

Abstract
Introduction:
Peripheral artery disease (PAD) is a major cause of cardiovascular morbidity and mortality, yet timely diagnosis is elusive. Larger genome-wide association studies (GWAS) have now provided the ability to evaluate whether genetic data, in the form of genome-wide polygenic risk scores (PRS), can help improve our ability to identify patients at high risk of having PAD.
Methods:
Using summary statistic data from the largest PAD GWAS from the Million Veteran Program, we developed PRSs with genome data from UK Biobank. We then evaluated the clinical utility of adding the best-performing PRS to a PAD clinical risk score.
Results:
A total of 487,320 participants (5759 PAD cases) were included in our final genetic analysis. Compared to participants in the lowest 10% of PRS, those in the highest decile had 3.1 higher odds of having PAD (95% CI, 3.06–3.21). Additionally, a PAD PRS was associated with increased risk of having coronary artery disease, congestive heart failure, and cerebrovascular disease. The PRS significantly improved a clinical risk model (Net Reclassification Index = 0.07, p < 0.001), with most of the performance seen in downgrading risk of controls. Combining clinical and genetic data to detect risk of PAD resulted in a model with an area under the curve of 0.76 (95% CI, 0.75–0.77).
Conclusion:
We demonstrate that a genome-wide PRS can discriminate risk of PAD and other cardiovascular diseases. Adding a PAD PRS to clinical risk models may help improve detection of prevalent, but undiagnosed disease.
Keywords: disease prediction model, peripheral artery disease (PAD), polygenic risk score, risk factors
Introduction
Peripheral artery disease (PAD) affects 200 million individuals world-wide and has an estimated annual health care cost of $21 billion in the United States alone.1,2 PAD confers a two- to six-fold increased risk of major adverse cardiovascular events (MACE) but often goes undiagnosed.3-5 Those who go undiagnosed are less likely to receive evidenced-based therapy to reduce cardiovascular and limb-related morbidity and mortality. Thus, strategies to improve diagnosis and secondary prevention measures may improve quality of life and life expectancy for this patient population.
Older age, smoking, hypertension, hyperlipidemia, and diabetes mellitus are known traditional risk factors for PAD.6,7 However, risk factor models using purely clinical risk assessment have historically achieved modest performance.8,9 This is likely because other factors such as genetic risk can significantly contribute to the development of PAD. Although PAD-specific genetic risk loci have been difficult to identify in prior smaller studies, a new genome-wide association study (GWAS), including over 30,000 individuals with PAD, identified multiple new PAD-specific risk loci.10 These findings may make it possible to improve our ability to better quantify genetic risk of PAD and improve diagnosis of prevalent and undiagnosed disease.
Similar to coronary artery disease, there are multiple genetic risk variants that have small but additive risk for PAD development.10-12 Previously, quantification of this additive risk involved linear combinations of a small number of risk variants to develop a polygenic risk score (PRS) with mixed results in other diseases.13-15 Now, algorithms such as PRSice and PRScs have been developed to allow for the quantification of genetic risk across the entire genome.16,17 These methods can incorporate risk attributes from millions of markers across the whole genome, and account for different effect size distributions and genetic architectures of disease, while remaining computationally scalable.18,19 Research has shown that these new algorithms can provide disease risk quantification similar to monogenic disease risk.
In this study, we aimed to: (1) develop a genome-wide PRS for PAD, (2) evaluate whether a PRS developed from PAD GWAS results was informative for other cardiovascular diseases, and (3) quantify the additive value of a PRS when added to a clinical risk model.
Methods
Study population
The study population of interest included 502,536 participants aged 40–69 years from the UK Biobank. All participants completed a computer-based assessment, underwent a standardized portfolio of clinical measurements, and provided written informed consent for the use of their data for research. Approval was obtained by the National Health Service’s National Research Ethics Service North West (11/ NW/0382). Detailed descriptions of the study cohort have been previously reported.20,21 This study was approved by our institutional review board (e-protocol 55133) and under the UK Biobank application ID 13721.
For this study, demographic and clinical variables such as chronological age, sex, race, blood pressure, smoking status, and total cholesterol were directly obtained from the UK Biobank data set, whereas other clinical variables such as diabetes mellitus, coronary artery disease (CAD), cerebrovascular disease (CVD), and chronic kidney disease (CKD) were defined based on previously published algorithms (online Supplemental Table 1).22
Definition of PAD
The exact definition for PAD is detailed in online Supplemental Table 2 and includes primary and secondary diagnosis and surgical codes from hospital episodes, as well as use of PAD-specific medications and self-reported PAD diagnoses and surgeries. This PAD phenotyping algorithm was chosen to align with the definition of PAD used by Klarin and colleagues to validate their PAD GWAS results using the UK Biobank cohort.10 In addition to this, we added medication codes for cilostazol, which are specific for PAD treatment, to broaden our capture of PAD cases. Our phenotype definition was validated by performing a PAD GWAS on our cohort. In doing so we replicated all previously reported PAD risk loci. We also performed sensitivity analysis to exclude less specific definitions of PAD such as arterial embolism, amputation, and generalized atherosclerosis and re-evaluated model performance.
