenes (BODIPY) and Applications in Fluorescent Thiol Probes. J. Org. Chem 2012, 77, 2192. [DOI] [PubMed] [Google Scholar]; (b) Jiang MJ; Gu XG; Lam JWY; Zhang YL; Kwok RTK; Wong KS; Tang BZ Two-photon AIE bio-probe with large Stokes shift for specific imaging of lipid droplets. Chem. Sci 2017, 8, 5440. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Qi Y; Huang Y; Li BW; Zeng F; Wu SZ Real-Time Monitoring of Endogenous Cysteine Levels In Vivo by near-Infrared Turn-on Fluorescent Probe with Large Stokes Shift. Anal. Chem 2018, 90, 1014. [DOI] [PubMed] [Google Scholar]; (d) Butkevich AN; Lukinavicius G; D’Este E; Hell SW Cell-Permeant Large Stokes Shift Dyes for Transfection-Free Multicolor Nanoscopy. J. Am. Chem. Soc 2017, 139, 12378. [DOI] [PubMed] [Google Scholar]
- (7).(a) Zhao LH; He X; Huang YB; Li JL; Li YL; Tao S; Sun Y; Wang XH; Ma PY; Song DQ A novel ESIPT-ICT-based near-infrared fluorescent probe with large stokes-shift for the highly sensitive, specific, and non-invasive in vivo detection of cysteine. Sens. Actuators, B 2019, 296, 126571. [Google Scholar]; (b) Zhu MQ; Xu YM; Sang LF; Zhao ZY; Wang LJ; Wu XQ; Fan FG; Wang Y; Li H An ICT-based fluorescent probe with a large Stokes shift for measuring hydrazine in biological and water samples. Environ. Pollut 2020, 256, 113427. [DOI] [PubMed] [Google Scholar]; (c) Dahal D; McDonald L; Pokhrel S; Paruchuri S; Konopka M; Pang Y A NIR-emitting cyanine with large Stokes shifts for live cell imaging: large impact of the phenol group on emission. Chem. Commun 2019, 55, 13223. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Peng XJ; Song FL; Lu E; Wang YN; Zhou W; Fan JL; Gao YL Heptamethine cyanine dyes with a large stokes shift and strong fluorescence: A paradigm for excited-state intramolecular charge transfer. J. Am. Chem. Soc 2005, 127, 4170. [DOI] [PubMed] [Google Scholar]; (e) Wu YY; Chen Y; Gou GZ; Mu WH; Lv XJ; Du ML; Fu WF Large Stokes Shift Induced by Intramolcular Charge Transfer in N,O-Chelated Naphthyridine-BF2 Complexes. Org. Lett 2012, 14, 5226. [DOI] [PubMed] [Google Scholar]; (f) Hu RR; Gomez-Duran CFA; Lam JWY; Belmonte-Vazquez JL; Deng CM; Chen SJ; Ye RQ; Pena-Cabrera E; Zhong YC; Wong KS; Tang BZ Synthesis, solvatochromism, aggregation-induced emission and cell imaging of tetraphenylethene-containing BODIPY derivatives with large Stokes shifts. Chem. Commun 2012, 48, 10099. [DOI] [PubMed] [Google Scholar]; (g) Greiner R; Schlucker T; Zgela D; Langhals H Fluorescent aryl naphthalene dicarboximides with large Stokes shifts and strong solvatochromism controlled by dynamics and molecular geometry. J. Mater. Chem. C 2016, 4, 11244. [Google Scholar]
- (8).(a) Aparin IO; Sergeeva OV; Mishin AS; Khaydukov EV; Korshun VA; Zatsepin TS Excimer-FRET Cascade in Dual DNA Probes: Open Access to Large Stokes Shift, Enhanced Acceptor Light up, and Robust RNA Sensing. Anal. Chem 2020, 92, 7028. [DOI] [PubMed] [Google Scholar]; (b) Conlon P; Yang CYJ; Wu YR; Chen Y; Martinez K; Kim YM; Stevens N; Marti AA; Jockusch S; Turro NJ; Tan WH Pyrene excimer signaling molecular beacons for probing nucleic acids. J. Am. Chem. Soc 2008, 130, 336. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kim TI; Jin HY; Bae J; Kim Y Excimer Emission-Based Fluorescent Probe Targeting Caspase-3. Anal. Chem 2017, 89, 10565. [DOI] [PubMed] [Google Scholar]