Genotyping and imputation
DNA samples of participants were genotyped using custom Affymetrix arrays (Thermo Fisher Scientific, Waltham, MA, USA) and the UK Biobank Axiom array (Thermo Fisher Scientific), which were designed to optimize imputation performance across the genome. A merged sample of UK10K sequencing and 1000 Genomes Project were used as reference panels for genotype imputation via an algorithm implemented in the IMPUTE2 program.23 Imputed data were provided by the UK Biobank research team. Other detailed information regarding genotyping and imputation has been provided in previous studies.20
Genotype quality control
Related individuals and those that failed the X-chromosome sex concordance check were removed before imputation. UK Biobank includes three main racial populations: European, African, and Asian. Genetic principal components accounting for population stratification were centrally computed. We excluded individuals for whom the available information on race was incompatible with their genetic information. Individuals with more than 5% missing data were also excluded. Regarding genetic data, we excluded ambiguous single nucleotide polymorphisms (SNPs), indel SNPs, and SNPs with more than 1% missing data or minor allele frequency (MAF) below 1%. Finally, 487,320 participants with both PAD status and genotyped data were used for the statistical analyses.
Polygenic risk score (PRS) calculation
An important component to PRS validity is that the disease of interest be heritable, and thus we evaluated this characteristic for PAD. Genome-wide Complex Trait Analysis (GCTA) was applied to estimate the variance explained by filtered SNPs. We first calculated the genetic relationship matrix (GRM), which contained a measure of the genetic correlation among individuals. Then, we estimated the proportion of trait variance explained by genetic variation using the restricted maximum likelihood (REML) approach, relying on the GRM estimated from all of the SNPs.24 PAD heritability in the UK Biobank was estimated with the REML method using sex and age as covariates. We then derived PRSs based on the weighted sum of risk alleles using summary statistics from the largest PAD GWAS to date (derived from the Million Veteran Program (MVP)).10 Details of our analysis pipeline are illustrated in Figure 1.
Figure 1.

Study design and flowchart for PAD. To select the optimal genetic data for a PAD genome-wide PRS, we evaluated the performance of PRSs derived from summary statistics from the Million Veteran Program PAD GWAS and two GWASs for coronary artery disease (CARDIoGRAM and CARDIoGRAMplusC4D). The best-performing PRS was applied to a nonoverlapping set of participants from the UK Biobank by 10-fold cross-validation. Lastly, we evaluated the performance of a clinical model in identifying risk of PAD and a clinical model combined with the PRS (i.e., the PRS-enhanced clinical risk model).
CARDIoGRAM, Coronary Artery Disease Genome-Wide Replication and Meta-Analysis; C4D, The Coronary Artery Disease consortium; GWAS, genome-wide association analysis; PAD, peripheral artery disease; PRS, polygenic risk score.
To identify the optimal algorithm for calculating a PAD PRS, we tested two methods: PRSice and PRScs. Both algorithms enable calculation of PRS but use different strategies to account for linkage disequilibrium (LD) as well as SNP effects. The PRSice (version 2.1.11) selects SNPs based on clumping and thresholding, and automatically selects these parameters by maximizing R2.16 In contrast, PRScs applies a Bayesian shrinkage parameter on effect size estimates for each SNP based on GWAS summary statistics.17 For the testing of different parameters to optimize PRScs model performance, we tuned two parameters: p-value threshold and global shrinkage parameter phi. Performance of each PRS was measured using area under the curve (AUC) adjusted for age, sex, and the first five principal components (PCs) of ancestry.
To select the optimal data for a PAD PRS, we evaluated performance of PRSs derived from summary statistics from the MVP PAD GWAS and two GWASs for CAD (Coronary Artery Disease Genome-Wide Replication and Meta-Analysis (CARDIoGRAM)25 and CARDIoGRAM plus The Coronary Artery Disease consortium (C4D)26) to see if general atherosclerotic risk also translated to high discrimination of PAD risk. The performance of the PRSs was validated using a nonoverlapping set of participants from the UK Biobank using 10-fold cross-validation.
Lastly, we evaluated the performance of a clinical model in identifying risk of PAD and a clinical model combined with the PRS (i.e., the PRS-enhanced clinical risk model). The clinical and PRS-enhanced models were compared in terms of AUC and reclassification metrics.
Statistical analysis
We compared demographic and clinical factors for those with and without PAD. A two-sided Student’s t-test was used for continuous variables and chi-squared test was used for categorical variables. Generalized linear models were used to estimate odds ratios (ORs) for risk of PAD across the three different PRSs (derived from the MVP PAD GWAS and CAD GWAS studies) as well as risk of CAD, CHF, and CVD across PAD PRS deciles. We also evaluated the odds of each disease (i.e., PAD, CAD, CHF, CVD) using a scaled PRS to enable direct comparison of risk of disease given 1 SD increase in the PRS.
We used the best performing PRS to evaluate the utility of a PAD-specific PRS in discriminating risk of PAD across the population. To this end, we divided the dataset into 100 groups based on the PRS score and evaluated the prevalence of PAD across percentiles. To assess the clinical usefulness of a PRS we combined the PRS with a clinical risk score and evaluated differences in model performance. The clinical risk score we used was developed by Duval and colleagues as a way to determine risk of prevalent PAD, which is our current use case.8 The clinical model described by Duval et al. evaluates risk of PAD using 10 variables: age, sex, race/ethnicity, diabetes, BMI, hypertension, smoking status, CAD, cerebrovascular disease, and CHF. In using this risk score, we calculated each individual’s nomogram score. Because PRS and the nomogram scores were on different scales, we used z-transformed PRS and z-transformed clinical risk nomogram scores to build a PRS-enhanced clinical risk model. Model discrimination performance was validated by performing 10-fold cross-validation using nonoverlapping 90%/10% splits (using 90% of the individuals to build the predictor and the remaining 10% of the individuals to test). To evaluate the additive effect of the PRS to the clinical model we evaluated the model goodness-of-fit by calculating C-statistics on models with an increasing number of variables, adding the PRS to the final model. We also calculated the net reclassification index (NRI) for the combined (clinical and PRS model) compared to the clinical model only. Model AUCs were compared using the DeLong test of significance, a nonparametric approach to comparing two or more correlated receiver operating characteristic curves.27 Lastly, we evaluated PRS-enhanced model calibration by plotting model predicted risk by observed risk. Patients with insufficient data to calculate their clinical risk score were excluded from this analysis. R version 3.6.3 (R Foundation for Statistical Computing, Vienna, Austria) was used for statistical analyses.
Results
Baseline characteristics of the study cohort
Of the total 487,320 individuals included in our final genetic analysis, 5759 had PAD. As outlined in Table 1, many baseline characteristics significantly differed between those with and without PAD. Those with PAD were more likely to be older than noncases, with a mean age of 61.1 years compared to 56.5 years (p < 0.001). There were also more men with PAD (65.7% vs 45.5%, p = 5.54 × 10−215), and a higher proportion of smokers (24.9 vs 10.3, p = 1.6 × 10−92) and those with first-degree relatives with a history of CAD (52.9% vs 43.7%, p = 1.39 × 10−47). On average, systolic blood pressure was significantly higher in PAD cases compared to controls (p = 3.13 × 10−74), and high-density lipoprotein (HDL) was lower in those with PAD (p = 4.21 × 10−155). Total cholesterol was lower in individuals with PAD (p = 4.31 × 10−283) and they were also more likely to be on a lipid-lowering medication compared to those without PAD (p < 0.001). Those with PAD also had higher prevalence of diabetes mellitus, CAD, cerebrovascular disease, and chronic kidney disease compared with those without PAD (p < 0.001).
Table 1.
Baseline characteristics of the individuals.
| PAD case (n = 5759) |
PAD control (n = 481,561) |
p-valuea | |
|---|---|---|---|
| Age, mean (SD), years | 61.08 (6.81) | 56.48 (8.09) | < 0.001 |
| Sex, male (%) | 3784 (65.71) | 219,307 (45.54) | 5.54 × 10−215 |
| Race | |||
| White, n (%) | 5497 (95.45) | 453,753 (94.23) | 1.91 × 10−5 |
| Black, n (%) | 52 (0.09) | 7592 (1.58) | |
| Asian, n (%) | 111 (1.93) | 10,809 (2.24) | |
| Others, n (%) | 99 (1.72) | 9407 (1.95) | |
| Systolic blood pressure, mean (SD), mmHg | 142.78 (20.36) | 137.76 (18.63) | 3.13 × 10−74 |
| Diastolic blood pressure, mean (SD), mmHg | 80.40 (10.71) | 82.24 (10.14) | 4.26 × 10−36 |
| Smoking status, current smoker (%) | 1432 (24.87) | 49,769 (10.33) | 1.60 × 10−92 |
| Total cholesterol, mean (SD), mmol/L | 5.06 (1.25) | 5.70 (1.14) | 4.31 × 10−283 |
| High-density lipoprotein, mean (SD), mmol/L | 1.30 (0.39) | 1.45 (0.38) | 4.21 × 10−155 |
| Diabetes mellitus, n (%) | 1652 (28.69) | 37,026 (7.69) | < 0.001 |
| CAD, n (%) | 2183 (37.91) | 26,561 (5.52) | < 0.001 |
| Cerebrovascular disease, n (%) | 642 (11.15) | 9405 (1.95) | < 0.001 |
| Chronic kidney disease, n (%) | 498 (8.65) | 5165 (1.07) | < 0.001 |
| CAD in a 1st degree relative, n (%) | 3048 (52.93) | 210,354 (43.68) | 1.39 × 10−47 |
| Statin usage, n (%) | 3060 (53.13) | 78,207 (16.24) | < 0.001 |
| Lipid-lowering medications, n (%) | 3120 (54.18) | 79,562 (16.52) | < 0.001 |
| Blood pressure-lowering medications, n (%) | 3260 (55.61) | 102,903 (21.37) | < 0.001 |
Statistical significance was determined by Student’s t-test for continuous variable, and chi-squared test for categorical variable.
CAD, coronary artery disease; PAD, peripheral artery disease.
Performance of PAD PRS
The calculated general genetic heritability of PAD in the UK Biobank was 11% (standard error, 0.06). The best performing PRS model adjusted for age, sex, and the first five PCs was derived from the largest PAD-specific GWAS with an AUC of 0.72 (95% CI 0.71–0.72; online Supplemental Tables 3 and 4). The PAD-specific PRS also produced higher odds of prevalent disease than PRSs generated from CARDIoGRAM or C4D GWAS data (online Supplemental Figure 1A-C). The distribution of PRS for PAD cases and noncases is illustrated in Figure 2A. As illustrated in Figure 2B, the prevalence of PAD increases as the PRS percentile increases. In the group with the highest PRS, the prevalence of PAD is 2.83%, which is a 4.5-fold difference compared with a PAD prevalence of 0.63% in the lowest PRS group.