- (9).(a) Sie EJ; McIver J; Lee YH; Fu L; Kong J; Gedik N Valley-selective optical Stark effect in monolayer WS2. Nat. Mater 2015, 14, 290. [DOI] [PubMed] [Google Scholar]; (b) Stokker-Cheregi F; Vinattieri A; Feltin E; Simeonov D; Levrat J; Carlin JF; Butte R; Grandjean N; Gurioli M Impact of quantum confinement and quantum confined Stark effect on biexciton binding energy in GaN/AlGaN quantum wells. Appl. Phys. Lett 2008, 93, 152105. [Google Scholar]
- (10).(a) Piatkevich KD; Malashkevich VN; Almo SC; Verkhusha VV Engineering ESPT Pathways Based on Structural Analysis of LSSmKate Red Fluorescent Proteins with Large Stokes Shift. J. Am. Chem. Soc 2010, 132, 10762. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Piatkevich KD; Hulit J; Subach OM; Wu B; Abdulla A; Segall JE; Verkhusha VV Monomeric red fluorescent proteins with a large Stokes shift. Proc. Natl. Acad. Sci. U. S. A 2010, 107, 5369. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Yang J; Wang L; Yang F; Luo HM; Xu LL; Lu JL; Zeng SQ; Zhang ZH mBeRFP, an Improved Large Stokes Shift Red Fluorescent Protein. PLoS One 2013, 8, e64849. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Chu J; Oh Y; Sens A; Ataie N; Dana H; Macklin JJ; Laviv T; Welf ES; Dean KM; Zhang FJ; Kim BB; Tang CT; Hu M; Baird MA; Davidson MW; Kay MA; Fiolka R; Yasuda R; Kim DS; Ng HL; Lin MZ A bright cyan-excitable orange fluorescent protein facilitates dual-emission microscopy and enhances bioluminescence imaging in vivo. Nat. Biotechnol 2016, 34, 760. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Shcherbakova DM; Hink MA; Joosen L; Gadella TWJ; Verkhusha VV An Orange Fluorescent Protein with a Large Stokes Shift for Single-Excitation Multicolor FCCS and FRET Imaging. J. Am. Chem. Soc 2012, 134, 7913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).(a) Sedgwick AC; Wu LL; Han HH; Bull SD; He XP; James TD; Sessler JL; Tang BZ; Tian H; Yoon J Excited-state intramolecular proton-transfer (ESIPT) based fluorescence sensors and imaging agents. Chem. Soc. Rev 2018, 47, 8842. [DOI] [PubMed] [Google Scholar]; (b) Zhao JZ; Ji SM; Chen YH; Guo HM; Yang P Excited state intramolecular proton transfer (ESIPT): from principal photo-physics to the development of new chromophores and applications in fluorescent molecular probes and luminescent materials. Phys. Chem. Chem. Phys 2012, 14, 8803. [DOI] [PubMed] [Google Scholar]
- (12).(a) Weller A Quantitative Untersuchungen der Fluoreszen-zumwandlung bei Naptholen. Zeitschrift Fur Elektrochemie 1952, 56, 662. [Google Scholar]; (b) Weller A Fast Reactions of Excited Molecules. Prog. React. Kinet. Mech 1961, 1, 187. [Google Scholar]
- (13).(a) Li CZ; Yang YG; Ma C; Liu YF Effect of amino group on the excited-state intramolecular proton transfer (ESIPT) mechanisms of 2-(2 ‘-hydroxyphenyl)benzoxazole and its amino derivatives. RSC Adv. 2016, 6, 5134. [Google Scholar]; (b) Padalkar VS; Tathe A; Gupta VD; Patil VS; Phatangare K; Sekar N Synthesis and Photo-Physical Characteristics of ESIPT Inspired 2-Substituted Benzimidazole, Benzoxazole and Benzothiazole Fluorescent Derivatives. J. Fluoresc 2012, 22, 311. [DOI] [PubMed] [Google Scholar]; (c) Li CZ; Ma C; Li DL; Liu YF Excited state intramolecular proton transfer (ESIPT) of 6-amino-2-(2 ‘-hydroxyphenyl)benzoxazole in dichloromethane and methanol: A TD-DFT