Figure 2.

Genome-wide PRS and the risk of PAD. (A) Density plot of PRS in PAD cases and noncases. (B) Prevalence of PAD across 100 groups based on PAD PRS percentiles.
PAD, peripheral artery disease; PRS, polygenic risk score.
The performance of PAD PRS in detecting risk of PAD and other cardiovascular diseases
To evaluate the association between a PAD-specific PRS and odds of PAD and other cardiovascular diseases, we categorized PAD PRS into deciles and calculated log odds of disease based on these score deciles. As expected, participants with the highest PAD PRS scores (90–100% PAD PRS percentile) showed the highest risk for PAD (OR = 3.1, 95% CI 3.06–3.21; Figure 3A) when compared to the lowest decile group (0–10% PAD PRS percentile) in this cross-section study design. When comparing the group in the highest decile and in the top 5th percentile to the rest of the population, the OR was 1.83 (95% CI 1.73–1.92; online Supplemental Figure 2A) and 1.91 (95% CI 1.80–2.01; online Supplemental Figure 2B), respectively. For cardiovascular-related diseases, we observed strong associations between the PAD PRS and prevalence of CAD and CHF (Figures 3B and 3C), and a moderate association with CVD (Figure 3D). When we scale the PRS for more direct comparisons across diseases, we observe a similar pattern of increased odds of disease for each standard deviation increase in the PRS score, with the relationship being more attenuated for CVD (Table 2).
Figure 3.
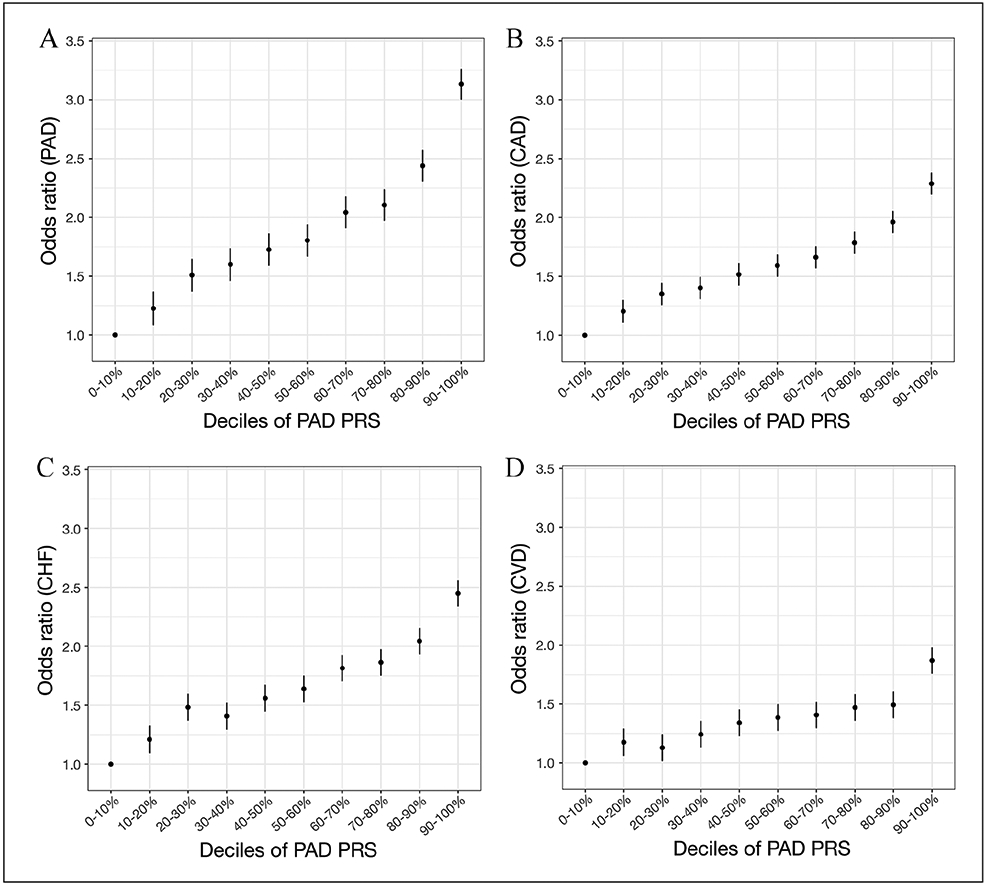
PAD genome-wide PRS is mainly predictive of vascular outcomes. A PAD PRS was calculated and categorized into deciles. (A–D) The PRS–outcome associations are shown for PAD and cardiovascular diseases (CAD: 28,744 cases, 458,576 controls; CHF: 11,849 cases, 475,471 controls; CVD: 10,047 cases, 477,273 controls).
Effect sizes and 95% CI are shown per decile per disease.
CAD, coronary artery disease; CHF, congestive heart failure; CVD, cerebrovascular disease; PAD, peripheral artery disease; PRS, polygenic risk score.
Table 2.
Odds of different cardiovascular diseases based on polygenic risk score.
| Cardiovascular disease | Odds ratio (95% CI)a | p-value |
|---|---|---|
| Peripheral artery disease | 1.44 (1.41, 1.46) | 1.81 × 10−113 |
| Coronary artery disease | 1.33 (1.32, 1.34) | 9.10 × 10−300 |
| Congestive heart failure | 1.34 (1.33, 1.36) | 7.10 × 10−140 |
| Cerebrovascular disease | 1.20 (1.19, 1.22) | 2.85 × 10−51 |
Polygenic risk scores (PRSs) scaled (center PRS −0.07 ± 0.83). Thus, for each 0.83 increase in PRS, the odds of peripheral artery disease or coronary artery disease increase by 44% and 33%, respectively. Odds ratios derived from logistic regression models of each disease, adjusting for age, sex, and first five principal components and using scaled PRS.
PRS-enhanced clinical risk model
We next evaluated whether adding the PRS to a clinical risk model improved overall model performance. By evaluating changes in model goodness-of-fit using the C-statistic metric, we found that a simple model with age and sex produced a C-statistic of 0.695, whereas adding clinical factors from the model described by Duval and colleagues increased the model performance to 0.797. The further addition of the PRS led to a modest increase in the C-statistic of 0.007 (AUC = 0.804; online Supplemental Figure 3A). We found our PRS-enhanced model to be well calibrated (online Supplemental Figure 3B).
After categorizing the clinical risk model scores and PRS-enhanced risk model scores into deciles, we plotted prevalence of PAD by decile. As shown in online Supplemental Figure 4, the PRS-enhanced model identified a higher prevalence of PAD cases at higher deciles of risk compared to the clinical risk model alone. The PRS-enhanced clinical risk model demonstrated an overall AUC of 0.76 in the UK Biobank cohort (Table 3) and when adding the PRS to the clinical model there was an NRI improvement of 0.073 (95% CI 0.05–0.085) (online Supplemental Table 5). Stratified analyses revealed that the PRS-enhanced clinical model performed better in men compared with women (NRImen = 0.10, NRIwomen = 0.04), in Europeans compared to nonEuropeans (NRIEuropean = 0.04; NRInonEuropean = 0.03), and in those older than 50 years of age (NRIage > 50 = 0.10). When evaluating reclassification metrics in detail, we found that the addition of the PRS downgraded risk in 14.7% of controls, and increased risk assessment in 8%. For cases, the PRS-enhanced clinical model increased risk estimates in 9.2% of cases and downgraded risk in 8.6% (online Supplemental Table 6).
Table 3.
Performance of clinical risk model and polygenic risk score-enhanced model.
| Group | Clinical risk model |
PRS-enhanced model |
||||
|---|---|---|---|---|---|---|
| AUCa | 95% CI | AUC | 95% CI | p-valueb | ||
| Sex | Men | 0.759 | (0.751, 0.767) | 0.769 | (0.761, 0.778) | 1.614 × 10−8 |
| Women | 0.688 | (0.674, 0.701) | 0.693 | (0.680, 0.707) | 4.254 × 10−3 | |
| Race | European | 0.755 | (0.748, 0.762) | 0.760 | (0.753, 0.768) | 4.371 × 10−8 |
| Non-European | 0.682 | (0.629, 0.735) | 0.686 | (0.635, 0.738) | 0.656 | |
| Age | Age > 50y | 0.751 | (0.743, 0.758) | 0.760 | (0.753, 0.768) | 5.5 × 10−12 |
| Age ⩽ 50y | 0.626 | (0.598, 0.653) | 0.636 | (0.609, 0.663) | 0.028 | |
| All individuals | 0.749 | (0.742, 0.760) | 0.757 | (0.750, 0.765) | 1.799 × 10−11 | |
AUC calculated using logistic regression (adjusted the first five principal components) in a validation dataset of 454,486 participants in the UK Biobank of which 5228 had been diagnosed with PAD.
Statistical significance of the improvement in AUC after adding PRS was calculated by the Delong test.
AUC, area under the curve; PRS, polygenic risk score.
Sensitivity analysis
In performing sensitivity analysis to restrict the definition of PAD, a total of 280 individuals were excluded. We found that our model AUC was similar at 0.756 for the clinical risk model alone and 0.762 for the PRS-enhanced model. NRI for the PRS-enhanced model ranged from 0.045 to 0.064.
Discussion
In this paper, we demonstrate that: (1) a PRS performs well in discriminating risk between PAD cases and noncases, (2) a PAD-specific PRS is associated with risk of other cardiovascular diseases, and (3) a PAD PRS can add to the performance of a clinical risk model. These results provide promising support that PRSs can improve accuracy in identifying risk of prevalent PAD.
Although certain areas of health care, such as oncology, have seen growth in the clinical application of precision health through the incorporation of genetic data, the progress has been slower for cardiovascular diseases. Though previous studies have found utility in incorporating PRS in determining risk of diseases such as CAD, such analysis has never been published for PAD. This is in large part due to the previous lack of PAD-specific risk loci identified by prior GWAS. In previous work, only four loci reached genome-wide significance for PAD, and one of these loci, 9p21, has been found to broadly increase risk of other cardiovascular diseases, not just PAD.28-31 One likely reason for the small number of risk loci is related to the relatively low number of PAD cases in each of these studies.