quantum chemical study. J. Lumin 2016, 172, 29. [Google Scholar]; (d) Chung MW; Lin TY; Hsieh CC; Tang KC; Fu H; Chou PT; Yang SH; Chi Y Excited-State Intramolecular Proton Transfer (ESIPT) Fine Tuned by Quinoline-Pyrazole Isomerism: pi-Conjugation Effect on ESIPT. J. Phys. Chem. A 2010, 114, 7886. [DOI] [PubMed] [Google Scholar]; (e) Liu B; Wang JF; Zhang G; Bai RK; Pang Y Flavone-Based ESIPT Ratiometric Chemodosimeter for Detection of Cysteine in Living Cells. ACS Appl. Mater. Interfaces 2014, 6, 4402. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Zheng DY; Zhang MZ; Zhao GJ Combined TDDFT and AIM Insights into Photoinduced Excited State Intramolecular Proton Transfer (ESIPT) Mechanism in Hydroxyl-and Amino-Anthraquinone Solution. Sci. Rep 2017, 7, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).(a) Leiderman P; Genosar L; Huppert D Excited-state proton transfer: Indication of three steps in the dissociation and recombination process. J. Phys. Chem. A 2005, 109, 5965. [DOI] [PubMed] [Google Scholar]; (b) Spry DB; Fayer MD Proton Transfer and Proton Concentrations in Protonated Nafion Fuel Cell Membranes. J. Phys. Chem. B 2009, 113, 10210. [DOI] [PubMed] [Google Scholar]; (c) Liu WM; Han FY; Smith C; Fang C Ultrafast Conformational Dynamics of Pyranine during Excited State Proton Transfer in Aqueous Solution Revealed by Femtosecond Stimulated Raman Spectroscopy. J. Phys. Chem. B 2012, 116, 10535. [DOI] [PubMed] [Google Scholar]; (d) Premont-Schwarz M; Barak T; Pines D; Nibbering ETJ; Pines E Ultrafast Excited-State Proton-Transfer Reaction of 1-Naphthol-3,6-Disulfonate and Several 5-Substituted 1-Naphthol Derivatives. J. Phys. Chem. B 2013, 117, 4594. [DOI] [PubMed] [Google Scholar]; (e) Han FY; Liu WM; Fang C Excited-state proton transfer of photoexcited pyranine in water observed by femtosecond stimulated Raman spectroscopy. Chem. Phys 2013, 422, 204. [Google Scholar]
- (15).(a) Sheng W; Nairat M; Pawlaczyk PD; Mroczka E; Farris B; Pines E; Geiger JH; Borhan B; Dantus M Ultrafast Dynamics of a “Super” Photobase. Angew. Chem., Int. Ed 2018, 57, 14742. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lahiri J; Moemeni M; Kline J; Borhan B; Magoulas I; Yuwono SH; Piecuch P; Jackson JE; Dantus M; Blanchard GJ Proton Abstraction Mediates Interactions between the Super Photobase FR0-SB and Surrounding Alcohol Solvent. J. Phys. Chem. B 2019, 123, 8448. [DOI] [PubMed] [Google Scholar]
- (16).(a) Wang WJ; Nossoni Z; Berbasova T; Watson CT; Yapici I; Lee KSS; Vasileiou C; Geiger JH; Borhan B Tuning the Electronic Absorption of Protein-Embedded All-trans-Retinal. Science 2012, 338, 1340. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sheng W; Nick ST; Santos EM; Ding XL; Zhang J; Vasileiou C; Geiger JH; Borhan B A Near-Infrared Photoswitchable Protein-Fluorophore Tag for No-Wash Live Cell Imaging. Angew. Chem., Int. Ed 2018, 57, 16083. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Santos EM; Berbasova T; Wang WJ; Salmani RE; Sheng W; Vasileiou C; Geiger JH; Borhan B Engineering of a Red Fluorogenic Protein/Merocyanine Complex for Live-Cell Imaging. ChemBioChem 2020, 21, 723. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Yapici I; Lee KSS; Berbasova T; Nosrati M; Jia XF; Vasileiou C; Wang WJ; Santos EM; Geiger JH; Borhan B “Turn-On” Protein Fluorescence: In Situ Formation of Cyanine Dyes. J. Am. Chem. Soc 2015, 137, 1073. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Ghanbarpour A; Nairat M; Nosrati M; Santos EM; Vasileiou C; Dantus M; Borhan B; Geiger JH Mimicking Microbial Rhodopsin Isomerization in a Single Crystal. J. Am. Chem. Soc 2019, 141, 1735. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Ghanbarpour A; Pinger C; Esmatpour Salmani R; Assar Z; Santos EM; Nosrati M; Pawlowski K; Spence D; Vasileiou C; Jin X; Borhan B; Geiger JH Engineering the hCRBPII Domain-Swapped Dimer into a New Class of Protein Switches. J. Am. Chem. Soc 2019, 141, 17125. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Nosrati M; Berbasova T; Vasileiou C; Borhan B; Geiger JH A Photoisomerizing Rhodopsin Mimic Observed at Atomic Resolution. J. Am. Chem. Soc 2016, 138, 8802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).(a) Diwu Z; Lu YX; Zhang CL; Klaubert DH; Haugland RP Fluorescent molecular probes 0.2. The synthesis, spectral properties and use of fluorescent solvatochromic Dapoxyl-(TM) dyes. Photochem. Photobiol 1997, 66, 424. [Google Scholar]; (b) Min J; Lee JW; Ahn YH; Chang YT Combinatorial dapoxyl dye library and its application to site selective probe for human serum albumin. J. Comb. Chem 2007, 9, 1079. [DOI] [PubMed] [Google Scholar]; (c) Ando Y; Homma Y; Hiruta Y; Citterio D; Suzuki K Structural characteristics and optical properties of a series of solvatochromic fluorescent dyes displaying long-wavelength emission. Dyes Pigm. 2009, 83, 198. [Google Scholar]
- (18).Storch J; Corsico B The emerging functions and mechanisms of mammalian fatty acid-binding proteins. Annu. Rev. Nutr 2008, 28,73. [DOI] [PubMed] [Google Scholar]
- (19).Berbasova T; Nosrati M; Vasileiou C; Wang W; Lee KSS; Yapici I; Geiger JH; Borhan B Rational Design of a Colorimetric pH Sensor from a Soluble Retinoic Acid Chaperone. J. Am. Chem. Soc 2013, 135, 16111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).(a) Lee KSS; Berbasova T; Vasileiou C; Jia XF; Wang WJ; Choi Y; Nossoni F; Geiger JH; Borhan B Probing Wavelength Regulation with an Engineered Rhodopsin Mimic and a C15-Retinal Analogue. ChemPlusChem 2012, 77, 273. [Google Scholar]; (b) Crist RM; Vasileiou C; Rabago-Smith M; Geiger JH; Borhan B Engineering a rhodopsin protein mimic. J. Am. Chem. Soc 2006, 128, 4522. [DOI] [PubMed] [Google Scholar]
- (21).(a) Gao F; Gao T; Zhou KC; Zeng WB Small Molecule-Photoactive Yellow Protein Labeling Technology in Live Cell Imaging. Molecules 2016, 21, 1163. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kumar N; Hori Y; Nishiura M; Kikuchi K Rapid no-wash labeling of PYP-tag proteins with reactive fluorogenic ligands affords stable fluorescent protein conjugates for long-term cell imaging studies. Chem. Sci 2020, 11, 3694. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Leng S; Qiao QL; Miao L; Deng WG; Cui JN; Xu ZC A wash-free SNAP-tag fluorogenic probe based on the additive effects of quencher release and environmental sensitivity. Chem. Commun 2017, 53, 6448. [DOI] [PubMed] [Google Scholar]; (d) Sun XL; Zhang AH; Baker B; Sun L; Howard A; Buswell J; Maurel D; Masharina A; Johnsson K; Noren CJ; Xu MQ; Correa IR Development of SNAP-Tag Fluorogenic Probes for Wash-Free Fluorescence Imaging. Chem-BioChem 2011, 12, 2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.