In contrast, Klarin and colleagues performed the largest GWAS to date using PAD diagnosis as the outcome variable. In this analysis, 19 PAD-specific risk loci reaching genome wide significance were identified, of which 18 were not previously reported.10 In addition to its size, their analysis included a racially and ethnically diverse cohort with 72% Caucasian, 20% African Americans, and 8% Hispanics. In our analyses, we were able to utilize summary statistics from 7,497,749 SNPs across the entire genome. And although we utilized a GWAS from a US population (MVP) to assess risk in a European population (UK Biobank), we still found the PRS to be clinically useful. Furthermore, our analyses demonstrate that a GWAS attuned to a specific type of cardiovascular disease – in this case PAD – can better quantify genetic risk than when using GWAS data from other cardiovascular diseases such as CAD.
Our PRS-enhanced model demonstrated excellent calibration and goodness-of-fit with a C-statistic of 0.804 (online Supplemental Figures 3A and B). Additionally, using 10-fold cross-validation, our PRS-enhanced model demonstrated a significant improvement in AUC compared to a clinical model alone. Because additions of biomarkers may only show small changes in AUC yet be clinically useful,32 we also evaluated the NRI of adding the PRS. This demonstrated that overall the addition of the PRS net improved PAD risk classification beyond the clinical risk model alone. In particular, the PRS-enhanced model primarily helped to downgrade risk of controls and increased risk category for cases, though to a smaller extent.
Our findings compare favorably to studies evaluating PRS for CAD risk discrimination. Elliott and colleagues developed a PRS for CAD using summary statistics from the CARDIoGRAMplusC4D GWAS meta-analyses.22 They found that a CAD PRS achieved a C-statistic of 0.61 and when combining clinical and genetic data their overall model performance improved to 0.78. In our adjusted models, we found that those in the highest PRS decile had 3.1 higher odds of having prevalent PAD than those in the lowest decile. Similarly, Wang et al. were able to develop a CAD PRS that improved clinical risk assessment in the South Asian population where they found those with the highest quintile PRS had an increased CAD risk of 2.16-fold compared to the lowest risk group.33
Stratified analysis revealed that a clinical risk score for PAD enhanced with the PRS performs less well in women than in men. Reasons for these findings could be explained by differences in environmental risk factors that may attenuate or exacerbate genetic risk. For instance, smoking is a well-known risk factor for PAD, and previous gene-environment studies have found genetic factors that interact with smoking to increase the risk of PAD.34,35 In this study, there were many more current smokers among men (12.5%) compared to women (8.9%) (p < 0.001). This may increase the discrimination performance of the PRS for men. Disease prevalence can also affect metrics such as AUC and women did demonstrate lower prevalence of disease in this cohort – only 34% of PAD cases were women. Conversely, there may be sex-specific protective factors that are present in the women but not seen in men.36,37 Furthermore, genetic differences in risk of disease may also play an important factor in differences in model performance between men and woman. Indeed, Huang and colleagues recently demonstrated that genetic risk scores for CAD risk differed in performance among men and women, and part of this difference was due to at least one genetic locus that demonstrated meaningful genetic–sex interactions for incident CAD.38 Our findings suggest that similar studies of sex differences in genetic risk of PAD may be warranted. Lastly, we posit that the lower clinical and PRS-enhanced model performance in the younger age cohort was due to low prevalence of PAD (0.45%), and the nature of cardiovascular diseases in general, that manifest after decades of smoking and high blood pressure, for instance. Even so, adding the PRS, slightly, but significantly, increased model performance in the younger cohort.
Study strengths and limitations
Although this study is strengthened by the use of two of the largest biobanks developed to date and the use of a genome-wide approach to develop and evaluate a PAD PRS, there are limitations. First, our models were developed to detect prevalent disease, rather than incident disease. Although it is helpful to identify prevalent PAD given the large number of undiagnosed individuals, another important use case would be to identify high–risk patients before development of PAD or in the earlier stages of disease to engage in aggressive risk management that may better improve long-term outcomes. Another limitation is the youthful age structure of UK Biobank, which leads to a low prevalence of PAD that limits the performance of a PRS-enhanced clinical model. Furthermore, previous work has shown that when different populations are used to develop and test genetic risk factors, these risk models can demonstrate decreased performance.39 The MVP cohorts consist of 72% European, 19.7% African, and 8.3% with Hispanic ancestries, whereas the UK Biobank has 96% Europeans, 1.4% African, and 2.6% Asians. Such racial and ethnic differences likely attenuate the performance of our PRS-enhanced risk model. This can be due to the differences in variant frequencies, as well as the linkage disequilibrium patterns between racial and ethnic populations. Thus, it is possible that a PRS tested in a US-based population would achieve better performance metrics given that the summary statistics used to generate the PRS are from a US cohort. Future studies should evaluate the performance of the PAD GWAS in larger, older US-based populations to determine differences in PRS performance across diverse populations and in higher risk cohorts. Another potential weakness of this study is that the ‘non-PAD’ controls could potentially have PAD but be undiagnosed. In this sense, the PRS score reported here may be more reflective of the PAD patients who were more sensitive to symptoms of lower-extremity atherosclerosis. Indeed, we did find that changing the definition of PAD did slightly change the AUC and NRI, and thus performance of a PRS in general is likely sensitive to cohort definitions. Furthermore, it is difficult to know for sure from the net reclassification statistics whether those controls deemed higher risk for PAD in the PRS-enhanced model may actually have the disease but be asymptomatic and undiagnosed. Further study could focus on a cohort for which a more objective test such as the ankle–brachial index is used to define cases and controls, regardless of symptoms. However, this information was not available to us in the UK Biobank cohort. We are also limited by the available genetic data related to risk of PAD. Using a REML approach, we found the heritability of PAD to be 11%; however, given previous estimates, the genetic risk of PAD may be as high as 21%.40 There are likely genetic risk variants that remain unaccounted for and warrant further study. For example, gene-environment studies that can identify risk variants that only become significant due to exposure to certain environmental risks may help improve our ability to quantify genetic risk of PAD. Li et al., for instance, found that GSTT1 interacts with smoking to increase the risk of lower-extremity arterial disease.34 Another approach entirely would be to perform whole-genome sequencing to better identify rare disease variants that can significantly amplify risk of PAD.41 Whole-genome sequencing is currently prohibitively expensive for clinical use, but just as costs related to genotyping have significantly decreased over the last decade, a similar cost-trajectory for whole-genome sequencing may make its routine use increasingly possible.
Conclusion
In conclusion, we demonstrate that a PRS can determine risk of prevalent PAD and that the PRS can enhance the performance of a clinical risk model. Furthermore, a PAD PRS is associated with the risk of CAD, CHF, and CVD. Validation of our PRS-enhanced model in a larger, older, and better aligned racial/ethnic cohort may improve performance.
Supplementary Material
Acknowledgements
This research has been conducted using the UK Biobank Resource under Application Number ‘13721’. Thank you to Dr. Erik Ingelsson, of the Ingelsson Lab, Stanford University School of Medicine, Stanford, California, USA, for your conceptual input. We would also like to thank all the supporting staff from the Stanford Research Computing Center, Dr Yu Liu from Stanford University School of Medicine, Stanford, California, USA, and Dr Stefan Gustafsson from Uppsala University Science for Life Laboratory, Uppsala, Sweden, for their technical computing and data management expertise.
Funding
Drs E Ross and N Leeper acknowledge support from the National Institutes of Health, National Heart, Lung, and Blood Institute (K01HL148639-02) (Ross) and 5R01HL125224-02 (Leeper), and the Society of University Surgeons (Ross). The funders were not involved in any aspect of this study.
Footnotes
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplementary material
The supplementary material is available online with the article.
References
- 1.Mahoney EM, Wang K, Cohen DJ, et al. One-year costs in patients with a history of or at risk for atherothrombosis in the United States. Circ Cardiovasc Qual Outcomes 2008; 1: 38–45. [DOI] [PubMed] [Google Scholar]
- 2.Fowkes FG, Rudan D, Rudan I, et al. Comparison of global estimates of prevalence and risk factors for peripheral artery disease in 2000 and 2010: A systematic review and analysis. Lancet 2013; 382: 1329–1340. [DOI] [PubMed] [Google Scholar]
- 3.McDermott MM, Kerwin DR, Liu K, et al. Prevalence and significance of unrecognized lower extremity peripheral arterial disease in general medicine practice*. J Gen Intern Med 2001; 16: 384–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Becker GJ, McClenny TE, Kovacs ME, et al. The importance of increasing public and physician awareness of peripheral arterial disease. J Vasc Interv Radiol 2002; 13: 7–11. [DOI] [PubMed] [Google Scholar]
- 5.Diehm C, Allenberg JR, Pittrow D, et al. Mortality and vascular morbidity in older adults with asymptomatic versus symptomatic peripheral artery disease. Circulation 2009; 120: 2053–2061. [DOI] [PubMed] [Google Scholar]
- 6.Meijer WT, Grobbee DE, Hunink MG, et al. Determinants of peripheral arterial disease in the elderly: The Rotterdam study. Arch Intern Med 2000; 160: 2934–2938. [DOI] [PubMed] [Google Scholar]
- 7.Joosten MM, Pai JK, Bertoia ML, et al. Associations between conventional cardiovascular risk factors and risk of peripheral artery disease in men. JAMA 2012; 308: 1660–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Duval S, Massaro JM, Jaff MR, et al. An evidence-based score to detect prevalent peripheral artery disease (PAD). Vasc Med 2012; 17: 342–351. [DOI] [PubMed] [Google Scholar]
- 9.Bali V, Yermilov I, Coutts K, Legorreta AP. Novel screening metric for the identification of at-risk peripheral artery disease patients using administrative claims data. Vasc Med 2016; 21: 33–40. [DOI] [PubMed] [Google Scholar]
- 10.Klarin D, Lynch J, Aragam K, et al. Genome-wide association study of peripheral artery disease in the Million Veteran Program. Nat Med 2019; 25: 1274–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Consortium CAD, Deloukas P, Kanoni S, et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet 2013; 45: 25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nikpay M, Goel A, Won HH, et al. A comprehensive 1,000 genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet 2015; 47: 1121–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Knowles JW, Zarafshar S, Pavlovic A, et al. Impact of a genetic risk score for coronary artery disease on reducing cardiovascular risk: A pilot randomized controlled study. Front Cardiovasc Med 2017; 4: 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Natarajan P, Young R, Stitziel NO, et al. Polygenic risk score identifies subgroup with higher burden of atherosclerosis and greater relative benefit from statin therapy in the primary prevention setting. Circulation 2017; 135: 2091–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Andersson C, Lukacs Krogager M, Kuhr Skals R, et al. Association of genetic variants previously implicated in coronary artery disease with age at onset of coronary artery disease requiring revascularizations. PLoS One 2019; 14: e0211690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Choi SW, O’Reilly PF. PRSice-2: Polygenic Risk Score software for biobank-scale data. Gigascience 2019; 8: giz082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ge T, Chen CY, Ni Y, et al. Polygenic prediction via Bayesian regression and continuous shrinkage priors. Nat Commun 2019; 10: 1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Loh PR, Tucker G, Bulik-Sullivan BK, et al. Efficient Bayesian mixed-model analysis increases association power in large cohorts. Nat Genet 2015; 47: 284–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khera AV, Chaffin M, Aragam KG, et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet 2018; 50: 1219–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bycroft C, Freeman C, Petkova D, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 2018; 562: 203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marchini J, O’Connell J, Delaneau O, et al. UK Biobank Phasing and Imputation Documentation, version 1.2, https://biobank.ctsu.ox.ac.uk/crystal/crystal/docs/impute_ukb_v1.pdf (2015, accessed May 17, 2019). [Google Scholar]
- 22.Elliott J, Bodinier B, Bond TA, et al. Predictive accuracy of a polygenic risk score-enhanced prediction model vs a clinical risk score for coronary artery disease. JAMA 2020; 323: 636–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet 2009; 5: e1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang J, Benyamin B, McEvoy BP, et al. Common SNPs explain a large proportion of the heritability for human height. Nat Genet 2010; 42: 565–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schunkert H, Konig IR, Kathiresan S, et al. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat Genet 2011; 43: 333–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coronary Artery Disease (C4D) Genetics Consortium. A genome-wide association study in Europeans and South Asians identifies five new loci for coronary artery disease. Nat Genet 2011; 43: 339–344. [DOI] [PubMed] [Google Scholar]
- 27.DeLong ER, DeLong DM, Clarke-Pearson DL. Comparing the areas under two or more correlated receiver operating characteristic curves: A nonparametric approach. Biometrics 1988; 44: 837–845. [PubMed] [Google Scholar]
- 28.Matsukura M, Ozaki K, Takahashi A, et al. Genome-wide association study of peripheral arterial disease in a Japanese population. PLoS One 2015; 10: e0139262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Murabito JM, White CC, Kavousi M, et al. Association between chromosome 9p21 variants and the ankle-brachial index identified by a meta-analysis of 21 genome-wide association studies. Circ Cardiovasc Genet 2012; 5: 100–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kullo IJ, Shameer K, Jouni H, et al. The ATXN2-SH2B3 locus is associated with peripheral arterial disease: An electronic medical record-based genome-wide association study. Front Genet 2014; 5: 166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sofer T, Emery L, Jain D, et al. Variants associated with the ankle brachial index differ by Hispanic/Latino ethnic group: A genome-wide association study in the Hispanic Community Health Study/Study of Latinos. Sci Rep 2019; 9: 11410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cook NR. Statistical evaluation of prognostic versus diagnostic models: Beyond the ROC curve. Clin Chem 2008; 54: 17–23. [DOI] [PubMed] [Google Scholar]
- 33.Wang M, Menon R, Mishra S, et al. Validation of a genome-wide polygenic score for coronary artery disease in South Asians. J Am Coll Cardiol 2020; 76: 703–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li R, Folsom AR, Sharrett AR, et al. Interaction of the glutathione S-transferase genes and cigarette smoking on risk of lower extremity arterial disease: The Atherosclerosis Risk in Communities (ARIC) study. Atherosclerosis 2001; 154: 729–738. [DOI] [PubMed] [Google Scholar]
- 35.Olshan AF, Li R, Pankow JS, et al. Risk of atherosclerosis: Interaction of smoking and glutathione S-transferase genes. Epidemiology 2003; 14: 321–327. [PubMed] [Google Scholar]
- 36.Kittnar O. Selected sex related differences in pathophysiology of cardiovascular system. Physiol Res 2020; 69: 21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gaudio A, Xourafa A, Rapisarda R, et al. Peripheral artery disease and osteoporosis: Not only age-related (Review). Mol Med Rep 2018; 18: 4787–4792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang Y, Hui Q, Gwinn M, et al. Sexual differences in genetic predisposition of coronary artery disease. Circ Genom Precis Med 2020; 14: e003147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Duncan L, Shen H, Gelaye B, et al. Analysis of polygenic risk score usage and performance in diverse human populations. Nat Commun 2019; 10: 3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kullo IJ, Leeper NJ. The genetic basis of peripheral arterial disease: Current knowledge, challenges, and future directions. Circ Res 2015; 116: 1551–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fan R, Huang CH, Lo SH, et al. Identifying rare disease variants in the Genetic Analysis Workshop 17 simulated data: A comparison of several statistical approaches. BMC Proc 2011; 5 (Suppl 9): S17. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.